101 - Pathology of Carcinoma of the Lung
Editors: Shields, Thomas W.; LoCicero, Joseph; Ponn, Ronald B.; Rusch, Valerie W.
Title: General Thoracic Surgery, 6th Edition
Copyright 2005 Lippincott Williams & Wilkins
> Table of Contents > Volume II > Section XVII - Other Tumors of the Lung > Chapter 118 - Benign Tumors of the Lung
function show_scrollbar() {}
Chapter 118
Benign Tumors of the Lung
Thomas W. Shields
Philip G. Robinson
Benign tumors of the lung are infrequently encountered. Martini and Beattie (1983) reported that less than 1% of the lung tumors resected at Memorial Sloan-Kettering Hospital were benign. Benign tumors may be derived from all cell types present in the lung and may be parenchymal or endobronchial in location. A review by Kuda and associates (1990) of 35 benign tumors removed at the Kyushu Cancer Center revealed 23 hamartomas, 4 sclerosing hemangiomas, 3 benign mesotheliomas (solitary fibrous tumors), and 1 each of the following: plasma cell granuloma, fibroma, lipoma, and pseudolymphoma. Sekine and co-workers (1998) reviewed 32 cases of rare benign and malignant pulmonary tumors from 1976 to 1995. Of the 32 cases, 12 were benign rare tumors, which accounted for 0.34% of pulmonary tumors, and they were composed of papilloma (3/12), leiomyoma (3/12), adenoma (1/12), fibroma (1/12), and meningioma (1/12). The various lesions are listed in Table 118-1, which includes the common, as well as the rare, benign pulmonary tumors.
HAMARTOMA
The most common benign tumor is the hamartoma. Arrigoni and associates (1970) reported that hamartomas account for 77% of all benign tumors of the lungs. Bateson (1973) described this lesion as a benign true neoplasm of fibrous connective tissue of the bronchi encased by a passively included lining of respiratory epithelium. Most often, hamartomas contain cartilage (Fig. 118-1), and fatty tissue is also a frequent component. The tumor has been referred to as a fibrolipochondroma.
Ninety percent of these lesions manifest as a solitary peripheral mass (Fig. 118-2A, B). Khouri and associates (1987) reported that hamartomas represent 4% of all solitary pulmonary nodules. Rarely, multiple lesions are observed, as one of us (TWS) encountered and as recorded by Bennett and associates (1985). The rare cystic pulmonary hamartoma has been reported in the literature at least nine times. A few of these reports were by Jackson (1956), Demos (1983), and K. Miura (1990) and their colleagues. The remaining hamartomas are endobronchial in location. Le Roux (1964) recorded an incidence of 8% of endobronchial hamartomas in 27 patients. Gjevre and associates (1996), however, noted an incidence of only 1.4% in 215 cases of hamartoma seen at the Mayo Clinic. The true incidencce undoubtedly lies somewhere between these two percentages.
Hamartomas are most common in the middle-aged adult, although no age group is exempt. Arrigoni and associates (1970) reported that pulmonary hamartomas are observed twice as often in men as in women. Ge and co-workers (1998) described 67 patients from 1970 to 1997 with pulmonary hamartomas. There were 38 men and 29 women, a male-to-female ratio of 1.3:1. The patient ages ranged from 21 to 82 years, with a mean age of 47 years. The peak incidence was between 40 and 60 years. Thirty-nine percent of the patients were symptomatic with hemoptysis, cough, phlegm, or chest pain. In contrast, Gjevre and associates (1996) found 215 patients with pulmonary hamartomas at the Mayo Clinic between 1976 and 1992. They noted that only 3% were symptomatic and that the ratio of men to women was 2:1. Slow growth of the lesion may be observed (see Fig. 118-2C, D), but the doubling time usually is well above that of malignant lesions (see Chapter 93). Hansen and colleagues (1992) reported that the size of the hamartoma could increase by an average of 3.2 2.6 mm per year. Rapid growth rarely may occur, as observed by Sagel and Ablow (1968).
Most patients with peripherally located lesions are asymptomatic. Only those few patients who have an endobronchial lesion have symptoms that include cough, hemoptysis, and, frequently, repeated or persistent pulmonary infection.
Radiographically, the peripheral lesion, most often located in the lower lung fields, appears as a smooth and well-circumscribed mass; at times, the margins appear lobulated or, more specifically, bosselated (Fig. 118-3). The usual size is 1 to 2 cm, but larger lesions occasionally are
P.1779
observed. Calcifications have been noted in 10% to 30% of these lesions. On computed tomographic (CT) examination, however, Ledor and associates (1981) found identifiable calcification in less than 5% of these tumors. When present, the calcification occurs most often in a diffuse or popcorn distribution. This calcification can be seen on a standard tomogram, but high-resolution computed tomographic (HRCT) scans may demonstrate it more readily (Fig. 118-4). Siegelman and colleagues (1984) also reported that fatty tissue was identified in 50% of the hamartomas evaluated by CT. In the areas of the fat content, the CT number [Hounsfield unit (HU)] is often low ( 40 to 120) and is of diagnostic significance when at least eight voxils are involved. Alternating areas of fat and calcification (HU >175) are also diagnostic. It is now accepted that the presence of a fat density identified by the HRCT scan (Fig. 118-5) in a peripheral solitary lesion is strong presumptive evidence that the lesion is a benign hamartoma, and excision can be deferred.
Table 118-1. Benign Tumors of the Lung | |
|---|---|
|
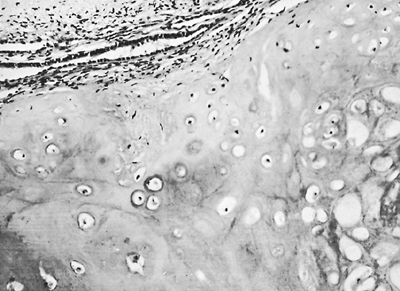 |
Fig. 118-1. Photomicrograph of a hamartoma. Note the predominance of cartilage cells. |
The endobronchial lesions are undetectable radiographically, except that distal parenchymal lung changes (e.g., atelectasis, obstructive pneumonia, or abscess formation) may suggest an obstructing, endobronchial lesion.
Bronchoscopy and biopsy are indicated in any patient with pulmonary symptoms: cough, hemoptysis, repeated pulmonary infection, or atelectasis. Endoscopy is not an essential diagnostic step in patients with a peripheral lesion.
Hamper and colleagues (1985) reported that percutaneous, transthoracic needle aspiration biopsy yields diagnostic information in 85% of hamartomas. Care must be taken when aspirating these peripheral lesions because of their firm consistency. The aforementioned authors reported a 50% incidence of postaspiration pneumothorax, which is twice the incidence after biopsy of other peripheral nodules. One may suspect that the lesion is a hamartoma when fibromyxomatous tissue, which stains metachromatically with Giemsa or Wright's stain, and fragments of low columnar epithelium are present. When fragments of cartilage are present cytologically, the aspiration is diagnostic of a hamartoma. Azua Blanco and colleagues (2001) performed a fine-needle aspiration (FNA) on a pulmonary hamartoma. They found spindle and stellate cells with a fibromyxoid background. These findings enabled them to establish a diagnosis before surgery. The histologic examination of the aspiration has a higher diagnostic yield than cytologic examination of the aspirated specimen. Cartilage is more often demonstrated by standard histologic examination of the aspirated material. Therefore, if the diagnosis of a hamartoma is suspected from the standard radiographic, tomographic, or CT studies, but doubt still remains as to the diagnosis, a needle aspiration biopsy is indicated, particularly in a patient who is a poor candidate for a major
P.1780
operation. A positive result negates the necessity of a thoracotomy.
 |
Fig. 118-2. Hamartoma demonstrated on posteroanterior (A) and lateral (B) radiographs of the chest, presenting as a peripheral mass in the middle lobe. Posteroanterior (C) and lateral (D) radiographs of the chest of the peripheral hamartoma show slow growth of the mass over a 4-year period. |
When the diagnosis was known in a patient with a peripherally located hamartoma, Nili and associates (1979) reported that the patient could be observed without surgical intervention. de Rooij and co-workers (1988) concur with this suggestion, but they believe that if the mass is larger than 2.5 cm, it should be removed. We believe that clinical judgment should be the final determinant as to whether lesions larger than 2.5 cm should be removed. Minimal growth over time may be noted. Unless the growth rate becomes excessive, excision remains unnecessary. In a patient
P.1781
in whom the diagnosis has been established by histologic evaluation of a needle biopsy, the necessity of resection, except under unusual circumstances, can be questioned.
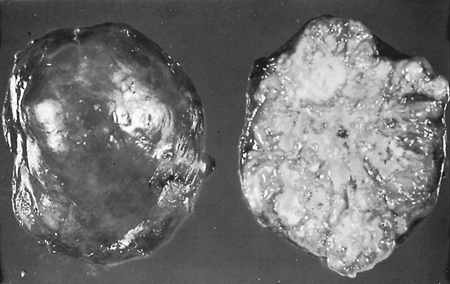 |
Fig. 118-3. Gross specimen of a resected hamartoma showing typical bosselated appearance. |
If a prethoracotomy diagnosis has not been made, video-assisted thoracoscopic removal of a peripherally located suspected hamartoma is an acceptable approach. Thoracotomy and excision may be indicated at times. The least possible amount of normal pulmonary tissue should be excised. At times, when a suspected hamartoma is palpated within the lung, the mass is readily moved and may be advanced to just beneath the visceral pleural surface. Incision of the pleura and enucleation of the mass can then be readily carried out. Any fixation of the mass within the parenchyma of the lung necessitates a standard resection (wedge resection), segmentectomy, or even at times a lobectomy. A pneumonectomy should be avoided if at all possible. Recurrence after excision of a hamartoma is practically unknown. A second, separate primary hamartoma rarely occurs later.
The treatment of endobronchial hamartomas has evolved over the years, and at present, most of these lesions are best managed by endoscopy and laser ablation, as recorded by Cosio and co-workers (2002). This approach was successful in 38 patients, most of whom (86.1%) had distal pulmonary obstruction; although four patients had subsequent local recurrence of the tumors, in three of whom a second laser ablation was curative. In another five patients of the total of 43 patients in the aforementioned series, standard thoracotomy and pulmonary resection were required for the removal of the tumor and the destroyed distal pulmonary parenchma, as one of us (TWS) and Lynn described in 1958.
There have been several reports of malignancy occurring in a hamartoma. Hayward and Carabasi (1967) presented a patient in whom an adenocarcinoma was believed to have developed from a hamartoma, and Basile and associates (1989) recorded a sarcoma that developed promptly at the site of a resected benign hamartoma. Neither of these events is convincing on close scrutiny, and no real evidence exists that either tumor actually arose from an underlying hamartoma. One may be as critical of these reports as were Hayward and Carabasi (1967) of the 12 cases of possible malignancy they reviewed in their own publication. However, of interest in this regard is the possible potential of malignant change. Okabayashi and colleagues (1993) reported a giant hamartoma associated with a high production of carbohydrate antigen 19 9. The significance of this is unknown, but the source of the antigen was demonstrated to be the epithelial component and not the mesenchymal component. Further investigations, such as those of Fletcher and co-workers (1991), might provide insight into any possible malignant potential of these two components of this normally benign biphasic tumor.
Karasik and associates (1980) reported that a bronchial carcinoma (synchronous or metachronous) was identified 6.3 times more often in patients with a hamartoma than would be expected in the normal population. They suggested an etiologic relationship was present. Van den Bosch and colleagues (1987), however, who identified six synchronous and five metachronous bronchial carcinomas in a series of 154 patients with a hamartoma (an incidence of 7%), believed the association was essentially coincidental. In a more recent series of 65 patients with a hamartoma, Ribet and associates (1994) recorded that three patients had an associated bronchial carcinoma, a 6.6-fold increase in the number of cases normally expected. These authors came to the same conclusion that there was an etiologic relationship present, as had Karasik and colleagues (1980). However, in nine other series of a total of 598 patients with hamartomas, the rate of occurrence of a lung cancer was only 5.8%. The question of the nature of the relationship between these two lesions remains unresolved.
OTHER SOLITARY BENIGN TUMORS
Benign tumors of epithelial, mesenchymal, or lymphoid origin are rare. Many of these tumors may be either endobronchial or peripheral in location but generally have a greater predilection for one of the two locations. The symptomatology depends on whether a bronchus is irritated or a bronchial lumen is occluded partially or completely by an endobronchial lesion. The peripherally located tumors usually are asymptomatic.
Primarily Endobronchial Tumors
Benign Endobronchial Fibrous Histiocytoma
A fibrous histiocytoma is a benign lung tumor that is composed of collagen, inflammatory cells, and mesenchymal cells. It has been called by a variety of names, the most common of which are inflammatory pseudotumor, plasma cell granuloma, and fibroxanthoma. We have chosen to discuss
P.1782
the endobronchial lesion separately from the peripheral lesion (see Inflammatory Pseudotumor, later in this chapter).
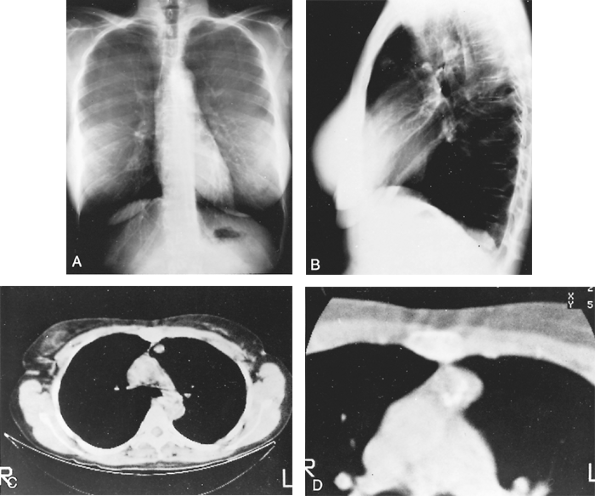 |
Fig. 118-4. A. Posteroanterior radiograph of calcified lesion overlying aortic knob on the left. B. Lateral radiograph shows the lesion to be in the anterior segment of the left lung. C. Computed tomographic scan showing calcification within the mass. D. Enhanced computed tomographic scan revealing popcornlike calcifications in the mass, typical of a hamartoma. |
Duncan and associates (1986) described a patient with endobronchial fibrous histiocytoma that revealed no clinical evidence of malignant behavior. This rare endobronchial lesion has been noted in at least 13 patients, one third of whom were children. The other patients were generally young adults. Tagge and co-workers (1991) described two cases of this lesion and advocated a combination of tumor resection with lavage and selective ventilation to salvage the distal lung as an alternative to pneumonectomy, even if the lung appears to be unsalvageable. Aisner and colleagues (1995) suggested that a wide local excision (i.e., a lobectomy or a bronchial sleeve lobectomy) should be carried out because of the low-grade malignant potential of these tumors. Bueno and co-workers (1996) at the Massachusetts General Hospital concur with this approach and have carried out bronchoplastic resections of five endobronchial fibrous histiocytomas.
Fibrous Polyps and Squamous Papillomas
Drennan and Douglas (1965) divided bronchial papillomas into three groups: (a) multiple papillomatosis, (b) inflammatory polyps, and (c) solitary squamous papilloma. Multiple papillomatosis is usually a disease of children in which the patients develop multiple papillomas of the vocal cords and trachea. These papillomas are thought to be caused by the human papillomavirus. This lesion is discussed in Chapter 78.
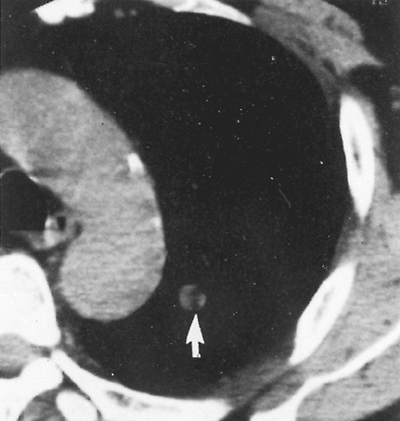 |
Fig. 118-5. Thin-section computed tomographic scan demonstrating a 1-cm pulmonary nodule with fat density material within (arrow). Lesion has appearance consistent with a hamartoma. From Swensen SJ, et al: An integrated approach to evaluation of the solitary pulmonary nodule. Mayo Clin Proc 65:173, 1990. With permission. |
P.1783
Fibrous polyps are in the second group and can be solitary or multiple. They are polypoid areas of bronchial mucosa that have a fibrous stalk and are covered by ciliated columnar epithelium with possible areas of squamous metaplasia. The stalk is usually composed of loose connective tissue with capillaries and an infiltrate of plasma cells, lymphocytes, and eosinophils. These polyps are thought to be secondary to a chronic inflammatory process. They are always benign, but they may cause bronchial obstruction. Arguelles and Blanco (1983) described an asthmatic 10-year-old boy who had multiple bronchial polyps.
Solitary squamous papillomas are in the third group. They are defined as a benign neoplasm of squamous epithelium. Histologically, they have a thin central fibrovascular core that is covered by stratified squamous epithelium, and they form multiple papillary fronds. They occur in adults as solitary lesions rather than the multiple lesions seen in children, although adults may have multiple lesions. H. Miura (1993) and Popper (1994) and their colleagues, as well as Katial and associates (1994), have all reviewed cases of bronchial squamous papillomas. The patients range in age from 22 to 85 years, with men being affected more commonly than women. The most common symptom is cough, but patients may have hemoptysis as well as symptoms secondary to obstruction. Chest radiography may show an area of postobstruction pneumonia or atelectasis. The papillomas are usually located in segmental or more proximal bronchi. They do not have a predilection for either the right or left lung. The human papillomavirus is probably the cause for most of these lesions. Popper and colleagues (1994) demonstrated that human papillomavirus types 11 and 6 were associated with benign papillomas, whereas types 16 or 18, sometimes in combination with type 31, 33, or 35, were found in papillomas of patients who developed squamous cell carcinoma.
Colby and colleagues (1995) described a transitional cell papilloma, which usually occurs in the upper aerodigestive tract but can occur in the bronchial tree. It resembles bladder transitional cell carcinoma, hence the name. On electron microscopy, it has a squamous phenotype and therefore represents a variant of nonkeratinizing squamous papilloma.
H. Miura and colleagues (1993) pointed out that treatment should be conservative, but patients may require further surgery if they develop a malignancy. They suggested that if the lesion is limited to a small area, it could be treated with photodynamic therapy, yttrium-aluminum-garnet laser, or both; these patients should be followed. These aforementioned lesions are usually removed endoscopically, or occasionally, a bronchotomy or sleeve resection may be necessary. When irreversible parenchymal damage distal to the lesion is present, surgical resection of the destroyed lung tissue also is required.
Granular Cell Tumors (Myoblastoma)
Granular cell tumors are rare, benign tumors that used to be called granular cell myoblastomas because they were thought to be derived from skeletal muscle. Fisher and Wechsler (1962) were among the first investigators to suggest that these tumors are of Schwann cell origin. Oparah and Subramanian (1976) and Lui and associates (1989) reviewed the features of the endobronchial granular cell tumors. Deavers and co-workers (1995) reviewed a series of 20 cases. The patients ranged in age from 20 to 57 years and were evenly divided between men and women. In approximately one half of the patients, the tumors were incidental findings. The other patients had symptoms secondary to obstruction, which included postobstructive pneumonia and atelectasis. A few patients had hemoptysis. The chest radiographs showed lobar infiltration, coin lesions, and lobar atelectasis. Some patients had findings caused by other diseases. Solitary lesions were present in 75% of the patients. Multiple pulmonary lesions were present in 10% (two patients). Three other patients (15%) had a solitary pulmonary lesion in addition to the presence of multiple skin tumors.
On gross examination, the tumors ranged in size from 0.3 to 5.0 cm. They are usually located in an endobronchial position in a large bronchus but occasionally may occur in the pulmonary parenchyma. The cut surfaces of the tumor may be tan-white, pink, or yellow. They are usually circumscribed, but not encapsulated. On microscopic examination, the tumors are composed of large cells with an abundant
P.1784
pink granular cytoplasm. Cutlan and Eltorky (2001) described three granular cell tumors that were associated with malignancies (mucoepidermoid carcinoma, squamous cell carcinoma, and adenocarcinoma). The treatment is conservative resection except in those few patients with an associated malignant lesion. Complete resection is curative, although these tumors may recur. Asymptomatic patients may be followed. Epstein and Mohsenifar (1993) used neodymium:yttrium-aluminum-garnet (Nd:YAG) laser to treat an obstructing granular cell tumor and suggested this might be an effective tool for treating these cases in certain instances. Surgical resection of the tumor and any associated damaged lung tissue may be required.
Mucous Gland Adenomas
Mucous gland adenoma, also known as mucous gland cystadenoma, adenomatous polyp, and adenoma of mucous gland type, is a benign tumor of the bronchus that is derived from the mucous glands of the bronchus. The tumor must be composed of cystic glands, be superficial to the cartilaginous plate, be in the bronchus, and have some normal bronchial seromucous glands. This rare tumor has been described by Weinberger and associates (1955), as well as by Gilman (1956), Weiss and Ingram (1961), Kroe and Pitcoc (1967), Emory and associates (1973), and Edwards and Matthews (1981). England and Hochholzer (1995) reported 10 additional cases. Their patients ranged in age from 25 to 67 years, with a mean of 52 years. Historically, this lesion tends to occur twice as often in men as in women, but these authors found a slight predominance in women. The symptoms are cough, fever, recurrent pneumonia, and hemoptysis. The chest radiograph may also show obstructive pneumonitis, postobstructive atelectasis, and on rare occasion, a solitary peripheral lesion. The lesion occurs equally between the right and left sides and more often is found in the major bronchi of the middle and lower lobes. On gross examination, the tumors varied in size from 0.8 to 6.8 cm, with a mean of 1.8 cm. The tumors projected into the lumen of the bronchus. They were usually encapsulated by a thin membrane and easily separated from the bronchus. The cut surface is cystic with mucus within the cystic space.
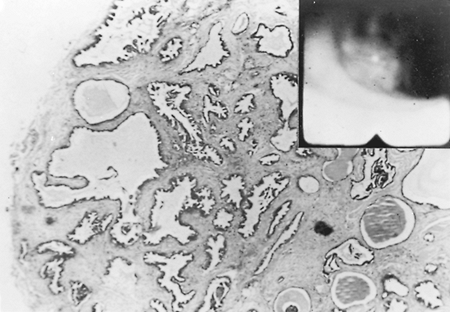 |
Fig. 118-6. Low-power photomicrograph of a mucous gland adenoma. Inset shows the endoscopic appearance. |
Endoscopically, they appear as firm pink masses with intact overlying epithelium (Fig. 118-6). Histologically, they are composed of numerous small mucus-filled cysts lined by well-differentiated mucous epithelium (Fig. 118-7). The major differential diagnosis is low-grade mucoepidermoid carcinoma. Even though these lesions rarely have a stalk, they can be completely removed endoscopically by curettage, cryotherapy, or laser ablation, as reported by Ishida and colleagues (1996). Thoracotomy and surgical resection are indicated only when distal lung has been destroyed or endoscopic removal is contraindicated or incomplete. Complete removal of these tumors endoscopically or surgically results in a permanent cure.
Lipomas
Lipomas arise most often from the wall of the tracheobronchial tree (80%). These lesions are more common in men than in women. They may cause obstruction with pulmonary complications. Bango (1993) and Yokozaki (1996) and their colleagues have urged CT examination to determine the extent of pulmonary involvement. They also suggest bronchoscopic laser vaporization of the tumor as the treatment of choice, although a local resection by bronchotomy or sleeve resection may be required.
Muraoka and associates (2003) reported 64 cases that had been recorded in the Japanese literature. Fifty patients were men and 14 were women. Forty of the lipomas were in the right lung and 23 in the left lung. Sixty-one of the lipomas were found in the first three subdivisions of the bronchial tree, and radiographic findings were present in 78% of the patients. Surgical procedures, including pneumonectomy (4), lobectomy (24), bilobectomy (8), and
P.1785
bronchotomy (4), were required in 57.9% and bronchoscopic removal by Nd:YAG laser (17) or electrosurgical (5) resection, or a combination of both (5), was carried out in the other 42% of patients. Bronchoscopic removal is preferred whenever possible.
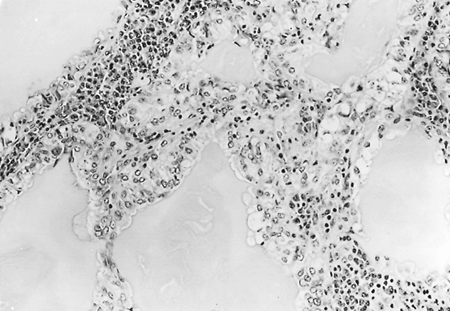 |
Fig. 118-7. High-power photomicrograph of a mucous gland adenoma consisting of cysts of various diameters lined by columnar mucous cells. A chronic inflammatory reaction separates the tubules. |
Pulmonary Chondroma
Pulmonary chondromas are rare pulmonary lesions and are true pulmonary mesenchymal tumors. These tumors should not be confused with hamartomas. According to Carter and associates (1998), most chondromas are endobronchial in location and occur most often in men. Extension beyond the bronchial wall may occur, and this may preclude endobronchial removal. Surgical resection is curative.
In addition to these rare endobronchial chondromas, there is a subgroup of pulmonary chondromas that are found in young women and only infrequently in men that are associated with gastric sarcomas [a gastrointestinal stromal tumor (GIST)] or an extraadrenal paraganglioma. This association has become known as Carney's triad. Unfortunately, this is often also referred to as Carney's syndrome, which is incorrect, as noted subsequently in this chapter. The location of these pulmonary chondromas in Carney's triad is primarily parenchymal, as noted by the radiologic description of these tumors in Carney's 1999 report. These chondromas also have a tendency to be or become multiple in number during the course of the patient's disease (multiple unilateral in 24% and bilateral in 13%). When multiple chondromas are initially discovered in the lung, the other features of Carney's triad should be sought by the appropriate studies, as noted by Yalcin and Kars (2000). As noted previously, these chondromas (although 58% contain calcified areas and occasionally even mature bone formation) should be differentiated from the more common pulmonary hamartomas. On microscopic examination, the most mature bone and cartilage are at the periphery of the lesion, with the central area usually showing degenerative changes. Yalcin and Kars (2000), as well as Kiryu and colleagues (1999), refer to these lesions as chondromatous hamartomas of the lung. It should be noted that Carney's triad is infrequently associated with a true hamartoma, such as in two cases of the total of 65 patients with hamartomas recorded by Ribet and associates (1994). Osteochondromas have also been observed to be infrequently associated with the triad.
Carney's Triad
Carney and co-workers (1977) first described a possibly inherited, familial association of a pulmonary chondroma, a gastric sarcoma (often a leiomyosarcoma located in the gastric antrum or lesser curvature) now referred to as a GIST, and an extraadrenal paraganglioma in seven patients. This condition came to be known as Carney's triad. This is not to be confused with Carney's syndrome, an inherited disorder transmitted as a mendelian autosomal trait, consisting of a melanotic schwannoma, multiple myomas, multiple areas of skin pigmentation, and one or more endocrine disorders, as described in Chapter 189. Carney and associates (1983) further elaborated on the subject and presented a total of 24 patients with the triad. In the most recent review of the triad by Carney (1999), 79 cases were analyzed 67 women and 12 men. The triad has not been observed in African Americans. Most patients were between the ages of 10 to 35 years at the time of diagnosis. Twenty-two percent had all three lesions, and 78% had two lesions. Gastric sarcoma was present in 99%. A combination of (a) a gastric lesion, a pulmonary chondroma, and an extra-adrenal paraganglioma; (b) a gastric lesion and a pulmonary chondroma; (c) a gastric lesion and an extraadrenal paraganglioma; and (d) a pulmonary chondroma and an extraadrenal paraganglioma was present in 22%, 53%, 24%, and 1% of subjects, respectively. The extraadrenal paragangliomas were located in the neck, thorax, or abdomen. Excess catecholamine secretion was noted in 13 patients. The paraganglioma was locally symptomatic in 10 patients. The triad, despite repeated resections (41% of the patients had one or more recurrences of the gastric lesion) is truly a chronic, persistent, indolent disease, with only 16% (13 patients) dying of the disease over a long period of follow-up. Management consists of repeated, judicious surgical resections of the respective tumors. A familial, inherited nature of the disease is yet to be confirmed.
Primarily Benign Parenchymal Tumors
Myoepithelioma and Adenomyoepithelioma
Myoepithelial cells are flat cells that lie between the epithelial cells of a gland and the basement membrane. They are usually found in the salivary glands and are thought to have contractile properties. Myoepitheliomas are benign tumors of these cells that are usually found in the salivary glands or the breasts. These tumors can be divided into two types: (a) myoepitheliomas that are composed of the myoepithelioma cells and (b) adenomyoepitheliomas (epithelial-myoepithelial tumors) that are composed of both epithelial cells and myoepithelial cells. Strickler and co-workers (1987) described a pulmonary myoepithelioma, and Tsuji (1995) and Pelosi (2001) and their associates described adenomyoepitheliomas, or as Pelosi and colleagues (2001) preferred to call them, pulmonary epithelial-myoepithelial tumor of unproven malignant potential (PEMTUMP). They prefer this term because, at present, these tumors appear benign, unlike the epithelial-myoepithelial carcinoma of the salivary gland, which is malignant.
The myoepithelioma described by Strickler and co-workers (1987) was an incidental finding on a chest radiograph in a man in his 60s. On gross examination, the lesion was 3.3 cm in greatest dimension, had tan-yellow-white surfaces, and had well-demarcated margins. On microscopic examination, it was composed of spindle cells that immunostained
P.1786
for S-100 and actin, but not keratin. On electron microscopy, the tumor contained filaments consistent with myofilaments.
Pelosi and associates (2001) reported a PEMTUMP and reviewed the six cases in the world's literature, including the one described by Tsuji and co-workers (1995). In the review, the patients ranged from 47 to 66 years of age, with an average age of 56 years. Tumors were present in four women and three men and ranged in size from 1.3 to 16 cm. The cut surfaces were tan-yellow-white, and the tumors were randomly distributed throughout both lungs. Radiographically, the lesions were solid nodules. On microscopic examination, the tumors showed a biphasic pattern that was composed of glands (epithelial cells) and spindle cells (myoepithelial cells). The epithelial cells stained strongly for keratin and weakly for S-100. In contrast, the myoepithelial cells stained strongly for S-100 and actin, but weakly for keratin. Overall, the patients did well, but some were lost to follow-up, and the longest follow-up was 36 months. None of these patients are known to have died from their disease. Surgery appears curative, but follow-up is advised.
Mucinous Cystadenoma
According to Colby and colleagues (1995), a mucinous cystadenoma is a unilocular cystic lesion whose fibrous wall is lined by well-differentiated, presumably benign columnar mucinous epithelium. This lesion was first described by Sambrook Gowar (1978) and later by Dail (1988). Kragel (1990), Dixon (1993), and Roux (1995) and their colleagues, as well as Graeme-Cook and Mark (1991) and Dail (1994), have further described this entity. These lesions occur in both men and women, who are usually in their 50s and 60s and are smokers. Most of these tumors are discovered as an asymptomatic mass on routine chest radiography. The mass is usually located at or toward the periphery of the lung.
On gross examination, the mass is a unilocular cyst filled with clear gelatinous material. On microscopic examination, a fibrous cyst wall is lined by mucinous epithelium. Occasionally, the wall is thinned, and the mucin extravasates into the adjacent pulmonary parenchyma. The cysts should be examined completely because they may have areas of borderline malignancy or adenocarcinoma. The differential diagnosis ranges from bronchogenic cysts to bronchoalveolar carcinoma. The treatment for these lesions is complete resection, and the prognosis of the benign tumors is excellent.
Mann and associates (2001) reported the local recurrence of a mucinous cystic tumor of borderline malignancy 4 years after its initial resection. They suggested that a mucinous cystic tumor with any histologic identification of evidence of even early malignant features should undergo a lobectomy as the procedure of choice rather than a more limited excision.
Alveolar Adenoma
Yousem and Hochholzer (1986) reported six patients with alveolar adenomas. These tumors are a proliferation of benign alveolar epithelium and septal mesenchyme. The patients were mostly women and ranged in age from 45 to 74 years. Most of the lesions were found on routine chest radiography. Radiographically, Fujimoto and colleagues (2002) describe alveolar adenomas as peripheral, well-circumscribed, solitary nodules. Magnetic resonance (MR) imaging shows the nodules to have a cystic space with fluid and thin rim enhancement. On excision, the tumors averaged 2 cm in diameter and were easily shelled out from the adjacent pulmonary parenchyma. Five of the patients were alive and well at the end of a 12-month follow-up period, with one patient lost to follow-up.
Since the description of Yousem and Hochholzer (1986), Bohm and colleagues (1997), as well as Oliveira and associates (1996), have each described an additional case. Both groups believe this is a distinct benign neoplasm of the lung. Burke and co-workers (1999) reviewed 17 cases and performed a variety of immunohistochemical stains on the tumors. They concluded that alveolar adenonamas were benign neoplasms consisting of an admixture of alveolar epithelium, mostly type 2 pneumocytes, and septal mesenchymal tissue, fibroblasts, or fibroblastlike cells. Oliveira and colleagues (1996) speculate that the lesion is derived from a primitive mesenchymal cell with the capacity to differentiate toward a type 2 pneumocyte lineage. In contrast, Bohm and associates (1997) believe the neoplasm is derived from a benign proliferation of both the type 2 pneumocytes and the septal mesenchyme. Both groups conclude that alveolar adenoma is a distinct, rare, benign pulmonary neoplasm.
Papillary Adenoma of Type 2 Cells (Clara Cell Adenoma)
Papillary adenoma of type 2 pneumocytes is a rare benign papillary neoplasm of type 2 pneumocytic origin that may have admixed Clara cells. This lesion may also be called a Clara cell adenoma, bronchiolar papilloma, or a papillary adenoma of type 2 pneumocytes. Spencer and colleagues (1980) described two cases. Fantone (1982) and Noguchi (1986) and their associates described papillary adenomas of the lung that had ultrastructural differentiation toward type 2 pneumocytes (lamellar bodies) and Clara cells (membrane-bound, electron-dense granules). Hegg (1992), Sanchez-Jimenez (1994), Fukuda (1992), Mori (1996), and Dessey (2000) and their colleagues have all added additional cases to the literature.
The patients range in age from 2 months to 60 years, and the tumor may occur in either gender. The lesions are usually detected in asymptomatic patients as a result of mass radiologic screening. On gross examination, the tumors are usually described as well-demarcated white nodules in the pulmonary parenchyma. Microscopic examination shows a
P.1787
papillary architecture with prominent fibrovascular cores. The epithelial cells, lining the cores, are predominantly cuboidal with basal nuclei and eosinophilic cytoplasm. Ultrastructurally, Clara cells and type 2 pneumocytes are identified. The differential diagnosis includes alveolar adenoma, papillary bronchioloalveolar carcinoma, sclerosing hemangioma, papillary variant of carcinoid tumor, and metastatic carcinoma. Resection appears to be curative, with all of the patients surviving for at least 2 to 10 years. Mori and co-workers (1996), using morphometry with 12-dimensional cluster analysis, found a resemblance of some of the cells to type 2 pneumocyte adenocarcinoma, but their patient was alive with no evidence of recurrence at 3 years. Dessy and colleagues (2000) described two cases of this lesion with infiltrative features. They also found two other cases in the literature with infiltrative features. They proposed changing the name to peripheral papillary tumor of undetermined malignant potential. Infiltrative lesions should be followed closely.
Leiomyoma
Leiomyomas account for about 2% of the benign tumors of the lung. They may occur in the trachea, bronchus, or pulmonary parenchyma (Table 118-2). As Hurt (1984), as well as Arrigoni (1970), Yellin (1984), and White (1985) and their colleagues, noted, the distribution is approximately equal between a tracheobronchial and a parenchymal location. The tumor is most often discovered in young and middle-aged adults and is more common in women than in men. In women who have had a uterine leiomyoma removed in the past, it is difficult, if not impossible, to differentiate a true pulmonary leiomyoma from a benign metastasizing leiomyoma from the original uterine tumor. Thus, in such patients, as noted by Sato and colleagues (1997), meticulous follow-up is necessary. The parenchymal lesions are solitary masses of varying size. Gotti and associates (1993) described one that was a multiloculated mass associated with a large pedunculated cyst that occupied the upper third of the left pleural space. These authors also noted other reports of leiomyomas associated with cyst formation, but most if not all of these were in patients with leiomyomatosis (see later in this chapter in the section, Multiple Benign Tumors). Surgical resection is the treatment of choice. In selected patients with endobronchial lesions without distal destroyed lung tissue, laser resection of the tumor may be possible. Archambeaud-Mouveroux and associates (1988) reported successful management of a benign bronchial leiomyoma by endoscopic use of a Nd:YAG laser. Endobronchial resection without the use of laser is also satisfactory. Kim and colleagues (1993) reported two such cases. Itoh and co-workers (2001) reported the resection of a peripheral leiomyoma by video-assisted thoracic surgery (VATS).
Table 118-2. Leiomyoma of the Lower Respiratory Tract | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
Pulmonary Myelolipoma
Myelolipoma, a benign tumor composed of an admixture of mature adipose tissue and hematopoietic cells (myeloid, erythroid, megakaryocytic, and, at times, lymphoid tissues) is a rare primary pulmonary parenchymal neoplasm. Sabate and Shahian (2002) reported a case, and from their review of the literature, fewer than 10 cases have been recorded. The tumor is recognized as a solid, spherical mass with a sharply defined smooth border on radiographic and CT examination. As noted by Kenney and colleagues (1998), the CT features are variable as a result of the different densities of the two components that compose the tumor as well as the relative proportions of either present. MR imaging of these tumors has been presented by Kammen and associates (1998). The benign nature and diagnosis of the lesion may be confirmed by FNA. The treatment may be expectant, although conservative surgical excision may be elected if major growth or symptoms occur.
Benign Neurogenic Tumors
Benign neurogenic tumors [neurilemoma (neurinoma, schwannoma) or neurofibroma] may occur in the lung, but nothing distinctive is evident in these tumors. Yamakawa and colleagues (1993) reviewed 20 examples of a neurilemoma in the Japanese literature. The sex distribution was equal and the tumor could occur at any age. Six were in a major bronchus and the other 14 were located within the lung parenchyma. Sugita and co-workers (1996) performed a sleeve resection on a neurilemoma to preserve lung capacity. McCluggage and Bharucha (1995) pointed out that the neurogenic tumor may be difficult to classify because of degenerative changes. In this case, a special stain for S-100 may be helpful because the neurogenic tumor stains positively.
In contrast, most neurofibromas occur in an endobronchial location, but these endobronchial neurofibromas are rare. In a recent review of the literature, Hsu and associates (2002) were only able to identify eight cases. Most were in young to middle-aged adults, only three were in women, and interestingly, all the tumors were located in the right bronchial tree. Surgical resection or endobronchial ablation is indicated as dictated by the extent and location of the tumor.
Simansky and associates (2000)* reported the occurrence of a psammomatous melanotic schwannoma in the
P.1788
lung. This unusual tumor is most commonly found in the costovertebral area of the thorax arising from a neurogenic structure. It may be associated with myxomas, skin pigmentation, and endocrine overactivity, as it was in the aforementioned case reported by Simansky and associates (2000).* This latter associated group of findings is known as Carney's syndrome (1990) and is transmitted as a mendelian autosomal dominant trait (see Chapter 189). This is not to be confused with Carney's triad. This rare pulmonary tumor most often is benign, but malignant behavior occasionally may be seen (see Chapter 119). On immunohistochemical studies, the psammomatous melanotic schwannoma is positive for S-100 protein, vimentin, and HMB-45 and negative for the presence of chromogranin, synaptophysin, and keratin. Treatment is surgical removal.
Cavernous Hemangioma
Yousem (1989) reviewed the pulmonary vascular lesions and believed that benign ones include cavernous hemangiomas, arteriovenous malformations (see Chapter 82), capillary hemangiomas, and pulmonary telangiectasia. Cavernous hemangiomas are rare lung tumors that in reality are pulmonary arteriovenous malformations. Early on, Wodehouse (1948) described four patients with pulmonary hemangiomas, and Forsee and associates (1950) described this tumor in a 20-year-old man with cyanosis. Galliani and colleagues (1992) described a cavernous hemangioma in a 10-week-old male infant. They emphasized that if the lesion is a true arteriovenous malformation, the possibility of hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber disease) should be considered. Silverman and colleagues (1994) studied patients with pulmonary venous malformations by MR imaging. They concluded that by using their criteria, MR imaging was an excellent noninvasive modality for evaluating these lesions. Pulmonary telangiectasia is usually associated with Rendu-Osler-Weber disease or is found in individuals with cirrhosis of the liver. Solitary cavernous hemangiomas are excised surgically. Paul and associates (1991) and Cohen and Kaschula (1992) have described capillary hemangiomas in infants. Paul and associates (1991) described how their patient developed a bronchial obstruction, and they performed a successful sleeve resection of the right main-stem bronchus.
Table 118-3. Pulmonary Disorders of the Lymphatic System | ||
|---|---|---|
|
Lymphatic Lesions
Lymphatic lesions of the lung and thorax are rare and, according to Faul and associates (2000), who have recently reviewed the subject, can be divided into four basic catagories (Table 118-3): (a) lymphangioma, (b) lymphangiectasis, (c) lymphangiomatosis, and (d) lymphatic dysplasia, as well as some additional categories that are not listed in the table. The occurrence of a solitary intrapulmonary lymphangioma is rare. The lesion is usually a small peripheral nodule, and no clue as to its nature is readily given in standard radiographs of the chest. However, HRCT usually shows the lesion to be a cystic lesion with a smooth border. The tumor, as a rule, is asymptomatic but occasionally may be associated with dyspnea, as in the case in a child reported by Kim and colleagues (1995), or with hemoptysis as reported by Holden and associates (1987). A pneumothorax may also be seen with these cystic lesions. Pathologically, the mass is composed of benign-appearing contiguous interconnecting lymphatic spaces, as described by Langston and Askin (1995). A small number of cases have been reported in the past several years (1994 to 2001) by Takemura (1995), Takahara (1998), and Wilson (2001) and their associates. Treatment consists of limited but complete surgical removal by one of the many appropriate surgical techniques. The outlook for success is excellent.
In the article of Faul and associates (2000), the authors clearly define the other three entities. Pulmonary lymphangiectasis is found in a primary and secondary form. The primary form is usually found in neonates, and it causes a severe respiratory failure that is usually fatal. The secondary form results from a pathologic process that impairs lymph drainage and increases lymph production. Histologically, the visceral pleura has a network of dilated capillaries. Lymphangiomatosis is the presence of multiple lymphangiomas. Histologically, the lesions resemble solitary lymphangiomas. This disease frequently presents in late childhood and is equally distributed between boys and girls. Patients with pulmonary involvement have a poor prognosis. The last group of diseases is the lymphatic dysplasia syndrome. This entity includes primary lymphedema syndromes, congenital chylothorax, idiopathic effusions, and the yellow nail syndrome. Lymphatic dysplasia consists of a primary lymphedema with prominent fibrous septation in the subcutaneous fat. The condition is more frequent in women than men. Clinically, patients develop recurrent chest infections, pleural effusions, and bronchiectasis. Their prognosis is related to the severity of the bronchiectasis.
P.1789
Pleomorphic Adenoma (Mixed Tumor)
Pleomorphic adenomas, mixed tumors, or salivary gland type tumors are benign tumors composed of epithelial and myoepithelial cells, usually set in a cartilaginous stroma. Sakamoto and colleagues (1991) reported one case of pleomorphic adenoma in the lung and reviewed six other reported cases. The patients ranged in age from 47 to 74 years, with an average age of 57 years. Both sexes were equally affected by these tumors. The clinical symptoms included pneumonia and cough; one patient was asymptomatic. Moran (1995) also summarized the findings in 16 patients with pleomorphic adenomas. His patients ranged in age from 35 to 74 years, and most were women. The tumors could be either endobronchial or parenchymal in location, but no predilection occurred for a particular lung or segment. On microscopic examination, Moran (1995) pointed out that the pulmonary tumors do not have as prominent a cartilaginous stroma as do the salivary gland tumors. Treatment is surgical excision. These tumors can also occur in the trachea.
Nodular Amyloid
Hui and colleagues (1986) reported 48 cases of amyloid involving the upper and lower respiratory tracts. They divided their cases into three types: (a) tracheobronchial, (b) nodular pulmonary, and (c) diffuse (interstitial) pulmonary. We focus on the nodular form. Nodular pulmonary amyloidosis is a focal collection of amyloid in the lung, usually with a surrounding giant cell reaction. It can occur as either a solitary nodule or multiple nodules. Hayes and Bernhardt (1969), as well as Desai (1979) and Laden (1984) and their colleagues, described cases of nodular pulmonary amyloidosis. The patients range in age from young to old, but most patients tend to be in their sixth and seventh decades of life, and both sexes are affected equally. Higuchi and associates (1997) collected 34 cases of primary nodular pulmonary amyloidosis from the Japanese literature and added one case of their own. Sixteen of the patients had single lesions, and 19 had multiple lesions. Most of the patients, as in the aforementioned series, were middle-aged or older adults. Patients are usually asymptomatic, and the amyloid tumors are discovered on incidental chest radiography. Surgical resection is considered curative. Multiple myeloma does not appear to be frequently associated with these lesions, but the patients should be evaluated for the possibility of this disease.
These patients with nodular pulmonary amyloidosis should undergo long-term follow-up because of the occasional association with macroglobulinemia and the occurrence of malignant lymphoma, as noted by Kyle and Garton (1987). Lim (2001) and Dacic (2000) and their co-workers addressed the literature relative to the link between nodular amyloid and lymphoma. Lim and associates (2001) concluded that marginal-zone lymphomas of the mucosa-associated lymphoid tissue (MALT) type could be found in association with nodular amyloid. Dacic and colleagues (2000) described histologic and immunohistologic observations that could be employed to separate nodular amyloid from malignant lymphoma. The lymphomas were identified by lymphatic tracking of the lymphocytic infiltrate, pleural infiltration, and sheetlike masses of plasma cells. Immunohistochemically, the lymphomas showed a dominant CD20+, CD79a+ B-cell population with light chain restriction and aberrant antigen expression of CD20/CD43.
Inflammatory Pseudotumor
Inflammatory pseudotumors are benign tumors that have a wide histologic spectrum. Over the years, they have been called fibroxanthoma, histiocytoma, xanthofibroma, xanthoma, xanthogranuloma, mast cell granuloma, plasma cell granuloma, and, incorrectly, a sclerosing hemangioma. Colby and co-workers (1995) divide the inflammatory pseudotumors into two major groups, according to their histology: (a) fibrohistiocytic and (b) plasma cell granuloma. Berardi and associates (1983) reviewed this subject and found that these tumors could occur in patients of any age, with an average age of 29.5 years. No predilection for either sex was evident. Most (74%) of the patients were asymptomatic. Patients may have obstructive symptoms if the lesion is located in the bronchus. Cohen and Kaschula (1992) described these lesions as the most common lung tumor of childhood. Matsubara and co-workers (1988) postulated that these tumors originate as an organizing intraalveolar pneumonia. Gomez-Roman and colleagues (2001) described human herpes virus-8 as being expressed in inflammatory pseudotumors, which raises the possibility that this virus may play an etiologic role in the pathogenesis of the tumor. Ishida and colleagues (1989) reported seven patients with this lesion and described its clinicopathologic features. Agrons and associates (1998) described the chest radiographs as showing a solitary, peripheral, sharply circumscribed mass, usually in one of the lower lobes. Diagnosis and treatment are often made by resection of the mass. Fine-needle aspiration may yield confusing results because many malignancies may be surrounded by reactive connective tissue. These lesions are best treated by a conservative surgical excision, but with the realization that they may recur if they are not completely excised. Doski and colleagues (1991) used corticosteroids to lessen infiltration in an unresectable plasma cell granuloma. Imperato and co-workers (1986) used radiation therapy to treat lesions that could not be completely resected. Gal and associates (1994) addressed the problem of tumor extension into the mediastinum, recurrence, and blood vessel invasion. They believed there was an intermediate form between malignant fibrous histiocytoma and inflammatory pseudotumors that could be locally recurrent.
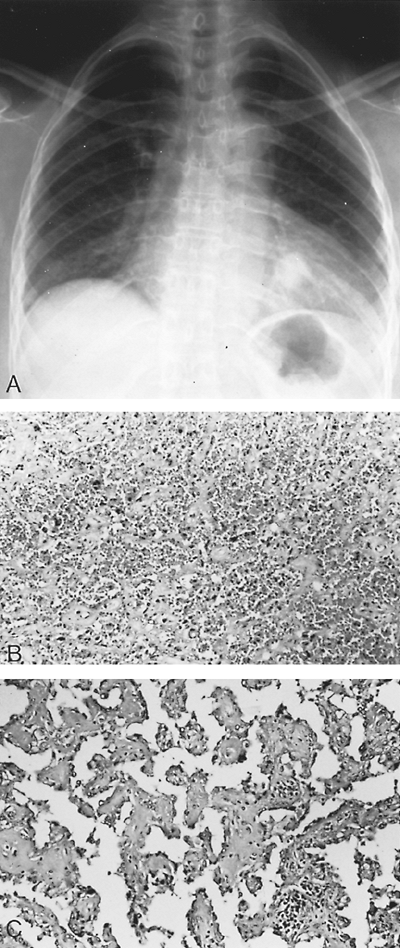 |
Fig. 118-8. A. Radiograph of the chest of an asymptomatic 63-year-old woman with a mass in the left lower lung field that had been present for more than 5 years. Recent growth was noted. Removal and histologic examination of the mass revealed a sclerosing hemangioma. B. Low-power photomicrograph of the sclerosing hemangioma. Dense solid areas contain typical polygonal cells. C. High-power photomicrograph shows papillary areas with the fibrovascular cores lined by cuboidal cells. |
P.1790
Sclerosing Hemangioma
Sclerosing hemangioma, originally described by Liebow and Hubbell (1956), is a benign lung tumor of undetermined histogenesis. Katzenstein and associates (1982) described 51 cases that Liebow and Hubbell saw after their initial description in 1956. These authors noted that the patients ranged in age from 15 to 69 years, with an average age of 42 years. Eighty-four percent of the patients were women. Most (78%) of the patients were asymptomatic. Those who were symptomatic complained of hemoptysis, vague chest pain, or both. On radiographic study, the sclerosing hemangioma appears as a solitary nodule that is found more often in one of the lower lobes (Fig. 118-8). Sugio and co-workers (1992) described an additional 10 cases of this tumor with similar clinical findings to those of Katzenstein and associates (1982). Most of these tumors are peripheral in location (95%), and the tumors are solitary in 96% of patients. In Devouassoux-Shisheboran and associates' (2000) review of 100 of these tumors, four were located in the pleura and one each endobronchially and in the mediastinum. Three-fourths of the tumors were less than 3 cm in size. The histologic pattern consisted of two cellular types: (a) surface cells that are cuboidal in shape, and (b) round (polygonal)-shaped cells within the surface layer. The architecture is made up of a combination of two or more intermixed four patterns: (a) papillary, (b) sclerotic, (c) solid, and (d) hemorrhagic. Calcifications are present in approximately one third of the tumors. A few are associated with a tumorlet, but no carcinoids have been described. Of all of the sclerosing hemangiomas reported (probably in the range of less than 200 cases), four patients (2%) have been reported to have had metastatic spread of the sclerosing hemangioma to one or several bronchial or hilar lymph nodes. These cases were reported by Tanaka (1986), Devouassoux-Shisheboran (2000), and Yano (2002) and their colleagues, as well as by Chan and Chan (2000). These tumors have been large in size (generally, >5 cm), and the lymph nodes that contain the metastatic disease are also increased in size. Nodal dissection is indicated when both of these findings are present.
The origin of the cells (surface or round) has been in doubt. However, Sugio and co-workers (1992) cited studies that suggest the tumor may be derived from type 2 alveolar pneumocytes. One of these studies was reported by Yousem and colleagues (1988). These latter investigators performed immunohistochemical studies on eight sclerosing hemangiomas and concluded that the tumor was of epithelial origin, with evidence of both bronchiolar and alveolar pneumocyte differentiation. Nagata and co-workers (1987) also supported the concept of epithelial origin from type 2 pneumocytes. Despite these aforementioned studies, Leong and associates (1995), from an extensive immunohistochemical and ultrastructural analysis of 25 sclerosing hemangiomas, concluded that although the tumor cells are of epithelial origin, they believe these cells do not clearly correspond
P.1791
to any cell currently recognized in the periphery of the lung. They strongly believe no good evidence exists of pneumocyte origin. In a subsequent publication, Leong and associates (1997) listed three reasons for their aforementioned conclusion: (a) by light microscopy, the pneumocytes are morphologically different than the tumor cells; (b) by electron microscopy, the pneumocytes are clearly separated from the underlying tumor cells, and (c) the immunophenotype expressed by pneumocytes differs from that of the tumor cells. These authors further opine that the name of this tumor is inappropriate as there is no evidence to implicate endothelial differentiation. As with any other controversial situation, progress does indeed occur. Recent reports in the year 2000 by Devouassoux-Shisherboran and associates and by Chan and Chan have shed further light on the subject. With the use of immunohistochemical studies, both groups have shown that both the cuboidal (surface) cells and the round (polygonal) cells stain positive for the epithelial membrane antigen (EMA) and the thyroid transcription factor-1 (TTF-1). The latter is expressed in thyroid and pulmonary epithelial cells, such as type 2 pulmonary and Clara cells. The cuboidal cells also stain positively for Clara cell antigen, but the round cells do not. The latter cells are also negative for the presence of surfactant protein A and B. These latter negative responses suggest that the round cells are, in truth, primitive respiratory epithelium. Chan and Chan (2000) have shown the cuboidal cells to be cytokeratin positive as well as surfactant aproprotein A positive, whereas the polygonal cells are negative to both. Why this discordancy is present remains unresolved. Nonetheless, it is reasonable to suggest that both cellular types arise from the same embryologic permordial cells.
Regardless of the controversy as to the cellular origin of a sclerosing hemangioma or as to the correctness of its name, one can reasonably recommend surgical excision of the lesion as the procedure of choice. Other than local lymph node dissection around the bronchus of origin of the tissue removed, a standard extensive nodal dissection is not indicated unless enlarged lymph nodes are present or calcium-containing nodes are present on the CT preoperative evaluation of the patient's lung.
Sugar Tumor (Clear Cell Tumor) of the Lung
Benign clear cell or sugar tumor of the lung is a benign lung lesion of unknown histogenesis. The tumor was first reported by Liebow and Castleman (1963), who later described a series of 12 cases (1971). Seventy-five percent of the lesions occurred in patients between 45 and 59 years of age. The tumors were equally distributed between men and women. All of the patients were asymptomatic. Similar age and gender distribution were noted in the 14 cases in the Japanese literature reviewed by K. Miura and associates (1993). Radiographically, the lesions were solitary and peripheral and ranged in size from 1.5 to 3 cm. Excision is curative. From their light microscopic, histochemical, and ultrastructural study of the tumor, Andrion and colleagues (1985) suggested it is derived from either epithelial nonciliated bronchiolar (Clara cells) epithelium or epithelial serous cells. Gaffey and co-workers (1990) studied eight clear cell tumors of the lung by electron microscopy and immunohistochemistry. They found some evidence of neuroendocrine differentiation in some of the tumor cells, but they could not be certain as to the cell of origin for this tumor. In a subsequent report, Gaffey and colleagues (1991) noted that the sugar tumor cells were uniformly negative for epithelial features but were strongly reactive for human melanin black or melanosome-associated protein (HMB-45) and positive for S-100 protein. Hashimoto and co-workers (2001) analyzed one clear cell tumor by special stains, immunohistochemistry, and electron microscopy. They concluded that the tumor cells had features of pericytes, melanocytes, and neuroendocrine cells.
Leong and Meredith (1997) not only have extensively discussed the possible histogenesis of the benign sugar tumor but also have stressed the necessity of differentiating these benign lesions from the numerous malignant clear cell tumors that can be encountered in the lung. Metastatic renal cell carcinoma or a primary clear cell carcinoma of the lung are the most troubling that are encountered. The aforementioned authors suggest that the immunohistochemical features are most important in distinguishing these various tumors (Table 118-4). Searching for an asymptomatic primary tumor is most often fruitless.
Pulmonary Paraganglioma
Primary pulmonary paraganglioma and chemodectoma are rare. Singh and colleagues (1977) reported one case and reviewed the literature, finding only 11 cases up to that time. Tanimura and associates (1993) found a total of 23 case reports and added one case of their own. More recently, Saeki and co-workers (1999) described a pulmonary pheochromocytoma that was 13 cm in greatest dimension with the patient alive and well with no evidence of disease 5 years after surgical resection. Kwekkeboom and associates (1993)
P.1792
have suggesed the use of octreotide scintigraphy for the detection of paragangliomas. Muros and colleagues (1998) report that indium 111 (111In) pentetreotide is superior to iodine 123 (123I) metaiodobenzylguanidine (MIBG) scintigraphy in the diagnosis and location of chemodectomas. Whether or not octreotide scintigraphy would be helpful in defining this rare lesion in the lung is unknown. Furthermore, the octreotide scintigraphy likewise would be picked up by a carcinoid tumor in the lung. This would unfortunately pose a problem because, histologically, one of the major problems with this tumor has been the inability to distinguish it from a carcinoid tumor. Tanimura and colleagues (1993) suggested the use of immunohistochemical staining techniques to aid in this differentiation. They noted that Googe and co-workers (1988) reported that paragangliomas had a positive reaction to S-100 protein and were negative for cytokeratin and serotonin, whereas carcinoid tumors were negative for S-100 protein but were positive for cytokeratin and serotonin. Therefore, results of these studies should be used when considering the cellular origin of a tumor that histologically simulates a possible paraganglioma. Rarely, a pulmonary paraganglioma, although its histologic features are those of a benign lesion, may metastasize to the regional lymph nodes; such a tumor must be considered malignant. Hangartner (1989) and Lemonick (1990) and their colleagues each reported such a case. Pulmonary paragangliomas should not be confused with the so-called minute chemodectoma of the lung, which are now known as minute meningotheliallike nodules, as described by Torikata and Mukai (1990). These are multiple minute tumors that are usually found incidentally on microscopic examination of the tissue. The origin of these latter tumors is obscure.
Table 118-4. Immunophenotype of Common Clear Cell Tumors of the Lung | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Pulmonary Gangliocytic Paraganglioma
Gangliocytic paraganlioma is a rare tumor that is usually found in the duodenum. The tumor is composed of three types of cells: (a) ganglionlike cells, (b) neuroendocrine cells, and (c) Schwann cells. It is thought to represent either a hyperplastic or neoplastic proliferation. Nearly all duodenal cases have a benign course despite the presence of lymph node metastasis. Hironaka and associates (2001) reported the first case of one of these tumors occurring in the lung. The patient was a 75-year-old man who complained of right anterior chest pain. The chest radiograph revealed atelectasis of the right middle and lower lobes of the lung. A CT scan showed a 1.5-cm endobronchial mass almost occluding the bronchus at the lower portion of the truncus intermedius. The patient underwent a bilobectomy of the right middle and lower lobes. There was no lymph node involvement. Grossly, the yellow polypoid tumor measured 1.6 cm in greatest dimension. Microscopically, the tumor consisted of three cell types. The first was a round endocrine cell with an oval nuclei, stippled chromatin, and eosinophilic cytoplasm. These cells had a Zellballen arrangement. The second was a large ganglionlike cell. The third was a spindle-shaped cell (Schwann cells). Immunohistochemically, the endocrine cells stained with cytokeratin (CAM 5.2), synaptophysin, and chromogranin. The ganglionlike cells stained with neurofilaments. The spindle-shaped cells stained with S-100 and neurofilaments. This staining pattern is consistent with the respective cells. The endocrine cells also stained for -human chorionic gonadotropin ( -HCG), calcitonin, and somatostatin. The ganglionlike cells stained with somatostatin. The treatment is surgical resection.
Glomus Tumor
According to Marchevsky (1995), a glomus tumor (glomangioma) is derived from the cells of a special arteriovenous shunt, the Suquet-Hoyer canal. They are generally located in the nail beds as well as the pads of the fingers and toes. They are involved in temperature control. On rare occasions, these tumors may be found in the lung as a solitary nodule. Koss (1998) and Gaertner (2000) and their co-workers reported a total of seven cases of glomus tumors of the lung. The patients ranged in age from 20 to 67 years, with an average age of 45 years. Six of the patients were men, with only one woman. For the most part, the patients were asymptomatic. Most of the lesions were in the right lung, but one was in the left main-stem bronchus, and one was in the parenchyma of the left lung. On chest radiography, pulmonary glomus tumors appear as solitary nodules. On gross examination, the tumors were well circumscribed. They ranged in size from 1.1 to 6.5 cm in greatest dimension and had a yellow-tan to white cut surface. Microscopically, the tumors demonstrated a combination of blood vessels, polygonal cells, and varying stroma. Immunohistochemically, all of the tumors stained strongly for smooth muscle actin and muscle-specific actin, and less strongly for vimentin. Several of the tumors stained for desmin. These tumors should be distinguished from hemangiopericytomas, carcinoids, paragangliomas, and smooth muscle tumors. All of the cases were treated surgically, with three patients having a lobectomy, three a wedge resection, and one a sleeve resection. Two patients were lost to follow-up, but the other five patients were followed for 6 months to 5 years, and all were free of disease.
Teratoma
Teratomas occur rarely as primary lung tumors, but several cases have been reported recently (see Reading References). More than 20 cases have been reported, but many of the early reports may have represented anterior mediastinal teratomas that had extended into the lung. Most that do occur in the lung are found in the anterior segment of the left upper lobe. Resection is curative.
P.1793
Pulmonary Meningioma
Meningiomas in the pulmonary parenchyma can be primary or metastatic. One of us (PGR) (1992), as well as Flynn and Yousem (1991) and Drlicek and co-workers (1991), described primary pulmonary meningiomas. Lockett (1997), Kaleem (1997), and Cesario (2002) and their co-workers have reviewed and described primary pulmonary meningiomas. Spinelli and associates (2000) have suggested that this tumor may arise from meningotheliallike nodules found in the lungs. Patients with this tumor are more often women (59%), and the age range is 24 to 74 years, with a mean age of 61 years. The patients generally present with an asymptomatic nodule on chest radiography. Grossly, the tumor is usually a well-circumscribed, round, gray-to-white nodule ranging in size from 1.7 to 6.0 cm in greatest dimension. Multiple lesions occur rarely, as reported by Ueno (1998) and de Perrot (1999) and their colleagues. Microscopically, the tumors are composed of meningothelial cells with psammoma bodies. Electron microscopically, the tumors contain interdigitating cell membranes and desmosomes. The tumor cells immunohistochemically stain consistently with vimentin and variably with epithelial membrane antigen, but not with keratin, S-100, or neuron-specific enolase. Moran and co-workers (1996) concluded that vimentin and epithelial membrane antigen are the most reliable immunologic markers of these tumors. The treatment is surgical excision, and the prognosis is excellent. Unfortunately, a primary pulmonary meningioma may be malignant. Two cases have been reported, one by Erlandson (1981) and one by Prayson and Farver (1999). The fate of the former patient was unknown, but the second patient had a rapid downhill course and had both local and distant disease within 5 months. Thus, of the 29 cases reviewed by Cesario and colleagues (2002), the incidence of possible malignancy is 6.8%.
Wende (1983), Miller (1985), and Kodama (1991) and their colleagues reported cases of metastatic meningioma from the cranium to the lung. A patient in whom a pulmonary meningioma is diagnosed should be examined to exclude an intracranial lesion.
Hyalinizing Granuloma
Pulmonary hyalinizing granuloma is a tumor of dense hyalinized connective tissue that occurs in the lung as a result of inflammatory or postinflammatory changes. Engleman and associates (1977) first described the lesion. Yousem and Hochholzer (1987) summarized 24 cases. The tumor occurs in patients between 24 and 77 years of age, with an average age of 42.3 years. The lesions are equally distributed between men and women. Patients may be asymptomatic or complain of cough, shortness of breath, chest pain, or weight loss. The lesions are nodular and vary from a few millimeters to 15 cm in greatest dimension. Many of the patients have multiple lesions, with most being bilateral. More than one half of the patients who develop hyalinizing granulomas have a history of an autoimmune disorder or a past history of fungal or mycobacterial disease. John and associates (1995) described a case in a patient with multiple sclerosis, and Kuramochi and colleagues (1991) described one in a patient with systemic idiopathic fibrosis.
Fibroma (Solitary Fibrous Tumor and Localized Fibrous Mesothelioma)
A fibroma is known by a variety of names. Colby and associates (1995) prefer the term intrapulmonary localized fibrous tumor, but this lesion has also been called solitary fibrous tumor, intraparenchymal localized fibrous mesothelioma, intrapulmonary fibrous mesothelioma, and inverted fibrous tumor of the pleura. These benign tumors have a histologic appearance that is identical to that of a localized fibrous tumor of the pleura (see Chapter 64). These tumors are most commonly found arising from the visceral pleura of the lung, but occasionally they are intrapulmonary. They also may be found in the mediastinum, retroperitoneum, and external surface of the stomach and small intestine. Rarely, as reported by Yousem and Flynn (1988), a fibrous tumor can occur in the lung tissue without any connection to the visceral pleural layer. Histologically, the tumors are composed of spindle cells with dense bundles of collagen. According to a review by Khalifa and associates (1997), the cells immunostain for vimentin and CD34. A malignant variant of this tumor exists. Adequate resection is curative.
Primary Pulmonary Thymoma
A primary thymoma may occur in the lung. In all such cases, the mediastinal thymus gland must be normal, and a thymoma in the mediastinal gland or the history of a previous resection of a thymoma must be absent. Kalish (1963) considered the primary intrapulmonary thymomas as either peripheral or hilar in location. In the latter instance, the thymoma must be inside the visceral pleura because ectopic mediastinal thymic tissue can be found in the aortopulmonary window as well as in the aortocaval groove (see Chapter 155). Twenty cases have been reported in the literature, with a preponderance of the cases in women. Two patients had myasthenia gravis.
The tumor is usually found in older individuals. The location of either the hilar or the peripheral tumors may be in either lung. The more recent cases have been described by James (1992), Moran (1995), and Veynovich (1997) and their colleagues. Fukayama (1988) and Veynovich (1997) and co-workers used immunohistochemical techniques to identify thymic T lymphocytes in the tumor to differentiate the tumor from lymphoepitheliallike carcinoma of the lung and from primary lymphomas of the lung. Moran and associates (1995) used different immunohistochemical studies
P.1794
to determine the nature of the epithelial cells in six of their eight cases. These cells had positive results for keratin and the epithelial membrane antigen, but negative results for vimentin, desmin, actin, and S-100 protein. Although these studies do not differentiate the cells from being squamous in origin, the benign appearance and the absence of mitotic activity of these epithelial cells rule out the possibility of an undifferentiated squamous cell tumor. When localized, as most of these primary thymomas of the lung are, surgical resection appears to be curative. When the tumor is extensive with involvement of the pleura and is thought not to be resectable, which is rare, therapeutic radiation therapy has resulted in long-term survival.
Multiple Benign Tumors
Benign Metastasizing Leiomyoma
Rare, multiple benign peripheral lesions, such as the benign metastasizing leiomyoma, may be seen. The exact nature of the lesions remains undetermined. Kayser and colleagues (2000) studied 10 women with benign metastasizing leiomyomas who had undergone hysterectomy for leiomyomas. They compared the metastasis with the original leiomyoma by using a variety of markers, such as Ki-67 and estrogen and progesterone receptors. They concluded that benign metastasizing leiomyomas are a slow-growing variant of uterine leiomyosarcoma. These tumors occur in young women and frequently are associated with a leiomyoma of the uterus. The lesions may grow. Radiographically, Abramson and colleagues (2001) thought that the disease is characterized by numerous well-defined randomly distributed pulmonary nodules. Regression of these tumors can occur after oophorectomy. Winkler and colleagues (1987) suggested resection of the pulmonary lesions when feasible. Gal and co-workers (1989) described a series of 12 patients with smooth muscle tumors of the lung. In five of these patients, the tumors were thought to be from the uterus, and four of these patients died of their disease.
Parenti and colleagues (1992) reported a case and reviewed the subject. These authors pointed out that no standard of treatment exists for benign metastasizing leiomyoma. Patients are usually managed with a combination of surgery, hormonal manipulation, and chemotherapy. Jautzke and colleagues (1996) tested five benign metastasizing leiomyomas for estrogen and progesterone receptors. All had a high content of progesterone receptors, and four of five had a high content of estrogen receptors. These findings give a scientific basis for treating patients with hormonal manipulation. Jacobson and associates (1995) treated a patient with goserelin, a luteinizing hormone releasing hormone analogue, and showed improvement in the patient's blood gas analyses and chest radiograph. The exact etiology of this disease is unknown, but Takemura and colleagues (1996) pointed out that this condition could be the result of multicentric benign leiomyomatous growths rather than metastases.
Pulmonary Lymphangioleiomyomatosis
Pulmonary lymphangioleiomyomatosis (LAM), as recorded by Bonetti and Chiodera (1997), was first reported by Lutenbacher (1918) in a woman with tuberous sclerosis (Fig. 118-9). As subsequently observed, the pulmonary involvement accompanying tuberous sclerosis was only seen in women. At times, there may be no other manifestation of tuberous sclerosis. When other organ involvement is absent, Valensi (1973), as well as others, consider LAM as a forme fruste of tuberous sclerosis.
The pulmonary disease (LAM) is seen almost exclusively in women during their reproductive years. Numerous series of women with LAM have been reported by Corrin (1975), Taylor (1990), Tazelaar (1993), Kitaichi (1995), and Urban (1999) and their colleagues. Single case reports abound in the literature. The common age of the women at the time of diagnosis is between 20 and 40 years, although a few cases have been reported in postmenopausal women by Sinclair (1985) and Baldi (1994) and their associates. Because of LAM's almost exclusive incidence in women during their reproductive years, it has been assumed that the disease has a hormonal basis as well as its association with tuberous sclerosis. The hormonal relationship is unclear. However, it has been observed that LAM does not develop in women with low levels of estrogen and that the disease process, as reported by Shen and co-workers (1987), is exacerbated by exogenous estrogens. Likewise, the disease is worsened during the menstrual periods, as well as during pregnancy; in fact, Urban and associates (1999) noted that 20% of cases had the onset during pregnancy.
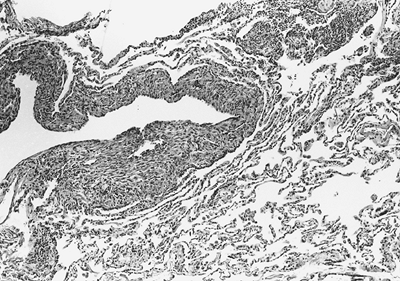 |
Fig. 118-9. Photomicrograph of a section of lung in a patient with lymphangioleiomyomatosis. Note the accumulation of muscle cells that appear immature and are clustered randomly in the alveolar walls and around the small bronchi and blood vessels. |
P.1795
Clinically, the patient frequently presents initially with an episode of spontaneous pneumothorax with marked dyspnea. The history, however, reveals antecedent shortness of breath for a variable period. Pulmonary hemorrhage with hemoptysis, chylothorax (present at presentation in up to 7% of patients and seen in 28% during the course of the disease according to Sullivan [1998]), and increasing respiratory insufficiency occurs over time. Intraabdominal tumors (i.e., angiomyolipomas of the kidneys) and chylous ascites may occur in these patients. The course of the disease is progressive lung involvement, but the pace of progression is variable. Kitaichi and colleagues (1995) recorded that four of six treated patients died within 71.3 25.4 months after the onset of symptoms.
The radiographic findings are those of a reticulonodular pattern throughout the lung fields, although, according to M ller (1990) and Kitaichi (1995) and their associates, the radiograph may be normal in 10% to 20% of the cases initially. In late cases, a honeycomb appearance may be, and in fact is usually, present. The CT findings have been described by Swensen (1995) and M ller (1990) and their colleagues, among others. The HRCT findings consist of numerous, variable-sized cysts surrounded by normal-appearing lung parenchyma. The cysts usually vary between 0.2 and 2 cm in size but may be as large as 6 cm. M ller and associates (1990) noted that, usually, the smaller the size of the cysts, the milder the disease. The cysts are distributed equally throughout the lung, but occasionally the lower lobes appear to be more involved.
Pathologically, Corrin and associates (1975) described the process to be an irregular, nodular or laminar irrational proliferation of smooth muscle within all portions of the lungs. The accumulation of the smooth muscle cells is progressive. As the smooth muscle cells proliferate, they produce secondary obstruction of the small airways, small veins, and lymphatics of the lung parenchyma. Cystic changes within the lung parenchyma occur as the process progresses. The cystic spaces throughout the lungs are characteristic CT features in advanced cases. Immunohistochemical studies, as noted by Bonetti and Chiodera (1997), reveal the smooth muscle cells in cases of LAM to have a distinctive phenotype (in addition to the presence of muscle deviation, such as muscle-specific actin). The cells are consistently positive for human melanin black (HMB-45), the marker for melanogenesis. Lacronique and Urban (1999) report that this monoclonal antibody labels the cells of the melanocytic line, LAM cells, and renal angiomyolipoma cells. They suggest the antigen carried by all these cells is probably gp-100 involved in melanogenesis. These authors discuss the two gene loci implicated in the pathogenesis of Bourneville tuberous sclerosis, one of which may be associated with both LAM and tuberous sclerosis. The European Chromosome 16 Tuberous Sclerosis Consortium (1993) noted two genes: TSC1 on chromosome 9 and TSC2 on chromosome 16, both of which act as tumor suppressor genes in patients with tuberous sclerosis. The TSC2 gene on chromosome 16 is also present in patients with LAM. The hypothesis is yet to be confirmed.
Hormonal therapy (i.e., the use of oophorectomy, antiestrogen agents, and progesterone therapy, alone or in combination) has been used with equivocal results. Banner (1981), Adamson (1985), and Urban (1992) and their associates, among others, have reported varying success rates with such medical intervention. A meta-analysis of many small series by Eliasson and colleagues (1989), however, noted that some stabilization of the disease process and longer survival were observed. It is generally agreed that, at present, medroxyprogesterone acetate is the drug of choice with or without oophorectomy. The role of tamoxifen is unsettled. In unresponsive patients, Klein and co-workers (1992) suggested the alternative use of oophorectomy, interferon-a2b, and tamoxifen. The value of this regimen remains to be shown. In the publication of Sullivan (1998), the status of medical therapy is covered extensively, as it also is in the review of Kalassian and associates (1997).
In patients who have failed to respond to the aforementioned medical regimens and have repeated pneumothoracies and pleural effusions have undergone pleurodesis. Urban and colleagues (1999) performed 40 such operations in their series of 69 patients. Occasionally, pleurectomy has been done, as in 2 of 12 patients reported by Roman and colleagues (2000). In those patients who progress to severe pulmonary insufficiency, bilateral or unilateral lung transplantation has been carried out. The early experience has been noted by Marchevsky and colleagues (1991). Wellens and associates (1985) successfully used combined heart and lung transplantation. Nine (1994) and O'Brien (1995) and their colleagues used single-lung transplantation. In each of the patients, however, LAM subsequently developed in the allograft; the donors for both allografts were men, which is of some interest. Thus, the use of single-lung transplantation in patients with end-stage LAM may not be a suitable solution. Nonetheless, transplantation remains the procedure of choice for the terminally ill patient. Urban and colleagues (1999) carried out transplantation in 13 patients, and after a relatively short period of follow-up (2.3 2.2 years), 9 patients were alive. Roman and associates (2000) reported that six of eight patients who underwent lung transplantation had satisfactory postoperative survival.
Pulmonary Capillary Hemangiomatosis
Pulmonary capillary hemangiomatosis is an aggressive benign tumor of the lung that, according to Tron and colleagues (1986), is formed by numerous cytologically benign thin-walled capillary-sized vessels proliferating diffusely through the pulmonary interstitium, in and around the pulmonary vessels and small airways. The clinical course is characterized by pulmonary hypertension. Hemoptysis may be present. The process is slowly progressive and leads to right-sided heart failure. Radiographic examination
P.1796
of the lung reveals a reticulonodular pattern. Fewer than 20 cases have been recorded in the literature. The only successful treatment has been bilateral lung transplantation, as reported by Faber and associates (1989). Eltorky and colleagues (1994) reported that single-lung transplantation was unsuccessful in this condition.
Cystic Fibrohistiocytic Tumor
Joseph and colleagues (1990) described two cases of cystic fibrohistiocytic tumors of the lungs. These tumors are histologically identical to benign fibrous histiocytomas (dermatofibromas) in the skin and other sites. Leroyer and colleagues (1993) described a similar lesion that they thought was better termed a mesenchymal cystic hamartoma. In the lung, the tumor proliferation is interstitial and is associated with microscopic and macroscopic cyst formation. The lesions, as described by Joseph and colleagues (1990), were bilateral and nodular, cystic, or both, in appearance. Slow growth of the lesions was observed. Clinically, one patient was asymptomatic, and the other experienced episodes of pneumothorax and shortness of breath. No specific treatment is indicated other than to control any complications that arise.
REFERENCES
Abramson S, et al: Benign metastasizing leiomyoma: clinical, imaging, and pathologic correlation. AJR Am J Roentgenol 176:1409, 2001.
Adamson D, et al: Successful treatment of pulmonary lymphangiomyomatosis with oophorectomy and progesterone. Am Rev Respir Dis 132:916, 1985.
Agrons GA, et al: Pulmonary inflammatory pseudotumor: radiologic features. Radiology 206:511, 1998.
Aisner SC, et al: Endobronchial fibrous histiocytoma. Ann Thorac Surg 60:710, 1995.
Andrion A, et al: Benign clear cell ( sugar ) tumor of the lung: a light microscopic, histochemical, and ultrastructural study with a review of the literature. Cancer 56:2657, 1985.
Archambeaud-Mouveroux F, et al: Bronchial leiomyoma. Report of a case successfully treated by endoscopic neodymium-yttrium aluminum garnet laser. J Thorac Cardiovasc Surg 95:536, 1988.
Arguelles M, Blanco I: Inflammatory bronchial polyps associated with asthma. Arch Intern Med 143:570, 1983.
Arrigoni MG, et al: Benign tumors of the lung: a ten-year surgical experience. J Thorac Cardiovasc Surg 60:589, 1970.
Azua Blanco J, et al: Cytologic features of pulmonary hamartoma. Report of a case diagnosed by fine needle aspiration cytology. Acta Cytol 45:267, 2001.
Baldi S, et al: Pulmonary lymphangioleiomyomatosis in postmenopausal women: report of two cases and review of the literature. Eur Respir J 7:1013, 1994.
Bango A, et al: Endobronchial lipomas. Respiration 60:297, 1993.
Banner AS, et al: Efficacy of oophorectomy in lymphangioleiomyomatosis and benign metastasizing leiomyoma. N Engl J Med 305:204, 1981.
Basile A, et al: Malignant change in a benign pulmonary hamartoma. Thorax 44:232, 1989.
Bateson EM: So-called hamartoma of the lung: a true neoplasm of fibrous connective tissue of the bronchi. Cancer 31:1458, 1973.
Bennett LL, Lesar MJ, Tellis CJ: Multiple calcified chondrohamartomas of the lung: CT appearance. J Comput Assist Tomogr 9:180, 1985.
Berardi RS, et al: Inflammatory pseudotumors of the lung. Surg Gynecol Obstet 156:89, 1983.
Bohm J, et al: Pulmonary nodule caused by an alveolar adenoma of the lung. Virchows Arch 430:181, 1997.
Bonetti F, Chiodera P: The lung in tuberous sclerosis. In Corrin B (ed): Pathology of Lung Tumors. New York: Churchill Livingstone, 1997, p. 225.
Bueno R, et al: Bronchoplasty in the management of low-grade airway neoplasms and benign bronchial stenoses. Ann Thorac Surg 62:824, 1996.
Burke LM, et al: Alveolar adenoma: a histochemical, immunohistochemical, and ultrastructural analysis of 17 cases. Hum Pathol 30:158, 1999.
Carney JA: The triad of gastric epithelioid leiomyosarcoma, functioning extra-adrenal paraganglioma, and pulmonary chondroma. Cancer 43: 374, 1979.
Carney JA: The triad of gastric epithelioid leiomyosarcoma, pulmonary chondroma, and functioning extra-adrenal paraganglioma: a five year review. Medicine 62:159, 1983.
Carney JA: Psammomatous melanotic schwannoma. A distinctive, heritable tumor with special associations, including cardiac myxoma and the Cushing syndrome. Am J Surg Pathol 14:206, 1990.
Carney JA: Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc 74:543, 1999.
Carney JA, et al: The triad of gastric epithelioid leiomyosarcoma, functioning extra-adrenal paraganglioma, and pulmonary chondroma. N Engl J Med 296:1517, 1977.
Carter D, Patchefaky AS, Mountain CF: Tumors and Tumor-like Lesions of the Lung. Philadelphia: WB Saunders, 1998, p. 390.
Cesario A, et al: Case report: unsuspected primary pulmonary meningioma. Eur J Cardiothorac Surg 21:553, 2002.
Chan AC, Chan JK: Pulmonary sclerosing hemangioma consistently expresses thyroid transcription factor-1 (TTF-1): a new clue to its histogenesis. Am J Surg Pathol 24:1531, 2000.
Cohen MC, Kaschula ROC: Primary pulmonary tumors in childhood: a review of 31 years experience and the literature. Pediatr Pulmonol 14:222, 1992.
Colby TV, Koss MN, Travis WD: Atlas of Tumor Pathology: Tumors of the Lower Respiratory Tract. Washington, DC: Armed Forces Institute of Pathology, 1995, pp. 61, 338.
Corrin B, Liebow AA, Friedman PJ: Pulmonary lymphangiomyomatosis. A review. Am J Pathol 79:348, 1975.
Cosio BG, et al: Endobronchial hamartoma. Chest 122:202, 2002.
Cutlan RT, Eltorky M: Pulmonary granular cell tumor coexisting with bronchogenic carcinoma. Ann Diagn Pathol 5:74, 2001.
Dacic S, Colby TV, Yousem SA: Nodular amyloidoma and primary pulmonary lymphoma with amyloid production: a differential diagnostic problem. Mod Pathol 13:934, 2000.
Dail DH: Uncommon tumors. In Dail DH, Hammar SP (eds): Pulmonary Pathology. New York: Springer, 1988, p. 847.
Dail DH: Uncommon tumors. In Dail DH, Hammar SP, Colby TV (eds): Pulmonary Pathology: Tumors. New York: Springer-Verlag, 1994, p. 1279.
Deavers M, et al: Granular cell tumors of the lung: clinicopathologic study of 20 cases. Am J Surg Pathol 19:627, 1995.
Demos TC, et al: Cystic hamartoma of the lung. J Can Assoc Radiol 34:149, 1983.
de Perrot M, et al: Primary pulmonary meningioma presenting as lung metastasis. Scand Cardiovasc J 33:121, 1999.
de Rooij PD, et al: Solitary hamartoma of the lung: is thoracotomy still mandatory? Neth J Surg 40:145, 1988.
Desai RA, et al: Pulmonary amyloidoma and hilar adenopathy. Rare manifestations of primary amyloidosis. Chest 76:170, 1979.
Dessy E, et al: Peripheral papillary tumor of type-II pneumocytes: a rare neoplasm of undetermined malignant potential. Virchows Arch 436: 289, 2000.
Devouassoux-Shisherboran M, et al: A clinicopathologic study of 100 cases of pulmonary sclerosing hemangioma with immunohistochemical studes: TTF-1 is expressed in both round and surface cells, suggesting an origin from primitive respiratory epithelium. Am J Surg Pathol 24:906, 2000.
Dixon AY, et al: Pulmonary mucinous cystic tumor: case report and review of the literature. Am J Surg Pathol 17:722, 1993.
Doski JJ, et al: Corticosteroids in the management of unresected plasma cell granuloma (inflammatory pseudotumor) of the lung. J Pediatr Surg 26:1064, 1991.
Drennan JM, Douglas AC: Solitary papilloma of a bronchus. J Clin Pathol 18:401, 1965.
Drlicek M, et al: Pulmonary meningioma: immunohistochemical and ultrastructural features. Am J Surg Pathol 15:455, 1991.
Duncan JD, et al: Benign fibrous histiocytoma: a rare endobronchial neoplasm. Int Surg 71:110, 1986.
P.1797
Edwards CW, Matthews HR: Mucous gland adenoma of the bronchus. Thorax 36:147, 1981.
Eliasson AH, Phillips YY, Tenholder MF: Treatment of lymphangioleiomyomatosis. A meta-analysis. Chest 96:1352, 1989.
Eltorky MA, et al: Pulmonary capillary hemangiomatosis: a clinicopathological review. Ann Thorac Surg 57:772, 1994.
Emory WB, Mitchell WT, Hatch HB Jr: Mucous gland adenoma of the bronchus. Am Rev Respir Dis 108:1407, 1973.
England DM, Hochholzer L: Truly benign bronchial adenoma. Report of 10 cases of mucous gland adenoma with immunohistochemical and ultrastructural findings. Am J Surg Pathol 19:887, 1995.
Engleman P, et al: Pulmonary hyalinizing granuloma. Am Rev Respir Dis 115:997, 1977.
Epstein LJ, Mohsenifar Z: Use of Nd:YAG laser in endobronchial granular cell myoblastoma. Chest 104:958, 1993.
Erlandson RA: Diagnostic Transmission Electron Microscopy of Human Tumors. New York: Masson Publishing, 1981, p. 125.
The European Chromosome 16 Tuberous Sclerosis Consortium. Identificaton and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75:1305, 1993.
Faber CN, et al: Pulmonary capillary hemangiomatosis. A report of three cases and a review of the literature. Am Rev Respir Dis 140:808, 1989.
Fantone JC, Geisinger KR, Appelman HD: Papillary adenoma of the lung with lamellar and electron dense granules: an ultrastructural study. Cancer 50: 2939, 1982.
Faul JL, et al: Thoracic lymphangiomas, lymphangiectasis, lymphangiomatosis and lymphatic dysplasia syndrome. Am J Respir Crit Care Med 161:1037, 2000.
Fisher ER, Wechsler H: Granular cell myoblastoma a misnomer. Electron microscopic and histochemical evidence concerning its Schwann cell derivation and nature. Cancer 15:936, 1962.
Fletcher JA, et al: Lineage-restricted clonality in biphasic solid tumors. Am J Pathol 138:1199, 1991.
Flynn SD, Yousem SA: Pulmonary meningiomas: a report of two cases. Hum Pathol 22:469, 1991.
Forsee JH, Mahon HW, James LA: Cavernous hemangioma of the lung. Ann Surg 131:418, 1950.
Fujimoto K, et al: Alveolar adenoma of the lung: computed tomography and magnetic resonance imaging findings. J Thorac Imaging 17:163, 2002.
Fukayama M, et al: Pulmonary and pleural thymoma. Diagnostic application of lymphocyte markers to the thymoma of unusual site. Am J Clin Pathol 89:617, 1988.
Fukuda T, et al: Papillary adenoma of the lung: histological and ultrastructural findings in two cases. Acta Pathol Jpn 42:56, 1992.
Gaertner EM, et al: Pulmonary and mediastinal glomus tumors report of five cases including a pulmonary glomangiosarcoma: a clinicopathlogic study with literature review. Am J Surg Pathol 24:1105, 2000.
Gaffey MJ, et al: Clear cell tumor of the lung: a clinicopathologic, immunohistochemical, and ultrastructural study of eight cases. Am J Surg Pathol 14:248, 1990.
Gaffey MJ, et al: Clear cell tumor of the lung. Immunohistochemical and ultrastructural evidence of melanogenesis. Am J Surg Pathol 15:644, 1991.
Gal AA, Brooks JS, Pietra GG: Leiomyomatous neoplasms of the lung: a clinical, histologic, and immunohistochemical study. Mod Pathol 2:209, 1989.
Gal AA, et al: Prognostic factors in pulmonary fibrohistiocytic lesions. Cancer 73:1817, 1994.
Galliani CA, Beatty JF, Grosfeld JL: Cavernous hemangioma of the lung in an infant. Pediatr Pathol 12:105, 1992.
Ge F, Tong F, Li Z: Diagnosis and treatment of pulmonary hamartoma. Chin Med Sci J 13:61, 1998.
Gilman RA, Klassen KP, Scarpelli DG: Mucous gland adenoma of the bronchus. Am J Clin Pathol 26:151, 1956.
Gjevre JA, Myers JL, Prakash UB: Pulmonary hamartomas. Mayo Clin Proc 71:14, 1996.
Gomez-Roman JJ, et al: Human herpesvirus-8 genes are expressed in pulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). Am J Surg Pathol 25:624, 2001.
Googe PB, et al: A comparison of paraganglioma, carcinoid tumor, and small-cell carcinoma of the larynx. Arch Pathol Lab Med 112:908, 1988.
Gotti G, et al: Pedunculated pulmonary leiomyoma with large cyst formation. Ann Thorac Surg 56:1178, 1993.
Graeme-Cook F, Mark EJ: Pulmonary mucinous cystic tumors of borderline malignancy. Hum Pathol 22:185, 1991.
Hamper UM, et al: Pulmonary hamartoma: diagnosis by transthoracic needle aspiration biopsy. Radiology 155:15, 1985.
Hangartner JRW, et al: Malignant primary pulmonary paraganglioma. Thorax 44:154, 1989.
Hansen CP, et al: Pulmonary hamartoma. J Thorac Cardiovasc Surg 104:674, 1992.
Hashimoto T, et al: Benign clear cell tumor of the lung. Ultrastruct Pathol 25:479, 2001.
Hayes WT, Bernhardt H: Solitary amyloid mass of the lung: report of a case with 6-year follow-up. Cancer 24:820, 1969.
Hayward RH, Carabasi RJ: Malignant hamartoma of the lung: fact or fiction? J Thorac Cardiovasc Surg 53:457, 1967.
Hegg CA, et al: Papillary adenoma of the lung. Am J Clin Pathol 97:393, 1992.
Higuchi M, et al: A case of primary nodular pulmonary amyloidosis. J Jpn Assoc Chest Surg 11:34, 1997.
Hironaka M, et al: Pulmonary gangliocytic paraganglioma: case report and comparative immunohistochemical study of related neuroendocrine neoplasms. Am J Surg Pathol 25:688, 2001.
Holden WE, et al: Adult intrapulmonary and mediastinal lymphangioma causing hemoptysis. Thorax 42:635, 1987.
Hsu H-S, et al: Endobronchial neurofibromas. Ann Thorac Surg 74:1704, 2002.
Hui AN, et al: Amyloidosis presenting in the lower respiratory tract: clinicopathologic, radiologic, immunohistochemical, and histochemical studies on 48 cases. Arch Pathol Lab Med 110:212, 1986.
Hurt R: Benign tumours of the bronchus and trachea: 1951 1981. Ann R Coll Surg Engl 66:22, 1984.
Imperato JP, et al: Treatment of plasma cell granuloma of the lung with radiation therapy. A report of two cases and a review of the literature. Cancer 57:2127, 1986.
Ishida T, et al: Inflammatory pseudotumor of the lung in adults: radiographic and clinicopathological analysis. Ann Thorac Surg 48:90, 1989.
Ishida T, et al: Mucous gland adenoma of the trachea resected with an endoscopic neodymium:yttrium aluminum garnet laser. Intern Med 35: 890, 1996.
Itoh H, et al: Solitary fibroleiomyomatous hamartoma of the lung in a patient without a pre-existing smooth-muscle tumor. Pathol Int 51:661, 2001.
Jackson RC, et al: Massive cystic hamartoma. A report of two cases. J Thorac Surg 31:504, 1956.
Jacobson TZ, Rainey EJ, Turton CWG: Pulmonary benign metastasizing leiomyoma: response to treatment with goserelin. Thorax 50:1225, 1995.
James CL, Iyer PV, Leong AS: Intrapulmonary thymoma. Histopathology 21:175, 1992.
Jautzke G, Muller-Ruchholtz E, Thalman U: Immunohistochemical detection of estrogen and progesterone receptors in multiple and well differentiated leiomyomatous lung tumors in women with uterine leiomyomas (so-called benign metastasizing leiomyomas). Pathol Res Pract 192:215, 1996.
John PG, Rahman J, Payne CB: Pulmonary hyalinizing granuloma: an unusual association with multiple sclerosis. South Med J 88:1076, 1995.
Joseph MG, et al: Multiple cystic fibrohistiocytic tumors of the lung: report of two cases. Mayo Clin Proc 65:192, 1990.
Kalassian KG, et al: Lymphangioleiomyomatosis: new insights. Am J Respir Crit Care Med 155:1183, 1997.
Kaleem Z, Fitzpatrick MM, Ritter JH: Primary pulmonary meningioma. Report of a case and review of the literature. Arch Pathol Lab Med 121:631, 1997.
Kalish PE: Primary intrapulmonary thymoma. NY State J Med 63:1705, 1963.
Kammen BF, et al: Extraadrenal myelolipoma: MR imaging findings. AJR Am J Roentgenol 171:721, 1998.
Karasik A, et al: Increased risk of lung cancer in patients with chondromatous hamartoma. J Thorac Cardiovasc Surg 80:217, 1980.
Katial RK, Ranlett R, Whitlock WL: Human papilloma virus associated with solitary squamous papilloma complicated by bronchiectasis and bronchial stenosis. Chest 106:1887, 1994.
Katzenstein AL, Gmelich JT, Carrington CB: Sclerosing hemangioma of the lung: a clinicopathologic study of 152 cases. Am J Surg Pathol 4:343, 1982.
Kayser K, et al: Benign metastasizing leiomyoma of the uterus: documentation of clinical, immunohistochemical and lectin-histochemical data of ten cases. Virchows Arch 437:284, 2000.
Kenney PJ, et al: Myelolipoma: CT findings and pathologic features. Radiology 208:87, 1998.
P.1798
Khalifa MA, et al: Solitary fibrous tumors: a series of lesions, some in unusual sites. South Med J 90:793, 1997.
Khouri NF, et al: The solitary pulmonary nodule. Assessment, diagnosis, and management. Chest 91:128, 1987.
Kim KH, Suh JS, Han WS: Leiomyoma of the bronchus treated by endoscopic resection. Ann Thorac Surg 56:1164, 1993.
Kim WS, et al: Cystic intrapulmonary lymphangioma: HRCT findings. Pediatr Radiol 25:206, 1995.
Kiryu T, et al: Multiple chondromatous hamartomas of the lung: a case report and review of the literature with special reference to Carney syndrome. Cancer 85:2557, 1999.
Kitaichi M, et al: Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med 151:527, 1995.
Klein MO, et al: Treatment of lymphangioleiomyomatosis by ovariectomy, interferon alpha 2b and tamoxifen a case report. Arch Gynecol Obstet 252:99, 1992.
Kodama K, et al: Primary and metastatic pulmonary meningioma. Cancer 67:1412, 1991.
Koss MN, Hochholzer L, Moran CA: Primary pulmonary glomus tumor: a clinicopathologic and immunohistochemical study of two cases. Mod Pathol 11:253, 1998.
Kragel PJ, et al: Mucinous cystadenoma of the lung. A report of two cases with immunohistochemical and ultrastructural analysis. Arch Pathol Lab Med 114:1053, 1990.
Kroe DJ, Pitcoc JA: Benign mucous gland adenoma of the bronchus. Arch Pathol Lab Med 84:539, 1967.
Kuda T, et al: Clinical analysis of benign tumors and tumor-like lesions of the lung [English abstract]. J Jpn Assoc Chest Surg 4:416, 1990.
Kuramochi S, et al: Multiple pulmonary hyalinizing granulomas associated with systemic idiopathic fibrosis. Acta Pathol Jpn 41:375, 1991.
Kwekkeboom DJ, et al: Octreotide scintigraphy for the detection of paragangliomas. J Nucl Med 34:873, 1993.
Kyle RA, Garton JP: The spectrum of IgM monoclonal gammopathy in 430 cases. Mayo Clin Proc 62:719, 1987.
Lacronique J, Urban T: Current aspects of lymphangioleiomyomatosis. Rev Pneumol Clin 55:263, 1999.
Laden SA, Cohen ML, Harley RA: Nodular pulmonary amyloidosis with extrapulmonary involvement. Hum Pathol 15:594, 1984.
Langston C, Askin FB: Pulmonary distress in the neonate, infant and child. In Thurlbeck WM, Churg AM (eds): Pathology of the Lung. 2nd Ed. New York: Thieme Medical Publishers, 1995, p. 151.
Ledor K, et al: CT diagnosis of pulmonary hamartomas. J Comput Assist Tomogr 5:343, 1981.
Lemonick DM, Pai PB, Hines GL: Malignant primary pulmonary paraganglioma with hilar metastasis. J Thorac Cardiovasc Surg 99:563, 1990.
Leong AS-Y, Chan KW, Leong FJ W-N: Sclerosing hemangioma. In Corrin B (ed): Pathology of Lung Tumors. New York: Churchill Livingstone, 1997, p. 175.
Leong AS, Chan KW, Seneviratne HS: A morphological and immunohistochemical study of 25 cases of so called sclerosing hemangioma of the lung. Histopathology 27:121, 1995.
Leong AS-Y, Meredith DJ: Clear cell tumors of the lung. In Corrin B (ed): Pathology of Lung Tumors. New York: Churchill Livingstone, 1997, p. 159.
Le Roux BT: Pulmonary hamartomas. Thorax 19:236, 1964.
Leroyer C, et al: Mesenchymal cystic hamartoma of the lung. Respiration 60:305, 1993.
Liebow AA, Castleman B: Benign clear cell tumors of the lung [Abstract]. Am J Pathol 43:13a, 1963.
Liebow AA, Castleman B: Benign clear cell ( sugar ) tumors of the lung. Yale J Biol Med 43:213, 1971.
Liebow AA, Hubbell DS: Sclerosing hemangioma (histiocytoma xanthoma) of the lung. Cancer 9:53, 1956.
Lim JK, et al: Pulmonary marginal zone lymphoma of MALT type as a cause of localised pulmonary amyloidosis. J Clin Pathol 54:642, 2001.
Lockett L, Chiang V, Scully N: Primary pulmonary meningioma: report of a case and review of the literature. Am J Surg Pathol 21:453, 1997.
Lui RC, et al: Primary endobronchial granular cell myoblastoma. Ann Thorac Surg 48:113, 1989.
Lutenbacher R: Dysembryomes metatypique des reins. Carcinose submiliare aigue poumon avec emphys me generalis et double pneumothorax. Ann Med Intern (Paris) 5:435, 1918.
Mann GN, et al: Recurrence of pulmonary mucinous cystic tumor of borderline malignancy. Ann Thorac Surg 71:696, 2001.
Marchevsky A, et al: Lung transplantation: the pathologic diagnosis of pulmonary complications. Mod Pathol 4:133, 1991.
Marchevsky AM: Lung tumors derived from ectopic tissues. Semin Diagn Pathol 12:172, 1995.
Martini N, Beattie EJ Jr: Less common tumors of the lung. In Shields TW (ed): General Thoracic Surgery. 2nd Ed. Philadelphia: Lea & Febiger, 1983, p. 780.
Matsubara O, et al: Inflammatory pseudotumors of the lung: progression from organizing pneumonia to fibrous histiocytoma or to plasma cell granuloma in 32 cases. Hum Pathol 19:807, 1988.
McCluggage WG, Bharucha H: Primary pulmonary tumours of nerve sheath origin. Histopathology 26:247, 1995.
Miller DC, et al: Benign metastasizing meningioma. Case report. J Neurosurg 62:763, 1985.
Miura H, et al: Asymptomatic solitary papilloma of the bronchus: review of occurrence in Japan. Eur Respir J 6:1070, 1993.
Miura K, et al: Cystic pulmonary hamartoma. Ann Thorac Surg 49:828, 1990.
Miura K, et al: Benign clear cell tumor of the lung: a case report [English abstract]. J Jpn Assoc Chest Surg 7:95, 1993.
Moran CA: Primary salivary gland-type tumors of the lung. Semin Diagn Pathol 12:106, 1995.
Moran CA, et al: Primary intrapulmonary thymoma. A clinicopathologic and immunohistochemical study of eight cases. Am J Surg Pathol 19:304, 1995.
Moran CA, et al: Primary intrapulmonary meningiomas. A clinicopathologic and immunohistochemical study of ten cases. Cancer 78:2328, 1996.
Mori M, et al: Papillary adenoma of type II pneumocytes might have malignant potential. Virchows Arch 428:195, 1996.
M ller NI, Chiles C, Kullnig P: Pulmonary lymphangiomyomatosis: correlation of CT with radiographic and functional findings. Radiology 175:335, 1990.
Muraoka M, et al: Endobronchial lipoma: review of 64 cases reported in Japan. Chest 123:293, 2003.
Muros MA, et al: 111In-pentetreotide scintigraphy is superior to 123I-MIBG scintigraphy in the diagnosis and location of chemodectoma. Nucl Med Commun 19:735, 1998.
Nagata N, et al: Sclerosing hemangioma of the lung. An epithelial tumor composed of immunohistochemically heterogenous cells. Am J Clin Pathol 88:552, 1987.
Nili M, et al: Multiple pulmonary hamartomas: a case report and review of the literature. Scand J Thorac Cardiovasc Surg 13:157, 1979.
Nine JS, et al: Lymphangioleiomyomatosis recurrence after lung transplantation. J Heart Lung Transplant 13:714, 1994.
Noguchi M, et al: Papillary adenoma of type 2 pneumocytes. Am J Surg Pathol 10:134, 1986.
O'Brien JD, et al: Lymphangioleiomyomatosis recurrence in the allograft after single-lung transplantation. Am J Respir Crit Care Med 151:2033, 1995.
Okabayashi K, et al: Giant hamartoma of the lung with a high production of carbohydrate antigen 19 9. Ann Thorac Surg 55:511, 1993.
Oliveira P, et al: Alveolar adenoma of the lung: further characterization of this uncommon tumour. Virchows Arch 429:101, 1996.
Oparah SS, Subramanian VA: Granular cell myoblastoma of the bronchus: report of 2 cases and review of the literature. Ann Thorac Surg 22:199, 1976.
Parenti DJ, Morley TF, Giudice JC: Benign metastasizing leiomyoma. A case report and review of the literature. Respiration 59:347, 1992.
Paul KP, et al: Capillary hemangioma of the right main bronchus treated by sleeve resection in infancy. Am Rev Respir Dis 143:876, 1991.
Pelosi G, et al: Pulmonary epithelial-myoepithelial tumor of unproven malignant potential: report of a case and review of the literature. Mod Pathol 14:521, 2001.
Popper HH, et al: Prognostic importance of human papilloma virus typing in squamous cell papilloma of the bronchus: comparison of in situ hybridization and the polymerase chain reaction. Hum Pathol 25:1191, 1994.
Prayson RA, Farver CF: Primary pulmonary malignant meningioma. Am J Surg Pathol 23:722, 1999.
Ribet M, Jaillard-Thery S, Nuttens MC: Pulmonary hamartoma and malignancy. J Thorac Cardiovasc Surg 107:611, 1994.
Robinson PG: Pulmonary meningioma: report of a case with electron microscopic and immunohistochemical findings. Am J Clin Pathol 97:814, 1992.
Roman A, et al: Linfangioleiomiomatosis: estudio de 15 pacientes [Lymphangioleiomyomatosis: study of 15 patients]. Med Clin (Barc) 115:98, 2000.
P.1799
Roux FJ, et al: Mucinous cystadenoma of the lung. Cancer 76:1540, 1995.
Sabate CJ, Shahian DM: Pulmonary myelolipoma. Ann Thorac Surg 74:573, 2002.
Saeki T, et al: An extremely large solitary primary paraganglioma of the lung: report of a case. Surg Today 29:1195, 1999.
Sagel SS, Ablow RC: Hamartoma: on occasion a rapidly growing tumor of the lung. Radiology 91:971, 1968.
Sakamoto H, et al: Pleomorphic adenoma in the periphery of the lung: report of a case and review of the literature. Arch Pathol Lab Med 115:393, 1991.
Sambrook Gowar FJ: An unusual mucous cyst of the lung. Thorax 33:796, 1978.
Sanchez-Jimenez J, et al: Papillary adenoma of type 2 pneumocytes. Pediatr Pulmonol 17:396, 1994.
Sato N, et al: Two cases of parenchymal pulmonary leiomyoma. J Jpn Assoc Chest Surg 11:565, 1997.
Sekine I, et al: Rare pulmonary tumors a review of 32 cases. Oncology 55:431, 1998.
Shen A, et al: Exacerbation of pulmonary lymphangioleiomyomatosis by exogenous estrogens. Chest 91:782, 1987.
Shields TW, Lynn TE: Endobronchial hamartoma: a case report. Arch Surg 76:358, 1958.
Siegelman SS, et al: CT of pulmonary hamartoma. Presented at the 84th Annual Meeting of the American Roentgen Ray Society, Las Vegas, NV, April 1984.
Silverman JM, et al: Magnetic resonance imaging evaluation of pulmonary vascular malformations. Chest 106:1333, 1994.
Simansky DA, et al: Psammomatous melanotic schwannoma: presentation of a rare primary lung tumor. Ann Thorac Surg 70:671, 2000.
Sinclair W, Wright JL, Churg A: Lymphangioleiomyomatosis presenting in postmenopausal women. Thorax 40:475, 1985.
Singh G, Lee RE, Brooks DH: Primary pulmonary paraganglioma: report of a case and review of the literature. Cancer 40:2286, 1977.
Spencer H, Dail DH, Arneaud J: Non-invasive bronchial epithelial papillary tumors. Cancer 45:1486, 1980.
Spinelli M, et al: Primary pulmonary meningioma may arise from meningothelial-like nodules. Adv Clin Pathol 4:35, 2000.
Strickler JG, et al: Myoepithelioma of the lung. Arch Pathol Lab Med 111:1082, 1987.
Sugio K, et al: Sclerosing hemangioma of the lung: radiographic and pathological study. Ann Thorac Surg 53:295, 1992.
Sugita M, et al: Sleeve superior segmentectomy of the right lower lobe for endobronchial neurinoma. Report of a case. Respiration 63:191, 1996.
Sullivan EJ: Lymphangioleiomyomatosis: a review. Chest 114:1689, 1998.
Swensen SJ, et al: An integrated approach to evaluation of the solitary pulmonary nodule. Mayo Clin Proc 65:173, 1990.
Swensen SJ, et al: Diffuse pulmonary lymphangiomatosis. CT findings. J Comput Assist Tomogr 19:348, 1995.
Tagge E, et al: Obstructing endobronchial fibrous histiocytoma: potential for lung salvage. J. Pediatr Surg 26:1067, 1991.
Takahara T, et al: Intrapulmonary cyst lymphangioma: report of a case. Surg Today 28:1310, 1998.
Takemura G, et al: Metastasizing uterine leiomyoma. A case with cardiac and pulmonary metastasis. Pathol Res Pract 192:622, 1996.
Takemura T, et al: Thoracoscopic resection of a solitary pulmonary lymphangioma: report of a case. Surg Today 25:651, 1995.
Tanaka I, et al: A case of pneumocytoma (so-called sclerosing hemangioma) with lymph node metstasis. Jpn J Clin Oncol 16:77, 1986.
Tanimura S, et al: Primary pulmonary paraganglioma: a report of a case and review of the literature [English abstract]. J Jpn Assoc Chest Surg 7:88, 1993.
Taylor JR, et al: Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J Med 323:1254, 1990.
Tazelaar HD, et al: Diffuse pulmonary lymphangiomatosis. Hum Pathol 24:1313, 1993.
Torikata C, Mukai M. So-called minute chemodectoma of the lung. An electron microscopic and immunohistochemical study. Virchows Arch Pathol Anat Histopathol 417:113, 1990.
Tron V, et al: Pulmonary capillary hemangiomatosis. Hum Pathol 17:1144, 1986.
Tsuji N, et al: Adenomyoepithelioma of the lung. Am J Surg Pathol 19:956, 1995.
Ueno M, et al: Cytology of primary pulmonary meningioma. Report of the first multiple case. Acta Cytol 42:1424, 1998.
Urban T, et al: Pulmonary lymphangiomyomatosis. Follow-up and long-term outcome with antiestrogen therapy: a report of eight cases. Chest 102:472, 1992.
Urban T, et al: Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d'Etudes et de Recherche sur les Maladies Orphelines Pulmonaires (GERM'O'P). Medicine (Baltimore) 78:321, 1999.
Valensi QJ: Pulmonary lymphangiomyoma, a probable forme fruste of tuberous sclerosis. A case report and survey of the literature. Am Rev Respir Dis 108:1411, 1973.
Van den Bosch JM, et al: Mesenchymoma of the lung (so-called hamartoma): a review of 154 parenchymal and endobronchial cases. Thorax 42:790, 1987.
Veynovich B, et al: Primary pulmonary thymoma. Ann Thorac Surg 64:1471, 1997.
Weinberger MA, Katz S, Davis EW: Peripheral bronchial adenoma of mucus gland type. J Thorac Cardiovasc Surg 29:626, 1955.
Weiss L, Ingram M: Adenomatoid bronchial tumors: a consideration of the carcinoid tumors and salivary tumors of the bronchial tree. Cancer 14:161, 1961.
Wellens F, et al: Combined heart-lung transplantation for terminal pulmonary lymphangioleiomyomatosis. J Thorac Cardiovasc Surg 89:872, 1985.
Wende S, et al: Lung metastasis of a meningioma. Neuroradiology 24:287, 1983.
White SH, et al: Leiomyomas of the lower respiratory tract. Thorax 40:306, 1985.
Wilson C, Askin FB, Heitmiller RF: Solitary pulmonary lymphangioma. Ann Thorac Surg 71:1337, 2001.
Winkler TR, Burr LH, Robinson CLN: Benign metastasizing leiomyoma. Ann Thorac Surg 43:100, 1987.
Wodehouse GE: Hemangioma of the lung: a review of four cases. J Thorac Cardiovasc Surg 17:408, 1948.
Yalcin S, Kars A: Multiple chondromatous hamartomas of the lung: a case report and review of the literature with special reference to Carney syndrome. Cancer 88:964, 2000.
Yamakawa T, et al: Intrapulmonary schwannoma: a case report [English summary]. J Jpn Assoc Chest Surg 7:165, 1993.
Yano M, et al: Sclerosing hemangioma with metastases to multiple nodal stations. Ann Thorac Surg 73:981, 2002.
Yellin A, Rosenman Y, Lieberman Y: Review of smooth muscle tumours of the lower respiratory tract. Br J Dis Chest 78:337, 1984.
Yokozaki M, et al: Endobronchial lipoma: a report of three cases. Jpn J Clin Oncol 26:53, 1996.
Yousem SA: Pulmonary vascular neoplasia. Prog Surg Pathol 10:27, 1989.
Yousem SA, Flynn DS: Intrapulmonary localized fibrous tumor: intraparenchymal so-called localized fibrous mesothelioma. Am J Clin Pathol 89:365, 1988.
Yousem SA, Hochholzer L: Alveolar adenoma. Hum Pathol 17:1066, 1986.
Yousem SA, Hochholzer L: Pulmonary hyalinizing granuloma. Am J Clin Pathol 87:1, 1987.
Yousem SA, et al: So-called sclerosing hemangiomas of the lung: an immunohistochemical study supporting a respiratory epithelial origin. Am J Surg Pathol 12:582, 1988.
*These authors incorrectly listed 25 cases of this tumor in a review of the literature, but all on further review were simply nonpigmented schwannomas. See Reading References.
READING REFERENCES
General
Carter D, Eggleston JC: Tumors of the Lower Respiratory Tract, Series 2. Washington, DC: Armed Forces Institute of Pathology, 1980, p. 221.
Churg A: Tumors of the lung. In Thurlbeck W, ed. Pathology of the Lung. New York: Thieme, 1988, p. 311.
Dunnill MS: Rare pulmonary tumors. In Dunnill MS (ed): Pulmonary Pathology. 2nd Ed. New York: Churchill Livingstone, 1987, p. 413.
Hasleton PS: Benign tumors and their malignant counterparts. In Hasleton PS (ed): Spencer's Pathology of the Lung. 5th Ed. New York: McGraw-Hill, 1996, p. 875.
Mackay B, Lukeman JM, Ordonez NG: Tumors of the Lungs. Philadelphia: WB Saunders, 1991.
Madewell JE, Feigin DS: Benign tumors of the lung. Semin Roentgenol 12:175, 1977.
P.1800
Marchesky AM (ed): Surgical Pathology of Lung Neoplasms. New York: Marcel Dekker, 1990.
Spencer H: Rare pulmonary tumors. In Spencer H (ed): Pathology of the Lung. 4th Ed. New York: Pergamon Press, 1985, p. 933.
Connective Tissue Tumors, Benign
Orlowski TM, Stasiak K, Kolodziej J: Leiomyoma of the lung. J Thorac Cardiovasc Surg 76:257, 1978.
Schraufnagel DE, Morin JE, Wang NS: Endobronchial lipoma. Chest 75:97, 1979.
Epithelial Tumor
Spencer H, Dail DH, Arneaud J: Noninvasive bronchial epithelial papillary tumors. Cancer 45:1486, 1986.
Fibrous Histiocytoma, Benign
Viguera JL, et al: Fibrous histiocytoma of the lung. Thorax 31:475, 1976.
Granular Cell Myoblastoma Granular Cell Tumor
O'Connell DJ, MacMahon H, DeMeester TR: Multicentric tracheobronchial and oesophageal granular cell myoblastoma. Thorax 33:596, 1978.
Valenstein SL, Thurer RJ: Granular cell myoblastoma of the bronchus: case report and literature review. J Thorac Cardiovasc Surg 76:465, 1978.
Hamartoma
Becker RM, ViLorio J, Chiu C: Multiple pulmonary myomatous hamartomas in women. J Thorac Cardiovasc Surg 71:631, 1976.
Bennett LL, Lesar MS, Tellis CJ: Multiple calcified chondrohamartomas of the lung: CT appearance. J Comput Assist Tomogr 9:180, 1985.
Hansen CP, et al: Pulmonary hamartoma. J Thorac Cardiovasc Surg 104:674, 1992.
Koutras P, Urschel HC Jr, Paulson DL: Hamartoma of the lung. J Thorac Cardiovasc Surg 61:768, 1971.
Minasian H: Uncommon pulmonary hamartomas. Thorax 32:360, 1977.
Petheram IS, Heard BE: Unique massive pulmonary hamartoma: case report with review of hamartoma treated at Brompton Hospital in 27 years. Chest 75:95, 1979.
Ramzy I: Pulmonary hamartomas: cytologic appearances of fine-needle aspiration biopsy. Acta Cytol 20:15, 1976.
Shah JP, et al: Hamartomas of the lung. Surg Gynecol Obstet 136:406, 1973.
Spencer H: Hamartomas, blastoma and teratoma of the lung. In Spencer H (ed): Pathology of the Lung. 4th Ed. New York: Pergamon Press, 1985, p. 1061.
Tomashefski JF Jr: Benign endobronchial mesenchymal tumors: their relationship to parenchymal pulmonary hamartomas. Am J Surg Pathol 6:531, 1982.
Lymphangioleiomyomatosis
Graham ML, et al: Pulmonary lymphangiomyomatosis: with particular reference to steroid-receptor assay studies and pathologic correlation. Mayo Clin Proc 59:3, 1984.
Luna CM, et al: Pulmonary LAM associated with tuberous sclerosis: treatment with tamoxifen and tetracycline pleurodesis. Chest 88:473, 1985.
Pseudotumors
Bahadori H, Liebow AA: Plasma cell granulomas of the lung. Cancer 31:191, 1973.
Graham ML, et al: Pulmonary lymphangiomyomatosis: with particular attention to steroid-receptor assay studies and pathologic correlation. Mayo Clin Proc 59:3, 1984.
Spencer H: The pulmonary plasma cell/histiocytoma complex. Histopathology 8:903, 1984.
Spoto G Jr, Rossi NP, Allsbrook WC: Tracheobronchial plasma cell granuloma. J Thorac Cardiovasc Surg 73:804, 1977.
Warter A, Satge D, Roeslin N: Angioinvasive plasma cell granuloma of the lung. Cancer 59:435, 1987.
Benign Schwannomas
Bosch X, et al: Primary intrapulmonary benign schwannoma. A case with ultrastructural and immunohistochemical confirmation. Eur Respir J 3:234, 1990.
Higashimoto Y, et al: A case of primary intrapulmonary benign schwannoma [in Japanese]. Nihon Kyobu Shikkan Gakkai Zasshi 29:360, 1991.
Michel JL, et al: Endo- and exo-bronchial schwannoma treated by resection-anastomosis of the left bronchial stump [in French]. Rev Mal Respir 12:628, 1995.
Nesbitt JC, et al: Cellular schwannoma of the bronchus. Ultrastruct Pathol 20:349, 1996.
Roviaro G, et al: Primary pulmonary tumours of neurogenic origin. Thorax 38:942, 1983.
Uchiyama Y, et al: A case of intrapulmonary schwannoma and a review of the Japanese literature [in Japanese]. Nippon Kyobu Geka Gakkai Zasshi 37:1238, 1989.
Umeki S, et al: A case of intrapulmonary schwannoma that should be distinguished from lung cancer [in Japanese]. Nippon Kyobu Shikkan Gakkai Zasshi 28:1635, 1990.
Teratomas, Benign
Ali MY, Wong PK: Intrapulmonary teratoma. Thorax 19:228, 1964.
Groeger AM, et al: Intrapulmonary teratoma associated with thymic tissue. Anticancer Res 20:3919, 2000.
Holt S, Peverall PB, Boddy JE: A teratoma of the lung containing thymic tissue. J Pathol 126:85, 1978.
Iwasaki T, et al: Intrapulmonary mature teratoma. Jpn J Thorac Cardiovasc Surg 48:468, 2000.
EAN: 2147483647
Pages: 203