12. The Hematologic System
Authors: Corwin, Elizabeth J.
Title: Handbook of Pathophysiology, 3rd Edition
Copyright 2008 Lippincott Williams & Wilkins
> Table of Contents > Unit IV - Oxygen Balance and Deficiencies > Chapter 12 - The Hematologic System
Chapter 12
The Hematologic System
The hematologic system includes all the blood cells, the bone marrow in which the cells mature, and the lymphoid tissue where the cells are stored when not in circulation. The hematologic system is designed to carry oxygen and nutrients, transport hormones, remove waste products, and deliver cells to prevent infection, stop bleeding, and promote healing. This system allows the body to feed and heal itself and to communicate between sites.
Physiologic Concepts
Composition of the Blood
The blood is made up of approximately 45% formed elements and 55% plasma. The formed elements are the red blood cells (erythrocytes), the white blood cells (leukocytes), and the platelets (thrombocytes). The red blood cells account for 99% of the formed elements; the white blood cells and platelets constitute the other 1%. The plasma is 90% water, with the other 10% made up of plasma proteins, electrolytes, dissolved gases, various waste products of metabolism, nutrients, vitamins, and cholesterol. The plasma proteins include albumin, the globulins, and fibrinogen. Albumin is the most abundant of the plasma proteins and helps maintain the plasma osmotic pressure and blood volume. The globulins bind insoluble hormones and other plasma constituents and make them soluble. This allows these important substances to be transported in the blood from their site of production to their site of action. Examples of substances carried bound to plasma proteins include thyroid hormone,
P.354
iron, phospholipids, bilirubin, steroid hormones, and cholesterol. Other globulin proteins, the immunoglobulins, are the antibodies that travel in the blood to fight infection. Fibrinogen is an important element in blood clotting.
Hematopoiesis
Red blood cells, white blood cells, and platelets are formed in the liver and spleen in the fetus, and in the bone marrow after birth. The process of blood cell formation is called hematopoiesis.
Hematopoiesis begins in the bone marrow with pluripotential (meaning many possible ) stem cells. Stem cells are the source of all blood cells. These cells continually self-renew and differentiate throughout a lifetime; their supply is endless and they are often described as immortal. After several stages of differentiation, a stem cell becomes committed to forming just one type of blood cell. This cell, called a progenitor cell, remains in the marrow and, when instructed by specific growth factors, differentiates into a red blood cell, a white blood cell, or a platelet. The development of the blood cells from original pluripotential stem cells to differentiated cells is shown in Figure 12-1.
Control of Progenitor Cell Differentiation
Progenitor cells are stimulated to proliferate and differentiate by a variety of hormones and locally produced agents that collectively are called hematopoietic growth factors. Each progenitor cell responds only to some of these growth factors, but many growth factors may be acting nonspecifically on several progenitor cells. Many hematopoietic growth factors are cytokines. Cytokines are released from immune and inflammatory cells and communicate to the progenitor cells the need for more cells to fight infection or help the body heal. Hematopoietic growth factors specific to the line of cells they stimulate are called colony-stimulating factors. For example, granulocyte colony-stimulating factor stimulates the production of white blood cells known as granulocytes (see later), whereas
P.355
monocyte-macrophage colony-stimulating factor increases the proliferation of monocytes and macrophages. An example of an important colony-stimulating growth factor for red blood cells is the hormone erythropoietin, which is produced by the kidney in response to low oxygen concentration in the blood. Other nonspecific cytokines may act on cells less differentiated than the progenitor cells, stimulating the production of a variety of blood cells.
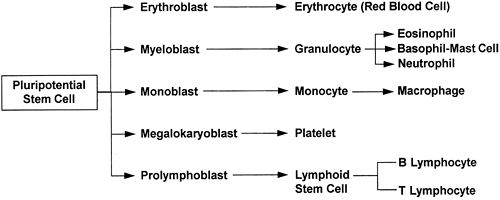 |
Figure 12-1. Blood cell maturation. |
The Red Blood Cell
The red blood cell (erythrocyte) contains no nucleus, mitochondria, or ribosomes. It cannot reproduce or undergo oxidative phosphorylation or protein synthesis. The red blood cell contains the protein hemoglobin, which carries oxygen from the lungs to all cells of the body. Hemoglobin takes up most of the red blood cell's intracellular space. Red blood cells are produced in the bone marrow in response to hemopoietic growth factors, especially erythropoietin, and require iron, folic acid, and vitamin B12 for their synthesis. Once a red blood cell nears maturity, it is released from the bone marrow, completes its maturation in the bloodstream, and lives out its approximately 120-day life span. It then disintegrates and dies. Dying red blood cells are replaced with new ones released from the bone marrow. If red blood cell death is excessive, a larger than normal number of immature red blood cells, called reticulocytes, will be released from the bone marrow; elevated levels of circulating reticulocytes are suggestive of certain types of anemia.
Characteristics of Red Blood Cells
Red blood cells are small, biconcave (two-sided) disks shaped like donuts without holes. Their high surface area allows for rapid diffusion of oxygen and carbon dioxide, while their small size (7 m in diameter) and relative flexibility allows them to squeeze through even the smallest of capillary beds without damage. In a blood sample, the percentage of the blood that is taken up by red blood cells is called the hematocrit, which usually ranges from approximately 36% to 52% depending on age and sex. The concentration of hemoglobin in a blood sample (grams per 100 mL) usually is approximately one third of the hematocrit. Red blood cells are described clinically by their size and by the amount of hemoglobin present in the cell. The suffix cytic refers to size, and the suffix chromic refers to the concentration of hemoglobin in the cell. The mean corpuscular volume (MCV) is a measure in cubic microns of the volume occupied by a single red cell. The MCV is the most commonly used index for identifying whether a cell is of normal, small, or large size and is used clinically to categorize an anemia.
Normocytic: cells of normal size (MCV 87 103 fL/red cell or m3/red cell)
Microcytic: cells too small in size (MCV < 87 m3/red cell)
Macrocytic: cells too large in size (MCV > 103 m3/red cell)
Hypochromic: cells with too little hemoglobin
Normochromic: cells with normal amounts of hemoglobin
Hyperchromic: cells with too dense hemoglobin
P.356
Red Blood Cell Antigens
The red blood cell has a variety of specific antigens present on its cell membrane that are not found on other cells. The most important of these antigens are known as A and B and Rh.
ABO Antigens
An individual carries two alleles (genes), each coding for the A antigen, the B antigen, or neither antigen, which is designated O. One allele is received from each parent. The A and B antigens are codominant. Individuals with both the A and B antigen (AB) will have AB blood. Those with two A antigens (AA), or one A and one O (AO), will have A blood. Those with two B antigens (BB), or one B and one O (BO), will have B blood. Individuals with neither antigen (OO) will have O blood. Individuals who have AB blood will accept A, B, or O blood. However, an immune response will develop if an individual without an A or a B antigen is exposed to that antigen in a blood transfusion.
Rh Antigens
Rh antigens are the other main antigen group on the red blood cell and are also passed as genes from each parent. The main Rh antigen is called the Rh factor. An individual who carries the Rh antigen is considered Rh positive (Rh+). An individual who lacks the Rh antigen is considered Rh negative (Rh-). The Rh-positive gene is dominant; therefore, an individual must have two negative Rh factors to be Rh negative. Individuals who are Rh positive will accept Rh-negative blood, but those who do not have the Rh antigen will develop an immune response if exposed to Rh-positive blood.
Universal Blood Recipients and Donors
Universal blood recipients are those with AB-positive blood because their immune system will recognize as self the A or B antigen and the Rh-positive antigen. Therefore, they will accept blood with any ABO or Rh profile. Universal blood donors are those with O-negative blood. Although their immune systems will attack blood containing the A or B antigen or the Rh factor, their blood can be given for transfusion to any recipient in an emergency. Other, weaker antigens on red blood cells exist, however, and may provoke an immune reaction in a recipient.
Hemoglobin
Hemoglobin consists of an iron-containing substance called heme and the protein globulin. There are approximately 300 hemoglobin molecules in each red blood cell. Each hemoglobin molecule contains four binding sites for oxygen. Oxygen bound to hemoglobin is called oxyhemoglobin. Hemoglobin in the red blood cell may be partially or completely bound with oxygen on all four sites. Fully
P.357
saturated hemoglobin is completely bound with oxygen, while partially saturated or deoxygenated hemoglobin is less than 100% saturated. Systemic arterial blood from the lungs is fully saturated with oxygen. Because hemoglobin releases oxygen to the cells, the hemoglobin saturation in venous blood is approximately 60%. The final job of the hemoglobin is to pick up carbon dioxide and hydrogen ions and carry them to the lungs, where they are exhaled to the air. There are at least 100 types of abnormal hemoglobin molecules that have been recognized in humans, resulting from a variety of different mutations. Most of these mutations cause the hemoglobin molecule to carry oxygen more poorly than normal.
Breakdown of the Red Blood Cell
When the red blood cell begins to disintegrate at the end of its lifetime, it releases hemoglobin into the circulation. Hemoglobin is broken down in the liver and spleen. The globulin molecule is converted into amino acids that are used again by the body. The iron is stored in the liver and spleen until it is reused. The rest of the molecule is converted to bilirubin, which is excreted in the stool as bile or in the urine. Normally, the rate of red cell breakdown is equal to the rate of synthesis. In certain conditions, either synthesis or breakdown may outpace the other.
White Blood Cells
White blood cells are formed in the bone marrow from committed progenitor cells. On further differentiation, the progenitor cells become non-granular-appearing T and B lymphocytes, monocytes, and macrophages, or granular-appearing neutrophils, basophils, and eosinophils. The job of the white blood cells is to recognize and fight microorganisms in immune reactions, and to assist in the inflammation and healing processes. The platelets, which are fragments of bone marrow cells, are essential in the control of bleeding. In addition, they often function with the white blood cells in the inflammatory and healing processes.
Types of White Blood Cells
B lymphocytes develop in the bone marrow and then circulate in the blood until they encounter the antigen to which they are programmed to respond. At this point, B lymphocytes mature further, become plasma cells, and begin secreting antibody, as described later.
T lymphocytes leave the bone marrow and develop during migration through the thymus. After leaving the thymus, they circulate in the blood or reside in lymphatic tissue until they encounter an antigen to which they are programmed to respond. Once stimulated by antigen, they produce chemicals to destroy the microorganism and alert other white blood cells that an infection is occurring, as described later.
Monocytes are formed in the bone marrow and enter the bloodstream in an immature form. At the site of injury or infection, the monocytes leave the blood and mature into macrophages in the tissues.
P.358
Macrophages may remain stored in the tissues, or may be used in an inflammatory reaction as soon as they mature.
Neutrophils, basophils, and eosinophils are granular-appearing white blood cells that assist in the inflammatory response. Macrophages, neutrophils, and eosinophils function as phagocytes, cells that destroy and digest microorganisms and accumulated cell debris. Although the exact function of the basophils is unclear, they appear to act like circulating mast cells (Chapter 5) that release vasoactive peptides that stimulate the inflammatory response.
The Spleen
The spleen is a small organ located in the upper left abdominal cavity. It is considered a secondary lymphoid organ, as opposed to the bone marrow and thymus, which are primary lymphoid organs. Like all lymphoid organs, the spleen is involved in the formation and storage of the blood.
The spleen is the site of hematopoiesis in the fetus. After birth, the spleen contains tissue macrophages and aggregates of lymphocytes. The spleen is well supplied with blood vessels that branch off from the splenic artery, itself a branch of the abdominal aorta. The intricate vasculature of the spleen circulates blood containing microorganisms, dead cells, and other debris past the macrophages and lymphocytes, where they can be acted on or destroyed. After flowing through the splenic capillary networks, the blood vessels rejoin into venules, and blood is delivered to the liver through the hepatic portal blood flow system.
As blood passes through the spleen, residing macrophages act as phagocytes to clear the blood of cell debris (including lysed red blood cells) and digest microorganisms. The macrophages present pieces of digested microorganisms to nearby B and T cells, initiating an immune response. Individuals who have lost their spleen (usually after trauma, although some individuals may have their spleen removed surgically when platelet count is low and cannot be corrected) are at a disadvantage in fighting certain infections compared to those with a functioning spleen.
The spleen also serves as a reservoir for blood, capable of holding a few hundred milliliters in the adult. With a decrease in blood pressure, the spleen can expel this blood into the venous circulation to help return pressure. It also serves as a storage site for iron released during the catabolism of hemoglobin. Iron is stored in splenic macrophages until required for production of new red blood cells. Iron deficiency may occur without the spleen. The spleen also stores senescent (aged) red blood cells.
Lymph Nodes
Lymph nodes are small capsules of lymphoid tissue interspersed throughout the lymphatic system, near the lymphatic veins. Lymph flowing in the lymphatic vessels is filtered through many nodes.
P.359
Lymph nodes contain many lymphocytes, monocytes, and macrophages. These cells proliferate in the nodes and some are released into the circulation during infection or inflammation. Residing white blood cells entrap and phagocytize microorganisms that are delivered by the lymph flow, cleansing the lymph before it is returned to the general circulation. The lymph node closest to an infection is exposed to the highest number of microorganisms, which causes the macrophages and lymphocytes to proliferate and the node to enlarge. An active node may become tender as it fights to contain infection.
Hemostasis
The human body experiences frequent small capillary tears and occasional large blood vessel cuts. While unable to control large vessel bleeding without external support, the body is able to stop small vessel bleeding. Control of bleeding occurs in two steps the formation of a platelet plug followed by the formation of a blood clot. These processes are interdependent and occur one after the other in rapid succession. The control of bleeding is called hemostasis.
Role of the Platelets in Hemostasis
Platelets play an important role in both steps of hemostasis. Platelets normally circulate throughout the bloodstream without sticking to vascular endothelial cells. However, within seconds after damage to a blood vessel, platelets are drawn to the area in response to exposed collagen on the subendothelial layers of the damaged blood vessel. Platelets attach to proteins (called von Willebrand factors) expressed on the damaged surface of a blood vessel, and release several vasoactive chemicals, including serotonin and adenosine diphosphate (ADP). Serotonin causes vasoconstriction, helping to reduce blood flow to the area and limit bleeding. Serotonin and other chemicals, including ADP, also cause the platelets to change shape and become sticky, beginning the formation of what is called a platelet plug inside the damaged blood vessel. Other platelets are drawn to the area and further build up the plug. Thromboxane A2 is produced by the platelets and contributes to the attraction of more platelets to the area. Fibrinogen, a circulating plasma protein, connects between exposed sites on the platelets, serving like a bridge to add stability to the plug. The platelet plug effectively seals the damaged area. Deficiencies in any of the involved factors will result in excessive bleeding of even small capillary tears.
Limits on Platelet Function
Unimpeded platelet aggregation could cause a prolonged decrease in blood flow to the tissue or result in a plug becoming so enlarged that it may break off from the original site and travel downstream as an embolus, blocking downstream flow. To prevent either of these occurrences from
P.360
happening, neighboring undamaged endothelial cells release other substances that limit the extent of platelet aggregation. The main substances released by neighboring endothelial cells to limit platelet aggregation are prostaglandin I2, also called prostacyclin, and nitric oxide, an important vasodilator.
Ultimately, the balance between pro-clotting and anti-clotting factors serves to keep the platelets active at the site of injury while preventing excessive platelet aggregation and spread of the platelet plug to uninjured vascular tissue. Platelet aggregation and the production of the platelet plug are shown in Figure 12-2.
Blood Clot
The platelet plug becomes a true clot as it enlarges and traps circulating red cells and macrophages. The entire clot is stabilized and strengthened by a network of fibrin strands, produced from the fibrinogen bridges mentioned earlier. The production of stabilized fibrin is the final step in the other essential component of hemostasis, the coagulation cascade.
Coagulation Reactions
Coagulation reactions involve a series of coagulation factors or proteins activated in domino fashion, leading to the coagulation (clotting) of the
P.361
blood. There are a total of 13 proteins involved in the coagulation pathways; some of these are activated in what is called the intrinsic pathway and some are activated in the extrinsic pathway. Under most physiologic conditions, coagulation occurs first through the extrinsic pathway; activation of the extrinsic pathway then turns on the more powerful intrinsic pathway. Both pathways ultimately merge and function by activating one protein, factor X; the merging of the intrinsic and extrinsic pathways at factor X is called the final common pathway. Factor X is responsible for converting the plasma protein prothrombin to thrombin. Thrombin is the key catalyst that drives the conversion of fibrinogen to fibrin and causes coagulation. Thrombin also acts in a positive feedback manner to stimulate the proteins involved in its own production, furthering the coagulation cascade. Both pathways are shown in Figure 12-3.
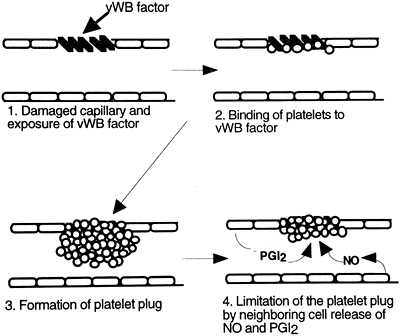 |
Figure 12-2. Steps involved in the formation of the platelet plug. The role of von Willebrand (vWB) factor, platelets, and prostaglandin I2 (PGI2) and nitric oxide (NO) released from neighboring endothelial cells is emphasized. |
The intrinsic pathway begins with the activation of the circulating coagulation factor, factor XII, also called the Hageman factor. Factor XII is activated when it comes into contact with damaged vascular tissue. Ultimately, activation of factor XII leads to the conversion of prothrombin to thrombin. Factors XI and IX are important intermediate steps in the cascade, and factors V and VIII are important cofactors. Lack of any of these factors could interfere with coagulation.
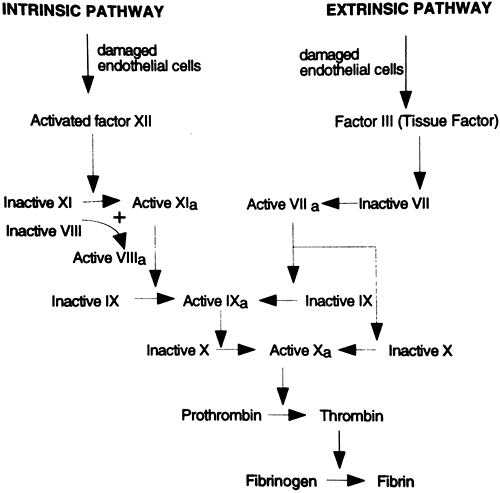 |
Figure 12-3. Simplified diagram of the intrinsic and extrinsic pathways of coagulation. Factor VII is highlighted to identify its pivotal role as a coenzyme in the intrinsic pathway. Letter a designates an activated factor. |
P.362
The extrinsic pathway, the usual way of stimulating coagulation, begins with the release into the circulation of factor III, also called tissue factor or thromboplastin, from damaged vascular endothelial cells. When tissue factor encounters another coagulation factor circulating in the plasma, factor VII (also called serum prothrombin conversion factor), the extrinsic cascade is stimulated, again resulting in the production of factor X. The extrinsic pathway also can turn on the intrinsic pathway through activation of factor IX.
The blood does not continually and excessively clot even though factors XII and VII are always present in the circulation because healthy endothelial cells are smooth and intact. Therefore, they do not directly activate factor XII or produce tissue factor and activate factor VII. Healthy vascular endothelium repels coagulation factors and platelets. It is only when the endothelium is damaged by trauma, infection, forces of chronic hypertension, or accumulation of fat and cholesterol (Chapter 13) that a clot begins to develop.
Because several coagulation factors are produced in the liver in reactions involving vitamin K, liver disease or vitamin K deficiency can impair the production of coagulation factors and cause bleeding.
Anticoagulants
Anticoagulants are present in the blood to prevent clots from developing. For example, antithrombin proteins are released by undamaged endothelial cells and function to inactivate thrombin. The most important of these, antithrombin III, is itself activated by heparin, an anticoagulant produced by mast cells and basophils in response to tissue injury and inflammation. Other substances, called tissue factor inhibitors, circulate in the plasma and bind to tissue factor (factor III), directly blocking its activation and interfering with the extrinsic pathway. Finally, as mentioned earlier, nondamaged endothelial cells secrete prostacyclin and nitric oxide, which limit platelet aggregation and thus reduce coagulation.
Anticoagulation drugs are available and include the prostaglandin inhibitor aspirin, which in low doses inhibits the production of thromboxane A2 but not prostaglandin I2, and oral anticoagulants such as warfarin. Other drugs are available that do not prevent clotting, but serve to break down previously formed clots. Examples of these drugs, called thrombolytic agents, include streptokinase and t-PA; thrombolytic agents play an important role in the early treatment of myocardial infarct and thrombotic stroke.
Laboratory Tests of the Blood
The Complete Blood Count with Differential and Platelet Count
The blood is frequently tested for adequacy of cell number and function. The most common test is the complete blood count (CBC), which provides information on the number, concentration, and physical characteristics of red blood cells, white blood cells, and platelets present in a venous blood sample. The CBC with differential is age dependent and, to a lesser extent, sex dependent. Exercise, reproductive status, and many drugs may cause
P.363
test deviations. The CBC with differential is used as part of well physical examinations, to screen for specific conditions, and to determine preoperative health. The CBC is also used to evaluate treatment success.
A description of red cell size as indicated by the mean corpuscular volume (MCV) and mean corpuscular hemoglobin concentration (MCHC) provides additional information when evaluating patients with anemia. Red cells also are described by the red cell size distribution width (RDW) in a blood sample. If the RDW is high, it means there is a wide range of RBC sizes in the sample. The RDW is useful in distinguishing between similar types of anemia. For example, a patient with microcytic (small) red cells who has a normal RDW may have a hemoglobin abnormality such as thalassemia, while a patient with similarly microcytic cells but a high RDW is more likely experiencing iron deficiency. Other combinations of red cell values provide different clues to the etiology of blood disorders.
Other common blood tests include blood typing of ABO and Rh antigens and tests to identify the presence of microorganisms and antibody titers. The erythrocyte sedimentation rate (SED rate) is a test that evaluates the tendency of red blood cells to settle out of unclotted blood in one hour. This test is based on the fact that inflammation and similar processes stimulate the liver to release an increased number of proteins into the blood, which cause red cells to aggregate together, becoming heavier and thus settling to the bottom of a container. Because of this, the SED rate is often increased nonspecifically with inflammatory disease.
The Normal CBC with Differential and Platelet Count (Adult)
Red blood cell count: 4.0 to 5.5 million/mL of blood
White blood cell count: 5,000 to 10,000/mL of blood
Platelet count: 140,000 to 400,000/mL of blood
Hematocrit (% of red blood cells): 42% to 52% for males; 36% to 48% for females
Hemoglobin: 14.0 to 17.5 grams/100 mL for males; 12.0 to 16.0 grams/100 mL for females
Neutrophils: 50% to 62%
Eosinophils: 0% to 3%
Basophils: 0% to 1%
Lymphocytes: 25% to 40%
Monocytes: 3% to 7%
Tests of Red Blood Cell Size and Hemoglobin (Adult)
MCV: 82 to 98 fL/red cells
MCHC: 32 to 36 g/dL
RDW: 11.5 to 14.5 coefficient of variation of red cell size
Sedimentation Rate
SED Rate: 0 to 20 mm/hour
P.364
Bleeding Time
Bleeding time refers to the length of time bleeding occurs after a standardized puncture wound to the skin. Bleeding time is measured in minutes and indicates the functioning status of the platelets, specifically the effectiveness of the platelet plug. Bleeding time should not exceed 15 minutes (normal: 3.0 to 9.5 minutes) for a forearm stick.
Partial Thromboplastin Time/Prothrombin Time
Partial thromboplastin time (PTT) and prothrombin time (PT) detect deficiencies in the activity of various clotting factors. Both tests evaluate clotting in a venous blood sample.
PTT especially demonstrates the effectiveness of the intrinsic pathway of coagulation and should not exceed 90 seconds (normal: 30 to 45 seconds). This test is important in determining the effectiveness and safety of heparin therapy.
PT demonstrates the effectiveness of the vitamin K dependent coagulation factors, especially the extrinsic and common pathways of coagulation. PT should not exceed 40 seconds, or 2 to 2.5 times a control level (normal: 11 to 13 seconds). PT is used to determine the effectiveness of warfarin (Coumadin) therapy.
![]() ediatric Consideration
ediatric Consideration
Newborn values of red blood cells, white blood cells, hemoglobin, and hematocrit are elevated compared with those of older infants, children, and some adults. High values begin to decrease within 2 weeks of birth, reaching a plateau after approximately 6 months. Levels reach adult values at approximately 18 years of age. Bleeding time, especially PT, is prolonged in infants because of a natural deficiency in vitamin K. At birth, infants in the United States are given injections of vitamin K to reduce the risk of bleeding.
Pathophysiologic Concepts
Anemia
Anemia is a decrease in the quantity of circulating red blood cells, an abnormality in the hemoglobin content of red blood cells, or both. Anemia can be caused by a disorder in red blood cell production or an elevated loss of red blood cells through chronic bleeding, sudden hemorrhage, or excessive lysis (destruction). All anemias result in decreased values for hematocrit and hemoglobin but may vary in values of MCV, MCHC, and RDW. For example, using the MCV as an index, a microcytic anemia has a MCV < 82 fL/red cell; a normocytic anemia has a MCV between 82 98 fL/red cell; and a macrocytic anemia has a MCV > 98 fL/red cell. The symptoms associated with anemia depend on its duration, its severity, and the host's age and prior health status.
P.365
All symptoms ultimately relate to a reduction in the delivery of oxygen to host cells and organs, thereby interfering with function and compromising health.
Anemia Caused by a Disorder in Red Cell Production
Anemias that result from a disorder in red cell production occur if there is inadequate or inaccessible iron, or a lack of folic acid, vitamin B12, or globulin. Red blood cell production may also be insufficient if there is bone marrow disease, as would occur in leukemia, after radiation exposure, or with other diseases of the marrow. A deficiency in erythropoietin, as would occur in renal failure, would also lead to a decrease in red cell production. Anemias due to disorders in RBC production may result in a red cell that is too small (microcytic) or too large (macrocytic), and hemoglobin content that is abnormally low (hypochromic).
Anemia Caused by Sudden or Chronic Hemorrhage or Lysis
Anemias caused by sudden hemorrhage, a slow chronic hemorrhage, or lysis result in a decrease in the total number of circulating red cells. This type of anemia may be associated with an increased percentage of circulating immature red cells (reticulocytes). Normal red blood cells live approximately 120 days. Red cell destruction or loss occurring before 100 days is abnormal.Common anemias, their causes, and laboratory profiles are shown in Table 12-1, and some are discussed more fully in the final section of this chapter.
Table 12-1. Common Anemias | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
P.366
Polycythemia
Polycythemia is an increase in the number of red blood cells. Primary polycythemia (polycythemia vera) is characterized by an increase in platelets and granulocytes as well as red blood cells, and is believed to be the result of a precursor cell abnormality.
Polycythemia may occur secondarily to chronic hypoxia. Chronic hypoxia causes increased release of the renal hormone erythropoietin, which stimulates the production of red blood cells. Individuals who live at high altitude or suffer from chronic lung disease frequently experience secondary polycythemia. Athletes who blood dope, meaning they accept self-transfusions of previously collected packed red cells, demonstrate polycythemia. And finally, polycythemia may be relative, rather than absolute. During dehydration, for instance, a decrease in plasma volume is reflected as an increase in the concentration of red cells. Polycythemia from any cause is associated with an increased risk of thrombus formation and an increase in the workload of the heart.
Leukopenia
Leukopenia is a decrease in the number of white blood cells. Leukopenia may be caused by a variety of conditions, including prolonged stress, viral infection, bone marrow disease or destruction, radiation, or chemotherapy. Severe systemic diseases such as lupus erythematosus, thyroid disease, and Cushing's syndrome may cause a decrease in white blood cells. All or one type of white cells may be affected. Leukopenia may predispose the individual to infection.
Leukocytosis
Leukocytosis is an increase in the number of circulating white blood cells. Leukocytosis is a normal response to infection or inflammation. It can be seen after emotional disturbance, after anesthesia or exercise, and during pregnancy. Abnormal leukocytosis is observed in certain malignancies and bone marrow disorders. Usually, only one type of white blood cell is affected. For example, allergic responses and asthma are specifically associated with increased numbers of eosinophils. Leukemia is characterized by an abnormally high level of one type of white cell and deficiencies in the others.
Shift to the Left
Shift to the left is a term used to describe an increased proportion of immature leukocytes (usually neutrophils) seen in the blood of an individual fighting an infection. In a shift to the left, neutrophils will be released from the bone marrow before their final maturation, when the demand for white blood cells is excessive. The immature neutrophils are frequently referred to as bands or stabs. As the infection or inflammation begins to recede, the release of immature
P.367
neutrophils stops and the blood is said to show a return shift to the right, as mature neutrophils again dominate a blood smear. The returning mature neutrophils are frequently referred to as segmented neutrophils.
Thrombocytopenia
Thrombocytopenia is a decrease in the number of circulating platelets. It is associated with increased risk of severe bleeding, even with small injuries or small spontaneous bleeds. Thrombocytopenia is characterized by small spots of subcutaneous bleeding, called petechiae, or larger areas of subcutaneous bleeding, called purpura. Ecchymosis (bruising) may also be present. Primary thrombocytopenia, also referred to as immune thrombocytopenic purpura, may occur idiopathically (for unknown reasons) or as a result of an autoimmune disorder characterized by antibodies built against the platelets. Secondary causes of thrombocytopenia include bone marrow damaging chemotherapeutic drugs and radiation, and certain viral infections, including HIV. Thrombocytopenia also develops in the serious condition disseminated intravascular coagulation (DIC), in which, after periods of extensive clotting, platelets begin to be consumed, leading to extensive bleeding and high mortality.
Thrombocythemia
Thrombocythemia is an increase in the number of circulating platelets. Thrombocythemia is associated with increased risk of thrombosis (clotting) in the vasculature. Depending on the site of clot formation or trapping, stroke, myocardial infarct, or respiratory distress may develop.
Primary thrombocythemia may occur with malignancy, polycythemia vera, and other diseases of the bone marrow. Secondary causes of thrombocythemia include acute infection, exercise, stress, and ovulation. Secondary thrombocythemia caused by these conditions is usually short-lived. However, prolonged secondary thrombocythemia may occur after removal of the spleen because this organ normally stores some platelets until they are needed in the circulation. Inflammatory diseases such as rheumatoid arthritis may also be associated with prolonged thrombocythemia.
Lymphadenopathy
Lymphadenopathy, or lymphoid hyperplasia, is the enlargement of the lymph nodes in response to a proliferation of B or T lymphocytes. Lymphadenopathy typically occurs after infection by a microorganism.
Regional lymphadenopathy indicates a localized infection. Generalized lymphadenopathy usually indicates a systemic infection such as AIDS or an autoimmune disorder such as rheumatoid arthritis or systemic lupus erythematosus. Occasionally, lymphadenopathy of either type may indicate a malignancy.
P.368
Splenomegaly
Splenomegaly is enlargement of the spleen. It is usually a result of lymphocyte proliferation in the spleen, caused by an infection elsewhere in the body. Splenomegaly caused by macrophage proliferation occurs if there are excessive numbers of dead cells (especially red blood cells) needing to be cleared from the circulation.
Splenomegaly may also occur as a result of engorgement of the spleen with blood. This is usually a complication of portal hypertension. Splenic tumors or cysts may also cause splenomegaly. Splenomegaly in response to an infection is usually associated with lymphadenopathy; other causes of splenomegaly are not.
Conditions of Disease or Injury
Aplastic Anemia
Aplastic anemia is a normocytic, normochromic anemia caused by dysfunction of the bone marrow such that dying blood cells are not replaced. Aplastic anemia is usually associated with a deficiency in red blood cells, white blood cells, and platelets, although rarely it may affect only the red cells.
There are many causes of aplastic anemia, including cancers of the bone marrow, autoimmune destruction of bone marrow, vitamin deficiency, ingestion of many different drugs or chemicals, and high-dose radiation or chemotherapy. Aplastic anemia may also develop after various viral infections, including mononucleosis, hepatitis, and AIDS. Frequently, the cause is unknown.
Clinical Manifestations
Classic systemic signs of anemia are common to all of the anemias described in this chapter and include the following:
Increased heart rate as the body attempts to deliver more oxygen to the tissues.
Increased respiratory rate as the body attempts to provide more oxygen to the blood.
Dizziness caused by decreased brain blood flow.
Fatigue caused by decreased oxygenation of various organs, including cardiac and skeletal muscles.
Skin pallor caused by decreased oxygenation.
Nausea caused by decreased gastrointestinal and central nervous system blood flow.
Decreased hair and skin quality.
In aplastic anemia, if platelets and white blood cells are involved, additional symptoms include:
Bleeding from the gums and teeth; easy bruising, including petechia and purpura.
Recurrent infection.
Poor healing of skin and mucosal sores.
P.369
Diagnostic Tools
CBC with differential and platelet count, MCV, and MCHC will diagnose anemia.
Bone marrow biopsy will determine involved cells.
Complications
Heart failure and death as a result of cardiac overload can occur with severe anemia.
Death from infection and hemorrhage if white blood cells or platelets are involved.
Treatment
Treat underlying disorder if known or remove causative agent.
Transfusions to reduce symptomatology.
Bone marrow transplant.
Immunosuppression if an autoimmune disease is involved.
Drugs to stimulate bone marrow function may be effective.
Hemolytic Anemia
Hemolytic anemia is a decrease in red blood cell number caused by excessive destruction of red cells. Remaining red cells are normocytic and normochromic. Red blood cell production in the bone marrow will increase to replace destroyed cells, and the advancement into the blood of immature red cells, or reticulocytes, will be accelerated.
Hemolytic anemia can occur from many different causes, such as a genetic defect in the red blood cell that accelerates its destruction, or the idiopathic development of autoimmune destruction of the cells. A severe burn, infection, incompatible blood exposure, or exposure to certain drugs or toxins may also cause hemolytic anemia. Depending on the cause, hemolytic anemia may occur once or repeatedly. Specific causes of hemolytic anemia that are discussed in detail include sickle cell anemia, malaria, hemolytic disease of the newborn, and transfusion reaction.
Sickle Cell Anemia
Sickle cell anemia is an autosomal recessive disorder caused by inheritance of two copies of a defective hemoglobin gene, one from each parent. The defective hemoglobin, called hemoglobin S (HbS), becomes rigid and elongated and forms a sickle shape when exposed to low oxygen. Oxidative stress also triggers the production of circulating advanced glycation end products that aggravate the vascular pathology in individuals with sickle cell anemia. The sickled red blood cell loses its ability to move easily
P.370
through narrow vascular spaces and becomes trapped in the microvasculature. This causes a blockage in blood flow to downstream tissues, leading to painful tissue ischemia. Although sickling is reversible if oxygen saturation of the hemoglobin returns, the sickled cells are especially fragile and many are destroyed in the microvasculature, leading to anemia. Destroyed cells are filtered and removed from the circulation in the spleen; this places an extra demand on splenic function. Scarring and sometimes infarction (cell death) of various organs, especially the spleen and bone, may occur. Multiorgan dysfunction is common after many years.
Stimuli for sickling include hypoxia, anxiety, fever, and exposure to the cold. Because the spleen is an important immune organ, infections, especially of bacterial origin, are common and frequently stimulate a sickle cell crisis.
At birth, signs of sickle cell anemia may not be apparent because all infants have a high level of a different type of hemoglobin, fetal (F) hemoglobin. Fetal hemoglobin does not sickle, but only lasts until approximately 4 months after birth. It is at this time that signs of disease become apparent. These signs include the classic symptoms of anemia, as well as signs related to the painful occlusions characteristic of the disorder.
Individuals with sickle cell anemia carry two defective genes and thus have only hemoglobin S. Individuals who are heterozygous for the sickle cell gene (carry one defective gene) are said to carry the sickle cell trait. Heterozygotes usually express hemoglobin S in approximately 30 to 40% of their red cells, with normal hemoglobin carried in the rest of the red cells. These individuals are typically asymptomatic unless exposed to low oxygen levels, especially while exercising.
The demographic roots of sickle cell anemia may be traced to malaria-endemic areas. The sickle cell trait has been shown to offer protection against the destruction of the red blood cell after an infection by the microorganism responsible for malaria. It is thought that this protection allowed the sickle cell gene to survive during evolution in areas with endemic (widespread) malaria, such as equatorial Africa. Thus, in the United States, sickle cell anemia primarily affects individuals who trace their ancestry to this area of Africa: up to approximately 10% of African Americans carry the trait, and approximately one child in every 375 African American live births has the disease. Figure 12-4 shows a chi square diagram of the genetic inheritance of sickle cell anemia.
Clinical Manifestations
Systemic signs of anemia.
Intense pain caused by vascular occlusion in a sickling episode.
Serious bacterial infections caused by inadequate splenic filtering of microorganisms.
Splenomegaly as the spleen removes the dead cells, sometimes leading to an acute crisis.
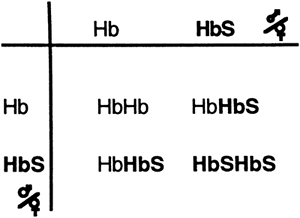 |
Figure 12-4. A chi square diagram showing the transmission of the sickle cell hemoglobin (HbS). |
P.371
Diagnostic Tools
In 1949, Linus Pauling suggested that sickle cell anemia was a molecular disease, an assumption he based on the differing electrophoretic mobilities of hemoglobin in the red blood cells of homozygous sickle cell (Hb SS) patients compared to those of normal hemoglobin A and heterozygous AS individuals. To this day, hemoglobin electrophoresis is used to identify the presence of sickle cell hemoglobin and confirm the disease. Electrophoretic screening for abnormal hemoglobin in a newborn is standard care in the United States and other countries following the birth of a high-risk infant.
Serial blood tests demonstrate decreased hematocrit, hemoglobin, and red cell count.
Prenatal testing identifies the presence of the homozygote state in the fetus.
Complications
Vaso-occlusive events leading to tissue infarct may cause intense pain and disability.
A sudden trapping of blood in the spleen, called splenic sequestration, may lead to hypovolemia, shock, and possibly death. The cause of splenic sequestration is unknown, but it may occur with fever and pain. Frequently the spleen will be removed after an occurrence. Loss of the spleen compromises the individual's subsequent responses to infection.
Stroke leading to weakness, seizures, or inability to speak may occur from occlusion of the cerebral vessels.
Aplastic crisis, during which the bone marrow temporarily stops erythropoiesis, may occur.
Avascular necrosis of the long bones of the leg or arm may occur from occlusion. Hip replacement is a common sequela of severe disability.
Priapism, prolonged and painful erection, may occur with vaso-occlusion of the vessels of the penis; this may lead to impotence in some circumstances.
P.372
Treatment
Newborn screening for sickle cell anemia has dramatically improved the prognosis of infants with the disease. All identified infants are given prophylactic antibiotics (penicillin or erythromycin) to prevent infections, from birth until at least 5 years of age.
If at any time a fever or other sign of infection develops, the child should be evaluated immediately and parenteral antibiotics should be provided. Most children with a fever should be admitted to the hospital.
Ibuprofen or acetaminophen should be administered to relieve minor pain, and more potent pain medication should be provided if needed.
All childhood immunizations should be administered on schedule, with the addition of the pneumococcal vaccine in the first 2 years of life and a booster dose at 5 years of age. This will reduce the main cause of mortality in children with sickle cell anemia: infection leading to sepsis.
Increased hydration by at least 1.5 to 2 times normal requirements may reduce the severity of a vaso-occlusive event.
Avoidance of low oxygen situations or oxygen-demanding activities.
Therapeutic drugs, including hydroxyurea (an S-phase cytotoxic drug), are available for use. Hydroxyurea has a direct effect on increasing cell volume and increasing fetal hemoglobin production. Not only does fetal hemoglobin not sickle, it also has an increased affinity for oxygen compared to adult hemoglobin. Other drugs with similar mechanisms of action are available as well.
Use of ion channel blockers, anti-adhesion and anti-inflammatory agents that interfere with injurious red cell endothelial interactions, limits the vascular damage associated with the disorder.
Red blood cell transfusions are frequently required but should be limited when possible to reduce the risk of transmission of infectious agents.
Bone marrow transplant with an HLA-matched donor may eliminate the production of sickled cells, but will not improve already damaged organs, and requires the subsequent lifelong use of immunosuppressants to block rejection.
Nitric oxide inhalation may be used to prevent pulmonary hypertension associated with sickle cell anemia.
Genetic counseling for families allows for future informed childbearing decisions.
Malaria
Malaria is a cause of hemolytic anemia related to an infection of the red blood cells by a protozoan of the genus Plasmodium that is transmitted to humans in the saliva of a mosquito. Malaria is endemic in tropical and subtropical environments of the world. It is an acute disease that can become chronic, with repeated episodes of debilitation. Infants and children are frequently affected.
P.373
The Plasmodium microorganism first infects the cells of the liver and then passes into the erythrocytes. Infection causes massive hemolysis of the red blood cells. At this point, more parasites are released into the circulation and subsequent cycles of infection occur. Red cell hemolysis leads to acute and chronic anemia. Cycles of infection typically occur approximately 72 hours apart. The host response to infection includes activation of the immune system, including production of various cytokines designed to increase the immune response. These cytokines, including tumor necrosis factor and interleukins 1 and 6, are key in fighting against the parasite, but also are responsible for most of the clinical manifestations of the disease, especially fever and myalgia (muscle aches). Individuals usually recover, but may relapse.
Clinical Manifestations
Systemic signs of anemia.
Cyclic (usually every 72 hours) fever spikes.
Chills and sweating with the fever.
Headache.
Myalgia.
Hepatomegaly and splenomegaly.
Jaundice may occur from excessive red blood cell lysis and release of bilirubin.
Diagnostic Tools
Blood analysis will demonstrate the occurrence of anemia and the presence of red blood cell parasites.
Complications
With severe disease, hypoglycemia, respiratory distress, shock, and coma may develop.
Some strains of the parasite are becoming resistant to traditional drug therapy.
Treatment
Prophylactic therapy against malaria is advised for travelers to endemic areas.
Prevention in endemic areas involves elimination of standing sources of water and the use of insecticides, mosquito nets, and repellents.
The placement of mosquito-repellent nets around the sleeping areas of all those living in endemic areas, especially children, is advised.
Antimalarial drugs are available to treat the disease if contracted, although resistance to all available drugs, including the chloroquine-related drugs, is high.
Blood transfusions are occasionally performed; however, this has resulted in transmission of HIV in endemic areas.
Vaccines against malaria are being developed, including DNA vaccines that may stimulate the immune response to infection. Some vaccines in use do not prevent infection by the parasite, but may reduce the severity of the disease.
The mosquito-killing fungi Beauveria bassiana and Metarhizium anisopliae are being investigated with the hope that they will become new, environmentally friendly weapons against malaria.
P.374
Hemolytic Disease of the Newborn
Hemolytic disease of the fetus and newborn is a normocytic, normochromic anemia seen in an Rh-positive fetus or infant born to an Rh-negative mother who has previously been exposed to Rh-positive blood, and has therefore developed antibodies to the Rh antigen. The development of antibodies usually occurs only after multiple maternal exposures to the antigen; these may occur during previous pregnancies, abortions, or miscarriages, or during amniocentesis. The maternal antibodies, usually IgG, are transferred to the fetus through the placenta and attack fetal red blood cells, leading to excessive red cell lysis and anemia. If the condition is mild, the maternal circulation effectively eliminates the waste products of hemoglobin metabolism, including bilirubin, for the fetus, and it suffers few ill effects in utero. Occasionally, maternal destruction of the fetal cells may be excessive, leading to a severe anemia and hydrops fetalis, a fatal condition characterized by massive edema and heart failure.
After delivery in the less affected infant, clinical signs of anemia may occur. More significant in the neonatal period is the development of severe jaundice, as the breakdown products of hemoglobin are ineffectively cleared by the infant's immature liver. A dramatic elevation in bilirubin can lead to a significant neurologic disorder, called kernicterus, as the unconjugated bilirubin precipitates out in the infant's brainstem, causing brain damage.
Hemolytic disease of the newborn in response to Rh incompatibility is uncommon and has become rarer with fewer pregnancies experienced by each woman and important prophylactic interventions (see later). As a result of these factors, the incidence of hemolytic disease of the newborn has dropped by at least 80% in the last few decades. More common than Rh incompatibility is ABO incompatibility. In this condition, maternal antibodies are produced as a result of ABO incompatibility, even during a first pregnancy. The presence of antibodies against the A or B antigens seldom leads to full-blown newborn hemolytic disease. Hemolytic disease of the newborn is described further in Chapter 17.
Clinical Manifestations
Mild hemolytic disease may be relatively asymptomatic, with slight hepatomegaly and minimally elevated bilirubin.
Moderate and severe disease manifest with pronounced signs of anemia.
Hyperbilirubinemia, resulting from excessive red cell lysis, may occur, leading to jaundice.
P.375
Complications
Kernicterus.
Severe anemia may cause heart failure.
Hydrops fetalis. Affected fetuses often abort spontaneously at approximately 17 weeks' gestation.
In one study, 10% of school-aged children who had received in utero transfusions for severe Rh incompatibility showed neurologic abnormalities, most likely related to asphyxia and anemia at birth.
Treatment
Prevention of Rh-induced hemolytic disease begins with the prenatal visit and documentation of a woman's Rh-negative status and the presence or absence of Rh antibodies. Women who are confirmed Rh negative and who do not show Rh-positive antibodies are administered an anti-Rh antibody preparation called RhoGAM at 28 weeks' gestation, or at the time of a miscarriage, abortion, or amniocentesis. If, after birth, the infant is deemed to be Rh positive and the woman is still Rh negative, she is again given RhoGAM within 72 hours. The RhoGAM injection provides passive immunity to the woman such that she does not develop her own antibodies against the Rh factor. A woman who is found to be Rh positive at any time is not given RhoGAM, but she and her fetus are observed closely during pregnancy and after delivery.
If a woman becomes Rh positive during pregnancy, the fetus is observed by serial amniocentesis to determine bilirubin level. Mildly affected fetuses are delivered at term; moderately affected fetuses may be delivered before term; severely affected fetuses may receive an intrauterine transfusion and be delivered before term.
In the newborn with hemolytic disease, exchange blood transfusions may be required. Transfusion is with Rh-positive blood not containing Rh antibody. Treatment should begin within 24 hours of birth and be repeated until twice the blood volume of the infant has been exchanged.
In mild cases, phototherapy to reduce the levels of unconjugated bilirubin may be sufficient.
Transfusion Reaction
A transfusion reaction is an immune-mediated destruction of incompatible red blood cells received in a blood transfusion. Transfusion reactions against donated white blood cells occur more frequently, but are typically mild. Although host and donor blood antigens are always identified (typed) for ABO and Rh compatibility before a transfusion is given, an
P.376
error in red blood cell typing or a mix-up in the blood supplies may occur. Transfusion reactions may also develop as a result of an immune reaction to bacteria transferred in contaminated blood products.
Clinical Manifestations
Immediate, life-threatening reactions occur with ABO incompatibility. Manifestations include:
Immediate flushing of the face.
A feeling of warmth in the vein receiving the blood.
Fever and chills.
Chest, flank, or low back pain.
Abdominal pain with nausea and vomiting.
Decreased blood pressure with increased heart rate.
Dyspnea (a sensation of breathing difficulty).
Transfusion reactions against white blood cells are milder and usually include fever and occasionally chills.
Complications
Renal failure may result from red blood cell casts and hemoglobin obstruction of the nephrons.
Treatment
The transfusion must be stopped immediately.
Fluids may be given to reduce the risk of renal damage.
Anaphylactic responses are treated by anti-inflammatory drugs, including antihistamines and steroids.
Leukocyte-cleansed blood is available, which will eliminate reactions to white blood cells.
Posthemorrhagic Anemia
Posthemorrhagic anemia is a normocytic, normochromic anemia that results from sudden loss of blood in an otherwise healthy individual. The hemorrhage may be obvious or hidden.
Blood pressure decreases with sudden hemorrhage. Reflex responses to decreased blood pressure and tissue hypoxia include increased activation of the sympathetic nervous system. This results in increased vascular resistance, heart rate, and stroke volume, all of which serve to return blood pressure toward normal. Respiratory rate increases to improve oxygenation. Renal responses to decreased blood pressure include decreased urine output and increased release of the hormone renin. Salt and water reabsorption in the kidney increase, serving to return blood pressure toward normal. Renal secretion of erythropoietin is stimulated, leading to increased red cell production.
P.377
Clinical Manifestations
Increased heart rate and respiratory rate, with a decrease in blood pressure. Consciousness may be impaired.
The cause of the hemorrhage will present with individual clinical manifestations.
Diagnostic Tools
Reduction in red cell count, hematocrit, and hemoglobin on the CBC as interstitial fluid moves into the vascular compartment in an attempt to increase blood volume.
Complications
Hypovolemic shock with the possibility of renal failure, respiratory failure, or death.
Treatment
Restore blood volume with intravenous infusion of plasma or type-matched whole blood (or O negative). Saline or albumin may also be infused.
Pernicious Anemia
Pernicious anemia is a megaloblastic anemia characterized by abnormally large red blood cells with immature (blastic) nuclei (see page C8 for illustrations). Pernicious anemia is caused by a deficiency of vitamin B12 in the blood. Vitamin B12 is essential for DNA synthesis in red blood cells and for neuronal functioning. It is provided in the diet and absorbed across the stomach into the blood. A gastric hormone, intrinsic factor, is essential for absorption of vitamin B12. Intrinsic factor is secreted by the parietal cells of the gastric mucosa. Most causes of pernicious anemia result from intrinsic factor deficiency, but dietary deficiency of vitamin B12 may occur. Usually, B12 deficiency is a slowly developing disorder, and frequently goes unnoticed until symptoms are severe. Typically, patients affected are elderly; it is rare for an individual younger than 30 to suffer pernicious anemia unless it is present at or soon after birth.
Intrinsic factor deficiency may occur congenitally or may develop after atrophy or destruction of the gastric mucosa as a result of chronic gastric inflammation or an autoimmune disease. There appears to be a genetic susceptibility to autoimmune causes. Surgical removal of all or part of the stomach will also result in intrinsic factor deficiency.
Clinical Manifestations
Systemic signs of anemia.
Dementia related to neurologic deterioration.
Ataxia (poor muscle coordination) and sensory loss resulting from myelin degeneration.
P.378
Diagnostic Tools
Blood analysis will demonstrate anemia characterized by macrocytic cells with normal hemoglobin (significantly elevated MCV > 103, normal MCHC).
A decrease in serum B12 will confirm the disease.
Complications
Severe anemia may cause heart failure, especially in the elderly.
Treatment
Lifelong intramuscular injections of vitamin B12.
![]() eriatric Consideration
eriatric Consideration
The elderly are most prone to suffer from a dietary deficiency of vitamin B12, as a result of poor diet. Any elderly person demonstrating fatigue, rapid heart rate, and mental and physical sluggishness should be evaluated for vitamin B12 deficiency.
Folate-Deficiency Anemia
Folate (folic acid) deficiency anemia is a megaloblastic anemia characterized by enlarged large red cells with immature nuclei. Folic acid deficiency is caused by a lack of the vitamin folate. Folate is essential for red blood cell production and maturation. It is also important for DNA and RNA synthesis and for the function of several DNA proofreading enzymes (Chapter 2). Folic acid is provided in the diet, but deficiency is relatively common, especially in young women, malnourished individuals, and alcohol abusers. Folic acid absorption occurs across the small intestine and does not require intrinsic factor. Because of widespread folic acid deficiency and its recognized importance in maintaining health, folic acid supplementation of cereals and other grains is soon to be initiated in the United States. Folic acid supplementation is especially important for pregnant women, as described later.
Clinical Manifestations
Systemic signs of anemia are present.
Diagnostic Tools
Blood analysis will demonstrate anemia characterized by macrocytic cells with normal hemoglobin (elevated MCV > 98, normal MCHC).
P.379
Typically, the MCV will be elevated less than in pernicious anemia, and there will be no vitamin B deficiency.
Complications
Maternal deficiencies in folic acid are associated with an increased risk of fetal malformations, especially neural tube defects.
Adult deficiency may be associated with an increased risk of cardiovascular disease.
Treatment
Administration of oral folate. Women intending to become pregnant should begin vitamin supplementation at least 3 months before conception. It is important not to confuse folate-deficiency anemia with pernicious anemia because treatment with folic acid is contraindicated in pernicious anemia.
Blood transfusions may be required in severe cases.
Iron-Deficiency Anemia
Iron-deficiency anemia is a microcytic-hypochromic anemia that results from a diet deficient in iron, or from the slow, chronic loss of blood. Iron is an essential component of the hemoglobin that makes up a large part of the red blood cell. Iron deficiency is a problem in toddlers and children with increased growth demands. Pregnant women are frequently iron-deficient because of the iron demands of the growing fetus. Menstruating women also tend to be iron deficient because of iron loss each month and diets that may be deficient in iron. Menstruating women who exercise are at increased risk because exercise increases the metabolic demands of muscle cells. In men, iron deficiency usually occurs with an ulcer or liver disease characterized by bleeding. Iron deficiency develops slowly. Decreased red blood cell numbers prompt the bone marrow to increase the release of abnormally small, hemoglobin-deficient red cells. See page C8 for illustrations.
Clinical Manifestations
In adults, systemic signs of anemia are present once hemoglobin decreases to less than 12 g/100 mL. Individuals usually do not seek treatment for symptoms until hemoglobin decreases to 8 g/100 mL or below.
In addition to the previously described systemic signs of anemia, pale palms, pale conjunctivae, and pale earlobes may also be present.
Diagnostic Tools
Blood analysis demonstrates anemia characterized by microcytic cells (MCV < 87) and decreased serum iron. Iron-binding capacity in the blood is high because proteins that bind iron are in less demand.
Stool test for occult blood may be positive, suggesting a GI bleed or carcinoma.
P.380
Complications
A hemoglobin value of less than 5 g/100 mL can lead to heart failure and death.
Treatment
An iron-rich diet containing red meat and dark green vegetables, such as spinach.
Oral iron supplementation.
Treat the cause of abnormal bleeding if known.
Sideroblastic Anemia
Sideroblastic anemia is a microcytic-hypochromic anemia characterized by the presence of abnormal red cells (sideroblasts) in the circulation and the bone marrow. Sideroblasts carry iron in the mitochondria rather than in the hemoglobin molecules, and thus are unable to transport oxygen to the tissues. There is no iron deficiency.
Poor transport of oxygen causes hypoxia. This is sensed by erythropoietin-secreting kidney cells. Erythropoietin stimulates new red cell production in the bone marrow, which causes the marrow to become congested and increases the production of sideroblasts, worsening the anemia.
Primary sideroblastic anemia can occur as a result of a rare genetic defect on the X chromosome (primarily seen in males) or may occur spontaneously, especially in the elderly. Secondary causes of sideroblastic anemia include certain drugs (e.g., some chemotherapeutic agents) and lead ingestion.
Clinical Manifestations
Systemic signs of anemia are present.
Iron accumulation results in hepatomegaly and splenomegaly.
Diagnostic Tools
Blood analysis demonstrates anemia characterized by microcytic hypochromic cells, with elevated plasma iron and normal iron-binding capacity.
A bone marrow examination demonstrates the presence of iron accumulations, sideroblasts, and phagocytic macrophages.
Complications
Some cases progress to myelodysplastic syndrome and acute myeloblastic leukemia.
P.381
Treatment
The cause of the disease, if related to a drug, is removed.
The drug pyridoxine may successfully treat the disease, especially in persons with no evidence of neutropenia or thrombocytopenia. Iron is not given.
Acute Infectious Mononucleosis
Mononucleosis is an acute infection of the B lymphocytes, usually caused by the Epstein-Barr virus or, less frequently, by the cytomegalovirus. Most adults were exposed to these viruses in early childhood and at that time successfully fought off active infection to gain lifelong immunity. Usually, individuals who develop mononucleosis are children who do not fight off the infection, or teenagers and young adults who are exposed to the virus for the first time. Transmission of the virus occurs primarily through oral secretions and appears to require repeated exposures. In the teenage to young adult years, the immune system may be less active or depressed by poor dietary and sleep habits, making those age groups especially susceptible to infection.
Clinical Manifestations
The classic triad of symptoms of mononucleosis includes: severe sore throat, fever, and swelling of the cervical lymph nodes.
Overwhelming or mild fatigue may be present.
Swelling of all lymphoid tissues, including spleen, tonsils, and other neck nodes, may be present. The lymph nodes are usually tender.
The liver and spleen may be enlarged and tender.
![]() eriatric Consideration
eriatric Consideration
Although mononucleosis is usually seen in young adults or children, the elderly may contract the disease. When infected, the elderly may be difficult to diagnose because of the absence of the classic triad of symptoms. Jaundice may be present.
Diagnostic Tools
The liver may be palpable and liver function tests are abnormal in 95% of cases. Jaundice is rare in young people.
Laboratory findings demonstrate a brief leukopenia followed by proliferation first of the infected B cells, and then immunoactive T cells. Many white cells are atypical.
Blood tests, especially the Monospot agglutination test, demonstrate antibodies to the Epstein-Barr virus.
P.382
Complications
Complications are rare, but may include hepatitis, meningitis, encephalitis, and Guillain-Barr syndrome. Rarely, Burkitt's lymphoma or B cell lymphoma may develop.
Treatment
Mononucleosis is usually self-limiting. Treatment is supportive and encourages adequate rest and hydration.
It is important to avoid contact sports so as not to injure or rupture the spleen.
Ibuprofen and acetaminophen may be given. Aspirin is not recommended because of its association with Reye's syndrome.
Leukemia
Leukemia is a cancer of one class of white blood cells in the bone marrow, which results in the proliferation of that cell type to the exclusion of other types.
Leukemia appears to be a clonal disorder, meaning one abnormal cancerous cell proliferates without control, producing an abnormal group of daughter cells. These cells prevent other blood cells in the bone marrow from developing normally, causing them to accumulate in the marrow. Because of these factors, leukemia is called an accumulation and a clonal disorder. Eventually, leukemic cells take over the bone marrow. This reduces blood levels of all nonleukemic cells, causing the many generalized symptoms of leukemia.
Types of Leukemia
Leukemia is described as acute or chronic, depending on the suddenness of appearance and how well differentiated the cancerous cells are. The cells of acute leukemia are poorly differentiated, whereas those of chronic leukemia are usually well differentiated.
Leukemia is also described based on the proliferating cell type. For instance, acute lymphoblastic leukemia, the most common childhood leukemia, describes a cancer of a primitive lymphocyte cell line. Granulocytic leukemias are leukemias of the eosinophils, neutrophils, or basophils. Leukemia in adults is usually chronic lymphocytic or acute myeloblastic. Long-term survival rates for leukemia depend on the involved cell type, but range to more than 75% for childhood acute lymphocytic leukemia, which is a remarkable statistic for what was once a nearly always fatal disease.
Risk Factors for Developing Leukemia
Risk factors for leukemia include a genetic predisposition coupled with a known or unknown initiator (mutating) event. Siblings of children with
P.383
leukemia are 2 to 4 times more likely to develop the disease than other children. Certain abnormal chromosomes are seen in a high percentage of patients with leukemia. Likewise, individuals with certain chromosomal abnormalities, including Down syndrome, have an increased risk of developing leukemia. Exposure to radiation, some drugs that depress the bone marrow, and various chemotherapeutic agents have been suggested to increase the risk of leukemia. Environmental agents such as pesticides and certain viral infections also have been implicated.
Previous illness with a variety of diseases associated with hematopoiesis (blood cell production) has been shown to increase the risk of leukemia. These diseases include Hodgkin lymphoma, multiple myeloma, polycythemia vera, sideroblastic anemia, and myelodysplastic syndromes. Chronic leukemia may sometimes transform into acute leukemia.
Clinical Manifestations
Acute leukemia has marked clinical manifestations. Chronic leukemia progresses slowly and may have few symptoms until advanced.
Pallor and fatigue from anemia.
Frequent infections caused by a decrease in white blood cells.
Bleeding and bruising caused by thrombocytopenia and coagulation disorders.
Bone pain caused by accumulation of cells in the marrow, which leads to increased pressure and cell death. Unlike growing pains, bone pain related to leukemia is usually progressive.
Weight loss caused by poor appetite and increased caloric consumption by neoplastic cells.
Lymphadenopathy, splenomegaly, and hepatomegaly caused by leukemic cell infiltration of these lymphoid organs may develop.
Central nervous system symptoms may occur.
Diagnostic Tools
Laboratory findings include alterations in specific blood cell counts, with overall elevation or deficiency in white blood cell count variable, depending on the type of cell affected.
Bone marrow tests demonstrate clonal proliferation and blood cell accumulation.
Cerebral spinal fluid is examined to rule out central nervous system involvement.
Complications
Children who survive leukemia have an increased risk of developing a new malignancy later on in life when compared to children who have never had leukemia, most likely related to the aggressiveness of chemotherapeutic (or radiological) regimens.
Treatment regimens, including bone marrow transplant, are associated with temporary bone marrow depression, and increase the risk of developing a severe infection that could lead to death.
Even with successful treatment and remission, leukemic cells may still persist, suggesting residual disease. Implications for prognosis and cure are unclear.
P.384
Treatment
Multiple drug chemotherapy.
Antibiotics to prevent infection.
Transfusions of red blood cells and platelets to reverse anemia and prevent bleeding.
Bone marrow transplant may successfully treat the disease. Blood products and broad spectrum antibiotics are provided during bone marrow transplant procedures to fight and prevent infection.
Immunotherapy, including interferons and other cytokines, is used to improve outcome.
Therapy may be more conservative for chronic leukemia.
The treatments described earlier may contribute to the symptoms by causing further bone marrow depression, nausea, and vomiting. Nausea and vomiting may be controlled or reduced by pharmacologic and behavioral intervention.
Anthocyanins (chemicals with known antioxidant and liver protecting properties) isolated from the plant Hibiscus sabdariffa are being studied as chemopreventive agents in that they cause cancer cell apoptosis (death) in human promyelocytic leukemia cells.
Hodgkin Lymphoma (Hodgkin's Disease)
Hodgkin lymphoma, formerly called Hodgkin's disease, is a cancer of the lymphoid tissue, usually the lymph nodes and spleen. It is one of the most common cancers in young adults, especially young males. There is a second peak in incidence in the 6th decade of life.
Hodgkin lymphoma is a clonal disorder, arising from one abnormal cell. The abnormal cell population appears to be derived from a B cell or, less frequently, a T cell or monocyte. Neoplastic cells of Hodgkin lymphoma are called Reed-Sternberg cells. These cells intersperse among normal lymph tissue present in the lymph organs. Because of its B-cell and clonal nature, in 2005, Hodgkin's disease was reclassified by the World Health Organization as a lymphoma and renamed Hodgkin lymphoma.
There are four major classifications of Hodgkin lymphoma, based on the cells involved and whether the neoplasms are nodular in form. Staging of Hodgkin lymphoma is important because it guides treatment and strongly influences outcome. The early stages of the disease, stages I and II, are usually curable. Cure rates for stages III and IV are approximately 75% and 60%, respectively.
P.385
The cause of Hodgkin lymphoma is unknown. However, individuals with the disease and in remission from it demonstrate reduced T-cell mediated immunity. In addition, sporadic case clusters suggest that a virus, perhaps one of the herpes strains, especially the Epstein-Barr virus, may be involved. There is likely a genetic tendency to develop the disease.
![]() eriatric Consideration
eriatric Consideration
Elderly patients with Hodgkin lymphoma have a poorer response to therapy than do young patients. This is largely because of the increased morbidity associated with aggressive chemotherapy. A more advanced stage of disease at the time of diagnosis also may be involved. Concurrent disease in the elderly (lung, cardiac, and renal) may affect the response to chemotherapy and radiation.
Clinical Manifestations
Painless enlargement of lymph nodes, especially in the neck and under the arms.
Evening fevers and night sweats may occur.
Weight loss accompanies advanced stages of the disease.
Diagnostic Tools
Lymph node biopsy can diagnose Hodgkin lymphoma.
Complications
Secondary malignancies and cardiotoxicity may develop after aggressive treatment. Because of these and other treatment complications, Hodgkin lymphoma patients have a higher chance of dying from acute and late treatment toxicities than from the disease itself.
Treatment
Multidrug chemotherapy.
Radiation therapy.
Bone marrow transplant.
Targeted biologically based therapies, such as the use of receptor-specific antibodies, inhibition of antiapoptotic pathways, and induction of specific cytotoxicity, may have better patient tolerability and fewer long-term complications.
Non-Hodgkin Lymphoma
Non-Hodgkin lymphomas are cancers of the lymph tissue that are not Hodgkin lymphoma. Non-Hodgkin lymphoma usually occurs in older adults and is typically discovered at a more advanced stage than Hodgkin lymphoma. Non-Hodgkin lymphoma is not confined to a single group of
P.386
lymph nodes as in Hodgkin lymphoma, but rather is diffusely spread throughout the lymphoid organs, including the lymph nodes, liver, spleen, and occasionally the bone marrow. Disease may also be found in the sinuses. Like Hodgkin lymphoma, non-Hodgkin disease is classified under several divisions, primarily related to whether the neoplastic tissue is nodular or diffuse.
Non-Hodgkin disease appears to develop primarily from a malignancy of the B cells, but T cells and macrophages may also be the original site of the cancer. Causes of non-Hodgkin lymphoma are unclear, but viral infection, including HIV infection, appears to be responsible for at least some cases. Overall, non-Hodgkin lymphoma has a poorer prognosis than Hodgkin lymphoma, but there are multiple types of this disease, with some aggressive and others less so; therefore, prognosis varies greatly.
Clinical Manifestations
Painless enlargement of lymph nodes.
Splenomegaly.
GI complications may occur.
Fever; fatigue.
Weight loss.
Back and neck pain with hyper-reflexia.
Diagnostic Tools
Lymph node biopsy can diagnose non-Hodgkin lymphoma.
Treatment
Aggressive chemotherapy is used for advanced disease. Diffuse disease usually requires even more aggressive therapy.
In current practice, a combination of drugs known as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) plus adjuvant radiotherapy is used. For patients less than 61 years old with localized large B cell lymphoma, an intensive regimen of another drug combination, ACVBP (doxorubicin, cyclophosphamide, vindesine, bleomycin, prednisone), appears superior to CHOP.
Conservative chemotherapy may be used for slow-growing lymphomas and for palliative treatment.
Radiotherapy is also used, as is surgery to remove large tumors.
Bone marrow transplant may be performed.
Multiple Myeloma
Multiple myeloma is a clonal disorder characterized by proliferation of one type of B lymphocyte, and plasma cells derived from that lymphocyte. These cells disperse throughout the circulation and deposit primarily in the bone, causing bone breakdown, inflammation, and pain. Antibodies produced by the plasma cells are
P.387
usually clonal IgG or IgA. Monoclonal fragments of these antibodies may be found in the urine of patients with the disease. These fragments are called Bence Jones proteins. The cause of multiple myeloma is unknown, but risk factors are believed to include occupational exposures to certain materials and gases, ionizing radiation, and possibly multiple drug allergies. Survival rate is generally low, although some patients may live a long time with this disease.
Clinical Manifestations
Bone pain and fracture may occur.
Weight loss and fatigue may occur.
Neurologic dysfunction resulting from high blood calcium levels is seen with bone breakdown.
Recurrent infections from reduced B-cell function are common.
Diagnostic Tools
Bone biopsy and blood analysis confirm the disease. Urine may also be diagnostic with the presence of Bence Jones proteins.
Hypercalcemia may be present when bones are involved.
Complications
Renal failure may develop as a result of Bence Jones proteins depositing in the renal tubules.
Patients may become severely anemic.
Treatment
Chemotherapy may prolong life. One type of chemotherapy involves the use of an old drug, thalidomide, which is an immunomodulator as well as an inhibitor of blood vessel development. Other drug therapies include proteasome inhibitors (bortezomib) and alkylating agents.
Radiation therapy is used to reduce the size of bone lesions and relieve pain.
Bone marrow transplant may be successful in some patients.
Hemophilia A
Hemophilia A, also called classic hemophilia, is an X-linked recessive disease resulting from an error in the gene coding for coagulation factor VIII. Classic hemophilia is the most common inherited coagulation disorder. It is seen in boys who inherit the defective gene on the X chromosome from their mother. The mother is usually heterozygous for the disorder and shows no symptoms. However, 25% of cases come from new X chromosome mutations. The defective gene may result from one of several different deletions or point mutations.
Without factor VIII, the intrinsic coagulation pathway is interrupted and extensive bleeding from small wounds or microvascular tears occurs.
P.388
Bleeding is frequently into the joints and can cause significant pain and disability.
Other Types of Hemophilia
Other forms of hemophilia exist. These hemophilias result from the absence of different coagulation factors. Hemophilia B is an X-linked disorder caused by a lack of factor IX. Hemophilia C is an autosomal disorder caused by a lack of factor XI. Von Willebrand disease is an autosomal-dominant disease resulting from an abnormality of von Willebrand factor (vWF). This factor is released from endothelial cells and platelets and is essential for the formation of the platelet plug. With a reduction of vWF factor, factor VIII levels are also reduced.
Clinical Manifestations of Classic Hemophilia
Spontaneous or excessive bleeding after a minor wound.
Joint swelling, pain, and degenerative changes.
Diagnostic Tools
Laboratory studies show a normal bleeding time, but prolonged PTT.
Measurement of factor VIII is reduced.
Prenatal testing for the gene is possible.
Complications
Intracranial hemorrhage may occur.
Infection with HIV was common before artificial production of factor VIII reduced the need for transfusions.
Treatment
Factor VIII replacement. Factor VIII may be from frozen plasma concentrate donated from the father of the affected boy or may be produced by monoclonal antibody techniques. Multiple donor plasma extracts of Factor VIII are no longer used because of the risk of transmission of viral infections such as HIV and hepatitis B and C.
Liver Disease and Vitamin K Deficiency
The liver is the site of synthesis for many coagulation factors, several of which are vitamin K dependent. Disease of the liver or inadequate plasma levels of vitamin K will interrupt the coagulation pathways. Vitamin K is a fat-soluble vitamin absorbed in the diet by means of bile. Because bile is produced in the liver, a healthy liver and a clear bile duct are required for successful coagulation. Vitamin K also is synthesized by bacteria in the gut. Newborns are vitamin K deficient because of a lack of vitamin K producing bacteria in the intestine and immature liver function.
P.389
Clinical Manifestations
Bleeding characterized by petechia (small hemorrhage spots on the skin) and purpura (purplish discoloration of the skin).
Treatment
Vitamin K is administered intramuscularly to the neonate and orally in children or adults.
Disseminated Intravascular Coagulation
Disseminated intravascular coagulation (DIC) is a unique condition characterized by the formation of multiple blood clots throughout the microvasculature. Eventually, the components of the blood clotting cascade and the platelets are used up, and hemorrhages begin to occur at all bodily orifices, at sites of injury or venous puncture, and throughout many organ systems.
DIC is never a primary condition. Instead, it occurs as a complication of major clinical incidents or trauma such as shock, widespread infection, major burn, myocardial infarct, or obstetric complication. Hypoxemia and acidemia develop, damaging the endothelial cells of the vasculature. Multiple endothelial cell injuries initiate extensive activation of the platelets and the intrinsic coagulation pathway, leading to microthrombi throughout the vascular system. Tissue damage, occurring as the precipitating event or after hypoxia and acidemia, causes the production of thromboplastin, which activates the extrinsic coagulation pathway. Clotting is extensive, with fibrin strands firming and holding the emboli.
As the coagulation cascades proceed, fibrinolytic processes (breaking down of fibrin strands) are accelerated. These processes result in the release of anticoagulation enzymes into the circulation. Eventually, clotting factors and platelets are used up and hemorrhage and oozing of blood into mucous membranes occur. The loop is completed with bleeding and clotting occurring simultaneously.
Clinical Manifestations
Hemorrhage from puncture sites, wounds, and mucous membranes in a patient with shock, obstetric complications, sepsis (widespread infection), or cancer. If bleeding is under the skin, vascular lesions will be apparent.
Altered consciousness indicates a cerebral thrombus.
Abdominal distention indicates a GI bleed.
Cyanosis and tachypnea (increased respiratory rate) caused by poor tissue perfusion and oxygenation are common. Mottling of the skin indicates tissue ischemia.
Hematuria (blood in urine) caused by hemorrhage or oliguria (decreased urine output) caused by poor renal perfusion.
P.390
Diagnostic Tools
Blood tests demonstrate accelerated clotting and decreased platelets.
Fibrin degradation products are elevated. Platelets and plasma fibrinogen levels are reduced.
Complications
The many clots cause obstruction to blood flow in all organs of the body. Widespread organ failure may occur. Mortality is greater than 50%.
Treatment
Treatment is difficult because of the combination of hemorrhage and clotting. Prevention of DIC and early identification of the condition is essential. Treatment is geared toward:
Removal of the precipitating event.
Heparin therapy may be initiated if organ failure caused by hypoxia is imminent. Heparin is not suggested when DIC is caused by sepsis or if central nervous system bleeding occurs.
Fluid replacement is important to maintain organ perfusion as high as possible.
Plasma containing factor VIII, red cells, and platelets may be administered.
Selected Bibliography
Armitage, J.O. (2005). Defining the stages of aggressive non-Hodgkin's lymphoma a work in progress. New England Journal of Medicine 352, 1250 1252.
Chang, Y.C., Huang, H.P., Hsu, J.D., Yang, S.F., & Wang, C.J. (2005). Hibiscus anthocyanins rich extract-induced apoptotic cell death in human promyelocytic leukemia cells. Toxicology and Applied Pharmacology 205, 201 212.
Dich, J., Zahm, S.H., Hanberg, A., & Adami, H.O. (1997). Pesticides and cancer. Cancer Causes and Control 8, 420 443.
Enserink, M. (2005). Mosquito-killing fungi may join the battle against malaria. Science 308, 1531, 1533.
Friebert, S.E., & Shurin, S.B. (1998). ALL: Diagnosis and outlook. Contemporary Pediatrics 15, 118 136.
Guyton, A.C., & Hall, J.E. (2006). Textbook of medical physiology (11th ed.) Philadelphia: W.B. Saunders.
Hickey, S.M., & Strasburger, V.C. (1997). What every pediatrician should know about infectious mononucleosis in adolescents. Pediatric Clinics of North America 44, 1541 1556.
Kyle, R.A., & Rajkumar, S.V. (2004). Multiple myeloma. New England Journal of Medicine 351, 1860 1873.
Lew, V.L., & Bookchin, R.M. (2005). Ion transport pathology in the mechanism of sickle cell dehydration. Physiological Reviews 85, 179 200.
Pauling, L., Itano, H. A., Singer, S. J., & Wells, I. C. (1949). Sickle cell anemia: a molecular disease. Science 110, 543 548.
Porth, C.M. (2005). Pathophysiology: Concepts of altered health states (7th ed.). Philadelphia: Lippincott Williams & Wilkins.
Re, D., Thomas, R.K., Behringer, K., & Diehl, V. (2005). From Hodgkin's disease to Hodgkin's lymphoma: Biologic insights and therapeutic potential. Blood 105, 4553 4560.
P.391
Reyes, F., Lepage, E., Ganem, G., Molina, T.J., Brice, P., Coiffier, B., et al. (2005). ACVBP versus CHOP plus radiotherapy for localized aggressive lymphoma. New England Journal of Medicine 352, 1197 1205.
Somjee, S.S., Warrier, R.P., Thomson, J.L., Ory-Ascani, J., & Hempe, J.M. (2005). Advanced glycation end-products in sickle cell anemia. British Journal of Haematology 128, 112 118.
Stuart, M.J., & Nagel, R.L. (2004). Sickle-cell disease. Lancet 364, 1343 1360.
Vander, A.J., Sherman, J., & Luciano, D. (2001). Human physiology (8th ed.). Boston: McGraw-Hill.
Werler, M.M. (1993). Periconceptual folic acid exposure and the risk of occurrent neural tube defects. Journal of the American Medical Association 269, 1257 1261.
EAN: 2147483647
Pages: 26