11 - Magnetic Resonance Imaging of the Thorax
Editors: Shields, Thomas W.; LoCicero, Joseph; Ponn, Ronald B.; Rusch, Valerie W.
Title: General Thoracic Surgery, 6th Edition
Copyright 2005 Lippincott Williams & Wilkins
> Table of Contents > Volume I - The Lung, Pleura, Diaphragm, and Chest Wall > Section IV - Diagnostic Procedures > Chapter 15 - Molecular Diagnostic Studies in Pulmonary Disease
function show_scrollbar() {}
Chapter 15
Molecular Diagnostic Studies in Pulmonary Disease
M ir n E. McMenamin
Massimo Loda
Tremendous progress has been made in new technologies involved in the manipulation of DNA, RNA, and proteins aimed at the study of the molecular biology of disease. The first drafts of the sequence of the human genome were published independently by the genome international sequencing consortium by McPherson (2001) and Venter (2001) and their co-workers. New technologies capable of dealing with a large amount of information originating from genomic, large-scale gene expression and proteomic techniques are being developed and are supported by tremendous developments in bioinformatics and computer software. Evaluation of the functional significance of gene expression by means of genome-wide study is facilitated by these technologies. Medical treatment is increasingly based on information regarding the mechanisms of action of key genes and disturbances in important cellular pathways that have been implicated in various disease processes. Small molecules, such as interfering RNA, or chemical compounds modeled on the protein structure of their targets are being actively developed and potentially are powerful therapeutic agents, owing to their ability to interfere with critical intracellular signaling pathways.
Discoveries in molecular pathogenetic mechanisms have had a major impact in the field of pulmonary disease, with important implications for the screening, diagnosis, and treatment of diverse pulmonary diseases, including genetic disorders, infections, and particularly cancer. Novel molecular screening techniques are being developed, and new therapeutic approaches are being actively investigated. Molecular biology has had profound effects on the development of new products, including vaccines, therapeutic agents, and novel gene therapies, and is the basis of pharmacogenomics, the study of genes or chromosomal loci that are involved in determining the responsiveness of an individual to a particular drug.
As a result of all these developments, those involved in patient care and treatment should have an understanding of molecular biology and related techniques to facilitate selection of the most appropriate investigation or therapy for their patients, to understand the novel agents and techniques that are under investigation in clinical trials, and to communicate adequately with their increasingly well-informed patients. In addition, it is imperative that institutions and departments bank frozen tissue on a routine basis with patient informed consent because DNA, RNA, and protein extracted from such material is optimal for evaluating genetic information.
This chapter provides an introduction to basic molecular biology principles and selected molecular diagnostic techniques and demonstrates their application in specific types of pulmonary disease, including genetic disease, infections, interstitial lung disease, and particularly cancer. We have entered an era of molecular classification of disease, and a brief overview of microarray technology is included. Animal models of disease are also briefly reviewed. Gene therapy and other targeted molecular therapy, including small interfering RNA (siRNA), are discussed, with particular reference to their application as prospective anticancer therapies.
PRINCIPLES OF MOLECULAR PATHOLOGY
General Principles
DNA and RNA are designed to retain, store, and express genetic information in the form of proteins and perhaps have a functional role in and of themselves. DNA comprises two antiparallel strands of nucleotide bases wound around each other in a right-handed double helix and aligned on a sugar-phosphate backbone. The negatively charged sugar-phosphate molecules are on the outside, and the planar bases of each strand, stacked one above the other, form the center of the helix. The strands are joined by
P.262
means of noncovalent hydrogen bonds between pairs of bases on opposite strands. The sequence of one DNA strand uniquely specifies the sequence of the other strand; this complementarity underlies the ability to perform in vitro manipulation of nucleic acids. DNA is organized into highly compact, regular units called chromosomes. DNA is an extraordinarily stable molecule and loses its normal conformational structure only at extremes of heat or pH or in the presence of destabilizing agents. DNA can be extracted from formalin-fixed archival paraffin blocks of tissue. RNA, in contrast, is much less stable than DNA because of its single-stranded, more random structure; its susceptibility to alkaline hydrolysis; and its rapid degradation by ubiquitous enzymes called RNAses. With current technology, useful amounts of RNA can be extracted from fresh tissue or fresh-frozen samples. It is difficult to retrieve good-quality amplifiable RNA from archival paraffin-embedded tissue, because of degradation of the RNA that is extracted. New extraction methodologies are being developed in an attempt to overcome some of the problems of RNA extraction from archival material.
A gene is a portion of DNA that contains the amino acid sequence code to create a protein. Protein synthesis begins with the activation of the appropriate gene. A copy of the gene is made by the process of transcription, which is the synthesis of complementary single-stranded messenger RNA (mRNA) and takes place in the nucleus. Before export to the cytoplasm, mRNA undergoes posttranscriptional modification. Pre-mRNA contains both amino acid coding sequences called exons and intervening noncoding sequences called introns. The introns are excised from mRNA by a process called splicing. mRNA is subsequently transported from the nucleus to the cytoplasm, where its linear sequence of nucleotide triplets or codons is translated on the ribosomes to form a linear sequence of amino acids. The final product of a gene is the assembly of the constituent amino acids into functional proteins.
Control of the process of gene expression ultimately influences the activity or amount of protein product. Gene regulation takes place at both transcriptional and translational levels. Before transcription can be initiated, RNA polymerases (the enzymes responsible for linking together the mRNA polymer) attach initially onto DNA at a sequence termed the promoter. Certain genes, called transcription factors, together with coactivators and corepressors, can influence mRNA production of other genes because they enable RNA polymerase to recognize the promoter. Transcription factors are regulated either by other upstream proteins or by other modifications of the protein, such as phosphorylation. RNA transcription is capable of significant amplification of the signal it encodes.
Most of the human genome is composed of noncoding DNA, the function of which has not been fully elucidated. Most of the variability among individuals occurs in noncoding DNA; this variability is termed genetic polymorphism. Repetitive noncoding sequences are present in multiple copies throughout the genome in tandem arrays called satellites or microsatellites, according to their length. In addition, single nucleotide changes can be polymorphic in the population. Single nucleotide polymorphisms (SNPs) are the most abundant form of DNA polymorphism in the human genome, as shown by Sachidanandam and colleagues (2001). SNP microarrays represent a new technology that has numerous potential applications, including scanning of loss of heterozygosity (LOH) of loci for tumor suppressor genes, evaluation of clonality, and population-based genetic analyses. Alterations of microsatellite sequences, a phenomenon known as microsatellite instability (MI), is often due to malfunction of DNA repair enzymes. MI occurs in several tumor types, including in some lung cancers, and is particularly important in a form of hereditary colon cancer. Restriction fragment length polymorphisms (RFLPs), microsatellites, and SNPs are well suited for genetic linkage studies in humans. Assessment of the statistical significance of linkage is made by calculating the ratio of the likelihood of linkage to the likelihood of random assortment.
Cystic fibrosis (CF) is an autosomal recessive single gene disorder with an incidence of about 1 in 2,000 white births that results from mutations in the CF transmembrane conductance regulator (CFTR) gene, recently reviewed by Ratjen and Doring (2003). Cystic fibrosis is characterized by abnormal mucus production, significant pulmonary morbidity in terms of recurrent pulmonary infections, and pancreatic insufficiency. Northern Europeans have a high carrier frequency of 1 in 25 to 1 in 30; the rate is lower in other ethnic and cultural groups. More than 900 different mutations in the CFTR gene have been reported. Whereas inheritance of the diseased gene can be traced by polymorphic markers, molecular testing, usually by polymerase chain reaction (PCR), and sequencing of the disease-associated mutations can be performed. A single mutation (delta F508) may account for 85% of CF in the northern European and North American white populations, simplifying screening techniques. Carrier testing can be performed by investigating a panel of the more common mutations.
Hybridization Assays
The ability to manipulate nucleic acids in vitro is based on molecular hybridization, which is the complementary pairing of bases between the target nucleic acid and a DNA or RNA probe. The process of hybridization can be accomplished on solid support, such as nitrocellulose or nylon membranes as in Southern blotting, in solution as in the PCR, or at the cellular or subcellular levels, as for in situ hybridization (ISH).
Probes may be labeled either with a radioisotope or, increasingly more commonly, with a variety of nonisotopic reporter molecules that can subsequently be revealed by means of a color reaction. Several different types of probes
P.263
can be used, including short, single-stranded, synthetic DNA oligonucleotides; single-stranded antisense and complementary RNA probes of intermediate size; and long, double-stranded DNA probes. DNA oligonucleotide probes are primarily used in PCR reactions and for viral detection by ISH. Some of the disadvantages inherent in using these probes include the narrow window between specific signal and loss of it and the limited number of reporter molecules that can be tagged onto these oligonucleotides. RNA probes, also known as riboprobes, are synthesized in vitro using RNA polymerase from either cloned DNA templates or PCR products. They are particularly suited for use in ISH and are sometimes used in Northern hybridization. DNA probes are especially suitable for use in solid support hybridization assays and in fluorescent in situ hybridization (FISH). To perform Southern, Northern, and solution-phase PCR assays, nucleic acid extraction is required, which necessitates the destruction of tissue architecture. In contrast, the architecture is preserved in ISH, which involves the localization of labeled RNA or DNA molecules that hybridize with complementary target DNA or RNA sequences in specific cells on tissue sections or cytologic preparations. A limitation of ISH is its failure to detect oncogenes that are activated by means other than dysregulated transcription, such as point mutations or post-transcriptional modifications.
ISH can be used to identify cells that contain RNA for a protein of interest. ISH is therefore useful in distinguishing nonspecific uptake of hormones by cells from genuine hormone synthesis that may be of diagnostic value. Small cell lung carcinoma (SCLC) frequently produces neuroendocrine markers, and Hamid and co-workers (1989) used ISH to study the expression of the human bombesin (gastrin-releasing peptide) gene at the cellular level in SCLC. ISH has also been used to detect infectious microorganisms. Specific viral nucleic acids can be detected within tissues and tumors [e.g., Epstein-Barr virus (EBV)] because such DNA or RNA is present in multiple copies within an infected cell. Fain and associates (1991) demonstrated that Legionella species infection can be rapidly diagnosed by ISH detection of bacterial DNA. Hanazawa and colleagues (2000) showed that Aspergillus fumigatus could be detected in tissue samples by ISH. ISH has also been used to detect overexpression of cellular DNA or RNA, as in oncogene amplification or unregulated protein transcription. Noguchi and colleagues (1990) showed heterogeneous amplification of the oncogene myc in SCLC using this technique.
FISH is a molecular cytogenetic technique that permits the identification of translocations, deletions, structural rearrangements, LOH, and gene amplification assessment in interphase nuclei using fluorochrome dyes. FISH is especially effective when performed on touch imprints but can also be performed on either frozen sections or formalin-fixed and paraffin-embedded tissue. Computerized, bar code controlled instruments are available that perform fully automated ISH, including FISH, immunohistochemistry, and a dual-staining combination of ISH and immunohistochemistry, and are becoming more commonly used in clinical practice. Capodieci and colleagues (1998) reported that with the use of this instrument, conditions could be tightly controlled, resulting in increased reproducibility, reduced hybridization time, and diminished labor.
In filter hybridization, probes target DNA or RNA sequences that have been previously bound to a solid substrate. Southern blot hybridization is a technique whereby DNA is cut at specific sites by enzymes called restriction endonucleases before gel electrophoresis and then transferred onto nitrocellulose or nylon filters. This permits determination of the size of the restriction fragment to which the probe hybridizes. This technique requires intact genomic DNA obtained from either fresh-frozen or snap-frozen tissue. Northern hybridization or blotting is a related technique that is used to detect RNA.
PCR involves the exponential in vitro amplification of a segment of DNA. It also involves a repetitive cycle of heat denaturation of DNA, annealing of sequence-specific oligonucleotide primers flanking the selected DNA fragment to be amplified, and synthesis of the target sequence by heat-stable DNA polymerases. The reaction results in the preferential amplification of the target sequence on the background of extraneous DNA, producing virtually unlimited amounts of the target. The scant availability of starting material [e.g., from bronchial brushings or bronchioloalveolar lavage (BAL) fluid] is thus less of an obstacle when this technique is used for diagnostic purposes. Short segments of DNA extracted from formalin-fixed, paraffin-embedded tissues can also be amplified using PCR. Reverse-transcriptase PCR (RT-PCR) is a technique that can quantify gene expression by using PCR amplification of target RNA that has been previously converted to complementary DNA (cDNA) by reverse transcriptase. RT-PCR offers several advantages over Northern blotting because only minute tissue samples are required (e.g., from laser microdissection of tissue) and the technique can be quantitative. Several modifications of the basic PCR technique can be useful for specific indications. Examples include competitive PCR for gene dosage assays, nested PCR to increase the sensitivity of the reaction when targeting low-copy-number genes, multiplex PCR in which multiple primer sets are used in the same reaction to amplify several targets simultaneously, and more recent technologies that allow accurate automated gene quantification studies. Quantitative PCR and RT-PCR have a variety of diagnostic applications that range from the assessment of oncogene overexpression to viral load in pneumonitis. DeMuth and colleagues (1998) used quantitative RT-PCR to describe a gene expression index combining the expression levels of C-myc, E2F-1, and p21 to discriminate all cultured and primary normal lung specimens from malignant lung carcinoma cell lines and tumor specimens. Kotsimbos and associates (1997) described an efficient, highly sensitive, quantitative PCR assay for human cytomegalovirus load detection in transbronchial biopsies, BAL specimens, and
P.264
peripheral blood leukocytes in lung transplant recipients. A rapid, quantitative PCR assay was developed by Larsen and associates (2002) for the identification and quantification of a subspecies of Pneumocystis carinii that is commonly used in research as a model of P. carinii pneumonia. Rose and colleagues (2002) used quantitative TaqMan RT-PCR assays to quantify vector-derived mRNA after delivery of naked DNA and DNA liposome formulations expressing human and murine CFTR to mouse airways.
Microarray Technologies
Oligonucleotide and cDNA Microarrays
New genomic and proteomic technologies are emerging that will facilitate a global overview of the complex changes implicit in disease states in terms of gene expression and protein complement, facilitating the dawning of an era of molecular classification of disease. Pease and colleagues (1994) demonstrated a novel technology that required merging molecular biology and computer technology involving oligonucleotide arrays or DNA chips that carried cDNA microarrays. To create a microarray, thousands of synthetic oligonucleotide or cDNA probes are constructed to portions of the target sequence on a support grid. The bound sequences are then hybridized to labeled nucleic acids extracted from the specimen of interest. After hybridization, a reading device scans the entire chip. DNA chips enable the expression level of hundreds of genes to be determined in a single experiment. There have been tremendous developments in gene-chip technology and in the supporting bioinformatics and computer software needed to analyze the enormous amounts of data that are yielded from these chips. Much can be learned about cell function by global assessment of gene expression, and novel biomarkers of disease can be discovered using these technologies.
In cancer research, microarray technology can identify differences in the transcriptional profile between tumor and normal tissue or between tumors. Microarray applications have been concerned mainly with gene expression profiling, but they can also be used for mutation screening and for polymorphism genotyping. Gingeras and associates (1998) used this technology to develop an assay to detect mutations in the rpoB gene that result in rifampicin resistance in Mycobacterium tuberculosis strains. Wen and colleagues (2000) demonstrated how microarray sequencing was more sensitive, more accurate, and faster than classical sequencing approaches in the evaluation of TP53 mutations. Microarrays were also used by Cline and associates (2002) to assess the synergistic activity of antiangiogenic agents by identifying divergent patterns of gene expression in treated endothelial cells. A complementary technique, serial analysis of gene expression (SAGE), provides quantitative information on cellular gene transcription.
Comparative Genomic Hybridization
Comparative genomic hybridization (CGH) is a molecular cytogenetic approach described by Kallioniemi and associates (1992) that results in an analysis of gene dosage on a genome-wide scale by comparing hybridization of test and reference genomic DNA. A cytogenetic pattern of genetic losses and gains is analyzed, and a quantitative change is obtained (compared to the normal reference sample). To increase the mapping resolution, array CGH has been developed, in which cloned DNA targets are arrayed as described by Pinkel and colleagues (1998). The DNA targets can be bacterial artificial chromosomes (BACs), yeast artificial chromosomes (YACs), cosmid DNA, or cDNA. With array CGH, mapping resolution reaches the kilobase level and can be used to measure gene copy number with high precision. Array CGH can be performed on DNA obtained from fresh or frozen tissue, but it is difficult to extract suitable DNA from paraffin-embedded tissue.
Single Nucleotide Polymorphism Microarrays
SNPs represent the most abundant form of DNA polymorphism in the human genome. SNP microarrays have several potential applications such as LOH analysis, disease susceptibility studies, and pharmacogenetic and toxicogenetic studies. Lindblad-Toh and associates (2000) used SNP microarrays for scanning LOHs in small cell lung cancer. Use of SNP microarrays could potentially pinpoint patients with different susceptibilities to particular treatments and identify different sets of SNPs that are associated with a given phenotype or molecular profile by enabling rapid and accurate detection of genetic polymorphism. This ability to identify patients who are resistant to a drug or susceptible to specific drug side effects will facilitate drug research and also disease management.
Tissue Microarrays
Kononen and colleagues (1998) developed an array-based, high-throughput technique called tissue microarray (TMA) that facilitates the analysis of gene expression at the cellular level synchronously in a large number of tumors. Up to 1,000 cylindrical tissue biopsies obtained from formalin-fixed paraffin embedded individual tumors can be distributed in a single TMA. Ideally, several samples (generally three) from each tumor are assessed simultaneously for optimum results as noted in the publication by Hoos and Cordon-Cardo (2001). Sections of the paraffin block containing the microarray enable parallel in situ detection of DNA, RNA, and protein targets in each specimen on the array, and consecutive sections allow the rapid analysis of hundreds of molecular markers in the same set of specimens. Analysis of large numbers of clinical specimens is still limited by the time-consuming interpretation of hundreds of specimens on each slide. Instruments allowing automated acquisition and analysis of
P.265
specimens are under development. Modified TMAs, called cryoarrays, as reviewed by Fejzo and colleagues (2001), have also been created for frozen tissue samples, allowing preservation of nucleic acids and proteins. Bremnes and associates (2002) created TMAs for 193 cases of non small cell lung carcinoma (NSCLC) and performed immunohistochemical staining for cadherins and catenins. They showed that reduced expression of E-cadherin and catenins was associated with tumor cell dedifferentiation, local invasion, regional metastasis, and decreased survival.
Proteomics
The proteome refers to the protein complement of cells, tissue, serum, or other material and reflects the effects of posttranscriptional regulation, and proteomics refers to the characterization of proteins, protein ligand interaction screening, and differential protein expression profiling. Proteome array-based methodologies are under active investigation in many fields, including pulmonary disease. Proteomic spectra are generated from the sample of interest by means of matrix-assisted laser desorption and ionization time-of-flight mass spectroscopy, and the spectra are subsequently analyzed by means of complex algorithms. Proteomics is a very powerful tool to search for markers of disease, particularly for early cancer detection. Petricoin and colleagues (2002) showed that serum protein profiles could differentiate patients with ovarian carcinoma from those without ovarian carcinoma with high sensitivity and specificity. Using similar methods, it may also be possible to identify distinct protein and peptide patterns in BAL fluid or serum between individuals with lung cancer and healthy individuals in the form of a high throughput and rapid test that was noted by Hanash (2003) and by Noel-Georis and colleagues (2001). Databases of protein expression in lung cancer are being established. In addition to proteomics being used as a screening test to detect cancer, it can also be used to evaluate the effects of treatment in pulmonary disease. Griese and associates (2001) demonstrated that there was reduced proteolysis of surfactant protein A and changes in the proteome of BAL fluid following inhalation of 1-protease inhibitor in patients with CF. Katsuma and colleagues (2001) used cDNA microarrays to evaluate gene expression changes in a murine model of bleomycin-induced pulmonary fibrosis and showed that this technique could be used to evaluate progression. There is increasing evidence that susceptibility to acute lung injury may also depend on genetic factors, as reported by Leikauf and colleagues (2002).
Laser Capture Microdissection
Most, if not all, lung tumors are heterogeneous in terms of their tissue composition. Laser capture microdissection (LCM) is a technique that allows pure populations of cells to be isolated rapidly from a tissue section for further study. Analysis of the isolated cells can be at the DNA, RNA, or protein level. Thus, LCM is a powerful technique that enables comparison of two distinct cell populations from a given section of tissue. LCM uses a focused laser beam to isolate and capture cells from glass slides for analysis. The time required to collect the cells is minimal, and the isolation of specific cellular subsets from heterogeneous tissue is more accurate than manual dissection. LCM can be used to obtain cells from both fixed and frozen material. Identification of pure populations of cells can be difficult in frozen sections because it can be difficult to distinguish reliably the different cells. A refinement of the technique was reported by Lindeman and colleagues (2002), who described combining LCM with a rapid immunostaining technique in frozen sections of tissue as a means of guiding the isolation of pure samples of cellular subsets to facilitate extraction of RNA. They isolated pure populations of basal cells and secretory cells in sections of prostate tissue for p27 mRNA quantitation by real-time RT-PCR. The authors initiated RNA extraction within 40 minutes. This technique of rapid immuno-LCM has wide applicability in the evaluation of gene expression in distinct cell populations in different tissues, including the subsequent use of microarray platforms to assess transcriptional profiles in pure populations of cells.
Activation-specific Antibodies
There have been significant advances in the development of antibodies that recognize activated forms of proteins for use in both Western blots and in immunohistochemistry. Activated forms of oncogene products are key elements in important cell signaling pathways. Phosphospecific antibodies can detect phosphorylated form of proteins (e.g., phospho HER-2/neu) and thus detect activation of the oncogenic protein. Antibodies that can detect proteins that are activated by acetylation are also under active development. The ability to detect activated forms of proteins has important implications for the evaluation of prognostic markers and the monitoring and development of targeted treatments.
RNA Interference (Small Interfering RNAs)
RNA interference, an evolutionary conserved cellular process of gene silencing first identified in plants, is a process whereby double-stranded RNA (dsRNA) binds to and results in the degradation of mRNA, based on complementary base pairing. The siRNAs are short dsRNAs (21 to 23 nucleotides in length) that function to control important signaling pathways in mammalian cells by blocking the transcription of proteins from coding RNA, as reported by Elbashir and colleagues (2001). The precise mechanism of action of gene silencing by siRNA has not yet been fully
P.266
elucidated, but there is no question that it is a very important mechanism used in the regulation of gene expression. RNA interference technology is being exploited for functional genomic analysis and for the development of highly specific targeted treatments; for example, siRNAs can suppress transcription of key oncogenes and can be used in the development of antiviral therapies, as reported by Kitabwalla and Ruprecht (2002). The technology is also being used for the creation of animal models of disease, for example, by blocking the transcription of key tumor suppressor genes. In contrast to antisense RNA, siRNAs do not appear to incite the same nonspecific immune responses when injected into the host and may thus hold promise as therapeutic tools. The technique of RNA interference has been used by Peng and colleagues (2002) to silence the expression of a protein kinase, thereby sensitizing human cells for radiation-induced damage. Synthetic siRNA can suppress the expression of mdm2 in mammalian cells, thus restoring wild-type p53 function, as demonstrated by Martinez and co-workers (2002). Both of these applications may have an effect on human lung tumors.
DISEASE ENTITIES
Cancer
Lung cancer is the leading cause of cancer-related death in the United States, as reported by Greenlee and co-workers (2001). Only 15% of cases of lung cancer are diagnosed when the tumor is organ confined, and a disappointing 50% 5-year survival is expected for this cohort. Extensive efforts are needed in the development of new screening and diagnostic tests, particularly for populations at risk, in addition to novel therapeutic approaches. Lung cancer is divided into two groups for therapeutic purposes, composed of the four main histologic types. NSCLC makes up about 75% of all lung tumors and includes squamous cell carcinoma, adenocarcinoma, and large cell carcinoma. The fourth type is SCLC, which constitutes the remaining 25%. The role of molecular pathology in cancer diagnosis includes the following: detection of mutations in oncogenes and loss or mutations in tumor suppressor genes (TSGs), both somatic and germline; determination of overexpression of oncogenes; assessment of cytogenetic abnormalities and of clonality; determination of susceptibility to cancer by means of linkage analysis; molecular profiling of lung cancer with identification of novel prognostic and therapeutic markers; and development of sensitive and specific screening tests.
Oncogenes and Tumor Suppressor Genes
Protooncogenes are highly conserved cellular genes that are important in cellular pathways involved in proliferation, apoptosis, and differentiation. Generally, only one of the two alleles in a cell needs to be mutated to confer dominant oncogenic properties by altering the normal structure, expression pattern of the protooncogene, or both. Thus, oncogenes are genes that, when mutated in a critical area, promote carcinogenesis in a dominant manner. The activation of an oncogene may be associated with increased expression of its protein products. Overexpression of an oncogene can occur for a number of reasons: genomic amplification; juxtaposition to strong viral or tissue-specific promoters controlling its transcription (e.g., after a translocation event); or constitutive deregulated expression that follows a mutational event. Rearrangements of chromosomes, resulting in truncations, insertions, or deletions, may also result in oncogene activation or inactivation of TSGs. Most oncogenes that have been identified are part of the cell's growth regulatory pathways, or are homologues thereof, and include growth factors, growth factor receptors, intracellular signal transducers, cell cycle proteins, and nuclear transcription factors.
Many common sites of LOH exist in lung cancer, both SCLC and NSCLC, suggesting the presence of alterations in TSGs at these sites of chromosome loss, as discussed by Balsara and Testa (2002). Several well-described TSGs are known to be altered in lung cancer and almost certainly play a role in their evolution. TSGs code for proteins whose loss of function results in transformation, in contrast to the activating mutations that generate oncogenic alleles from protooncogenes. Both alleles of a TSG must be either lost, mutated, or inactivated by epigenetic means to result in inactivation of the gene, although haploinsufficiency of certain TSGs, such as PTEN and p27, can result in greater susceptibility to cancer in the experimental setting. When one allele is inactivated in the germline, such a genotype is transmitted to all somatic cells, rendering them more susceptible to a second mutational event that can affect the remaining functional allele. This results in a greater predisposition to cancer in families carrying such germline mutations.
Inherited Predisposition to Lung Cancer
Less is probably known regarding the contribution of hereditary factors to lung cancer development than for any of the other common solid tumors. No distinct familial forms of the common types of lung cancers have been described as have been defined for colon and breast cancer, and specific genetic loci responsible for a predisposition to lung cancer development have therefore not been elucidated. Lung cancer has been documented in some genetic syndromes that have an increased predisposition to other cancers. Li and associates (1988) showed that lung cancer occurs with increased frequency in the Li-Fraumeni syndrome that results from a germline mutation in the p53 TSG on chromosome 17. Donehower and colleagues (1992) showed that p53 knock-out mice are predisposed to develop lung adenocarcinoma. Lung cancers have also been described in carriers of inactivating mutations in the retinoblastoma TSG on chromosome 13.
P.267
Mattson and associates (1987) showed that only 10% of smokers develop lung cancer. Evidence from Sellers and colleagues (1990) and others show an important role for genetic predisposition in determining risk for lung cancer development among smokers. A growing body of data exists linking genetic differences in the capacity to metabolize tobacco carcinogens with the risk for developing lung cancer. Certain enzymes influence lung cancer risk by catalyzing detoxification reactions that enhance the elimination of the toxic products, and genetic polymorphisms of these enzymes appear to relate to lung cancer susceptibility, as discussed by Benhamou (2002), W. J. Lee (2002), and To-Figueras (2001) and their colleagues. The existing PCR assays for detecting the genetic status of each important metabolizing enzyme require refinement. A genetic profile of each of the factors might define a significant indicator of risk status in a person who smokes.
Genetic Alterations in Sporadic Lung Cancer
K-ras
Bos (1989), as well as Clements (1995) and Salgia and Skarin (1998) and their co-workers, showed that K-ras mutations occur in 30% to 50% of lung adenocarcinomas using sensitive PCR-based methods for detection. Although K-ras mutations are more common in adenocarcinomas than in squamous cell carcinomas, they are very rare in the bronchioloalveolar subtype of lung adenocarcinoma. K-ras mutations have not been described in SCLC. Ras proteins have been implicated in the transduction of growth and differentiation signals from activated transmembrane receptors to downstream protein kinases. When ras is mutated, the major defect is a marked decrease in its ability to interact with the guanosine triphosphatase (GTPase)-activating protein, RAS-GAP. Hence, the Ras protein remains in the GTP-bound or active state, and regulation of cell growth is disturbed. Lacal and associates (1986) showed that K-ras mutation could transform cells, thus establishing ras as an oncogene. Mutations in K-ras predominantly occur in codons 12 (85% of all mutations), 13, and 61 and result in constitutive activation of the extracellular signal [regulated kinase (Erk) signaling pathway in the absence of external stimuli]. Westra and colleagues (1996) demonstrated K-ras mutations in early-stage lung tumors, and it is believed that these mutations are involved in tumor initiation and early progression. Interestingly, Zhang and colleagues (2001) demonstrated that wild-type K-ras inhibits mouse lung carcinogenesis and tumorigenic properties of lung tumor cell lines, providing evidence for a tumor suppressor function for K-ras in its nonactivated form. Mutated K-ras gene has been shown to confer a worse prognosis to lung adenocarcinomas that were surgically treated with curative intent. However, the prognostic significance of specific K-ras mutations requires additional study. K-ras mutations have been detected in atypical adenomatous hyperplasia, according to work by Sagawa and colleagues (1998) and others. Mao and associates (1994) looked for mutations of K-ras in sputum from adult smokers. Six of the eight patients in whom K-ras mutations were present in their lung tumors had detectable K-ras mutations in sputum 1 to 4 months before clinical detection of the tumor, and no mutations were found in sputum of patients with no mutations in their subsequent lung tumors. Yakubovskaya (1995) and Mills (1995) and their colleagues identified K-ras mutations in sputum or BAL fluid in about 38% of patients with NSCLC but also found mutations in occasional patients without cancer. Sozzi and colleagues (1995) showed that K-ras mutations could be used to distinguish recurrent lung tumors from second primary tumors, the former having identical K-ras mutations. Thus, K-ras mutation detection may be useful in the early detection of lung adenocarcinoma, perhaps as part of a panel of informative biomarkers. New therapeutic approaches include inhibiting K-ras by means of small molecules that block posttranslational modifications required for K-ras activation and have been reviewed by Giaccone (2002).
C-myc
C-myc has oncogenetic properties at several levels, including the promotion of cell cycle progression. The CDK inhibitor p27KIP1 is downregulated by myc. C-myc induces cyclin D1/D2 expression and accumulation of cyclin D-CDK4 complex, which sequesters p21CIP1 and p27KIP1 and leads to the activation of cyclin E-CDK2 and progression of the cell cycle. Osada and Takahashi (2002) recorded that amplification of one of the members of the Myc gene family (C-myc, N-myc, or L-myc) is detectable in 25% to 30% of SCLC cell lines and in 5% to 15% of primary tumor specimens. Amplification of C-myc occurs most commonly and appears to be a negative prognostic factor in SCLC, possibly owing to modification of tumor response to certain treatment modalities. Myc gene amplification occurs in about 30% of cell lines of NSCLC and in 8% of tumor specimens, but overexpression of the protein occurs more frequently (about 70% of cell lines and 50% of tumor specimens).
Growth Factors and Their Receptors
c-erb-b2/HER-2/neu
The erb-b family of receptor-type tyrosine kinases includes four distinct members: HER-1 (epidermal growth factor receptor (EGFR)/c-erb-b1), HER-2 (neu/c-erb-b2), HER-3 (c-erb-b3), and HER-4 (c-erb-b4). Homology is high among the four members in the tyrosine kinase domain, but larger differences exist in the extracellular and intracellular C-terminal domain, the latter responsible for the diversified stimulation of downstream signal transduction pathways. HER-2 lacks a specific ligand but forms a heterodimer with other members of erb-b family and enhances
P.268
their signaling. Several growth factors and ligands have been identified. Epidermal growth factor (EGF), transforming growth factor- (TGF- ), and amphiregulin are specific for EGFR; neuregulins and heregulins bind to HER-3 or HER-4 and insulin to HER-3. Bunn and Franklin (2002) discussed the interaction of these ligands and receptors. They stimulate several signaling pathways, resulting in mediation of growth-stimulating signals (RAS-RAF-MAPK, PI3K-AKT, and PLC-PKC). In lung cancer, EGFR is overexpressed without gene amplification in 70% of squamous cell carcinomas and in 40% of adenocarcinomas. According to a metaanalysis reported by Meert and associates (2002), expression of EGFR in NSCLC is a negative prognostic indicator, although the impact is small. Yi and colleagues (1997) showed that HER-3 overexpression is detectable in 20% of NSCLC, showing an association with poor prognosis. Several EGFR inhibitors are being developed as therapeutic agents, both small molecules that inhibit the tyrosine kinase activity and monoclonal antibodies directed at the extracellular domain of the protein.
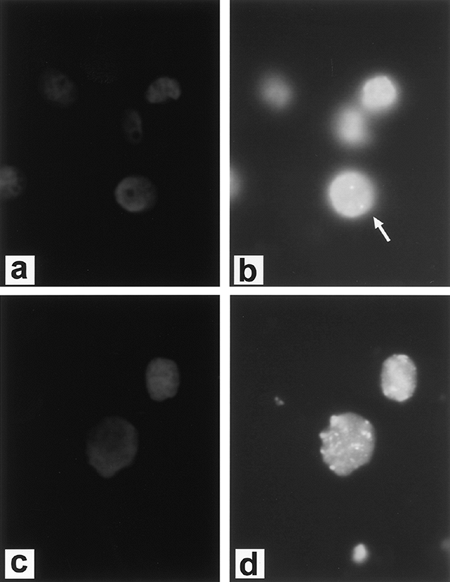 |
Fig. 15-1. Fluorescent in situ hybridization (FISH) demonstrating diploid complement of c-erb-b2/HER-2/neu in normal cells. A. DAPI (4',6-diamidino-2-phenylidole*2HCl) staining of cells. B. FISH (arrow designates diploid signal) and cancer cell line with amplification of c-erb-b2/HER-2/neu. C. DAPI staining of cells. D. FISH. (See Color Fig. 15-1.) Courtesy of Dr. Paula Capodieci. |
Unlike the other members of the HER family, HER-2 is not strictly a receptor tyrosine kinase because no high-affinity endogenous ligand has been identified. HER-2 acts as a signaling network coordinator and amplifier when it heterodimerizes with other HER family members. Overexpression of HER-2 has been found in many different human tumors, including NSCLC, and appears to be important in human carcinogenesis. Overexpression of HER-2 is present in up to 30% of lung cancers, being slightly more common in adenocarcinomas than squamous cell carcinomas, although gene amplification is rare (about 3%), as noted by Osada and Takahashi (2002). Persons (1993) and Capodieci (1998) and their colleagues reported that FISH could be used to detect HER-2 and C-myc amplification in formalin-fixed, paraffin-embedded tissue. Figure 15-1 (see Color Fig. 15-1) shows gene amplification of HER-2 by FISH in a
P.269
tumor cell line, compared with normal gene complement in the control normal cells. Giaccone (2002) reported that HER-2 is frequently overexpressed in NSCLC and appears to be associated with drug resistance, increased metastatic potential, increased production of vascular endothelial growth factor (VEGF), and poor prognosis. Figure 15-2 (see Color Fig. 15-2) shows immunohistochemical staining of HER-2 in a lymph node metastasis of NSCLC. Zhou and colleagues (2001) showed that HER-2 mediated resistance to DNA-damaging agents requires the activation of Akt, which phosphorylates murine double minute 2 (MDM2) and therefore enhances MDM2-mediated ubiquitination and degradation of p53. Blocking the Akt pathway mediated by HER-2 increases the cytotoxic effect of DNA-damaging drugs in tumour cells with wild-type p53.
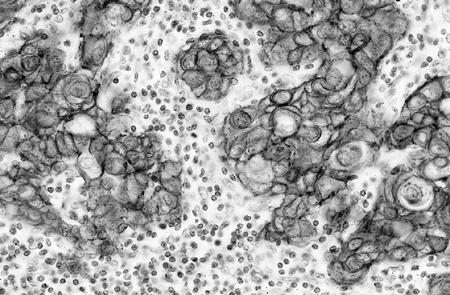 |
Fig. 15-2. Metastatic squamous cell carcinoma to a lymph node with positive staining of HER-2 by immunohistochemistry. (See Color Fig. 15-2.) |
Trastuzumab (Herceptin) is a chimerized monoclonal antibody against HER-2. Preliminary results from ongoing phase II clinical trials of trastuzumab as a single agent or in combination with chemotherapy in patients with stage IIIB or IV NSCLC were reviewed by Azzoli (2002) and Zinner (2002) and their colleagues. Combinations of trastuzumab and chemotherapy are well tolerated, with response rates of 21% to 40%. One trial showed that patients whose tumors highly overexpressed HER-2 (3+) by immunohistochemistry or evidence of amplification by FISH showed a good response. It appears that highly overexpressing HER-2 cases of NSCLC (3+ by immunohistochemistry), although relatively infrequent (3% to 9%), may show benefit with treatment with trastuzumab, and further clinical studies in such patients are warranted.
Mitogen-activated Protein Kinase Phosphatases
Because many protooncogenes are tyrosine kinases, the function of such proteins can be modulated by reversible phosphorylation by phosphatases. Several oncogene products, including HER-2 and EGFR, transmit signals through the protooncogene ras, leading to the activation by phosphorylation of mitogen-activated protein (MAP) kinases. MAP kinase activation is thus a central step in tumor growth stimulation by the most widely expressed and activated human oncogenes. The family of MAP kinase phosphatases (MKPs) functions to inactivate MAP kinase by dephosphorylation. The human prototype of MKP, MKP-1, is the product of immediate early genes induced by the same genes that, in fact, regulate MAP kinase activity. One of us (M.L.) and associates (1996) have shown that MKP-1 is overexpressed early in the carcinogenic pathway in lung cancer and also in indolent lung tumors, such as bronchioloalveolar carcinomas, and that progressive loss of expression occurs with increasing grade and stage. Figure 15-3 shows coexpression of EGFR by immunohistochemistry and expression of MKP-1 by ISH in a bronchioloalveolar carcinoma. Because MKP-1 has been shown to inhibit
P.270
apoptosis by inhibition of the c-Jun N-terminal kinase (JNK) pathway, its overexpression may result in inhibition of programmed cell death with resultant advantages to tumor cell growth.
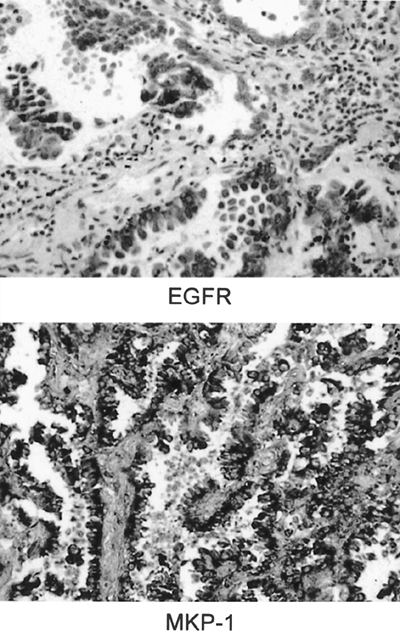 |
Fig. 15-3. A. Bronchioalveolar carcinoma stained by immunostain to epidermal growth factor receptor (EGFR). B. In situ hybridization with antisense probe for mitogen-activated protein kinase phosphatase-1 (MKP-1) demonstrating positive signal in the tumor cells. |
TP53
The most well-defined TSG that is altered in lung cancer is p53 gene mutation. Loss of p53 gene function in lung cancers appears to be the major correlate to the very frequent LOH of chromosome 17p13.1 in all subtypes of lung carcinoma. Mutation of p53 represents the single most common genetic alteration observed in human cancer, often occurring late in carcinogenesis. The p53 protein is normally expressed at low levels, and because it is unstable (rapidly degraded), it is therefore not normally detectable by immunohistochemistry. When the gene is mutated (most commonly by a missense mutation), the p53 protein has a longer half-life, accumulating to levels detectable by immunohistochemistry. However, although most of the tumors with missense mutations of p53 stain positively by immunohistochemistry, tumors with nonsense mutations, deletions or insertions in the coding regions, and splicing abnormalities are generally negative by immunohistochemical analysis. Normal growth control function may also be inactivated as a result of binding of p53 by proteins of DNA viruses (e.g., human papillomaviruses) and subsequent degradation by the ubiquitin proteosome pathway, as noted by Rossi and one of us (M.L.) (2002). Mutations of p53 occur in about 50% of NSCLC cases and in 70% to 100% of SCLC cases. Most of the mutations (80%) occur in the central, evolutionarily conserved region of exons 5 to 8. Nearly 500 unique mutations of the gene have been found, and the diversity is greater in lung cancer than in other cancers, possibly reflecting exposure to a wide array of carcinogens in cigarette smoke. Denissenko and colleagues (1996) found that benzo[a]pyrenediolepoxide, a tobacco carcinogen, directly binds to the hot spots for the p53 mutations found in lung carcinomas. Genetic alterations of p53 occur relatively early during carcinogenesis but follow genetic loss (deletions and loss of heterozygosity) on chromosome 3p. The occurrence of p53 and K-ras alterations has been used to identify tumor recurrence in pleural fluids, as shown by Dai and colleagues (2000). It appears that the weight of evidence currently suggests that p53 alteration is associated with a worse prognosis in NSCLC, according to a review by Campling and el-Deiry (2003). Husgafvel-Pursiainen and associates (1997) used an enzyme-linked immunosorbent assay for estimating serum levels of mutant p53 protein in a cohort of individuals with workplace exposure to asbestos or silica and subsequent lung cancers or mesotheliomas and compared serum p53 levels with p53 mutations and tissue accumulations of the mutant protein. They suggested that serum p53 levels are reasonably accurate in reflecting tissue alterations in p53 at either the gene level, protein level, or both and can thus serve as early biomarkers of disease risk. Roth (1996) and Zou (1998) and their associates reported the use of gene therapy to restore p53 wild-type function in lung cancers; the former group used a retrovirus that expressed wild-type p53 that was injected intratumorally, and the second group used liposome complexes. Additional work is needed before it is known whether this translates into survival benefit for patients. DNA oligonucleotide microarrays have been used by Kannan and colleagues (2001) to identify target genes that are regulated by p53 in a lung cancer cell line H1299 that had been modified to show temperature-sensitive expression of p53. Primary targets include cell cycle genes (p21, TGF-b, Cyclin E), genes involved in apoptosis (Fas, Bak), cell adhesion (Thymosin), and signaling (H-Ras), as well as transcription, neuronal growth, and DNA repair. These target genes may be exploited as potential therapeutic targets.
Retinoblastoma Gene
The retinoblastoma gene (RB) is located on chromosome region 13q14 and plays a critical role in cell cycle control. Kaye (2002) noted that the RB TSG is altered in nearly all SCLC tumors and in 30% to 40% of NSCLC. Studies have failed to show an association between Rb status and prognosis in NSCLC, but replacement of function of the gene may be a potential therapy to be explored for SCLC. Recently, DeSalle and associates (2001) reported that the de-ubiquitinating enzyme, ubiquitous nuclear protein (Unp), reported to be an oncogene in lung cancer, associates with Rb. These findings are provocative because Unp, although not directly affecting Rb, may target some other Rb-associated proteins important for cell cycle regulation.
Putative Tumor Suppressor Genes on Chromosome 3p
LOH of chromosome 3p occurs in 90% to 100% of SCLC cases and in 50% to 80% of NSCLC cases. Loss of material from chromosome 3p has been shown in most hyperplastic and dysplastic lesions of bronchial epithelium in smokers with and without lung cancer, indicating that a putative TSG in this region is altered early in lung carcinogenesis. Intense efforts are being undertaken to identify TSGs in the regions 3p21.2 22, and 3p14.
The FHIT (fragile histidine triad) gene encodes a diadenosine triphosphate hydrolase and is a TSG encompassing the most frequent fragile site at 3p14.2. The balance of evidence to date suggests that it is important in lung cancer development. FHIT inhibits tumor growth when it is introduced into cancer cells with an FHIT gene alteration, by the induction of apoptosis and cell cycle arrest. In addition to structural disruption of the gene, epigenetic mechanisms have also been suggested by Osada and Takahashi (2002) and Pekarsky and colleagues (2002) to result in loss of expression of the protein. Burke and co-workers (1998) demonstrated that loss of FHIT was a negative prognostic indicator in NSCLC, independent of tumor stage, size, differentiation, and p53 mutation status. Loss of FHIT expression is present in virtually all SCLC cases and in a
P.271
proportion of NSCLC cases, more frequent in squamous cell carcinoma than adenocarcinoma. Several other candidate TSGs with rare genetic changes but frequent epigenetic changes exist in the chromosome region 3p21.3.
p16 (CDKN2A)
Cytogenetic abnormalities of 9p are frequent in NSCLC. This site is the locus of the cyclin-dependent kinase inhibitor p16 (CDKN2A). The p16 gene functions to inhibit the kinase activity of the complex formed by the association of cyclin-dependent protein kinase 4 (CDK4) and cyclin D. This occurs through a reversible non covalent-binding interaction that blocks the ability of CDK4 to phosphorylate Rb. Absence of p16 inhibition of CDK4 cyclin D kinase activity contributes to uncontrolled growth of tumor cells. The mechanisms of inactivation of p16 in lung cancer include most commonly biallelic deletions, but also intragenic mutations and reduced mRNA expression owing to methylation of CpG dinucleotides in the p16 promoter region. The p16 gene is deleted in 20% and mutated in 10% of NSCLC cases, undetectable mRNA without gene mutation is seen in 28% of NSCLC cases, and undetectable protein is seen in 51% of NSCLC cases. Abnormalities of p16 are present in a minority of SCLC cases. Loss of p16 gene function occurs frequently through homozygous deletion in cell culture lines and has also been demonstrated in microdissected primary NSCLC. Transcriptional silencing associated with abnormal DNA methylation of the transcription start site region is another mechanism of loss of p16 function. Prospective studies are needed to evaluate the prognostic role of p16 in NSCLC. Absence of expression of the p16 gene product is also commonly observed in mesothelioma tumors and cell lines.
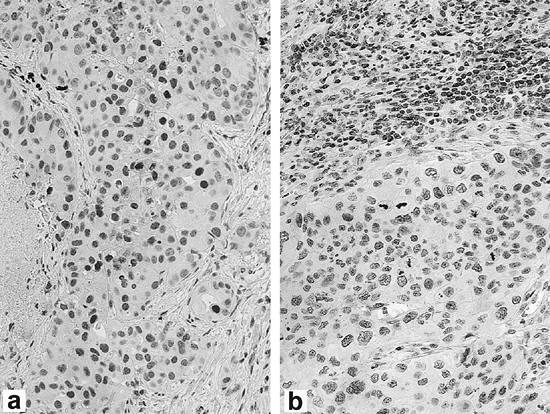 |
Fig. 15-4. Expression of p27 by immunohistochemistry in non small cell lung carcinoma. (A) High expression of p27 in tumor cells. (B) Low expression of p27 in tumor cells with lymphocytes serving as positive internal control. (See Color Fig. 15-4.) Courtesy of Dr. Michael Murphy. |
p27Kip1
Mutations of p27, another cyclin-dependent kinase inhibitor, are very rare in human tumors. Because of gene regulation occurring predominantly at the protein level, detection of loss of p27 by immunohistochemistry is the method of choice for assessing its status in lung cancers. Esposito and co-workers (1997) demonstrated that p27 loss was the result of tumor-specific degradation of p27, as we have previously demonstrated in colorectal carcinomas. Figure 15-4 (see Color Fig. 15-4) illustrates an example of an NSCLC with high p27 expression and another with low p27 expression, as assessed by immunohistochemistry. Catzavelos and colleagues (1999) found reduced levels of p27 in 86% of NSCLC cases by immunohistochemistry and showed a significant inverse correlation between p27 level and tumor grade. They found that ras mutations, found exclusively in adenocarcinomas in their study, showed no relationship to p27 levels. Esposito (1997), Yatabe (1998), Tsukamoto (2001), and Takahashi (2002) and their colleagues showed that loss of p27 expression was associated with an unfavorable prognosis in NSCLC. The ubiquitin-proteosome pathway is an important mechanism for irreversible elimination of critical cell regulatory pathways, including tumor suppressors, cell cycle proteins, transcription factors, and tyrosine kinase receptors. Proteolysis of many of these regulators is controlled by ubiquitin ligases, the substrate specificity of which is determined by different F-box proteins that act as substrate recognition factors. Substrates must be phosphorylated as specific sites to enable recognition and subsequent binding. Skp2 is the F-box protein that targets phosphorylated p27 for degradation, as shown by Carrano and colleagues (1999); therefore, novel
P.272
therapeutic strategies aimed at modifying skp2 may represent a valid therapeutic option in lung cancer.
Wilms' Tumor Susceptibility Gene 1 (WT1)
Malignant mesothelioma is one of the few extrarenal neoplasms in which the Wilms' TSG (WT1) expression is lost. Kleymenova and colleagues (1998) identified a site in intron 1 of WT1 in rat mesothelioma cell lines that is frequently methylated. Amin and associates (1995) showed that detection of WT1 mRNA or protein can provide a specific molecular or immunohistochemical marker for differentiating mesothelioma from adenocarcinoma. Hecht and colleagues (2002) showed that nuclear expression of the WT1 protein by immunohistochemistry was present in 100% of malignant mesothelioma cells in cytology preparations of pleural fluid.
Cytogenetics and Cancer
For cytogenetic analysis, fresh tissue is sent for short-term culture, from which metaphase chromosome spreads are plated on glass slides. Specific chromosomal abnormalities can be detected in these preparations. Alternatively, some cytogenetic abnormalities can be detected in interphase nuclei by FISH. This can be accomplished in paraffin-embedded tissue. Spectral karyotyping (SKY) is a 24-color multiple chromosomal painting assay that permits visualization of all human chromosomes and assessment of complex cytogenetic changes. The technique is reviewed by Bayani and Squire (2002) in relation to its application in cancer diagnosis and research. Distinct translocations in hematologic and solid tumors result either in the activation of protooncogene products or, more frequently, in the translation of novel tumor-specific fusion proteins. Because these fusion proteins are specific markers for individual subtypes of neoplasia, they are potential targets for therapy. Genetic changes in carcinomas are generally very complex, more so than those seen in many lymphoid or mesenchymal tumors.
Ewing's Sarcoma (Peripheral Neuroectodermal Tumor)
Ewing's sarcoma, or peripheral neuroectodermal tumor, comprises a family of tumors characterized by a specific and reproducible reciprocal chromosome translocation t(11;22) (q24;q12) involving the Ewing's sarcoma gene (EWS) on chromosome 22 with its most frequent translocation partner, the FLI-1 gene on chromosome 11. The resultant fusion transcript EWS-FLI-1 functions as both a transcriptional activator and repressor. Arvand and colleagues (2001) showed that both the DNA binding domain of FLI-1 and also the carboxy-terminal group are necessary for full tumorigenesis and transcriptional modulation. Askin and colleagues (1979) originally described the clinical subset of peripheral neuroectodermal lesions that arise on the chest wall, often with involvement of the ribs, pleura, and lung. All peripheral neuroectodermal tumors show immunohistochemical cytoplasmic membranous positivity for the MIC-2 protein, best demonstrated by the antibodies HBA-71 or O-13, in addition to variable evidence of neuroectodermal differentiation. Demonstration of the cloned fusion gene products of this translocation by the use of RT-PCR now allows accurate diagnosis at the molecular level. Rearrangement of the EWS gene can also be demonstrated by FISH.
Pleuropulmonary-based Synovial Sarcomas
Pleuropulmonary-based synovial sarcomas are being increasingly recognized, as reported by Essary and co-workers (2002), but these tumors can cause diagnostic confusion with other malignant tumors at this site, including lung carcinoma. Because of their distinct cytogenetic abnormality of t(X;18), confirmation of the diagnosis can be made by conventional cytogenetics, FISH, or RT-PCR to detect the fusion transcript.
Molecular Profiling of Cancer
Ross and associates (2000) analyzed gene expression profiles in various cancer cell lines, including lung cancer, and found that they clustered together according to the organ of origin. Traditional lung carcinoma classification has also been supported by gene expression profiling in work reported by Garber (2001), Bhattacherjee, (2001), and Virtanen (2002) and their colleagues. However, new data are emerging, indicating that new subclasses of lung cancer can also be revealed by gene expression studies, as was previously demonstrated in novel classification of leukemia, reported by Golub and associates (1999). Anbazhagan and colleagues (1999) analyzed gene expression in SCLC, bronchial carcinoids, and bronchial epithelial cells. SCLC and bronchial epithelial cells displayed a similar genetic profile, but bronchial carcinoids rather surprisingly clustered with astrocytic brain tumors rather than with SCLC, that is, tumors derived from the neural crest. These data suggest that SCLC may reflect more primitive cells in the same lineage as NSCLC. This explains why hybrid tumors composed of both SCLC and NSCLC exist, but tumors with both carcinoid and SCLC and NSCLC have not been described. It has been shown that normal epithelial cells cultured from various organs tend to be basaloid in appearance and capable of subsequent differentiation. New pulmonary adenocarcinoma subclasses were identified by Bhattacherjee and colleagues (2001) based on gene expression, including a group with neuroendocrine differentiation and another with differentiation toward cells with surfactant properties, and preliminary data suggest that those with neuroendocrine differentiation may have a worse prognosis. Figure 15-5 (see Color Fig. 15-5) is a figure from their paper depicting gene expression profiling that illustrates the new subclasses of adenocarcinoma. It is anticipated that expression
P.273
P.274
profiling of tumors will identify new targets for therapy, as evidenced by information on alteration of gene expression in tumors by Heighway (2002) and Wikman (2002) and their colleagues.
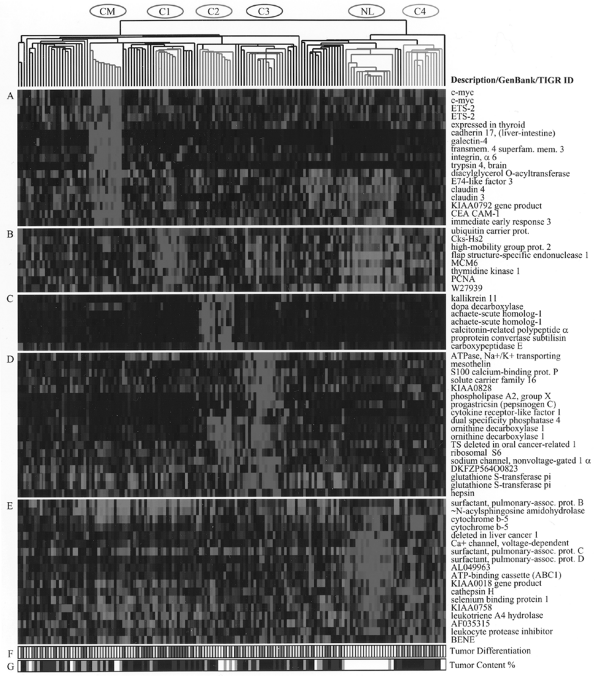 |
Fig. 15-5. Gene expression clusters and histologic differentiation within lung adenocarcinoma subclasses. Genes expressed at high levels in specific subsets of adenocarcinomas. (A) Colon metastases. (B) Proliferation-related gene expression (C1). (C) Neuroendocrine gene expression (C2). (D) Ornithine decarboxylase 1 and surfactant gene expression (C3 and C2). (E) Type II pneumocyte gene expression (C4, C3, and normal lung). (F) Histopathologic degree of differentiation. Red, poor; yellow, moderate; green, well; white, not available or irrelevant. (G) Estimated nucleated tumor content: white, not determined or irrelevant; gray, 30 to 40%; blue, 40 to 70%; black, greater than 70%).(See Color Fig. 15-5.). Reproduced with kind permission from Bhattacharjee A, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A 98:13790, 2001. |
Rihn and associates (2000) compared the gene expression profile of mesothelial cells to malignant mesothelioma and found that many of the differentially expressed genes were involved in cellular resistance to chemical and physical noxious agents, cell migration, cell division, and adhesion. Mohr and Rihn (2001) showed that malignant mesothelioma cells have overexpression of genes that are involved in DNA repair and in bleomycin and fluorouracil metabolism, helping to explain the resistance of tumor cells to radiation therapy and to chemotherapy. Combining genetic manipulation of signaling pathways with DNA microarray analysis can be used for addressing transcriptional alterations that occur downstream of the genetic modification in a given pathway. Microarray technology can be useful in pharmacogenomics and can be used to monitor responses to treatment, assess resistance, and facilitate generation of novel targets.
Screening and Molecular Markers
In a review of recent advances in the molecular diagnosis of lung cancer, Mao (2002) broke down the changes in lung cancer into four areas that could be exploited for screening purposes: (a) genetic abnormalities that are usually clonal and irreversible, such as mutations affecting well-defined oncogenes or loss of TSG; (b) epigenetic changes, which are often clonal but may be reversible under certain conditions (e.g., aberrant promoter DNA hypermethylation); (c) abnormal presence or amount of gene products, which may be variable under different conditions (e.g., EGFR), expression of neuroendocrine factors such as gastrin-releasing peptide and bombesinlike peptides and their receptors, and telomerase activity; and (d) abnormal autoantibodies. These changes could be used in the establishment of screening tests and panels of biomarkers for early diagnosis of lung cancer. Genetic abnormalities occur in the early carcinogenic process in lung tissue and have been detected in morphologically normal tissues that have suffered chronic carcinogen insult. Belinsky and associates (1998) and Soria and colleagues (2002) showed that aberrant hypermethylation may occur in preneoplastic stages in carcinogen-damaged lungs. Hypermethylation has been observed in several well-characterized candidate TSGs in early-stage lung cancer, including p16 as reported by Nuovo and colleagues (1999) and FHIT as demonstrated by Zochbauer-Muller and associates (2001). Alternative splicing of pre-mRNA is another mechanism to control gene expression. Aberrant splicing has been demonstrated in various cancers, including lung. Aberrant splicing has been described for FHIT in lung cancer by Sozzi and associates (1995) and by Tseng and co-workers (1999).
Progression from metaplasia to dysplasia and carcinoma in situ in SCLC is associated with a number of genetic changes, including loss of genetic material on 3p, 5q, 9p, 13q, and 17p. Wistuba (1997) and Mao (1997) and their associates, as well as Mao (2002), describe loss at 3p and 9p in normal-appearing bronchial mucosa from smokers and former smokers but not in nonsmokers. When the most discriminatory genetic biomarkers are identified, traditional diagnostic tests for lung cancer, such as sputum analysis, that are used for early diagnosis can be refined to incorporate detection of these markers in addition to cytologic assessment. Mao and co-workers (1994) reported the detection of p53 and ras gene mutations in sputum DNA months to years before clinical signs of tumor were reported, and Mao and colleagues (1997) showed microsatellite alterations in cytologically negative sputum from patients with lung cancer. Chen and associates (1996) demonstrated microsatellite alterations in plasma DNA of patients with SCLC. Liloglou and associates (2001), using four cancer-specific markers of microsatellite alteration in BAL samples from patients with and without lung cancer, achieved 74% detection sensitivity and 77% specificity. The relatively low specificity is believed to be due to microsatellite alterations in bronchial epithelial cells that have been damaged by carcinogens. Whether the cancer-free patients with such microsatellite alterations are at high risk for invasive lung cancer is not known because adequate follow-up is lacking. Somers and associates (1998) detected K-ras mutation up to 4 years before clinical diagnosis in about 50% of patients whose tumors harbored such mutations. BAL is more representative than sputum but requires bronchoscopy and has been used for molecular analysis by several groups, including Scott (1996) and Kersting (2000) and their associates. Nakajima and associates (2001) detected K-ras mutations in sputum of patients with K-ras mutation negative cancers and in individuals with no clinically diagnosable lung cancer.
Palmisano and colleagues (2000) used a PCR approach to detect methylated DNA sequences in sputum and found that aberrant methylation of the p16 and/or O6-methyl-guanine-DNA methyltransferase (MGMT) promoters can be detected in 100% of patients with squamous cell carcinoma, up to 3 years before clinical diagnosis. However, about 10% of sputum from cancer-free high-risk patients also contained these methylated sequences. Ahrendt and colleagues (1999a, 1999b) combined three PCR-based tests (tumor-specific oncogene mutations, CpG island methylation, and MI) and examined BAL samples but only detected half of the patients with lung cancer. Detection of molecular alterations is therefore not synonymous with tumor, and careful interpretation of such results is warranted. In the future, it is possible that germline DNA changes that reflect an increased risk for lung cancer development might also play a role in population screening.
The timing of genetic events in lung carcinogenesis requires additional study and will help to define genes responsible
P.275
for tumor initiation, providing targets to facilitate early lung cancer screening, and identifying genes involved in genetic predisposition to lung cancer. Proteomics may also be used in population screening to enable detection of early lung cancers, either in serum or sputum analysis.
Chromosomal Instability DNA Mismatch Repair Genes
Chromosomal abnormalities can be divided into numerical (i.e., aneuploidy) and structural (e.g., deletions, translocations, homogeneously staining regions, and double minutes) alterations. Mis-segregation of chromosomes in lung cancer can result from various causes, including defects of mitotic spindle checkpoint, abnormal centrosome formation, and failure of cytokinesis, whereas structural alterations of chromosomes may be caused by failure in repair of DNA double-strand breaks due to the impairment of DNA damage checkpoints and double-strand break repair systems as described by Masuda and Takahashi (2002). Telomere erosion may also be involved.
Highly conserved DNA repair genes play an important role in the maintenance of genetic integrity in normal cells. Inactivating mutations in DNA repair genes leads to an increased rate of mutations in a variety of cellular genes in affected cells, including protooncogenes and TSGs. The accumulation of mutations in these growth-regulatory genes appears to be the rate-limiting step in tumorigenesis; therefore, inactivation of genes involved in DNA damage recognition and repair greatly accelerates tumor progression. Microsatellites are highly polymorphic, short, tandem repeat sequences dispersed throughout the genome. Instability of these repeat sequences at multiple genetic loci reflects and is used to detect and monitor mismatch repair errors. The fundamental role of mismatch repair enzymes in human cancer is underscored by two findings: (a) that inherited cancer syndromes such as the autosomal dominant hereditary nonpolyposis colon cancer have been found to harbor defects in mismatch repair genes and are characterized by alteration of microsatellite sequences or MI; and (b) the frequency with which MI is demonstrated in several human cancers of diverse tissue origin. MI is observed in a subset of NSCLC cases, and the replication error phenotype is more common in clinically advanced disease and metastases.
Merlo and colleagues (1994) detected alterations of microsatellite loci consisting of deletions or expansions of (CA)n dinucleotide repeats in 45% of primary SCLC cases. Cancers that showed MI contained widespread allelic loss and had a uniformly poor prognosis. Fong and associates (1995) demonstrated MI in only 6.5% of NSCLC cases studied and found it to be associated with extensive, concurrent molecular changes, including K-ras and p53 mutations and frequent LOH at chromosomal regions 5q, 8p, 9p, 11p, and 17p. Wieland and colleagues (1996) detected MI at one or more loci in 29% of NSCLC cases. They found LOH of the mismatch repair gene hMLH1 at 3p21 in 82% of the tumors with MI, suggesting that defects in this gene may be important. Benachenhou and co-workers (1998) also revealed frequent allelic losses at the DNA mismatch repair locus hMSH3 in addition to hMLH1 in NSCLC.
Clonality
The detection of gene rearrangements and chromosomal translocations has been used to mark clonality. The identification of such genetic events provides additional clinically relevant data for the diagnosis and classification of lymphoid neoplasms beyond the knowledge that can be obtained with immunophenotyping. The immunoglobulin genes, T-cell receptor genes, or both rearrange at the DNA level before clonal expansion in lymphoid cells. Because a given rearrangement is unique to a committed cell and its derivative clone, Southern blotting or PCR techniques are used to detect clonal populations in either fresh or archival tissues, otherwise undetectable by immunohistochemical methods. This is particularly useful for the analysis of T-cell neoplasms because no immunophenotypic markers are available for assessment of clonality. Even though clonality is most often associated with cancer, clonal populations of lymphoid cells can be detected in patients with immune deficiencies, such as after organ transplantation immunosuppression or acquired immunodeficiency syndrome (AIDS), in the absence of overt malignancy. In the interpretation of results of gene rearrangement studies, analyzing the results in the context of morphologic, immunophenotypic, and clinical data is extremely important. Assessment of clonality is especially useful when evaluating borderline lesions, such as atypical lymphoid proliferations in the lung (especially in transbronchial biopsies), and the nature of a pleural effusion in a patient with a history of lymphoproliferative disease when flow cytometry is unhelpful, as in the case of recurrent or concurrent T-cell lymphoma. In these settings, a polyclonal population of cells indicates benign polyclonal hyperplasia, whereas clonal cells point to a neoplastic process. As mentioned previously, many methods have been used to demonstrate clonality in tissues: (a) analysis of karyotypic abnormalities; (b) gene rearrangements or translocations; (c) point mutations in oncogenes and TSGs; (d) viral sequences, such as the terminal repeat episomal sequence of EBV; (e) MI; (f) LOH; and (g) dosage compensation. Dosage compensation is a process in which the level of X-linked gene expression is equalized between the sexes by methylation of either the maternal or paternal allele. X-chromosome inactivation assays (which can be used only in females) use methylation-dependent RFLPs and probes to certain genes that are located on the X chromosome, including glucose-6-phosphate dehydrogenase, phosphoglycerate kinase-1, and androgen receptor genes. To be clinically useful, the choice of a gene in the assessment of clonality must take into account the degree of polymorphism of the locus in the population. Signoretti and colleagues (1999)
P.276
described a nonradioactive PCR single-strand conformational polymorphism (SSCP) technique to assess T-cell clonality in paraffin-embedded tissues of hyperplastic and neoplastic lymphoid populations. Consensus primers, generated against variable and joining regions, are used for T-cell receptor- (TCR- ) gene rearrangement amplification. The SSCP imprint of PCR products that are generated is specific for each TCR- gene rearrangement and may be used to evaluate recurrent lesions in the same patient. Murphy and colleagues (1999) describe how the technique can be adapted for cytology specimens, such as pleural fluid.
Infectious Diseases Affecting the Lungs
Diagnostic molecular microbiology applies the principles of nucleic acid hybridization to the detection and characterization of pathogenetic microorganisms associated with infectious disease. PCR-based methods are replacing media-based biologic amplification. Fastidious organisms or viruses that cannot be cultured are now targets of molecular techniques. In addition, new organisms are being identified by molecular methods, and their associations with human diseases are being elucidated. Weber and associates (2002) recently described a novel method to detect previously unidentified microorganisms in tissues using a computational subtraction approach by filtering a set of human tissue derived sequences against the human genome. Molecular techniques have contributed to our understanding of the biology of pulmonary opportunists. For instance, Pneumocystis species described by Kovacs and colleagues (2001), human immunodeficiency virus (HIV) reported by Clarke (2002), and human herpesvirus-8 (HHV-8) published by Tamm and colleagues (1998) have been detected by various molecular techniques in the lungs, sputum, or bronchial washings of immunocompromised patients.
Molecular identification of microorganisms is especially useful in several ways: (a) to avoid misinterpretation of false-positive serology due to the presence of cross-reacting antibodies to host antigens or to antigens of related organisms; (b) to identify pathogenic organisms before seroconversion; (c) to follow response to therapy; (d) to identify mutations responsible for drug resistance; (e) to subtype different strains; and (f) for the rapid and accurate identification of organisms that require elaborate and long media-based cultures. Although detailed discussions of molecular microbiology are beyond the scope of this chapter, mycobacterial infections and EBV are discussed briefly.
Mycobacterial Infections
Mycobacterial infections have significant clinical impact and have a mortality rate of 60% if untreated. The frequency of mycobacterial infections in immunocompromised patients has increased. Inadequate treatment of many patients has led to an increase in the proportion of patients with drug-resistant strains of M. tuberculosis, recently reviewed by Fisher (2002). Microscopic diagnosis of acid-fast bacilli is time consuming, not species specific, and insensitive, especially in treated patients. Culture of such organisms requires weeks, which can result in delays in therapy or inappropriate treatment of unaffected individuals. Molecular diagnostics can play an important role in adequate and timely treatment and in patient follow-up, as pointed out by Drobniewski and associates (2003). Species-specific PCR for mycobacteria can be performed on a variety of samples, including sputum and paraffin-embedded tissue. PCR is sensitive and specific when results are compared with bacteriologic and clinical data. These tests can be performed in as few as 4 to 5 hours, permitting same-day reporting of results.
Strain-specific differentiation of M. tuberculosis is complex, but it may be important to differentiate between whether an individual infection represents reactivation or exogenous reinfection or whether an unexpected positive culture result was due to possible contamination in the laboratory. IS6110 is one of a number of mycobacterial mobile genetic elements. IS6110 is a naturally occurring transposable genetic element that appears to be detectable only in species belonging to the M. tuberculosis complex. Each strain contains a different number of identical copies of this transposable element. The number of copies of IS6110 elements and the size of the restriction fragments containing these elements vary such that no two unrelated strains produce identical patterns when hybridized with a labeled IS6110 probe. This forms the basis for a unique genetic fingerprint. Thus, molecular epidemiology can identify numerous suspected and unsuspected transmission links. Another promising technology for clinical tuberculosis laboratories involves oligonucleotide arrays or DNA chips. Gingeras (1998) and Troesch (1999) and their associates demonstrated that DNA arrays could be used for genotyping and species identification and for the detection of rifampicin resistance (mutation in the rpoB gene encoding the -subunit of the RNA polymerase). Time-consuming culture-based methods may be complemented by faster techniques that facilitate a larger number of genetic tests. Fisher and associates (2002) used microarrays and real-time PCR to analyze the global transcriptional response of M. tuberculosis to low pH in vitro, which may mimic conditions experienced by phagocytosed mycobacteria, and showed that 81 genes were differentially expressed more than 1.5 fold, including many genes that are involved in fatty acid metabolism. Sassetti and colleagues (2001) describe a technique (transposon site hybridization) that permits rapid functional characterization by identifying the complete set of genes needed for growth under different conditions. The technique combines high-density insertional mutagenesis with microarray mapping of pools of mutants. This method could be applied to other microorganisms. Behr and associates (1999) used comparative genomic
P.277
hybridization experiments on DNA microarrays to show genomic composition differences between M. tuberculosis, Mycobacterium bovis, and various bacille Calmette-Gu rin (BCG) daughter strains. Hendrickson and colleagues (2000) used serologic proteome analysis in conjunction with mass spectrometry to identify and sequence a novel protein MTB 81 that may be useful for serodiagnosis of tuberculosis, especially for patients who are coinfected with HIV when other assays may fail. A better understanding of the gene expression profile and the proteome of M. tuberculosis and the genetic differences among mycobacteria should yield rational approaches to the design of improved diagnostic techniques and vaccines.
Epstein-Barr Virus Infections
EBV is a double-stranded DNA herpesvirus that can persist in latent form in epithelial cells and B lymphocytes. In most cases, EBV infection results in a self-limiting, transient lymphoproliferative disorder. In an immunosuppressed state, however, such as HIV infection or therapeutic immunosuppression after organ transplantation, reactivation of latent infection may contribute to the development of full-blown malignancies. EBV immortalizes the cell and blocks differentiation, thus committing the cell to indefinite T-cell independent proliferation. EBV has been implicated in the pathogenesis of several neoplasms, including lymphoepithelioma-like carcinomas that have been described at many sites, including the lung, and also in pyothorax-associated lymphoma (primary effusion lymphoma), recently reviewed by Nakatsuka and associates (2002). As is the case for most microorganisms, a variety of techniques, including ISH and PCR, can be used to assess the presence of the actively replicating virus. The terminal repeat regions of the virus have been used as markers of clonality in angiocentric immunoproliferative lesions. Nonisotopic ISH for early EBV RNA can be performed in paraffin-embedded tissues using oligonucleotide probes. Evidence is growing of a link between EBV and idiopathic pulmonary fibrosis. Kelly and associates (2002) reported finding a rearranged form of EBV DNA in lungs from patients with idiopathic pulmonary fibrosis.
Interstitial Lung Disease
Diffuse interstitial lung diseases are triggered by diverse factors in an individual with a genetic predisposition. Certain genetic traits are emerging as risk factors for the development of pulmonary fibrosis, and identification of patients with genetic susceptibility would theoretically allow improved monitoring of disease progression. Pathogenesis of pulmonary fibrosis is linked to induction of specific cytokines, such as: (a) tumor necrosis factor- (TNF- ), (b) TGF- , (c) interleukin-1B (IL-1B), and (d) platelet-derived growth factor (PDGF). TNF- is considered to be a key cytokine because it stimulates growth of fibroblasts and increases collagen deposition; anti TNF- antibody prevents development of bleomycin or silica-induced interstitial pneumonitis in mice. TNF- transgenic mice that overexpress TNF- only in the lungs develop interstitial pneumonitis resembling idiopathic pulmonary fibrosis in humans. TNF- transgenic mice have been studied by Sueoka and colleagues (1998) to analyze the sequential process of interstitial pneumonitis serving as a model of the inflammatory stage of idiopathic pulmonary fibrosis in humans. The authors showed that the pattern of cytokine gene expression and synthesis as detected by semiquantitative RT-PCR and Northern blot analysis changed with the progression of histologic features. Because TNF- and TGF- have been implicated in the pathogenesis of interstitial fibrosis, early diagnosis might result in the initiation of early and more effective therapy, including anticytokine therapy. Inhibiting interactions between lung cells and extracellular matrix that lead to aberrant activation of signal transduction may also prove beneficial. Katsuma and associates (2001) used microarray analysis in a murine model of bleomycin-induced fibrosis as an approach to understand the changing gene expression profiles, and Johnston and colleagues (2002) used microarray analysis to evaluate chemokine responses during development of radiation-induced fibrosis. Leikauf and co-workers (2002) evaluated the progression of acute lung injury in inbred mouse strains using cDNA microarrays in an attempt to assess the progression of acute lung injury, the temporal expression of genes, and expressed sequence tags and found evidence that genetic determinants related to specific chromosomal loci may account for survival differences in the mice.
Although the etiology of sarcoidosis is still unknown, it is suspected to be the result of an antigen-driven process, most likely an infective agent. Mycobacterium species have long been suspected in the etiology of sarcoidosis. Popper (1994), Ghossein (1994), Ikonomopoulos (1999), and Li (1999) and their colleagues detected M. tuberculosis DNA by PCR in lesions from different sites in a subset of patients with sarcoidosis, and Klemen and associates (2000) also detected mycobacterial DNA in recurrent sarcoidosis in the transplanted lung. More recently, Propionibacterium species have been implicated in the pathogenesis of sarcoidosis. Ishige (1999) and Eishi (2002) and their collaborators detected genomes of either Propionibacterium acnes or Propionibacterium granulosum by quantitative real-time PCR in almost 100% of samples of lymph nodes that were involved by sarcoidosis, significantly higher than that detected in controls or in samples from patients with tuberculosis.
ANIMAL MODELS
Animal models can prove invaluable for clarifying genetic determinants of human disease where little is known about the initial molecular steps underlying the disease process.
P.278
Current mouse models for lung cancer include strains that are susceptible to spontaneous and chemically induced lung tumor development, and several transgenic strains that express viral and cellular oncogenes have been discussed by Tuveson and Jacks (1999) and Zhao and co-workers (2000). Alternatively, suspected oncogenes and TSGs can be studied by reverse genetics, whereby such genes are overexpressed or interfered with (e.g., with siRNAs) in normal cells with the intent of inducing tumorigenicity.
Transgenic animals are defined as those in which foreign DNA, usually a human gene, is introduced into fertilized cells and is subsequently inherited in the progeny of the host. By virtue of the way in which the construct is designed, it can be overexpressed in all tissues when transcription is driven by a strong constitutive promoter; equally, its expression can be restricted by tissue-specific or developmentally regulated promoters. Various transgenic mice serve as models of both neoplastic and nonneoplastic pulmonary disease and are important for investigating new treatment approaches for pulmonary disease. Lubet and colleagues (2000) developed transgenic mice that expressed mutant p53, and they were more susceptible to various carcinogens and developed larger lung tumors when exposed to carcinogens than control mice. Orthotopic metastatic models of metastatic lung cancer have been created by injecting human lung cancer fragments into the lungs of immunocompromised mice that subsequently grow and metastasize, as discussed by Hoffman (1999). Such models serve as useful models for investigating new treatments. Examples of transgenic mice models that give rise to lung tumors include: (a) mice expressing SV40 large T antigen under the control of promoters specific to pulmonary epithelium, such as human surfactant protein C; (b) mice expressing activated forms of H-ras from the SV40 early promoter and protooncogenic H-ras under control of its own promoter; and (c) mice transgenic for mutant p53. Mice have also been created that have germline disruptions of p53, p16INK4a, and Rb, but the incidence of lung cancer is not increased in these mice. Transgenic overexpression of TNF- and TGF- was shown by Miyazaki (1995) and Lee (1995) and their colleagues to induce interstitial fibrosis, confirming the role of these growth factors in the pathogenesis of interstitial fibrosis.
New approaches include creating conditionally targeted alleles of genes that are mutated in human lung tumors. Johnson (2001) and Jackson (2001) and their colleagues have developed mouse models of lung carcinogenesis based on K-ras activation that more closely mirrors spontaneous oncogene activation as seen in human lung carcinogenesis than traditional transgenic strategies. They studied conditional expression of oncogenic K-ras using a Lox-Stop-Lox K-ras conditional mouse strain and also somatic activation of the gene by spontaneous recombination in mouse strains carrying latent oncogenic alleles of K-ras. The lung tumors in double mutant mice created by crossing mice carrying the latent K-ras oncogenic allele with p53 mutant mice showed more malignant histologic features, demonstrating that p53 mutation cooperates in the progression of lung cancer.
GENE THERAPY
The poor overall survival of patients with lung cancer treated with standard therapies (chemotherapy, radiation therapy, and surgery) mandates novel treatment approaches. It is hoped that additional therapeutic options can be offered to patients resistant to conventional therapies. Gene therapy is the delivery of functional genes to the cells of an individual for the purpose of treating disease. Gene therapy aims at inducing or abolishing the expression of proteins (depending on the target) by introducing or targeting the destruction of their coding genes in tumor cells. Normal function of a gene can be restored by inserting a wild-type gene in the case of a TSG that has been inactivated, or to switch off a gene that is inappropriately activated by a mutation in the case of an oncogene (e.g., by using siRNAs). Molecular techniques are identifying novel targets involved in intracellular signal transduction pathways or cell cycle regulation for the development of new treatment approaches for an expanding range of pulmonary diseases.
Delivery systems for therapeutic genes include naked DNA or RNA; DNA or RNA attached to other agents, such as cationic liposomes or agents; viral vectors (particularly retroviruses, adenoviruses, adeno-associated viruses, herpesviruses, and lentiviruses); and anaerobic, genetically modified, clostridial organisms. The effectiveness of these treatments depends on selection of the appropriate cellular target, high-enough stable expression of the introduced gene, refinements in delivery vectors, and suppression of host immune response. Molecular diagnostic tests are used with increasing frequency to monitor novel genetic approaches. Potential applications of gene therapy include the treatment of single-gene defects (such as cystic fibrosis) and more complex conditions, including cancer and inflammatory lung conditions. Only 6% to 10% of cells need to be corrected to counter the transepithelial chloride transport defect in CF, and only 5% of normal CFTR mRNA needs to be expressed to increase chloride transport markedly. Results in gene therapy of CF and of cancer have been hampered by vector problems and development of resistance, as noted by Ferrari (2002) and Griesenbach (2002) and their associates. Development of new vectors is an active area of research.
Genetic modification of critical elements of the inflammatory process may prove beneficial in complex pulmonary conditions characterized by lung inflammation, including adult respiratory distress syndrome, chronic obstructive pulmonary disease, and asthma. Examples of possible approaches include overexpression of genes encoding antiinflammatory cytokines, such as IL-10, and inhibiting inflammatory transcription factors.
P.279
Cancer gene therapies make up a large subset of approved gene therapy protocols in clinical trials because conventional therapies have yielded poor results. Various strategies can be used in cancer gene therapy: genes that enhance the host's natural immune attack on transfected malignant cells (genes for certain cytokines or alloantigens that stimulate T lymphocytes, natural killer lymphocytes, and macrophages); genes that result in death of malignant cells (pro-apoptotic genes that sensitize cells to chemotherapy or radiotherapy; and TSGs or siRNAs targeting oncogenes. Cells can also be driven to express thymidine kinase (a herpesvirus protein) in a tissue-specific manner, and subsequent treatment with antiviral agents, such as ganciclovir, results in tumor cell death. Some of the genetic changes that occur in lung cancers also serve as potential therapeutic targets. Roth and colleagues (1996) demonstrated regression of lung tumors after injection with a retrovirus expressing wild-type p53. In gene therapy, the identification of specific genes critical to the development of carcinogenesis has offered the opportunity to target these genes or their products for treatment. One possible gene therapy strategy that has been pursued in phase I and II lung cancer clinical trials is to replace nonfunctional tumor suppressor genes such as mutated or deleted p53 genes with wild-type p53 genes by adenoviral gene transfer (Ad-p53). Transduction of the tumors has been accomplished with both direct intratumoral injection and BAL. Many clinical phase I and II studies have been completed in lung cancer with adenoviral-mediated transfer of wild-type p53 (Ad-p53) either alone or in combination with chemotherapy or radiation therapy. Clinical phase I and II gene therapy trials with Ad-p53 in NSCLC have demonstrated that the treatment is associated with low toxicity and evidence of antitumoral activity at the locoregional site following either intratumoral delivery or BAL, as reviewed by Swisher and Roth (2002). A potential role for radiosensitization of previously radiation-resistant local tumors has been identified by combining Ad-p53 with radiation or possibly chemoradiation. Zou and associates (1998) reported the feasibility of in vivo p53 gene replacement in endobronchial precancerous and cancerous cells in transgenic mice that lacked p53 using a regionally administered cationic liposome-p53 formulation (DP3-p53) DP3-p53 was administered intratracheally into two nude mouse models of endobronchial human lung cancer created by using H358 (p53-null) and H322 (p53-mutant) cells and resulted in a twofold survival advantage and significantly inhibited lung tumor formation compared with untreated mice and mice treated with the DP3 empty vector. H. Y. Lee and colleagues (2002) reported the inhibition of oncogenic K-ras signaling by aerosolized gene delivery in a mouse model of human lung cancer. Sterman and co-workers (1998) demonstrated that intrapleural administration of adenoviral-mediated herpes simplex virus thymidine kinase and ganciclovir gene therapy in patients with mesothelioma resulted in detectable gene transfer that was well tolerated by patients. Frizelle and colleagues (1998) demonstrated that adenovirus-mediated reexpression of p16 in mesothelioma cells resulted in tumor regression. Hallahan and associates (1995) used a radiation-inducible promoter ligated to TNF- and demonstrated increased tumor cell death after irradiation. Dachs and colleagues (1997) demonstrated that transfection of cells in vitro with a hypoxia response element could be used to regulate transcriptionally gene expression in vivo under hypoxic conditions. Potentially, this approach allows the conversion of a nontoxic prodrug to its toxic metabolite selectively in tumors with low oxygen levels. Certain species of the genus Clostridium can selectively germinate and grow in the hypoxic regions of solid tumors after intravenous injection of spores, as demonstrated by Malmgren and Flanigan (1955). Lemmon and colleagues (1997) genetically engineered Clostridium beijerinkii by introducing a plasmid that expressed nitroreductase that can convert CB1954 into a potent bifunctional alkylating agent. After injection of the spores into mice, nitroreductase protein activity could be detected in tumors but not in normal tissue in the mice.
Gene therapeutic approaches in lung cancer have been delivered through intratumoral, intrabronchial, and intrapleural injections, but aerosol is a potentially more efficacious approach for nonmetastatic tumors because it can reach a larger target surface area of bronchial epithelium and lacks the toxic effects of systemic injection [see review by Albelda and colleagues (2002)]. The efficacy of aerosolized gene transfer has been limited by the vector system used. Future gene therapy research is required to develop systemic delivery systems to treat disseminated disease. Randomized phase III studies are needed to determine the potential of gene therapy to improve overall survival of patients with NSCLC and to determine whether gene therapy will play a therapeutic role as a novel agent to complement conventional chemotherapy, radiation therapy, and surgical treatment strategies in the treatment of NSCLC.
OTHER TARGETED MOLECULAR THERAPIES
There has been tremendous research and investment in the development of small molecules that target key proteins in cell signaling pathways that are aberrantly altered in disease, particularly in carcinogenesis. For instance, receptor tyrosine kinases (RTKs) serve as potential therapeutic targets in several solid tumors, including lung cancer. The RTK c-kit is highly expressed in SCLC (although it is not mutated), and this has led to clinical trials with the specific c-KIT inhibitor (STI-571, Glivec, Novartis), alone and in combination therapy. HER-2 acts as a signaling network coordinator and amplifier because it heterodimerizes with other HER family members. Preliminary evidence suggests that only cases that show strong immunohistochemical staining in a membranous pattern (3+) may demonstrate some benefit from treatment with trastuzumab in addition to chemotherapy. Antibodies against the angiogenic factor
P.280
VEGF and small molecules against VEGF receptors, such as SU5416, which is an inhibitor of Flk-1 receptor, are being tested in NSCLC and other tumor types. More recently, modification of gene expression using siRNAs has the promise of being the most powerful tool yet.
CONCLUSION
Elucidation of the molecular events that underlie disease pathogenesis is creating a fundamental change in the approach to disease. Complete understanding of the molecular basis of pulmonary disease, however, stills evades us. The functions of many potentially significant human genes that have been identified as a result of new technologies have not been fully elucidated. The clinical application of molecular diagnostic techniques has allowed a more precise and rapid assessment of both neoplastic and nonneoplastic pulmonary disease. Molecular biology is increasingly being used to elucidate the various steps involved in tumor initiation, progression, and metastasis. In addition, technical advances in recombinant DNA technology have resulted in the identification of novel biomarkers that are of prognostic importance and may aid in early diagnosis and present potential molecular targets for novel therapeutic strategies. Monitoring of gene therapy will require refined molecular diagnostic techniques to guide therapy. In this era of genomics and proteomics, there is an expectation of significant advances in early diagnosis of pulmonary disease, the elucidation of new relevant categories of disease, and the development of more targeted therapy. Specific disease profiles may be generated for individual patients to assist in the selection of therapy, and individualized therapy may be present in the future. Understanding the molecular events underlying pathogenic processes will hopefully translate into improved patient survival and ultimately prevention of pulmonary disease.
REFERENCES
Ahrendt SA, et al: Rapid p53 sequence analysis in primary lung cancer using an oligonucleotide probe array. Proc Natl Acad Sci U S A 96: 7382, 1999a.
Ahrendt SA, et al: Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. J Natl Cancer Inst 91:332, 1999b.
Albelda SM, Wiewrodt R, Sterman DH: Gene therapy for lung neoplasms. Clin Chest Med 23:265, 2002.
Amin KM, et al: Wilms' tumor 1 susceptibility (WT1) gene products are selectively expressed in malignant mesothelioma. Am J Pathol 146:344, 1995.
Anbazhagan R, et al: Classification of small cell lung cancer and pulmonary carcinoid by gene expression profiles. Cancer Res 59:5119, 1999.
Arvand A, et al: The COOH-terminal domain of FLI-1 is necessary for full tumorigenesis and transcriptional modulation by EWS/FLI-1. Cancer Res 61:5311, 2001.
Askin FB, et al: Malignant small cell tumor of the thoracopulmonary region in childhood: a distinctive clinicopathologic entity of uncertain histogenesis. Cancer 43:2438, 1979.
Azzoli CG, et al: Trastuzumab in the treatment of non-small cell lung cancer. Semin Oncol 29:59, 2002.
Balsara BR, Testa JR: Chromosomal imbalances in human lung cancer. Oncogene 21:6877, 2002.
Bayani JM, Squire JA: Applications of SKY in cancer cytogenetics. Cancer Invest 20:373, 2002.
Behr MA, et al: Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284:1520, 1999.
Belinsky SA, et al: Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci U S A 95:11891, 1998.
Benachenhou N, et al: High resolution deletion mapping reveals frequent allelic losses at the DNA mismatch repair loci hMLH1 and hMSH3 in non-small cell lung cancer. Int J Cancer 77:173, 1998.
Benhamou S, et al: Meta- and pooled analyses of the effects of glutathione S-transferase M1 polymorphisms and smoking on lung cancer risk. Carcinogenesis 23:1343, 2002.
Bhattacharjee A, et al: Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A 98:13790, 2001.
Bos JL: Ras oncogenes in human cancer: a review. Cancer Res 49:4682, 1989.
Bremnes RM, et al: High-throughput tissue microarray analysis used to evaluate biology and prognostic significance of the E-cadherin pathway in non-small-cell lung cancer. J Clin Oncol 20:2417, 2002.
Bunn PA Jr, Franklin W: Epidermal growth factor receptor expression, signal pathway, and inhibitors in non-small cell lung cancer. Semin Oncol 29:38, 2002.
Burke L, et al: Allelic deletion analysis of the FHIT gene predicts poor survival in non-small cell lung cancer. Cancer Res 58:2533, 1998.
Campling BG, el-Deiry WS: Clinical implications of p53 mutations in lung cancer. Methods Mol Med 75:53, 2003.
Capodieci P, et al: Automated in situ hybridization: diagnostic and research applications. Diagn Mol Pathol 7:69, 1998.
Carrano AC, et al: SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1:193, 1999.
Catzavelos C, et al: Reduced expression of the cell cycle inhibitor p27Kip1 in non-small cell lung carcinoma: a prognostic factor independent of Ras. Cancer Res 59:684, 1999.
Chen XQ, et al: Microsatellite alterations in plasma DNA of small cell lung cancer patients. Nat Med 2:1033, 1996.
Clarke JR: Molecular diagnosis of HIV. Expert Rev Mol Diagn 2:233, 2002.
Clements NC Jr, et al: Analysis of K-ras gene mutations in malignant and nonmalignant endobronchial tissue obtained by fiberoptic bronchoscopy. Am J Respir Crit Care Med 152:1374, 1995.
Cline EI, et al: Prediction of in vivo synergistic activity of antiangiogenic compounds by gene expression profiling. Cancer Res 62:7143, 2002.
Dachs GU, et al: Targeting gene expression to hypoxic tumor cells. Nat Med 3:515, 1997.
Dai Y, et al: Application of the p53 and K-ras gene mutation patterns for cytologic diagnosis of recurrent lung carcinomas. Cancer 90:258, 2000.
DeMuth JP, et al: The gene expression index c-myc x E2F-1/p21 is highly predictive of malignant phenotype in human bronchial epithelial cells. Am J Respir Cell Mol Biol 19:18, 1998.
Denissenko MF, et al: Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hot spots in p53. Science 274:430, 1996.
DeSalle LM, et al: The de-ubiquitinating enzyme Unp interacts with the retinoblastoma protein. Oncogene 20:5538, 2001.
Donehower LA, et al: Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature 356:215, 1992.
Drobniewski FA, et al: Modern laboratory diagnosis of tuberculosis. Lancet Infect Dis 3:141, 2003.
Eishi Y, et al: Quantitative analysis of mycobacterial and propionibacterial DNA in lymph nodes of Japanese and European patients with sarcoidosis. J Clin Microbiol 40:198, 2002.
Elbashir SM, et al: Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494, 2001.
Esposito V, et al: Prognostic role of the cyclin-dependent kinase inhibitor p27 in non-small cell lung cancer. Cancer Res 57:3381, 1997.
Essary LR, et al: Primary pleuropulmonary synovial sarcoma: reappraisal of a recently described anatomic subset. Cancer 94:459, 2002.
Fain JS, et al: Rapid diagnosis of Legionella infection by a nonisotopic in situ hybridization method. Am J Clin Pathol 95:719, 1991.
P.281
Fejzo MS, Slamon DJ: Frozen tumor tissue microarray technology for analysis of tumor RNA, DNA, and proteins. Am J Pathol 159:1645, 2001.
Ferrari S, Geddes DM, Alton EW: Barriers to and new approaches for gene therapy and gene delivery in cystic fibrosis. Adv Drug Deliv Rev 54: 1373, 2002.
Fisher M: Diagnosis of MDR-TB: a developing world problem on a developed world budget. Expert Rev Mol Diagn 2:151, 2002.
Fisher MA, et al: Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J Bacteriol 184:4025, 2002.
Fong KM, Zimmerman PV, Smith PJ: Microsatellite instability and other molecular abnormalities in non-small cell lung cancer. Cancer Res 55: 28, 1995.
Frizelle SP, et al: Re-expression of p16INK4a in mesothelioma cells results in cell cycle arrest, cell death, tumor suppression and tumor regression. Oncogene 16:3087, 1998.
Garber ME, et al: Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A 98:13784, 2001.
Ghossein RA, et al: A search for mycobacterial DNA in sarcoidosis using the polymerase chain reaction. Am J Clin Pathol 101:733, 1994.
Giaccone G: Clinical impact of novel treatment strategies. Oncogene 21: 6970, 2002.
Gingeras TR, et al: Simultaneous genotyping and species identification using hybridization pattern recognition analysis of generic Mycobacterium DNA arrays. Genome Res 8:435, 1998.
Golub TR, et al: Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286:531, 1999.
Greenlee RT, et al: Cancer statistics, 2001. CA Cancer J Clin 51:15, 2001.
Griese M, et al: Reduced proteolysis of surfactant protein A and changes of the bronchoalveolar lavage fluid proteome by inhaled alpha 1-protease inhibitor in cystic fibrosis. Electrophoresis 22:165, 2001.
Griesenbach U, et al: Gene therapy progress and prospects: cystic fibrosis. Gene Ther 9:1344, 2002.
Hallahan DE, et al: Spatial and temporal control of gene therapy using ionizing radiation. Nat Med 1:786, 1995.
Hamid QA, et al: Detection of human probombesin mRNA in neuroendocrine (small cell) carcinoma of the lung. In situ hybridization with cRNA probe. Cancer 63:266, 1989.
Hanash S: Disease proteomics. Nature 422:226, 2003.
Hanazawa R, Murayama SY, Yamaguchi H: In-situ detection of Aspergillus fumigatus. J Med Microbiol 49:285, 2000.
Hecht JL, et al: The value of Wilms tumor susceptibility gene 1 in cytologic preparations as a marker for malignant mesothelioma. Cancer 96: 105, 2002.
Heighway J, et al: Expression profiling of primary non-small cell lung cancer for target identification. Oncogene 21:7749, 2002.
Hendrickson RC, et al: Mass spectrometric identification of mtb81, a novel serological marker for tuberculosis. J Clin Microbiol 38:2354, 2000.
Hoffman RM: Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: a bridge to the clinic. Invest New Drugs 17: 343, 1999.
Hoos A, Cordon-Cardo C: Tissue microarray profiling of cancer specimens and cell lines: opportunities and limitations. Lab Invest 81:1331, 2001.
Husgafvel-Pursiainen K, et al: Mutations, tissue accumulations, and serum levels of p53 in patients with occupational cancers from asbestos and silica exposure. Environ Mol Mutagen 30:224, 1997.
Ikonomopoulos JA, et al: Multiplex polymerase chain reaction for the detection of mycobacterial DNA in cases of tuberculosis and sarcoidosis. Mod Pathol 12:854, 1999.
Ishige I, et al: Quantitative PCR of mycobacterial and propionibacterial DNA in lymph nodes of Japanese patients with sarcoidosis. Lancet 354:120, 1999.
Jackson EL, et al: Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15:3243, 2001.
Johnson L, et al: Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410:1111, 2001.
Johnston CJ, et al: Radiation-induced pulmonary fibrosis: examination of chemokine and chemokine receptor families. Radiat Res 157:256, 2002.
Kallioniemi A, et al: Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258:818, 1992.
Kannan K, et al: DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene 20:2225, 2001.
Katsuma S, et al: Molecular monitoring of bleomycin-induced pulmonary fibrosis by cDNA microarray-based gene expression profiling. Biochem Biophys Res Commun 288:747, 2001.
Kaye FJ: RB and cyclin dependent kinase pathways: defining a distinction between RB and p16 loss in lung cancer. Oncogene 21:6908, 2002.
Kelly BG, et al: A rearranged form of Epstein-Barr virus DNA is associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 166:510, 2002.
Kersting M, et al: Differential frequencies of p16(INK4a) promoter hypermethylation, p53 mutation, and K-ras mutation in exfoliative material mark the development of lung cancer in symptomatic chronic smokers. J Clin Oncol 18:3221, 2000.
Kitabwalla M, Ruprecht RM: RNA interference a new weapon against HIV and beyond. N Engl J Med 347:1364, 2002.
Klemen H, et al: Mycobacterial DNA in recurrent sarcoidosis in the transplanted lung-a PCR-based study on four cases. Virchows Arch 436:365, 2000.
Kleymenova EV, et al: Identification of a tumor-specific methylation site in the Wilms tumor suppressor gene. Oncogene 16:713, 1998.
Kononen J, et al: Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 4:844, 1998.
Kotsimbos AT, et al: Quantitative detection of human cytomegalovirus DNA in lung transplant recipients. Am J Respir Crit Care Med 156: 1241, 1997.
Kovacs JA, et al: New insights into transmission, diagnosis, and drug treatment of Pneumocystis carinii pneumonia. JAMA 286:2450, 2001.
Lacal JC, et al: Ras p21 proteins with high or low GTPase activity can efficiently transform NIH/3T3 cells. Cell 44:609, 1986.
Larsen HH, et al: Development of a rapid real-time PCR assay for quantitation of Pneumocystis carinii f. sp. carinii. J Clin Microbiol 40:2989, 2002.
Lee HY, et al: Inhibition of oncogenic K-ras signaling by aerosolized gene delivery in a mouse model of human lung cancer. Clin Cancer Res 8: 2970, 2002.
Lee M-S, et al: Accumulation of extracellular matrix and developmental dysregulation in the pancreas by transgenic production of transforming growth factor-1. Am J Pathol 147:42, 1995.
Lee WJ, et al: Microsomal epoxide hydrolase polymorphisms and lung cancer risk: a quantitative review. Biomarkers 7:230, 2002.
Leikauf GD, et al: Acute lung injury: functional genomics and genetic susceptibility. Chest 121:70S, 2002.
Lemmon ML, et al: Anaerobic bacteria as a gene delivery system that is controlled by the tumor microenvironment. Gene Ther 4:791, 1997.
Li FP, et al: A cancer family syndrome in twenty kindreds. Cancer Res 48: 5358, 1988.
Li N, et al: Identification of mycobacterial DNA in cutaneous lesions of sarcoidosis. J Cutan Pathol 26:271, 1999.
Liloglou T, et al: Cancer-specific genomic instability in bronchial lavage: a molecular tool for lung cancer detection. Cancer Res 61:1624, 2001.
Lindblad-Toh K, et al: Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nat Biotechnol 18:1001, 2000.
Lindeman N, et al: Gene transcript quantitation by real-time rt-PCR in cells selected by immunohistochemistry-laser capture microdissection. Diagn Mol Pathol 11:187, 2002.
Loda M, et al: Expression of mitogen-activated protein kinase phosphatase-1 in the early phases of human epithelial carcinogenesis. Am J Pathol 149:1553, 1996.
Loda M, et al: Increased proteosome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med 3:231, 1997.
Lubet RA, et al: Use of p53 transgenic mice in the development of cancer models for multiple purposes. Exp Lung Res 26:581, 2000.
Malmgren RA, Flanigan CC: Localization of the vegetative form of Clostridium tetani in mouse tumors following intravenous spore administration. Cancer Res 15:473, 1955.
Mao L: Recent advances in the molecular diagnosis of lung cancer. Oncogene 21:6960, 2002.
Mao L, et al: Detection of oncogene mutations in sputum precedes a diagnosis of lung cancer. Cancer Res 54:1634, 1994.
Mao L, et al: Clonal genetic alterations in the lungs of current and former smokers. J Natl Cancer Inst 89:857, 1997.
P.282
Martinez LA, et al: Synthetic small inhibiting RNAs: efficient tools to inactivate oncogenic mutations and restore p53 pathways. Proc Natl Acad Sci U S A 99:14849, 2002.
Masuda A, Takahashi T: Chromosome instability in human lung cancers: possible underlying mechanisms and potential consequences in the pathogenesis. Oncogene 21:6884, 2002.
Mattson ME, Pollack ES, Cullen JW: What are the odds that smoking will kill you? Am J Public Health 77:425, 1987.
McPherson JD, et al: A physical map of the human genome. Nature 409: 934, 2001.
Meert AP, et al: The role of EGF-R expression on patient survival in lung cancer: a systematic review with meta-analysis. Eur Respir J 20:975, 2002.
Merlo A, et al: Frequent microsatellite instability in primary small cell lung cancer. Cancer Res 54:2098, 1994.
Mills NE, et al: Detection of K-ras oncogene mutations in bronchoalveolar lavage fluid for lung cancer diagnosis. J Natl Cancer Inst 87:1056, 1995.
Miyazaki Y, et al: Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J Clin Invest 96:250, 1995.
Mohr S, Rihn B: Gene expression profiling in human mesothelioma cells using DNA microarray and high-density filter array technologies. Bull Cancer 88:305, 2001.
Murphy M, et al: Detection of concurrent/recurrent non-Hodgkin's lymphoma in effusions by PCR. Hum Pathol 30:1361, 1999.
Nakajima E, et al: K-ras mutation in sputum of primary lung cancer patients does not always reflect that of cancerous cells. Int J Oncol 18:105, 2001.
Nakatsuka S, et al: Pyothorax-associated lymphoma: a review of 106 cases. J Clin Oncol 20:4255, 2002.
Noel-Georis I, et al: Proteomics as the tool to search for lung disease markers in bronchoalveolar lavage. Dis Markers 17:271, 2001.
Noguchi M, et al: Heterogenous amplification of myc family oncogenes in small cell lung carcinoma. Cancer 66:2053, 1990.
Nuovo GJ, et al: In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proc Natl Acad Sci U S A 96:12754, 1999.
Osada H, Takahashi T: Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene 21:7421, 2002.
Palmisano WA, et al: Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res 60:5954, 2000.
Pease AC, et al: Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proc Natl Acad Sci U S A 91:5022, 1994.
Pekarsky Y, et al: FHIT: from gene discovery to cancer treatment and prevention. Lancet Oncol 3:748, 2002.
Peng Y, et al: Silencing expression of the catalytic subunit of DNA-dependent protein kinase by small interfering RNA sensitizes human cells for radiation-induced chromosome damage, cell killing, and mutation. Cancer Res 62:6400, 2002.
Persons DL, et al: Interphase molecular cytogenetic analysis of epithelial ovarian carcinomas. Am J Pathol 142:733, 1993.
Petricoin EF, et al: Use of proteomic patterns in serum to identify ovarian cancer. Lancet 359:572, 2002.
Pinkel D, et al: High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 20:207, 1998.
Popper HH, Winter E, Hofler G: DNA of Mycobacterium tuberculosis in formalin-fixed, paraffin-embedded tissue in tuberculosis and sarcoidosis detected by polymerase chain reaction. Am J Clin Pathol 101:738, 1994.
Ratjen F, Doring G: Cystic fibrosis. Lancet 361:681, 2003.
Rihn BH, et al: Differential gene expression in mesothelioma. FEBS Lett 480:95, 2000.
Rose AC, et al: Optimisation of real-time quantitative RT-PCR for the evaluation of non-viral mediated gene transfer to the airways. Gene Ther 9:1312, 2002.
Ross DT, et al: Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet 24:227, 2000.
Rossi S, Loda M: The role of the ubiquitination-proteasome pathway in breast cancer: use of mouse models for analyzing ubiquitination processes. Breast Cancer Res 5:16, 2002.
Roth JA, et al: Retrovirus wild p53 gene transfer to tumors of patients with lung cancer. Nat Med 2:985, 1996.
Sachidanandam R, et al: A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928, 2001.
Sagawa M, et al: K-ras point mutation occurs in the early stage of carcinogenesis in lung cancer. Br J Cancer 77:720, 1998.
Salgia R, Skarin AT: Molecular abnormalities in lung cancer. J Clin Oncol 16:1207, 1998.
Sassetti CM, Boyd DH, Rubin EJ: Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci U S A 98: 12712, 2001.
Scott FM, et al: Peptide amidating activity in human bronchoalveolar lavage fluid. Lung Cancer 14:239, 1996.
Sellers TA, et al: Evidence for mendelian inheritance in the pathogenesis of lung cancer. J Natl Cancer Inst 82:1272, 1990.
Signoretti S, et al: Detection of clonal T-cell receptor gamma gene rearrangements in paraffin-embedded tissue by polymerase chain reaction and nonradioactive single-strand conformational polymorphism analysis. Am J Pathol 154:67, 1999.
Somers VA, et al: Detection of K-ras point mutations in sputum from patients with adenocarcinoma of the lung by point-EXACCT. J Clin Oncol 16:3061, 1998.
Soria JC, et al: Aberrant promoter methylation of multiple genes in bronchial brush samples from former cigarette smokers. Cancer Res 62:351, 2002.
Sozzi G, et al: Genetic evidence for an independent origin of multiple preneoplastic and neoplastic lung lesions. Cancer Res 55:135, 1995.
Sterman DH, et al: Adenovirus-mediated herpes simplex virus thymidine kinase/ganciclovir gene therapy in patients with localized malignancy: results of a phase I clinical trial in malignant mesothelioma. Hum Gene Ther 9:1083, 1998.
Sueoka N, et al: Molecular pathogenesis of interstitial pneumonitis with TNF-alpha transgenic mice. Cytokine 10:124, 1998.
Swisher SG, Roth JA: Clinical update of Ad-p53 gene therapy for lung cancer. Surg Oncol Clin North Am 11:521, 2002.
Takahashi S, et al: Relationship between postoperative recurrence and expression of cyclin E, p27 and KI-67 in non-small cell lung cancer without lymph node metastases. Int J Clin Oncol 7:349, 2002.
Tamm M, et al: Diagnosis of pulmonary Kaposi's sarcoma by detection of human herpes virus 8 in bronchoalveolar lavage. Am J Respir Crit Care Med 157:458, 1998.
To-Figueras J, et al: Lung cancer susceptibility in relation to combined polymorphisms of microsomal epoxide hydrolase and glutathione S-transferase P1. Cancer Lett 173:155, 2001.
Troesch A, et al: Mycobacterium species identification and rifampin resistance testing with high-density DNA probe arrays. J Clin Microbiol 37: 49, 1999.
Tseng JE, et al: Loss of Fhit is frequent in stage I non-small cell lung cancer and in he lungs of chronic smokers. Cancer Res 59:4798, 1999.
Tsukamoto S, et al: Reduced expression of cell-cycle regulator p27(Kip1) correlates with a shortened survival in non-small cell lung cancer. Lung Cancer 34:83, 2001.
Tuveson DA, Jacks T: Modeling human lung cancer in mice: similarities and shortcomings. Oncogene 18:5318, 1999.
Venter JC, et al: The sequence of the human genome. Science 291:1304, 2001.
Virtanen C, et al: Integrated classification of lung tumors and cell lines by expression profiling. Proc Natl Acad Sci U S A 99:12357, 2002.
Weber G, et al: Identification of foreign gene sequences by transcript filtering against the human genome. Nat Genet 30:131, 2002.
Wen WH, et al: Comparison of TP53 mutations identified by oligonucleotide microarray and conventional DNA sequence analysis. Cancer Res 60:2716, 2000.
Westra WH, et al: K-ras oncogene activation in atypical alveolar hyperplasias of the human lung. Cancer Res 56:2224, 1996.
Wieland I, et al: Microsatellite instability and loss of heterozygosity at the hMLH1 locus on chromosome 3p21 occur in a subset of non-small cell lung carcinomas. Oncol Res 8:1, 1996.
Wikman H, et al: Identification of differentially expressed genes in pulmonary adenocarcinoma by using cDNA array. Oncogene 21:5804, 2002.
Wistuba II, et al: Deletions of chromosome 3p are frequent and early events in the pathogenesis of uterine cervical carcinoma. Cancer Res 57: 3154, 1997.
P.283
Yakubovskaya MS, et al: High frequency of K-ras mutations in normal appearing lung tissues and sputum of patients with lung cancer. Int J Cancer 63:810, 1995.
Yatabe Y, et al: p27KIP1 in human lung cancers: differential changes in small cell and non-small cell carcinomas. Cancer Res 58:1042, 1998.
Yi ES, et al: High c-erbB-3 protein expression is associated with shorter survival in advanced non-small cell lung carcinomas. Mod Pathol 10: 142, 1997.
Zhang Z, et al: Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat Genet 29:25, 2001.
Zhao B, et al: Transgenic mouse models for lung cancer. Exp Lung Res 26:567, 2000.
Zhou BP, et al: HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol 3:973, 2001.
Zinner RG, Kim J, Herbst RS: Non-small cell lung cancer clinical trials with trastuzumab: their foundation and preliminary results. Lung Cancer 37:17, 2002.
Zochbauer-Muller S, et al: 5' CpG island methylation of the FHIT gene is correlated with loss of gene expression in lung and breast cancer. Cancer Res 61:3581, 2001.
Zou Y, et al: Effective treatment of early endobronchial cancer with regional administration of liposome-p53 complexes. J Natl Cancer Inst 90:1130, 1998.
Zou Y, et al: Development of cationic liposome formulations for intratracheal gene therapy of early lung cancer. Cancer Gene Ther 7:683, 2000.
Reading References
Hensel CH, et al: Altered structure and expression of the retinoblastoma susceptibility gene in small cell lung cancer. Cancer Res 50:3067, 1990.
McLaren R, et al: The relationship of p53 immunostaining to survival in carcinoma of the lung. Br J Cancer 66:735, 1992.
Merlo A, et al: 5' CpG island methylation is associated with transcriptional silencing of the tumor suppressor p16/CDKN2/MTS1 in human lung cancers. Nat Med 1:686, 1995.
Mohr S, et al: Microarrays as cancer keys: an array of possibilities. J Clin Oncol 20:3165, 2002.
Niimi S, et al: Resistance to anticancer drugs in NIH3T3 cells transfected with c-myc and/or c-H-ras genes. Br J Cancer 63:237, 1991.
Slebos RJ, et al: K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N Engl J Med 323:561, 1990.
Srivastava M, Eidelman O, Pollard HB: Pharmacogenomics of the cystic fibrosis transmembrane conductance regulator (CFTR) and the cystic fibrosis drug CPX using genome microarray analysis. Mol Med 5:753, 1999.
Xu HJ, et al: Absence of retinoblastoma protein expression in primary non-small cell lung carcinomas. Cancer Res 51:2735, 1991.
Yokota J, et al: Loss of heterozygosity of chromosome 3 in adenocarcinoma of the lung. Proc Natl Acad Sci U S A 84:9252, 1987.
EAN: 2147483647
Pages: 203