11- Systemic Hypertension
Tierney, Lawrence M., McPhee, Stephen J., Papadakis, Maxine A.
Current Medical Diagnosis & Treatment, 45th Edition
document.getElementById('working').innerHTML = ''; function hide_scrollbar() { var e = document.body; e.style.scrollbarBaseColor = '#fff'; e.style.scrollbarArrowColor = '#fff'; e.style.scrollbarDarkShadowColor = '#fff'; e.style.scrollbarShadowColor = '#fff'; } function show_scrollbar() { var e = document.body; e.style.scrollbarBaseColor = '#ccc'; e.style.scrollbarArrowColor = '#000'; e.style.scrollbarDarkShadowColor = '#000'; e.style.scrollbarShadowColor = '#666'; } function toggle_CA(e) { var src; src = window.parent.getSrc(e); if (src.nextSibling.style.display == 'block') { src.nextSibling.style.display='none'; src.innerHTML='View Answer'; } else { src.nextSibling.style.display='block'; src.innerHTML='Hide Answer'; } } function resolve_link(e,setid,locator,dbid,toan) { e.href = "ovidweb.cgi?S=IDNJHKKKJGEPJK00D;FTS+Link+Set+Ref=" + setid + "|" + locator + "|" + dbid + "|" + toan; return true; }
15
Liver, Biliary Tract, & Pancreas
Lawrence S. Friedman MD
See http://www.cmdtlinks.com
JAUNDICE (ICTERUS)
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Results from accumulation of bilirubin in the body tissues; cause may be hepatic or nonhepatic.
Hyperbilirubinemia may be due to abnormalities in the formation, transport, metabolism, and excretion of bilirubin.
Total serum bilirubin is normally 0.2–1.2 mg/dL; jaundice may not be recognizable until levels are about 3 mg/dL.
Evaluation of obstructive jaundice begins with ultrasonography and is usually followed by cholangiography.
General Considerations
Jaundice results from the accumulation of bilirubin— a reddish pigment product of heme metabolism—in the body tissues; the cause may be hepatic or nonhepatic. Hyperbilirubinemia may be due to abnormalities in the formation, transport, metabolism, and excretion of bilirubin. Total serum bilirubin is normally 0.2–1.2 mg/dL (mean levels are higher in men than women and higher in whites and hispanics than blacks), and jaundice may not be recognizable until levels are about 3 mg/dL.
Jaundice is caused by predominantly unconjugated or conjugated bilirubin in the serum (Table 15-1). Unconjugated hyperbilirubinemia may result from overproduction of bilirubin because of hemolysis; impaired hepatic uptake of bilirubin due to certain drugs; or impaired conjugation of bilirubin by glucuronide, as in Gilbert's syndrome, due to mild decreases in glucuronyl transferase, or Crigler-Najjar syndrome, caused by moderate decreases or absence of glucuronyl transferase. In the absence of liver disease, hemolysis rarely elevates the serum bilirubin level to more than 7 mg/dL. Predominantly conjugated hyperbilirubinemia may result from impaired excretion of bilirubin from the liver due to hepatocellular disease, drugs, sepsis, hereditary disorders such as Dubin-Johnson syndrome, or extrahepatic biliary obstruction. Features of some hyperbilirubinemic syndromes are summarized in Table 15-2. The term “cholestasis” denotes retention of bile in the liver, and the term “cholestatic jaundice” is often used when conjugated hyperbilirubinemia results from impaired bile flow.
Manifestations of Diseases Associated with Jaundice
A. UNCONJUGATED HYPERBILIRUBINEMIA
Stool and urine color are normal, and there is mild jaundice and indirect (unconjugated) hyperbilirubinemia with no bilirubin in the urine. Splenomegaly occurs in hemolytic disorders except in sickle cell anemia. Abdominal or back pain may occur with acute hemolytic crises.
B. CONJUGATED HYPERBILIRUBINEMIA
1. Hereditary cholestatic syndromes or intrahepatic cholestasis
The patient may be asymptomatic; intermittent cholestasis is often accompanied by pruritus, light-colored stools, and, occasionally, malaise.
2. Hepatocellular disease
Malaise, anorexia, lowgrade fever, and right upper quadrant discomfort are frequent. Dark urine, jaundice, and, in women, amenorrhea occur. An enlarged tender liver, vascular spiders, palmar erythema, ascites, gynecomastia, sparse body hair, fetor hepaticus, and asterixis may be present, depending on the cause, severity, and chronicity of liver dysfunction.
C. BILIARY OBSTRUCTION
There may be right upper quadrant pain, weight loss (suggesting carcinoma), jaundice, dark urine, and light-colored stools. Symptoms and signs may be intermittent if caused by stone, carcinoma of the ampulla, or cholangiocarcinoma. Pain may be absent early in pancreatic cancer. Occult blood in the stools suggests cancer of the ampulla. Hepatomegaly and a palpable gallbladder (Courvoisier's sign) are characteristic, but neither specific nor sensitive, of pancreatic
P.650
head tumor. Fever and chills are far more common in benign obstruction and associated cholangitis.
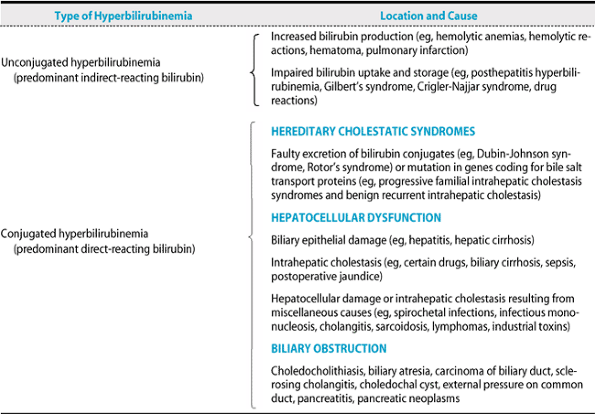 |
Table 15-1. Classification of jaundice. |
Diagnostic Methods for Evaluation of Liver Disease & Jaundice (Table 15-3)
A. LABORATORY STUDIES
Serum alanine and aspartate aminotransferase (ALT and AST) levels correlate with body mass index and possibly with mortality from liver disease and inversely with caffeine consumption. Elevated ALT and AST levels result from hepatocellular necrosis or inflammation, as in hepatitis; ALT is more specific for the liver than AST, but an AST level at least twice that of the ALT is typical of alcoholic liver injury. In contrast, the ALT level is greater than the AST level in nonalcoholic fatty liver disease prior to the development of cirrhosis. In addition to signaling underlying liver disease, an isolated elevation of the serum ALT level may be the only clue to the diagnosis of celiac disease. Elevated alkaline phosphatase levels are seen in cholestasis or infiltrative liver disease (such as tumor or granuloma). Alkaline phosphatase elevations of hepatic rather than bone, intestinal, or placental origin are confirmed by concomitant elevation of γ-glutamyl transpeptidase or 5′-nucleotidase levels. The differential diagnosis of any liver test elevation includes toxicity caused by drugs, herbal remedies, and toxins.
B. LIVER BIOPSY
Percutaneous liver biopsy is the definitive study for determining the cause and histologic severity of hepatocellular dysfunction or infiltrative liver disease. In patients with suspected metastatic disease or a hepatic mass, it is performed under ultrasound or CT guidance. A transjugular route can be used in patients with coagulopathy or ascites.
C. IMAGING
Demonstration of dilated bile ducts by ultrasonography or CT scan indicates biliary obstruction (90–95% sensitivity). Ultrasonography, CT scan, and MRI may also demonstrate hepatomegaly, intrahepatic tumors, and portal hypertension. Spiral arterial-phase and multislice CT scanning, in which the liver is imaged during peak hepatic enhancement while the patient holds one or two breaths, improves diagnostic accuracy. Multiphasic spiral or multislice CT, CT arterial portography, in which imaging follows intravenous contrast infusion via a catheter placed in the superior mesenteric artery, MRI with use of gadolinium or ferumoxides as contrast agents, and intraoperative ultrasonography are the most sensitive techniques for detection of individual small hepatic lesions in
P.651
patients eligible for resection of metastases. Use of color Doppler ultrasound or contrast agents that produce microbubbles increases the sensitivity of transcutaneous ultrasound for detecting small neoplasms. MRI is the most accurate technique for identifying isolated liver lesions such as hemangiomas, focal nodular hyperplasia, or focal fatty infiltration and for detecting hepatic iron overload. Because of its much lower cost (amount charged), ultrasonography
P.652
($500) is preferable to CT ($1200–$1400) or MRI ($2000) as a screening test. Ultrasonography can detect gallstones with a sensitivity of 95%.
Table 15-2. Hyperbilirubinemic disorders. | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Table 15-3. Liver function tests: Normal values and changes in two types of jaundice. | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Endoscopic retrograde cholangiopancreatography (ERCP) or percutaneous transhepatic cholangiography (PTC) identifies the cause, location, and extent of biliary obstruction. Magnetic resonance cholangiopancreatography (MRCP) appears to be a sensitive, noninvasive method of detecting bile duct stones, strictures, and dilation; however, it is less reliable than ERCP for distinguishing malignant from benign strictures. ERCP requires a skilled endoscopist and may be used to demonstrate pancreatic or ampullary causes of jaundice, to carry out papillotomy and stone extraction, or to insert a stent through an obstructing lesion. Complications of ERCP include pancreatitis (5%) and, less commonly, cholangitis, bleeding, or duodenal perforation after papillotomy. Risk factors for post-ERCP pancreatitis include female gender, prior post-ERCP pancreatitis, suspected sphincter of Oddi dysfunction, and a difficult or failed cannulation. Severe complications of PTC occur in 3% and include fever, bacteremia, bile peritonitis, and intraperitoneal hemorrhage. Endoscopic ultrasonography is the most sensitive test for detecting small lesions of the ampulla or pancreatic head and for detecting portal vein invasion by pancreatic cancer. It is also accurate in detecting or excluding bile duct stones.
Adams PC et al: Screening for liver disease: report of an AASLD clinical workshop. Hepatology 2004;39:1204.
Christensen M: Complications of ERCP: a prospective study. Gastrointest Endosc 2004;60:721.
Freeman ML et al: Prevention of post-ERCP pancreatitis: a comprehensive review. Gastrointest Endosc 2004;59:845.
Kim HC et al: Normal serum aminotransferase concentration and risk of mortality from liver diseases: prospective cohort study. BMJ 2004;328:983.
Ruhl CE et al: Coffee and caffeine consumption reduce the risk for elevated serum alanine aminotransferase activity in the United States. Gastroenterology 2005;128:24.
Zucker SD et al: Serum bilirubin levels in the U.S. population: gender effect and inverse correlation with colorectal cancer. Hepatology 2004;40:827.
DISEASES OF THE LIVER
VIRAL HEPATITIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Prodrome of anorexia, nausea, vomiting, malaise, aversion to smoking.
Fever, enlarged and tender liver, jaundice.
Normal to low white cell count; abnormal liver tests, especially markedly elevated aminotransferases early in the course.
Liver biopsy shows hepatocellular necrosis and mononuclear infiltrate but is rarely indicated.
General Considerations
Hepatitis can be caused by many drugs and toxic agents as well as by numerous viruses, the clinical manifestations of which may be quite similar. Viruses causing hepatitis are (1) hepatitis A virus (HAV), (2) hepatitis B virus (HBV), (3) hepatitis C virus (HCV), (4) hepatitis D virus (HDV) (delta agent), and (5) hepatitis E virus (HEV) (an enterically transmitted hepatitis seen in epidemic form in Asia, North Africa, and Mexico). The designation hepatitis G virus (HGV) applies to a virus that rarely, if ever, causes frank hepatitis. A DNA virus designated the TT virus (TTV) has been identified in up to 7.5% of blood donors and found to be transmitted readily by blood transfusions, but an association between this virus and liver disease has not been established. A related virus known as SEN-V has been found in 2% of US blood donors, is transmitted by transfusion, and may account for some cases of transfusion-associated non-ABCDE hepatitis. In immunocompromised and rare immunocompetent persons, cytomegalovirus, Epstein-Barr virus, and herpes simplex virus should be considered in the differential diagnosis of hepatitis. Severe acute respiratory syndrome (SARS) may be associated with marked serum aminotransferase elevations. As yet unidentified pathogens account for a small percentage of cases of apparent acute viral hepatitis.
A. HEPATITIS A
Figure 15-1 shows the typical course of HAV infection. HAV is a 27-nm RNA hepatovirus (in the picornavirus
P.653
family) causing epidemics or sporadic cases of hepatitis. The virus is transmitted by the fecal-oral route, and its spread is favored by crowding and poor sanitation. Common source outbreaks result from contaminated water or food, including inadequately cooked shellfish. A large outbreak among patrons of a restaurant in Monaca, Pennsylvania, in 2003 was traced to contaminated green onions from Mexico. The incubation period averages 30 days. HAV is excreted in feces for up to 2 weeks before clinical illness but rarely after the first week of illness. The mortality rate for hepatitis A is low, and fulminant hepatitis A is uncommon save for rare instances in which it occurs in a patient with chronic hepatitis C. Chronic hepatitis A does not occur, and there is no carrier state. Clinical illness is more severe in adults than in children, in whom it is typically asymptomatic. It is the only viral hepatitis causing spiking fevers. Rare cases of acute cholecystitis during the course of acute hepatitis A have been described.
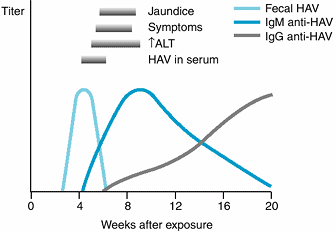 |
Figure 15-1. The typical course of acute type A hepatitis. (HAV, hepatitis A virus; anti-HAV, antibody to hepatitis A virus; ALT, alanine aminotransferase.) (Reprinted from Koff RS: Acute viral hepatitis. In: Handbook of Liver Disease. Friedman LS, Keeffe EB [editors], 2nd ed. 2004, with permission from Elsevier.) |
Antibody to hepatitis A (anti-HAV) appears early in the course of the illness. Both IgM and IgG antiHAV are detectable in serum soon after the onset. Peak titers of IgM anti-HAV occur during the first week of clinical disease and disappear within 3–6 months. Detection of IgM anti-HAV is an excellent test for diagnosing acute hepatitis A. Titers of IgG anti-HAV rise after one month of the disease and may persist for years. IgG anti-HAV indicates previous exposure to HAV, noninfectivity, and immunity. In the United States, about 30% of the population have serologic evidence of previous infection.
B. HEPATITIS B
Figure 15-2 shows the typical course of HBV infection. HBV is a 42-nm hepadnavirus with a partially double-stranded DNA genome, inner core protein (hepatitis B core antigen, HBcAg) and outer surface coat (hepatitis B surface antigen, HBsAg). There are eight different genotypes (A–H), which may influence the course of infection and responsiveness to antiviral therapy. HBV is usually transmitted by inoculation of infected blood or blood products or by sexual contact and is present in saliva, semen, and vaginal secretions. HBsAg-positive mothers may transmit HBV at delivery; the risk of chronic infection in the infant is as high as 90%.
HBV is prevalent in men who have sex with men and in injection drug users, but the greatest number of cases result from heterosexual transmission; the incidence has decreased by 75% since the 1980s. Groups at risk include patients and staff at hemodialysis centers, physicians, dentists, nurses, and personnel working in clinical and pathology laboratories and blood banks. Half of all patients with acute hepatitis B in the United States have previously been incarcerated or treated for a sexually transmitted disease. The risk of HBV infection from a blood transfusion is less than 1 in 60,000 units transfused in the United States. The incubation period of hepatitis B is 6 weeks to 6 months (average 12–14 weeks).
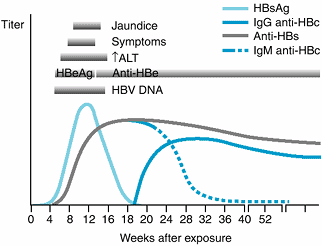 |
Figure 15-2. The typical course of acute type B hepatitis. (HBsAg, hepatitis B surface antigen; anti-HBs, antibody to HBsAg; HBeAg, hepatitis Be antigen; anti-HBe, antibody to HBeAg; anti-HBc, antibody to hepatitis B core antigen; ALT, alanine aminotransferase.) (Reprinted from Koff RS: Acute viral hepatitis. In: Handbook of Liver Disease. Friedman LS, Keeffe EB [editors], 2nd ed. 2004, with permission from Elsevier.) |
The onset of hepatitis B is more insidious and the aminotransferase levels are higher than in HAV infection. The risk of fulminant hepatitis is less than 1%, with a mortality rate of up to 60%. Following acute hepatitis B, HBV infection persists in 1–2% of immunocompetent adults but in a higher percentage of immunocompromised adults or children. Persons with chronic hepatitis B, particularly when HBV infection is acquired early in life and viral replication persists, are at substantial risk of cirrhosis and hepatocellular carcinoma (up to 25–40%). Men are more at risk than women. Infection caused by HBV may be associated with serum sickness, glomerulonephritis, and polyarteritis nodosa.
There are three distinct antigen-antibody systems that relate to HBV infection and circulating markers that are useful in diagnosis. Interpretation of common serologic patterns is shown in Table 15-4.
1. HBsAg
The appearance of HBsAg is the first evidence of infection, appearing before biochemical evidence of liver disease, and persists throughout the clinical illness. Persistence of HBsAg after the acute illness may be associated with clinical and laboratory evidence of chronic hepatitis for variable periods of time. The detection of HBsAg establishes infection with HBV and implies infectivity.
2. Anti-HBs
Specific antibody to HBsAg (anti-HBs) appears in most individuals after clearance of HBsAg and after successful vaccination against hepatitis B. Disappearance of HBsAg and the appearance of anti-HBs signals recovery from HBV infection, noninfectivity, and immunity.
Table 15-4. Common serologic patterns in hepatitis B virus infection and their interpretation. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
P.654
3. Anti-HBc
IgM anti-HBc appears shortly after HBsAg is detected. (HBcAg alone does not appear in serum.) Its presence in the setting of acute hepatitis indicates a diagnosis of acute hepatitis B, and it fills the serologic gap in patients who have cleared HBsAg but do not yet have detectable anti-HBs. IgM anti-HBc can persist for 3–6 months or longer. IgM anti-HBc may also reappear during flares of previously inactive chronic hepatitis B. IgG anti-HBc also appears during acute hepatitis B but persists indefinitely, whether the patient recovers (with the appearance of anti-HBs in serum) or develops chronic hepatitis B (with persistence of HBsAg). In asymptomatic blood donors, an isolated anti-HBc with no other positive HBV serologic results may represent a falsely positive result or latent infection in which HBV DNA is detectable only by polymerase chain reaction testing.
4. HBeAg
HBeAg is a soluble protein found only in HBsAg-positive serum. It is a secretory form of HBcAg appearing during the incubation period shortly after the detection of HBsAg. HBeAg indicates viral replication and infectivity. Persistence of HBeAg in serum beyond 3 months indicates an increased likelihood of chronic hepatitis B. Its disappearance is often followed by the appearance of anti-HBe, signifying diminished viral replication and decreased infectivity.
5. HBV DNA
The presence of HBV DNA in serum generally parallels the presence of HBeAg, although HBV DNA is a more sensitive and precise marker of viral replication and infectivity. Very low levels of HBV DNA, detectable only by polymerase chain reaction testing, may persist in serum and liver long after a patient has recovered from acute hepatitis B, but the HBV DNA in serum is bound to IgG and is rarely infectious. In some patients with chronic hepatitis B, HBV DNA is present at high levels without HBeAg in serum because of a mutation that prevents synthesis of HBeAg in infected hepatocytes. This “pre-core mutant” appears during the course of chronic wild-type HBV infection, presumably as a result of immune pressure. When additional mutations in the core gene are present, the pre-core mutant enhances the severity of HBV and increases the risk of cirrhosis.
C. HEPATITIS D (DELTA AGENT)
HDV is a defective RNA virus that causes hepatitis only in association with hepatitis B infection and specifically only in the presence of HBsAg; it is cleared when the latter is cleared.
HDV may coinfect with HBV or may superinfect a person with chronic hepatitis B, usually by percutaneous exposure. When acute hepatitis D is coincident with acute HBV infection, the infection is generally similar in severity to acute hepatitis B alone. In chronic hepatitis B, superinfection by HDV appears to carry a worse short-term prognosis, often resulting in fulminant hepatitis or severe chronic hepatitis that progresses rapidly to cirrhosis.
In the 1970s and early 1980s, HDV was endemic in some areas, such as the Mediterranean countries, where up to 80% of HBV carriers were superinfected with it. In the United States, HDV occurred primarily among injection drug users. However, new cases of hepatitis D are now infrequent (for reasons that are not entirely clear), and cases seen today are usually from cohorts infected years ago who survived the initial impact of hepatitis D and now have inactive cirrhosis. These patients have a threefold increased risk of hepatocellular carcinoma. Diagnosis is made by detection of antibody to hepatitis D antigen (anti-HDV) or, where available, HDV RNA in serum.
D. HEPATITIS C
Figure 15-3 shows the typical course of HCV infection. HCV is a single-stranded RNA virus (hepacivirus) with properties similar to those of flavivirus. At least six major genotypes of HCV have been identified. In the past, HCV was responsible for over 90% of cases of posttransfusion hepatitis, yet only 4% of cases of hepatitis C were attributable to blood transfusions.
P.655
Over 50% of cases are transmitted by injection drug use. Intranasal cocaine use, body piercing, and hemodialysis also are risk factors. The risk of sexual and maternal-neonatal transmission is low and may be greatest in a subset of patients with high circulating levels of HCV RNA. Having multiple sexual partners may increase the risk of HCV infection. Transmission via breast-feeding has not been documented. An outbreak of hepatitis C in patients with immune deficiencies occurred in some recipients of intravenous immune globulin, and nosocomial transmission via multidose vials of saline used to flush Portacaths and between hospitalized patients on a liver unit has occurred. Coinfection with HCV is found in at least 30% of persons infected with HIV; HIV infection leads to more rapid progression of chronic hepatitis C to cirrhosis, whereas HCV increases the hepatotoxicity of highly active antiretroviral therapy. Covert transmission during bloody fisticuffs has even been reported. In many patients, the source of infection is unknown. There are more than 2.7 million HCV carriers in the United States and another 1.3 million previously exposed persons who have cleared the virus.
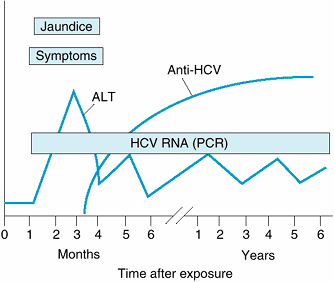 |
Figure 15-3. The typical course of acute and chronic hepatitis C. (ALT, alanine aminotransferase; Anti-HCV, antibody to hepatitis C virus by enzyme immunoassay; HCV RNA [PCR], hepatitis C viral RNA by polymerase chain reaction.) |
The incubation period averages 6–7 weeks, and clinical illness is often mild, usually asymptomatic, and characterized by waxing and waning aminotransferase elevations and a high rate (> 80%) of chronic hepatitis. In pregnant patients, serum aminotransferase levels frequently normalize despite persistence of viremia, only to increase again after delivery. HCV is a pathogenetic factor in mixed cryoglobulinemia and membranoproliferative glomerulonephritis and may be related to lichen planus, autoimmune thyroiditis, lymphocytic sialadenitis, idiopathic pulmonary fibrosis, sporadic porphyria cutanea tarda, monoclonal gammopathies, and probably lymphoma. Hepatitis C may induce insulin resistance (which in turn increases the risk of hepatic fibrosis), and the risk of type 2 diabetes mellitus is increased in persons with chronic hepatitis C. Hepatic steatosis is a particular feature of infection with HCV genotype 3 and may also occur in patients with risk factors for fatty liver (see below).
Diagnosis of hepatitis C is based on an enzyme immunoassay that detects antibodies to HCV. AntiHCV is not protective, and in patients with acute or chronic hepatitis its presence in serum generally signifies that HCV is the cause. Limitations of the enzyme immunoassay include moderate sensitivity (false-negatives) for the diagnosis of acute hepatitis C early in the course and in healthy blood donors and low specificity (false-positives) in some persons with elevated gamma globulin levels. In these situations, a diagnosis of hepatitis C may be confirmed by using an assay for HCV RNA and, in some cases, a supplemental recombinant immunoblot assay (RIBA) for anti-HCV. Most RIBApositive persons are potentially infectious, as confirmed by use of polymerase chain reaction–based tests to detect HCV RNA. Occasional persons are found to have anti-HCV in serum, confirmed by RIBA, without HCV RNA in serum, suggesting recovery from HCV infection in the past. Testing donated blood for HCV has helped reduce the risk of transfusion-associated hepatitis C from 10% in 1990 to about 1 case per 2 million units today.
E. HEPATITIS E
HEV is a 29to 32-nm RNA virus similar to calicivirus and responsible for waterborne hepatitis outbreaks in India, Burma, Afghanistan, Algeria, Mexico, and recently Sudan and Iraq. It is rare in the United States but should be considered in patients with acute hepatitis after a trip to an endemic area. Illness is selflimited (no carrier state), with a high mortality rate (10–20%) in pregnant women and an increased risk of hepatic decompensation in patients with underlying chronic liver disease.
F. HEPATITIS G
The designation HGV has been applied to a flavivirus that is percutaneously transmitted and associated with chronic viremia lasting at least 10 years. HGV has been detected in 1.5% of blood donors, 50% of injection drug users, 30% of hemodialysis patients, 20% of hemophiliacs, and 15% of patients with chronic hepatitis B or C, but it does not appear to cause important liver disease or affect the response of patients with chronic hepatitis B or C to antiviral therapy. HGV coinfection may improve survival in patients with HIV infection.
Clinical Findings
The clinical picture of viral hepatitis is extremely variable, ranging from asymptomatic infection without jaundice to a fulminating disease and death in a few days.
P.656
A. SYMPTOMS
1. Prodromal phase
The onset may be abrupt or insidious, with general malaise, myalgia, arthralgia, easy fatigability, upper respiratory symptoms, and anorexia. A distaste for smoking, paralleling anorexia, may occur early. Nausea and vomiting are frequent, and diarrhea or constipation may occur. Serum sickness may be seen early in acute hepatitis B. Fever is generally present but is low-grade save in occasional cases of hepatitis A. Defervescence and a fall in pulse rate often coincide with the onset of jaundice.
Abdominal pain is usually mild and constant in the right upper quadrant or epigastrium, often aggravated by jarring or exertion, and rarely may be severe enough to simulate cholecystitis.
2. Icteric phase
Jaundice occurs after 5–10 days but may appear at the same time as the initial symptoms. In many patients, jaundice never develops. With the onset of jaundice, there is often worsening of the prodromal symptoms, followed by progressive clinical improvement.
3. Convalescent phase
There is an increasing sense of well-being, return of appetite, and disappearance of jaundice, abdominal pain and tenderness, and fatigability.
4. Course and complications
The acute illness usually subsides over 2–3 weeks with complete clinical and laboratory recovery by 9 weeks in hepatitis A and by 16 weeks in hepatitis B. In 5–10% of cases, the course may be more protracted, but less than 1% will have a fulminant course. In some cases of acute hepatitis A, clinical, biochemical, and serologic recovery may be followed by one or two relapses, but recovery is the rule. A protracted course of hepatitis A has been reported to be associated with HLA DRB1*1301. Hepatitis B, D, and C (and G) may become chronic (see below). Rarely, aplastic anemia may complicate the course of non-A–E hepatitis.
B. SIGNS
Hepatomegaly—rarely marked—is present in over half of cases. Liver tenderness is usually present. Splenomegaly is reported in 15% of patients, and soft, enlarged lymph nodes—especially in the cervical or epitrochlear areas—may occur. Systemic toxicity is most often encountered in hepatitis A. Slight neurocognitive impairment has been described in patients with chronic hepatitis C.
C. LABORATORY FINDINGS
The white blood cell count is normal to low, especially in the preicteric phase. Large atypical lymphocytes may occasionally be seen. Rarely, aplastic anemia follows an episode of acute hepatitis not caused by any of the known hepatitis viruses. Mild proteinuria is common, and bilirubinuria often precedes the appearance of jaundice. Acholic stools are often present during the icteric phase. Strikingly elevated AST or ALT occurs early, followed by elevations of bilirubin and alkaline phosphatase; in a minority of patients, the latter persist after aminotransferase levels have normalized. Cholestasis is occasionally marked in acute hepatitis A. Marked prolongation of the prothrombin time in severe hepatitis correlates with increased mortality.
Differential Diagnosis
The differential diagnosis includes other viral diseases such as infectious mononucleosis, cytomegalovirus infection, and herpes simplex virus infection; spirochetal diseases such as leptospirosis and secondary syphilis; brucellosis; rickettsial diseases such as Q fever; druginduced liver disease; and shock liver (ischemic hepatitis). Occasionally, autoimmune hepatitis (see below) may have an acute onset mimicking acute viral hepatitis. Rarely, metastatic cancer of the liver may present with a hepatitis-like picture.
The prodromal phase of viral hepatitis must be distinguished from other infectious disease such as influenza, upper respiratory infections, and the prodromal stages of the exanthematous diseases. Cholestasis may mimic obstructive jaundice.
Prevention
Strict isolation of patients is not necessary, but hand washing after bowel movements is required. Thorough hand washing by medical staff who may contact contaminated utensils, bedding, or clothing is essential. Careful handling of disposable needles—including not recapping used needles—is required for medical personnel. Screening of donated blood for HBsAg, anti-HBc, and anti-HCV has reduced the risk of transfusion-associated hepatitis markedly. All pregnant women should undergo testing for HBsAg. HBVand HCV-infected persons should practice safe sex, but there is little evidence that HCV is spread easily by sexual contact. Vaccination against HAV (after prescreening for prior immunity) and HBV is recommended for patients with chronic hepatitis C, and vaccination against HAV is recommended for patients with chronic hepatitis B.
A. HEPATITIS A
Immune globulin should be given to all close (eg, household) personal contacts of patients with hepatitis A and should be considered in persons who consume food prepared by an infected food handler. The recommended dose of 0.02 mL/kg intramuscularly is protective if administered during incubation. Two effective inactivated hepatitis A vaccines are available and recommended for persons living in or traveling to endemic areas (including military personnel), patients with chronic liver disease upon diagnosis, persons with clotting-factor disorders who are treated with concentrates, homosexual and bisexual men, animal handlers, illicit drug users, sewage workers, food handlers, and children and caregivers in day care centers and institutions. Routine vaccination of all children has been recommended
P.657
in states with an incidence of hepatitis A at least twice the national average. HAV vaccine is also effective in the prevention of secondary spread to household contacts of primary cases. The recommended dose for adults is 1 mL (1440 ELISA units) of Havrix (GlaxoSmithKline) or 0.5 mL (50 units) of Vaqta (Merck) intramuscularly, followed by a booster dose at 6–12 months. A combined hepatitis A and B vaccine (Twinrix, GlaxoSmithKline) is available. HIV infection impairs the response to the HAV vaccine, especially in persons with a CD4 count less than 200/mcL.
B. HEPATITIS B
Hepatitis B immune globulin (HBIG) may be protective—or may attenuate the severity of illness—if given in large doses within 7 days after exposure (adult dose is 0.06 mL/kg body weight) followed by initiation of the HBV vaccine series (see below). This approach is currently recommended for persons exposed to HBsAgcontaminated material via mucous membranes or through breaks in the skin and for individuals who have had sexual contact with persons with HBV infection (irrespective of the presence or absence of HBeAg in the source). HBIG is also indicated for newborn infants of HBsAg-positive mothers followed by initiation of the vaccine series (see below).
The currently used vaccines are recombinant-derived. Initially, the vaccine was targeted to persons at high risk, including renal dialysis patients and attending personnel, patients requiring repeated transfusions, spouses of HBsAg-positive persons, men who have sex with men, injection drug users, newborns of HBsAg-positive mothers, beginning medical and nursing students, and all medical technologists. Because this strategy failed to lower the incidence of hepatitis B, the CDC recommended universal vaccination of infants and children in the United States. Over 90% of recipients of the vaccine mount protective antibody to hepatitis B; immunocompromised persons respond poorly. Reduced response to the vaccine may have a genetic basis and has also been associated with age over 40 years and celiac disease. The standard regimen for adults is 10–20 mcg initially (depending on the formulation) repeated again at 1 and 6 months, but alternative schedules have been approved. For greatest reliability of absorption, the deltoid muscle is the preferred site. Vaccine formulations free of the mercury-containing preservative thimerosal are given in infants less than 6 months of age. When documentation of seroconversion is considered desirable, postimmunization anti-HBs titers may be checked. Protection appears to be excellent even if the titer wanes—at least for 15 years—and booster reimmunization is not routinely recommended but is advised for immunocompromised persons in whom anti-HBs titers fall below 10 mIU/mL. For vaccine nonresponders, three additional vaccine doses may elicit seroprotective anti-HBs levels in 30–50% of persons. Universal vaccination of neonates in countries endemic for HBV reduces the incidence of hepatocellular carcinoma.
Treatment
Bed rest is recommended only if symptoms are marked. If nausea and vomiting are pronounced or if oral intake is substantially decreased, intravenous 10% glucose is indicated. Encephalopathy or severe coagulopathy indicates impending acute hepatic failure, and hospitalization is mandatory (see below).
Dietary management consists of palatable meals as tolerated, without overfeeding; breakfast is usually best tolerated. Strenuous physical exertion, alcohol, and hepatotoxic agents are avoided. Small doses of oxazepam are safe, as metabolism is not hepatic; morphine sulfate is avoided.
Corticosteroids have no benefit in patients with viral hepatitis, including those with fulminant disease. Treatment of acute hepatitis C patients with interferon alfa or peginterferon (see later) for 6–24 weeks appreciably decreases the risk of chronic hepatitis. Because 20% of patients with acute hepatitis C clear the virus without such treatment, reserving it for patients in whom serum HCV RNA levels fail to clear after 3–4 months may be advisable. Ribavirin may be added if HCV RNA fails to clear after 3 months of interferon alfa or peginterferon. Spontaneous viral clearance is much more likely in symptomatic patients than in asymptomatic patients. Antiviral therapy is generally unnecessary in patients with acute hepatitis B. Liver transplantation should be considered in patients with acute liver failure (see below).
Prognosis
In most patients, clinical recovery is complete in 3–6 weeks. Laboratory evidence of liver dysfunction may persist for a longer period, but most patients recover completely. The overall mortality rate is less than 1%, but the rate is reportedly higher in older people.
Hepatitis A does not cause chronic liver disease, although it may persist for up to 1 year, and clinical and biochemical relapses may occur before full recovery. The mortality rate is less than 0.2%. The mortality rate for acute hepatitis B is 0.1–1%, but higher with superimposed hepatitis D. Fulminant hepatitis C is rare in the United States. For unknown reasons, the mortality rate for hepatitis E is especially high in pregnant women (10–20%).
Chronic hepatitis, characterized by elevated aminotransferase levels for more than 6 months, develops in 1–2% of immunocompetent adults with acute hepatitis B but in as many as 90% of infected neonates and infants and a substantial proportion of immunocompromised adults. Chronic hepatitis, which progresses very slowly in many cases, develops in as many as 80% of all persons with acute hepatitis C. Ultimately, cirrhosis develops in up to 30% of those with chronic hepatitis C and 40% of those with chronic hepatitis B; the risk of cirrhosis is even higher in patients coinfected with both viruses or with HIV. Patients with cirrhosis are at risk for hepatocellular carcinoma at a rate of 3–5% per year.
P.658
Even in the absence of cirrhosis, patients with chronic hepatitis B—particularly those with active viral replication—are at increased risk.
Fiore AE: Hepatitis A transmitted by food. Clin Infect Dis 2004;38:705.
Ganem D et al: Hepatitis B virus infection—natural history and clinical consequences. N Engl J Med 2004;350:1118.
Medina J et al: Review article: hepatitis C virus-related extra-hepatic disease—aetiopathogenesis and management. Aliment Pharmacol Ther 2004;20:129.
Nomura H et al: Short-term interferon-alfa therapy for acute hepatitis C: a randomized controlled trial. Hepatology 2004;39:1201.
Strader DB et al: Diagnosis, management, and treatment of hepatitis C. Hepatology 2004;39:1147.
Tran TT et al: Hepatitis B: epidemiology and natural history. Clin Liver Dis 2004;8:255.
Vallet-Pichard A et al: Hepatitis viruses and human immunodeficiency virus co-infection: pathogenesis and treatment. J Hepatol 2004;41:156.
ACUTE HEPATIC FAILURE
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
May be fulminant or subfulminant; both forms carry a poor prognosis.
Acetaminophen and idiosyncratic drug reactions are the most common causes.
General Considerations
Acute hepatic failure may be fulminant or subfulminant. Fulminant hepatic failure is characterized by the development of hepatic encephalopathy within 8 weeks after the onset of acute liver disease. Coagulopathy is invariably present. Subfulminant hepatic failure occurs when these findings appear between 8 weeks and 6 months after the onset of acute liver disease and carries an equally poor prognosis.
Acetaminophen toxicity is now the most common cause of acute hepatic failure in the United States, as it has been in England for some time, accounting for up to 40% of cases. Other causes include idiosyncratic drug reactions (now the second most common cause), viral hepatitis, poisonous mushrooms, shock, hyperthermia or hypothermia, Budd-Chiari syndrome, malignancy (most commonly lymphomas), Wilson's disease, Reye's syndrome, fatty liver of pregnancy and other disorders of fatty acid oxidation, autoimmune hepatitis, and parvovirus B19 infection. The risk of acute hepatic failure is increased in patients with diabetes. Herbal and dietary supplements are thought to be contributory to acute hepatic failure in a substantial portion of cases, regardless of cause. Acute hepatic failure may rarely complicate grand mal seizures.
In the past, about 70% of all cases of acute hepatic failure in the United States were caused by acute viral hepatitis. Viral hepatitis now accounts for only 12% of all cases. The decline of viral hepatitis as the principal cause of acute hepatic failure is due to a decline in the contribution of hepatitis B to acute hepatic failure, possibly because of a policy of universal vaccination of infants and children. In endemic areas, hepatitis E is an important cause of acute hepatic failure. Hepatitis C appears to be a rare cause of acute hepatic failure in the United States, but acute hepatitis A or B superimposed on chronic hepatitis C is associated with a high risk of fulminant hepatitis.
Clinical Findings
In acute hepatic failure due to drug toxicity or hepatitis, extensive necrosis of large areas of the liver gives a typical pathologic picture of acute liver atrophy. A systemic inflammatory response, gastrointestinal symptoms, and hemorrhagic phenomena are common. Adrenal insufficiency often complicates acute hepatic failure. However, the value of glucocorticoid therapy is uncertain. Jaundice may be absent or minimal early, but laboratory tests show severe hepatocellular damage. In acute hepatic failure due to microvesicular steatosis (eg, Reye's syndrome), serum aminotransferase elevations may be modest (< 300 units/L).
Treatment
The treatment of acute hepatic failure is directed toward correcting metabolic abnormalities. These include coagulation defects, electrolyte and acid-base disturbances, renal failure, hypoglycemia, and encephalopathy. Prophylactic antibiotic therapy decreases the risk of infection, observed in up to 90%, but has no effect on survival and is not routinely recommended. For suspected sepsis, broad coverage is indicated. Early administration of acetylcysteine (140 mg/kg orally followed by 70 mg/kg orally every 4 hours for an additional 17 doses) is indicated for acetaminophen toxicity and improves cerebral blood flow and oxygenation in patients with fulminant hepatic failure due to any cause. (Acetylcysteine treatment can prolong the prothrombin time, leading to the erroneous assumption that liver failure is worsening.) Subclinical seizure activity is common in patients with acute liver failure, but the value of prophylactic phenytoin is uncertain. Early transfer to a liver transplantation center is essential. Extradural sensors may be placed to monitor intracranial pressure for impending cerebral edema.
Mannitol, 100–200 mL of a 20% solution by intravenous infusion over 10 minutes, may decrease cerebral edema but should be used with caution in patients with renal failure. Preliminary experience suggests that intravenously administered hypertonic saline to induce hypernatremia also may reduce intracranial hypertension. Hypothermia to a temperature of 33.1 C may reduce intracranial pressure when
P.659
other measures have failed and may improve survival long enough to permit liver transplantation. The value of hyperventilation and intravenous prostaglandin E1 is uncertain. Hepatic-assist devices using living hepatocytes, extracorporeal whole liver perfusion, hepatocyte transplantation, and liver xenografts have shown promise experimentally and may reduce mortality in patients with acute hepatic failure superimposed on chronic liver disease. They also serve as a “bridge” to liver transplantation. However, a meta-analysis of trials (involving small numbers of patients) of the molecular adsorbent recirculating system (MARS), which is based on extracorporeal albumin dialysis, showed no significant survival benefit when compared with standard medical therapy in patients with acute liver failure.
Prognosis
The mortality rate of fulminant hepatitis with severe encephalopathy is as high as 80%. The outlook is especially poor in patients younger than 10 and older than 40 years of age and in those with an idiosyncratic drug reaction. Spontaneous recovery is less likely for hepatitis B than for hepatitis A. Other adverse prognostic factors are a serum bilirubin level > 18 mg/dL, INR > 6.5, onset of encephalopathy more than 7 days after the onset of jaundice, and a low factor V level (< 20% of normal). For acetaminophen-induced fulminant hepatic failure, indicators of a poor outcome (which is less common than for other causes) are acidosis (pH < 7.3), INR > 6.5, and azotemia (serum creatinine ≥ 3.4 mg/dL), whereas an elevated serum alpha-fetoprotein level predicts a favorable outcome. Hyperphosphatemia (> 1.2 mmol/L) and an elevated blood lactate level (> 3.5 mmol/L) also predict poor survival outcomes. Emergency liver transplantation is considered for patients with stage II to stage III encephalopathy and is associated with a 70% survival rate at 5 years.
Bernuau J: Acute liver failure: avoidance of deleterious cofactors and early specific medical therapy for the liver are better than late intensive care for the brain. J Hepatol 2004;4:152.
Bhatia V et al: Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004;41:89.
Jalan R et al: Moderate hypothermia in patients with acute liver failure and uncontrolled intracranial hypertension. Gastroenterology 2004;127:1338.
Jalan R et al: Prospects for extracorporeal liver support. Gut 2004;53:890.
Khuroo MS et al: Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transplant 2004;10:1099.
Lee WM: Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology 2004;40:6.
Murphy N et al: The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver failure. Hepatology 2004;39:464.
Schmidt LE et al: Alpha-fetoprotein is a predictor of outcome in acetaminophen-induced liver injury. Hepatology 2005;41:26.
CHRONIC VIRAL HEPATITIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Defined by chronic infection (HBV, HCV, HDV) for greater than 6 months.
Diagnosis is usually made by antibody tests and viral nucleic acid in serum.
General Considerations
Chronic hepatitis is defined as chronic necroinflammation of the liver of more than 3–6 months' duration, demonstrated by persistently abnormal serum aminotransferase levels and characteristic histologic findings. In many cases, the diagnosis of chronic hepatitis may be made on initial presentation. The causes of chronic hepatitis include HBV, HCV, and HDV as well as autoimmune hepatitis, chronic hepatitis associated with certain medications (particularly isoniazid), Wilson's disease, and α1-antiprotease deficiency. Traditionally, chronic hepatitis has been categorized histologically as chronic persistent hepatitis and chronic active hepatitis. However, with improved serologic and autoimmune markers, more specific categorization is possible, based on etiology; the grade of portal, periportal, and lobular inflammation (minimal, mild, moderate, or severe); and the stage of fibrosis (none, mild, moderate, severe, cirrhosis).
Clinical Findings & Diagnosis
A. CHRONIC HEPATITIS B
Chronic hepatitis B afflicts nearly 400 million people worldwide and 1.25 million (predominantly males) in the United States. It may be noted as a continuum of acute hepatitis or diagnosed because of persistently elevated aminotransferase levels.
Early in the course, HBeAg and HBV DNA are present in serum, indicative of active viral replications and necroinflammatory activity in the liver. These persons are at risk for progression to cirrhosis (at a rate of 2–5.5% per year) and for hepatocellular carcinoma (at a rate of > 2% per year in those with cirrhosis). Low-level IgM anti-HBc is also present in about 70%. In some patients, clinical and biochemical improvement coincides with disappearance of HBeAg and reduced HBV DNA levels (< 105 copies/mL) in serum, appearance of anti-HBe, and integration of the HBV genome into the host genome in infected hepatocytes. If cirrhosis has not yet developed, such persons with the inactive HBsAg carrier state are at a low risk for cirrhosis and hepatocellular carcinoma. As noted, infection
P.660
by a pre-core mutant of HBV or spontaneous mutation of the pre-core or pre-core promoter region of the HBV genome during the course of chronic hepatitis caused by wild-type HBV (HBeAg-negative chronic hepatitis B) may result in particularly severe chronic hepatitis with rapid progression to cirrhosis (at a rate of 8–10% per year), particularly when additional mutations in the core gene of HBV are present. HIV coinfection is also associated with an increased frequency of cirrhosis when the CD4 count is low.
B. HEPATITIS D
Acute hepatitis D infection superimposed on chronic HBV infection may result in severe chronic hepatitis, which may progress rapidly to cirrhosis and may be fatal. Patients with long-standing chronic hepatitis D and B often have inactive cirrhosis. The diagnosis is confirmed by detection of anti-HDV in serum.
C. CHRONIC HEPATITIS C
Chronic hepatitis C develops in up to 85% of patients with acute hepatitis C. It is clinically indistinguishable from chronic hepatitis due to other causes and may be the most common. Worldwide, 170 million people are infected with HCV, with 1.8% of the US population infected. In approximately 40% of cases, serum aminotransferase levels are persistently normal. The diagnosis is confirmed by detection of anti-HCV by enzyme immunoassay (EIA). In rare cases of suspected chronic hepatitis C but a negative EIA, HCV RNA is detected by polymerase chain reaction testing. Progression to cirrhosis occurs in 20% of affected patients after 20 years, with an increased risk in men, those who drink more than 50 g of alcohol daily, and possibly those who acquire HCV infection after age 40. Immunosuppressed persons—including patients with hypogammaglobulinemia, HIV infection with a low CD4 count, or organ transplants receiving immunosuppressants—appear to progress more rapidly to cirrhosis than immunocompetent persons with chronic hepatitis C. Affected persons with persistently normal serum aminotransferase levels usually have mild chronic hepatitis with slow or absent progression to cirrhosis; however, cirrhosis is present in 10% of these patients.
Treatment (See Chapter 37)
A. CHRONIC HEPATITIS B
Patients with active viral replication (HBeAg and HBV DNA [≥ 105 copies/mL] in serum; elevated aminotransferase levels) may be treated with recombinant human interferon alfa-2b in a dose of 5 million units a day or 10 million units three times a week intramuscularly for 4 months. About 40% of treated patients will respond with sustained normalization of aminotransferase levels, disappearance of HBeAg and HBV DNA from serum, appearance of anti-HBe, and improved survival. A response is most likely in patients with a low baseline HBV DNA level and high aminotransferase levels. Moreover, over 60% of these responders may eventually clear HBsAg from serum and liver, develop anti-HBs in serum, and thus be cured. Relapses are uncommon in such complete responders. Patients with HBeAg-negative chronic hepatitis B (pre-core mutant) have a durable sustained response rate of only 15–25% after 12 months of interferon therapy. The response to interferon is poor in patients with HIV coinfection. Preliminary experience suggests that peginterferon is more effective than interferon and lamivudine, but peginterferon is not yet approved for the treatment of chronic hepatitis B.
The nucleoside analog lamivudine, 100 mg orally daily as a single dose, may be used instead of interferon for the treatment of chronic hepatitis B and is much better tolerated. This agent reliably suppresses HBV DNA in serum, improves liver histology in 60% of patients, and leads to normal ALT levels in over 40% and HBeAg seroconversion in 20% of patients after 1 year of therapy. However, by the end of 1 year, 15–30% of responders experience a relapse (and occasionally frank decompensation) as a result of a mutation in the polymerase gene (the YMDD motif) of HBV DNA that confers resistance to lamivudine. Moreover, hepatitis activity may recur when the drug is stopped. Therefore, despite a high rate of resistance (up to 70% by 5 years), long-term—perhaps indefinite—treatment may be required to suppress the disease when HBeAg seroconversion does not ensue. Rates of complete response increase with longer duration of therapy. In patients with advanced fibrosis or cirrhosis, continuous treatment with lamivudine reduces the risk of hepatic decompensation and hepatocellular carcinoma. The drug is well tolerated even in patients with decompensated cirrhosis (for whom the treatment threshold is an HBV DNA level ≥ 103 copies/mL) and may be effective in patients with rapidly progressive hepatitis B (“fibrosing cholestatic hepatitis”) following organ transplantation. Although lamivudine therapy leads to biochemical, virologic, and histologic improvement in patients with HBeAg-negative chronic hepatitis B (pre-core mutant) and baseline HBV DNA levels ≥ 104 copies/mL, relapse is frequent when therapy is stopped, and long-term treatment is associated with a high rate of viral resistance. Combined use of interferon and lamivudine offers no advantage over the use of either drug alone.
A nucleotide analog, adefovir dipivoxil, has activity against wild-type and lamivudine-resistant HBV. The standard dose is 10 mg orally once a day for at least 1 year. Resistance to this drug is rare, and the drug is effective in patients who have become resistant to lamivudine. As with lamivudine, only a small number of patients achieve sustained suppression of HBV replication with adefovir, and long-term suppressive therapy is often required. Patients with underlying renal dysfunction are at risk of nephrotoxicity from adefovir. Tenofovir, a drug used for HIV infection, also has activity against HBV. Other antiviral agents such as emtricitabine, entecavir, clevudine, and telbivudine
P.661
are under study, and strategies using multiple drugs are likely to be studied. Nucleoside analogs are also recommended for inactive HBV carriers prior to the initiation of immunosuppressive therapy or cancer chemotherapy to prevent reactivation.
B. CHRONIC HEPATITIS C
Treatment of chronic hepatitis C is generally considered in patients under age 70 with more than minimal fibrosis on liver biopsy. Because of high response rates to treatment in patients infected with HCV genotype 2 or 3, treatment may be initiated in these patients without a liver biopsy. In the past, standard therapy was with a combination of interferon alfa and ribavirin. A combination of “pegylated” interferon (peginterferon) and ribavirin is now standard therapy.
Recombinant human interferon alfa-2b or alfa-2a in a dose of 3 million units three times a week for 24 weeks and “consensus” interferon (a synthetic recombinant interferon derived by assigning the most commonly observed amino acid at each position of several alfa interferon subtypes) in a dose of 9 mcg three times a week were previously shown to induce biochemical, virologic, and histologic improvement in 30–50% of patients—return of ALT to normal, loss of HCV RNA from serum for at least 6 months after therapy is stopped, decrease in hepatic inflammation, and occasionally regression of fibrosis. More prolonged treatment (eg, for 12–18 months) was shown to increase the durability of remission. The use of higher doses of interferon alfa-2b (eg, 6 million units three times a week) increases toxicity and does not appear to increase the rate of sustained responses substantially.
Slow-release, long-acting pegylated interferon (peginterferon) taken only once a week is more effective than standard interferon—presumably because of sustained high blood levels—and is the preferred form of interferon for treatment of chronic hepatitis C. Two formulations are available: Pegintron (peginterferon alfa-2b), with a 12-kD polyethylene glycol (PEG), and Pegasys (peginterferon alfa2a), with a 40-kD PEG. Whether there are clinically important differences between the two formulations is unclear. With peginterferon alfa-2a administered in a dose of 180 mcg once per week for 48 weeks, a sustained biochemical and virologic response was achieved in 38% of patients with chronic hepatitis C compared with 17% of those treated with standard interferon. Peginterferon alfa-2b yields similar results but is given according to the patient's weight in a dose of 1.5 mcg/kg weekly.
Addition of the nucleoside analog ribavirin, 1000–1200 mg daily in two divided doses, results in higher sustained response rates than interferon or peginterferon alone in previously untreated patients with chronic hepatitis C or patients who have had a relapse after an initial response to interferon alfa alone. Longterm response rates are 40% and 50%, respectively, following combination therapy consisting of standard interferon and ribavirin. Rates with a combination of peginterferon and ribavirin are as high as 55% (and up to 80% for HCV genotypes 2 or 3). Low levels of HCV RNA may persist in the liver, lymphocytes, and macrophages of successfully treated (“cured”) patients, but the significance of this finding is uncertain. Response rates are lower in patients with advanced fibrosis, high levels of viremia, alcohol consumption, HIV coinfection, and severe steatosis and lower in blacks than in whites, in part because of a higher rate of genotype 1 among infected black patients. For nonresponders to standard interferon and ribavirin, sustained response rates to re-treatment with peginterferon and ribavirin are only 10–15%. When used with peginterferon alfa-2b, the dose of ribavirin is also based on the patient's weight and may range from 800 mg to 1400 mg daily in two divided doses. When used with peginterferon alfa-2a, the daily ribavirin dose is 1000 mg or 1200 mg depending on whether the patient's weight is less than or greater than 75 kg. Patients infected with genotype 1a or 1b are treated for 48 weeks if there has been a decrease in the serum HCV RNA level of at least 2 logs by 12 weeks. Patients with cirrhosis or a high viral level in serum (> 800,000 IU/mL), including those infected with genotype 3, may also require 48 weeks of treatment. Those infected with genotype 2 or 3 (without cirrhosis and with low levels of viremia) are treated for 24 weeks and require a ribavirin dose of only 800 mg; preliminary findings suggest that for patients infected with these genotypes who clear the virus within 4 weeks, a total treatment duration of only 14 weeks may be sufficient. For patients infected with HCV genotype 1 and without any fibrosis on liver biopsy, expectant management and a repeat liver biopsy in 3–5 years is often recommended.
Peginterferon alfa with ribavirin may be beneficial in the treatment of cryoglobulinemia associated with chronic hepatitis C. “Chronic HCV carriers” with normal serum aminotransferase levels respond just as well to treatment as do patients with elevated aminotransferase levels. Patients with both HCV and HIV infections may benefit from treatment of HCV if the CD4 count is not low. Moreover, in HCV/HIV-coinfected persons, long-term liver-related mortality increases as mortality from HIV infection is reduced by highly active antiretroviral therapy. Treatment with peginterferon alfa plus ribavirin is costly ($12,000 for a 24week supply), and side effects, which include flu-like symptoms, are almost universal; more serious toxicity includes psychiatric symptoms (irritability, depression), thyroid dysfunction, and bone marrow suppression. A blood count is obtained at weeks 1, 2, and 4 after therapy is started and monthly thereafter. Interferon is contraindicated in patients with decompensated cirrhosis, profound cytopenias, severe psychiatric disorders, and autoimmune diseases. Patients taking ribavirin must be monitored for hemolysis, and, because of teratogenic effects in animals, men and women taking the drug must practice strict contraception until 6 months after conclusion of therapy. Ribavirin
P.662
should be avoided in persons over age 65 and in others in whom hemolysis could pose a risk of angina or stroke. Rash, itching, headache, cough, and shortness of breath also occur with the drug. Lactic acidosis is a concern in patients also taking highly active antiretroviral therapy for HIV infection. Erythropoietin (epoetin alfa) and granulocyte colony-stimulating factor may be used to treat therapy-induced anemia and leukopenia. Interferon is generally contraindicated in heart, lung, and renal transplant recipients because of an increased risk of organ rejection. Selected liver transplant recipients with recurrent hepatitis C may be treated with peginterferon and ribavirin, but response rates are low.
In nonresponders to interferon-based therapy, long-term “maintenance” peginterferon therapy as a strategy to prevent liver fibrosis and reduce the risk of cirrhosis and hepatocellular carcinoma is under study. Ribavirin analogues that cause little hemolysis are also under study.
C. CHRONIC HEPATITIS D
Recombinant interferon alfa-2a (9 million units three times a week for 48 weeks) may lead to normalization of serum aminotransferase levels, histologic improvement, and elimination of HDV RNA from serum in about 50% of patients with chronic hepatitis D, but relapse is common after therapy is stopped. Lamivudine and adefovir are not effective in treating chronic hepatitis D.
Prognosis
The course of chronic viral hepatitis is variable and unpredictable. The sequelae of chronic hepatitis secondary to hepatitis B include cirrhosis, liver failure, and hepatocellular carcinoma. The 5-year mortality rate is 0–2% in those without cirrhosis, 14–20% in those with compensated cirrhosis, and 70–86% following decompensation. Antiviral treatment may improve the prognosis in responders. Chronic hepatitis C is an indolent, often subclinical disease that may lead to cirrhosis and hepatocellular carcinoma after decades. Indeed, the mortality rate from transfusion-associated hepatitis C may be no different from that of an age-matched control population. Nevertheless, mortality rates clearly rise once cirrhosis develops, and mortality from cirrhosis and hepatocellular carcinoma due to hepatitis C is expected to triple in the next 10–20 years. Peginterferon plus ribavirin appears to have a beneficial effect on survival and quality of life, is cost-effective, appears to retard and even reverse fibrosis, and in responders may reduce the risk of hepatocellular carcinoma.
Aggarwal R et al: Preventing and treating hepatitis B infection. BMJ 2004;329:1080.
Chung RT et al: Peginterferon alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIVcoinfected persons. N Engl J Med 2004;351:451.
Davila JA et al: Hepatitis C infection and the increasing incidence of hepatocellular carcinoma: a population-based study. Gastroenterology 2004;127:1372.
Farci P et al: Long-term benefit of interferon a therapy of chronic hepatitis D: regression of advanced hepatic fibrosis. Gastroenterology 2004;126:1740.
Keeffe EB et al: A treatment algorithm for the management of chronic hepatitis B virus infection in the United States. Clin Gastroenterol Hepatol 2004;2:87.
Keeffe EB et al: HBV viral kinetics and clinical management: key issues and current perspectives. Proceedings of a scientific and clinical expert panel meeting. Chicago, Illinois, USA, October 18, 2003. Semin Liver Dis 2004;24(suppl 1):1.
Liaw YF et al: Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med 2004;351:1521.
Shiffman ML: Chronic hepatitis C. Semin Liver Dis 2004;24 (Suppl 2):1.
Shiffman ML et al: Peginterferon alfa-2a and ribavirin in patients with chronic hepatitis C who have failed prior treatment. Gastroenterology 2004;126:1015.
AUTOIMMUNE HEPATITIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Usually young to middle-aged women.
Chronic hepatitis with high serum globulins.
Positive antinuclear antibody (ANA) and/or smooth muscle antibody in most common type.
Responds to corticosteroids.
Clinical Findings
A. SYMPTOMS AND SIGNS
Although autoimmune hepatitis is usually seen in young women, it can occur in either sex at any age. Affected younger persons are often positive for HLA-B8 and HLA-DR3; in older patients, HLADR4. The principal susceptibility allele among white Americans and northern Europeans is HLA DRB1*0301; HLA DRB1*0401 is a secondary but independent risk factor. The onset is usually insidious, but up to 40% present with an acute attack of hepatitis and some cases follow a viral illness such as hepatitis A, Epstein-Barr infection, or measles or exposure to a drug or toxin such as nitrofurantoin. Exacerbations may occur postpartum. Twenty percent of patients are anicteric. Typically, examination reveals a healthy-appearing young woman with multiple spider nevi, cutaneous striae, acne, hirsutism, and hepatomegaly. Amenorrhea may be a presenting feature. Extrahepatic features include arthritis, Sj gren's syndrome, thyroiditis, nephritis, ulcerative colitis, and Coombs-positive hemolytic anemia.
P.663
B. DIAGNOSTIC TESTS
Serum aminotransferase levels may be > 1000 units/L, and the total bilirubin is usually increased. In classic (type I) autoimmune hepatitis, ANA or smooth muscle antibody (either or both) is detected in serum. Serum gamma globulin levels are typically elevated (up to 5–6 g/dL). In patients with the latter, the EIA for antibody to HCV may be falsely positive. Other antibodies, including atypical perinuclear antineutrophil cytoplasmic antibodies (ANCA) and antibodies to histones, may be found. A second type, seen more often in Europe, is characterized by circulating antibody to liver-kidney microsomes (anti-LKM1)—directed against cytochrome P450 2D6—without anti-smooth muscle antibody or ANA. In some cases, anti-liver cytosol type 1, directed against formiminotransferase cyclodeaminase, is detected. This type of autoimmune hepatitis can be seen in patients with autoimmune polyglandular syndrome type 1. A third variant is characterized by antibodies to soluble liver antigen–liver pancreas (antiSLA/LP) and may represent a variant of type I autoimmune hepatitis characterized by severe disease, a high relapse rate after treatment, and absence of the usual antibodies (ANA and smooth muscle antibody). AntiSLA/LP appears to be directed against a transfer RNA complex responsible for incorporating selenocysteine into peptide chains. Concurrent primary biliary cirrhosis or primary sclerosing cholangitis has been recognized in up to 15% of patients with autoimmune hepatitis. Liver biopsy is indicated to help establish the diagnosis, evaluate disease severity, and determine the need for treatment.
Treatment
Prednisone with or without azathioprine improves symptoms; decreases the serum bilirubin, aminotransferase, and gamma globulin levels; and reduces hepatic inflammation. Symptomatic patients with aminotransferase levels elevated tenfold (or fivefold if the serum globulins are elevated at least twofold) are optimal for therapy, and asymptomatic patients with modest enzyme elevations may be considered for therapy depending on the clinical circumstances.
Prednisone or an equivalent drug is given initially in doses of 30 mg orally daily with azathioprine or mercaptopurine, 50 mg/d orally, which are generally well tolerated and permit the use of lower corticosteroid doses. Blood counts are monitored weekly for the first 2 months of therapy and monthly thereafter because of the small risk of bone marrow suppression. The dose of prednisone is lowered from 30 mg/d after 1 week to 20 mg/d and again after 2 or 3 weeks to 15 mg/d. Ultimately, a maintenance dose of 10 mg/d is achieved. While symptomatic improvement is often prompt, biochemical improvement is more gradual, with normalization of serum aminotransferase levels after several months in many cases. Histologic resolution of inflammation may require 18–24 months, the time at which repeat liver biopsy is recommended. Failure of aminotransferase levels to normalize invariably predicts lack of histologic resolution.
The response rate to therapy with prednisone and azathioprine is 80%. Fibrosis may reverse with therapy and rarely progresses after apparent biochemical and histologic remission. Once remission is achieved, therapy may be withdrawn, but the subsequent relapse rate is 50–90%. Relapses may again be treated in the same manner as the initial episode, with the same remission rate. After successful treatment of a relapse, the patient may be kept indefinitely on azathioprine (up to 2 mg/kg) and the lowest dose of prednisone needed to maintain aminotransferase levels as close to normal as possible—although another attempt at withdrawing therapy may be considered in patients remaining in remission long term (eg, ≥ 4 years). Budesonide, a corticosteroid with less toxicity than prednisone, does not appear to be effective in maintaining remission. Prednisone can be used to treat rare flares during pregnancy, and maintenance azathioprine does not have to be discontinued.
Nonresponders to prednisone and azathioprine may be considered for a trial of cyclosporine, tacrolimus, or methotrexate. Mycophenolate mofetil is an effective alternative to azathioprine in patients who cannot tolerate or do not respond to it. Bone density should be monitored—particularly in patients being maintained on corticosteroids—and measures undertaken to prevent or treat osteoporosis. Liver transplantation may be required for treatment failures, and the disease has been recognized to recur in up to 40% of transplanted livers (and rarely to develop de novo) as immunosuppression is reduced.
Bridoux-Henno L et al: Features and outcome of autoimmune hepatitis type 2 presenting with isolated positivity for antiliver cytosol antibody. Clin Gastroenterol Hepatol 2004;2: 825.
Czaja AJ et al: Progressive fibrosis during corticosteroid therapy of autoimmune hepatitis. Hepatology 2004;39:1631.
Medina J et al: Review article: immunopathogenic and therapeutic aspects of autoimmune hepatitis. Aliment Pharmacol Ther 2003;17:1.
ALCOHOLIC LIVER DISEASE
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Chronic alcohol intake usually exceeds 80 g/d in men and 30–40 g/d in women.
Fatty liver is often asymptomatic.
Alcoholic hepatitis may present as fever, right upper quadrant pain, tender hepatomegaly, and jaundice, but the patient may also be asymptomatic.
AST is usually elevated but rarely above 300
P.664
units/L; AST is greater than ALT, usually by a factor of 2 or more.Often reversible but it is the most common precursor of cirrhosis in the United States.
General Considerations
Excessive alcohol intake can lead to fatty liver, hepatitis, and cirrhosis. Alcoholic hepatitis is characterized by acute or chronic inflammation and parenchymal necrosis of the liver induced by alcohol. While alcoholic hepatitis is often a reversible disease, it is the most common precursor of cirrhosis in the United States and is associated with four to five times the number of hospitalizations and deaths as hepatitis C, which is the second most common cause of cirrhosis.
The frequency of alcoholic cirrhosis is estimated to be 10–15% among persons who consume over 50 g of alcohol (4 oz of 100-proof whiskey, 15 oz of wine, or four 12-oz cans of beer) daily for over 10 years (although the risk of cirrhosis may be lower for wine than for a comparable intake of beer or spirits). The risk of cirrhosis is lower (5%) in the absence of other cofactors such as chronic viral hepatitis. Genetic factors may also account in part for differences in susceptibility, and there are associations with polymorphisms of the genes encoding for tumor necrosis factor-a and cytochrome P450 2E1. Women appear to be more susceptible than men, in part because of lower gastric mucosal alcohol dehydrogenase levels. Although alcoholic hepatitis may not develop in many patients even after several decades of alcohol abuse, it appears in a few individuals within a year after onset of excessive drinking. In general, over 80% of patients with alcoholic hepatitis have been drinking 5 years or more before any symptoms that can be attributed to liver disease develop; the longer the duration of drinking (10–15 or more years) and the larger the alcoholic consumption, the greater the probability of developing alcoholic hepatitis and cirrhosis. In individuals who drink alcohol excessively, the rate of ethanol metabolism can be sufficiently high to permit the consumption of large quantities without raising the blood alcohol level over 80 mg/dL.
The role of deficiencies in vitamins and calories in the development of alcoholic hepatitis or in the progression of this lesion to cirrhosis remains controversial but is at least contributory. Ethanol-induced elevation of circulating endotoxin levels is thought to play a critical role in the pathogenesis of alcoholic liver disease by inducing release of tumor necrosis factor-a and other cytokines by Kupffer cells. Many of the adverse effects of alcohol on the liver are thought to be mediated by the oxidative metabolite acetaldehyde, which contributes to lipid peroxidation and induction of an immune response following covalent binding to proteins in the liver. Concurrent HBV or HCV infection and heterozygosity for the HFE gene mutation for hemochromatosis increase the severity of alcoholic liver disease.
Clinical Findings
A. SYMPTOMS AND SIGNS
The clinical presentation of alcoholic liver disease can vary from an asymptomatic patient who may have an enlarged liver to a critically ill individual who dies quickly or a patient with end-stage cirrhosis. A recent period of heavy drinking, complaints of anorexia and nausea, and the demonstration of hepatomegaly and jaundice strongly suggest the diagnosis. Abdominal pain and tenderness, splenomegaly, ascites, fever, and encephalopathy may be present.
B. LABORATORY FINDINGS
In patients with steatosis, laboratory findings may be normal except for mild liver enzyme elevations. Anemia (usually macrocytic) may be present. Leukocytosis with shift to the left is common in patients with severe alcoholic hepatitis. Leukopenia is occasionally seen and disappears after cessation of drinking. About 10% of patients have thrombocytopenia related to a direct toxic effect of alcohol on megakaryocyte production or to hypersplenism.
AST is usually elevated but rarely above 300 units/L. AST is greater than ALT, usually by a factor of 2 or more. Serum alkaline phosphatase is generally elevated, but seldom more than three times the normal value. Serum bilirubin is increased in 60–90% of patients. Serum bilirubin levels greater than 10 mg/dL and marked prolongation of the prothrombin time (≥ 6 seconds above control) indicate severe alcoholic hepatitis with a mortality rate as high as 50%. The serum albumin is depressed, and the gamma globulin level is elevated in 50–75% of individuals, even in the absence of cirrhosis. Increased transferrin saturation and hepatic iron stores are found in many alcoholic patients due to sideroblastic anemia. Folic acid deficiency may coexist.
C. LIVER BIOPSY
Liver biopsy, if done, demonstrates macrovesicular fat and, in patients with alcoholic hepatitis, polymorphonuclear infiltration with hepatic necrosis, Mallory bodies (alcoholic hyaline), and perivenular and perisinusoidal fibrosis. Micronodular cirrhosis may be present as well. The findings are identical to those of nonalcoholic steatohepatitis.
D. OTHER STUDIES
Imaging studies are used to exclude other diagnoses; they can detect moderate to severe steatosis reliably but not inflammation or fibrosis. Ultrasound helps exclude biliary obstruction and identifies subclinical ascites. CT scanning with intravenous contrast or MRI may be indicated in selected cases to evaluate patients for collateral vessels, space-occupying lesions of the liver, or concomitant disease of the pancreas.
P.665
Differential Diagnosis
Alcoholic hepatitis may be closely mimicked by cholecystitis and cholelithiasis and by drug toxicity. Other causes of hepatitis or chronic liver disease may be excluded by serologic or biochemical testing, by imaging studies, or by liver biopsy.
Treatment
A. GENERAL MEASURES
Abstinence from alcohol is essential. Fatty liver is quickly reversible with abstinence. Every effort should be made to provide sufficient amounts of carbohydrates and calories in anorectic patients to reduce endogenous protein catabolism, promote gluconeogenesis, and prevent hypoglycemia. Nutritional support (40 kcal/kg with 1.5–2 g/kg as protein) improves survival in patients with malnutrition. Use of liquid formulas rich in branched-chain amino acids does not improve survival beyond that achieved with less expensive caloric supplementation. The administration of vitamins, particularly folic acid and thiamine, is indicated, especially when deficiencies are noted; glucose administration increases the vitamin B1 requirement and can precipitate WernickeKorsakoff syndrome if thiamine is not coadministered.
B. CORTICOSTEROIDS
Methylprednisolone, 32 mg/d for 1 month or the equivalent, may reduce short-term mortality in patients with alcoholic hepatitis and either encephalopathy or a greatly elevated bilirubin concentration and prolonged prothrombin time (specifically, when a discriminant function defined by the patient's prothrombin time minus the control prothrombin time times 4.6 plus the total bilirubin in mg/dL is > 32). Failure of the serum bilirubin level to decline after 7 days of treatment predicts nonresponse and poor long-term survival. No benefit has been demonstrated in patients with concomitant gastrointestinal bleeding.
C. EXPERIMENTAL THERAPIES
Pentoxifylline—an inhibitor of tumor necrosis factor— 400 mg orally three times daily for 4 weeks, may reduce 1-month mortality rates in patients with severe alcoholic hepatitis, primarily by decreasing the risk of hepatorenal syndrome. Other experimental therapies include propylthiouracil, oxandrolone, S-adenosyl-l-methionine, infliximab, and extracorporeal liver support.
Prognosis
A. SHORT-TERM
When the prothrombin time is short enough to permit liver biopsy (< 3 seconds above control), the 1-year mortality rate is 7%, rising to 20% if there is progressive prolongation of the prothrombin time during hospitalization. Individuals in whom the prothrombin time prohibits liver biopsy have a 42% mortality rate at 1 year. Other unfavorable prognostic factors are a serum bilirubin greater than 10 mg/dL, hepatic encephalopathy, azotemia, lack of response to corticosteroid therapy, and possibly little steatosis on a liver biopsy specimen and reversal of portal blood flow by Doppler ultrasound. In addition to the discriminant function discussed above, the Model for End-Stage Liver Disease (MELD) score used for cirrhosis (see later) correlates with mortality from alcoholic hepatitis.
B. LONG-TERM
In the United States, the 3-year mortality rate of persons who recover from acute alcoholic hepatitis is ten times greater than that of control individuals of comparable age. Histologically severe disease is associated with continued excessive mortality rates after 3 years, whereas the death rate is not increased after the same period in those whose liver biopsies show only mild alcoholic hepatitis. Complications of portal hypertension (ascites, variceal bleeding, hepatorenal syndrome), coagulopathy, and severe jaundice following recovery from acute alcoholic hepatitis also suggest a poor long-term prognosis.
The most important prognostic consideration is continued excessive drinking. A 6-month period of abstinence is generally required before liver transplantation is considered, although this requirement has been questioned.
Dunn W et al: MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005;41:353.
Duvoux C et al: Low-grade steatosis and major changes in portal flow as new prognostic factors in steroid-treated alcoholic hepatitis. Hepatology 2004;40:1370.
Naveau S et al: A double-blind randomized controlled trial of infliximab associated with prednisone in acute alcoholic hepatitis. Hepatology 2004;39:1390.
Tome S et al: Review article: current management of alcoholic liver disease. Aliment Pharmacol Ther 2004;19:707.
DRUG& TOXIN-INDUCED LIVER DISEASE
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Drug-induced liver disease can mimic viral hepatitis, biliary tract obstruction, or other types of liver disease.
Clinicians must inquire about the use of many widely used therapeutic agents, including overthe-counter “natural” and “herbal” products, in any patient with liver disease.
General Considerations
The continuing synthesis, testing, and introduction of new drugs into clinical practice has resulted in an
P.666
increase in toxic reactions of many types. Many widely used therapeutic agents, including over-thecounter “natural” and “herbal” products, may cause hepatic injury. The medications most commonly implicated are nonsteroidal anti-inflammatory drugs, analgesics, and antibiotics because of their widespread use. Drug-induced liver disease can mimic viral hepatitis, biliary tract obstruction, or other types of liver disease. In any patient with liver disease, the clinician must inquire carefully about the use of potentially hepatotoxic drugs or exposure to hepatotoxins. In some cases, coadministration of a second agent may increase the toxicity of the first (eg, isoniazid and rifampin, acetaminophen and alcohol). Drug toxicity may be categorized on the basis of pathogenesis or histologic appearance.
Direct Hepatotoxic Group
The liver lesion caused by this group of drugs is characterized by (1) dose-related severity, (2) a latent period following exposure, and (3) susceptibility in all individuals. Examples include acetaminophen (toxicity is enhanced by fasting and chronic alcohol use because of depletion of glutathione and induction of cytochrome P450 2E1), alcohol, carbon tetrachloride, chloroform, heavy metals, mercaptopurine, niacin, plant alkaloids, phosphorus, tetracyclines, valproic acid, and vitamin A.
Idiosyncratic Reactions
Except for acetaminophen, most severe hepatotoxicity is idiosyncratic. Reactions of this type are (1) sporadic, (2) not related to dose, and (3) occasionally associated with features suggesting an allergic reaction, such as fever and eosinophilia. In some, toxicity results directly from a metabolite that is produced only in certain individuals on a genetic basis. Examples include amiodarone, aspirin, carbamazepine, chloramphenicol, diclofenac, flutamide, halothane, isoniazid, ketoconazole, lamotrigine, methyldopa, oxacillin, phenytoin, pyrazinamide, quinidine, streptomycin, troglitazone (withdrawn from the market in the United States), and less commonly other thiazolidinediones, and perhaps tacrine.
Cholestatic Reactions
A. NONINFLAMMATORY
The following drugs have a direct effect on bile secretory mechanisms: azathioprine, estrogens, or anabolic steroids containing an alkyl or ethinyl group at carbon 17, indinavir, mercaptopurine, methyltestosterone, and cyclosporine.
B. INFLAMMATORY
The following drugs cause inflammation of portal areas with bile duct injury (cholangitis), often with allergic features such as eosinophilia: amoxicillin-clavulanic acid, azithromycin, chlorothiazide, chlorpromazine, chlorpropamide, erythromycin, penicillamine, prochlorperazine, semisynthetic penicillins (eg, cloxacillin), and sulfadiazine.
Acute or Chronic Hepatitis
Medications that may result in acute or chronic hepatitis that is histologically—and in some cases clinically—indistinguishable from autoimmune hepatitis include aspirin, isoniazid (increased risk in HBV carriers), methyldopa, minocycline, nitrofurantoin, nonsteroidal anti-inflammatory drugs, and propylthiouracil. Hepatitis also can occur in patients taking cocaine, ecstasy, efavirenz, nevirapine (increased risk in HBV and HCV carriers), ritonavir, sulfonamides, troglitazone (withdrawn from the market in the United States), and zafirlukast as well as a variety of alternative remedies (eg, chaparral, germander, jin bu huan, skullcap, kava).
Other Reactions
A. FATTY LIVER
1. Macrovesicular
Alcohol, amiodarone, corticosteroids, methotrexate.
2. Microvesicular (often resulting from mitochondrial injury)
Didanosine, stavudine, tetracyclines, valproic acid, zidovudine.
B. GRANULOMAS
Allopurinol, quinidine, quinine, phenylbutazone, phenytoin.
C. FIBROSIS AND CIRRHOSIS
Methotrexate, vitamin A.
D. PELIOSIS HEPATIS (BLOOD-FILLED CAVITIES)
Anabolic steroids, azathioprine, oral contraceptive steroids.
E. NEOPLASMS
Oral contraceptive steroids, estrogens (hepatic adenoma but not focal nodular hyperplasia); vinyl chloride (angiosarcoma).
Chalasani N et al: Patients with elevated liver enzymes are not at higher risk for statin hepatotoxicity. Gastroenterology 2004;126:1287.
Rubenstein JH et al: Systematic review: the hepatotoxicity of nonsteroidal anti-inflammatory drugs. Aliment Pharmacol Ther 2004;20:373.
Rumack BH: Acetaminophen misconceptions. Hepatology 2004;40:10.
Suissa S et al: Newer disease-modifying antirheumatic drugs and the risk of serious hepatic adverse events in patients with rheumatoid arthritis. Am J Med 2004;117:87.
P.667
NONALCOHOLIC FATTY LIVER DISEASE
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Often asymptomatic.
Elevated aminotransferase levels and/or hepatomegaly.
Macrovesicular and/or microvesicular steatosis on liver biopsy.
General Considerations
Ethanol can cause hepatic steatosis (fatty liver) in the absence of malnutrition, although inadequate diets— specifically those deficient in choline, methionine, and protein—can contribute to liver damage caused by ethanol (see earlier). Causes of nonalcoholic steatosis, or nonalcoholic fatty liver disease (NAFLD), are obesity (present in ≥ 40%), diabetes mellitus (in ≥ 20%), hypertriglyceridemia (in ≥ 20%), corticosteroids, amiodarone, diltiazem, tamoxifen, highly active antiretroviral therapy, poisons (carbon tetrachloride and yellow phosphorus), endocrinopathies such as Cushing's syndrome and hypopituitarism, hypobetalipoproteinemia and other metabolic disorders, starvation and refeeding syndrome, and total parenteral nutrition. Steatosis is nearly universal in obese alcoholics and is a hallmark of insulin resistance syndrome, which is characterized by obesity, diabetes, hypertriglyceridemia, and hypertension. In addition to macrovesicular steatosis, histologic features may include focal infiltration by polymorphonuclear neutrophils and Mallory's hyalin, a picture indistinguishable from that of alcoholic hepatitis and referred to as nonalcoholic steatohepatitis (NASH). In patients with NAFLD, older age, obesity, and diabetes are risk factors for advanced hepatic fibrosis and cirrhosis. Cirrhosis caused by NASH appears to be uncommon in African Americans.
Microvesicular steatosis is seen with Reye's syndrome, valproic acid toxicity, high-dose tetracycline, or acute fatty liver of pregnancy and may result in fulminant hepatic failure.
There are several factors, presumably resulting from impaired responsiveness of fat cells to insulin, that are responsible for the accumulation of fat in the liver in patients with NAFLD: (1) increased mobilization of fatty acids from peripheral adipose depots, (2) decreased β oxidation of fatty acids by the liver, (3) increased hepatic production of very-low-density lipoprotein and apolipoprotein B and thus triglyceride synthesis, (4) decreased secretion of fat from the liver, and (5) decreased lipoprotein lipase activity in fat and skeletal muscle and consequent reduced clearance of triglyceride-rich lipoproteins.
High serum leptin levels in obese persons may contribute to hepatic steatosis by promoting insulin resistance and altering insulin signaling in hepatocytes. There is controversy over whether leptin also stimulates proinflammatory cytokines and thereby influences progression from hepatic steatosis to steatohepatitis. Women in whom fatty liver of pregnancy develops often have a defect in fatty acid oxidation due to reduced long-chain 3-hydroxyacyl-CoA dehydrogenase activity.
The exact stimulus that causes progression of steatosis to steatohepatitis and fibrosis (“second hit”) is unclear. The leading possibility is lipid peroxidation and oxidative stress. Some patients with NAFLD have hepatic iron overload, and some of these patients are heterozygous for the C282Y gene for hemochromatosis (HFE); increased hepatic iron as well as severe steatosis (both attributed to insulin resistance) have been associated with hepatic fibrosis in some studies. Hepatic concentrations of cytochrome P450 2E1 are increased in patients with nonalcoholic steatohepatitis, as they are in alcoholic liver disease, and patients with nonalcoholic steatohepatitis appear to replenish depleted hepatic adenosine triphosphate (ATP) stores more slowly than healthy subjects after an ATP-depleting challenge. Increased circulating levels of tumor necrosis factor-a due to obesity and to impaired macrophage function and low serum adiponectin levels may contribute to the development of nonalcoholic steatohepatitis. Mitochondrial structural abnormalities are associated with nonalcoholic steatohepatitis but not NAFLD alone.
Clinical Findings
A. SYMPTOMS AND SIGNS
Most patients are asymptomatic or have mild right upper quadrant discomfort. Hepatomegaly is present in up to 75% of patients with NAFLD, but the stigmas of chronic liver disease are uncommon. Rare instances of subacute liver failure caused by previously unrecognized nonalcoholic steatohepatitis have been described.
B. LABORATORY FINDINGS
Laboratory studies may show mildly elevated aminotransferase and alkaline phosphatase levels; however, laboratory values may be normal in up to 80% of persons with hepatic steatosis. In contrast to alcoholic liver disease, the ratio of ALT to AST is almost always greater than 1 in NAFLD, but it decreases to less than 1 as advanced fibrosis and cirrhosis develop. An AST/ ALT ratio > 2.0 is highly suggestive of alcoholic liver disease. Antinuclear or smooth muscle antibodies may be detected in one-fourth of patients with nonalcoholic steatohepatitis.
C. IMAGING
Macrovascular steatosis may be demonstrated on ultrasound, CT, or MRI. However, imaging does not distinguish steatosis from steatohepatitis.
P.668
D. LIVER BIOPSY
Percutaneous liver biopsy is diagnostic and is the only way to assess the degree of inflammation and fibrosis. The risks of the procedure must be balanced against the impact of the added information on management decisions and assessment of prognosis. The histologic spectrum includes fatty liver, isolated portal fibrosis, nonalcoholic steatohepatitis, and cirrhosis.
Treatment
Treatment consists of removing or modifying the offending factors. Weight loss, dietary fat restriction, and exercise often lead to improvement in liver tests and steatosis in obese patients with fatty liver, but the benefit of such measures is less clear in patients with steatohepatitis. Vitamin E (to reduce oxidative stress) is of uncertain benefit. Betaine (a methyl donor) may have some benefit and is under study. Metformin has been shown to reduce insulin resistance and reverse fatty liver in obese leptin-deficient mice and—in some preliminary clinical trials (but not others)—in humans. Thiazolidinediones also reverse insulin resistance and appear to improve serum aminotransferase levels and histologic features of steatohepatitis but lead to weight gain. Pentoxifylline, which inhibits tumor necrosis factor-α, improves liver biochemical test levels but is associated with a high rate of side effects, particularly nausea. Ursodeoxycholic acid, 13–15 mg/ kg/d, improved liver function test results and liver histologic features in preliminary studies but not in a larger randomized trial of patients with nonalcoholic steatohepatitis. Hepatic steatosis due to total parenteral nutrition may be ameliorated—and perhaps prevented—with supplemental choline. Other agents under study include orlistat, an inhibitor of gastrointestinal lipases, and losartan, an angiotensin IIreceptor antagonist. Gastric bypass may be considered in patients with a body mass index > 35.
Prognosis
Fatty liver is readily reversible with discontinuation of alcohol or treatment of other underlying conditions. Otherwise, the course is variable. Nonalcoholic steatohepatitis may be associated with hepatic fibrosis in 40% of cases; cirrhosis develops in 10–15%; and decompensated cirrhosis occurs in 2–5% of patients. The course may be more aggressive in diabetic persons than in nondiabetic persons. Instances of hepatocellular carcinoma in patients with cirrhosis caused by nonalcoholic steatohepatitis have been reported. Nonalcoholic steatohepatitis may account for a substantial percentage of cases labeled as cryptogenic cirrhosis and can recur following liver transplantation. Central obesity is an independent risk factor for death from cirrhosis.
Abrams GA et al: Portal fibrosis and hepatic steatosis in morbidly obese subjects: a spectrum of nonalcoholic fatty liver disease. Hepatology 2004;40:475.
Browning JD et al: Prevalence of hepatic steatosis in an urban population in the United States: impact on ethnicity. Hepatology 2004;40:1387.
Charlton M: Nonalcoholic fatty liver disease: a review of current understanding and future impact. Clin Gastroenterol Hepatol 2004;2:1048.
Contos MJ: The histologic spectrum of nonalcoholic fatty liver disease. Clin Liver Dis 2004;8:481.
Dixon JB et al: Nonalcoholic fatty liver disease: improvement in liver histological analysis with weight loss. Hepatology 2004;39:1647.
Donati G et al: Increased prevalence of fatty liver in arterial hypersensitive patients with normal liver enzymes: role of insulin resistance. Gut 2004;53:1020.
Hoppin AG: Obesity and the liver. Semin Liver Dis 2004;24:333.
Tolman KG et al: Narrative review: hepatobiliary disease in type 2 diabetes mellitus. Ann Intern Med 2004;141:946.
Younossi ZM et al: Nonalcoholic fatty liver disease in patients with type 2 diabetes. Clin Gastroenterol Hepatol 2004;2:262.
CIRRHOSIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
End result of injury that leads to both fibrosis and nodular regeneration.
May be reversible if cause is removed.
The clinical features result from hepatic cell dysfunction, portosystemic shunting, and portal hypertension.
General Considerations
Cirrhosis is the end result of hepatocellular injury that leads to both fibrosis and nodular regeneration throughout the liver. Cirrhosis is a serious and generally irreversible disease and is the tenth leading cause of death in the United States. The clinical features result from hepatic cell dysfunction, portosystemic shunting, and portal hypertension.
The most common histologic classification divides cirrhosis into micronodular, macronodular, and mixed forms. These are descriptive terms rather than separate diseases, and each form may be seen in the same patient at different stages of the disease. In micronodular cirrhosis—typical of alcoholic liver disease (Laennec's cirrhosis)—the regenerating nodules are no larger than the original lobules, ie, approximately 1 mm in diameter or less.
Macronodular cirrhosis is characterized by larger nodules, which can measure several centimeters in diameter and may contain central veins. This form corresponds more or less to postnecrotic (posthepatitic) cirrhosis but does not necessarily follow episodes of massive necrosis and stromal collapse.
P.669
Clinical Findings
A. SYMPTOMS AND SIGNS
Cirrhosis may cause no symptoms for long periods. The onset of symptoms may be insidious or, less often, abrupt. Weakness, fatigability, disturbed sleep, muscle cramps, and weight loss are common. In advanced cirrhosis, anorexia is usually present and may be extreme, with associated nausea and occasional vomiting. Abdominal pain may be present and is related either to hepatic enlargement and stretching of Glisson's capsule or to the presence of ascites. Menstrual abnormalities (usually amenorrhea), impotence, loss of libido, sterility, and gynecomastia in men may occur. Hematemesis is the presenting symptom in 15–25%.
In 70% of cases, the liver is enlarged, palpable, and firm if not hard and has a sharp or nodular edge; the left lobe may predominate. Skin manifestations consist of spider nevi (invariably on the upper half of the body), palmar erythema (mottled redness of the thenar and hypothenar eminences), and Dupuytren's contractures. Evidence of vitamin deficiencies (glossitis and cheilosis) is common. Weight loss, wasting, and the appearance of chronic illness are present. Jaundice—usually not an initial sign—is mild at first, increasing in severity during the later stages of the disease. Ascites, pleural effusions, peripheral edema, and ecchymotic lesions are late findings. Encephalopathy characterized by day-night reversal, asterixis, tremor, dysarthria, delirium, drowsiness, and ultimately coma also occur late except when precipitated by an acute hepatocellular insult or an episode of gastrointestinal bleeding. Fever may be a presenting symptom in up to 35% of patients and usually reflects associated alcoholic hepatitis, spontaneous bacterial peritonitis, or intercurrent infection. Splenomegaly is present in 35–50% of cases. The superficial veins of the abdomen and thorax are dilated, reflecting the intrahepatic obstruction to portal blood flow, as do rectal varices. The veins fill from below when compressed.
B. LABORATORY FINDINGS
Laboratory abnormalities are either absent or minimal in latent or quiescent cirrhosis. Anemia, a frequent finding, is often macrocytic; causes include suppression of erythropoiesis by alcohol as well as folate deficiency, hemolysis, hypersplenism, and insidious or overt blood loss from the gastrointestinal tract. The white blood cell count may be low, reflecting hypersplenism, or high, suggesting infection; thrombocytopenia is secondary to alcoholic marrow suppression, sepsis, folate deficiency, or splenic sequestration. Prolongation of the prothrombin time may result from failure of hepatic synthesis of clotting factors.
Blood chemistries reflect hepatocellular injury and dysfunction, manifested by modest elevations of AST and alkaline phosphatase and progressive elevation of the bilirubin. Serum albumin is low; gamma globulin is increased and may be as high as in autoimmune hepatitis. The risk of diabetes mellitus is increased in patients with cirrhosis, particularly when associated with HCV infection, alcoholism, hemochromatosis, and NAFLD. Patients with alcoholic cirrhosis may have elevated serum cardiac troponin I and brain natriuretic peptide levels. Blunted cardiac inotropic and chronotropic responses to exercise, stress, and drugs, as well as reduced ventricular function and prolongation of the QT interval, are common in cirrhosis of all causes, but overt heart failure is rare in the absence of alcoholism.
Liver biopsy may show inactive cirrhosis (fibrosis with regenerative nodules) with no specific features to suggest the underlying cause. Alternatively, there may be additional features of alcoholic liver disease, chronic hepatitis, or other specific causes of cirrhosis. Combinations of serum markers of hepatic fibrosis (eg, hyaluronic acid, amino-terminal propeptide of type III collagen, tissue inhibitor of matrix metalloproteinase 1) are under study as alternatives to liver biopsy for the diagnosis or exclusion of cirrhosis.
C. IMAGING
Plain films of the abdomen are seldom helpful. Barium studies of the upper gastrointestinal tract may reveal the presence of esophageal or gastric varices, although endoscopy is more sensitive. Ultrasound is helpful for assessing liver size and detecting ascites or hepatic nodules, including small hepatocellular carcinomas. Together with Doppler studies, it may establish patency of the splenic, portal, and hepatic veins. Hepatic nodules are characterized further by contrastenhanced CT scan or MRI. Nodules suspicious for malignancy may be biopsied under ultrasound or CT guidance.
D. SPECIAL EXAMINATIONS
Esophagogastroduodenoscopy confirms the presence of varices and detects specific causes of bleeding in the esophagus, stomach, and proximal duodenum. Liver biopsy may be performed by laparoscopy or, in patients with coagulopathy and ascites, by a transjugular approach. In selected cases, wedged hepatic vein pressure measurement may establish the presence and cause of portal hypertension.
Differential Diagnosis
Determining the cause of cirrhosis is important prognostically and therapeutically. The most common causes of cirrhosis are chronic hepatitis C and B and alcohol. Many cases of cirrhosis are “cryptogenic,” in which unrecognized NAFLD may play a role. Mutations in the keratin 8 gene have been associated with some cases of cryptogenic cirrhosis. In advanced cases, hemochromatosis may be associated with bronzing of the skin, arthritis, heart failure, and diabetes; however, most patients have no symptoms or signs. Diagnostic features include greater than 60% saturation of serum
P.670
transferrin or serum ferritin level above the upper limit of normal, detection of the mutated HFE gene (see below), and increased staining for iron and quantitation of the iron on liver biopsy. Other metabolic diseases that may lead to cirrhosis include Wilson's disease and α1-antiprotease (α1-antitrypsin) deficiency. Primary biliary cirrhosis occurs more frequently in women and is associated with pruritus, significant elevation of alkaline phosphatase, elevated immunoglobulin (IgM) and hypercholesterolemia, and antimitochondrial antibody. Secondary biliary cirrhosis may result from chronic biliary obstruction due to a stone, stricture, or neoplasm and is not associated with antimitochondrial antibody. Congestive heart failure and constrictive pericarditis may lead to hepatic fibrosis (“cardiac cirrhosis”) complicated by ascites and may be mistaken for other causes of cirrhosis. Hereditary hemorrhagic telangiectasia can lead to portal hypertension because of portosystemic shunting and nodular transformation of the liver.
Complications
Upper gastrointestinal tract bleeding may occur from varices, portal hypertensive gastropathy, or gastroduodenal ulcer (see Chapter 14). Hemorrhage may be massive, resulting in fatal exsanguination or encephalopathy. Varices may also result from portal vein thrombosis. Liver failure may be precipitated by alcoholism, surgery, and infection. The risk of carcinoma of the liver is increased in patients with cirrhosis. Hepatic Kupffer cell (reticuloendothelial) dysfunction and decreased opsonic activity lead to an increased risk of systemic infection.
Treatment
A. GENERAL MEASURES
The most important principle of treatment is abstinence from alcohol. The diet should be palatable, with adequate calories (25–35 kcal/kg body wt/d in those with compensated cirrhosis and 35–40 kcal/kg/d in those with malnutrition) and protein (1–1.2 g/kg/d in those with compensated cirrhosis and 1.5 g/kg/d in those with malnutrition) and, if there is fluid retention, sodium restriction. In the presence of hepatic encephalopathy, protein intake should be reduced to 60–80 g/d. The benefit of using specialized supplements containing branched-chain amino acids to prevent or treat hepatic encephalopathy or delay progressive liver failure is uncertain. Vitamin supplementation is desirable.
B. TREATMENT OF COMPLICATIONS
1. Ascites and edema
Diagnostic paracentesis is indicated for new ascites. It is rarely associated with serious complications such as bleeding, infection, or bowel perforation even in patients with severe coagulopathy. In addition to a cell count and culture, the ascitic albumin level should be determined; a serum-ascites albumin gradient (serum albumin minus ascitic albumin) > 1.1 suggests portal hypertension. An elevated ascitic adenosine deaminase level is suggestive of tuberculous peritonitis, but the sensitivity of the test is reduced in patients with portal hypertension. Occasionally, cirrhotic ascites is chylous (rich in triglycerides); other causes of chylous ascites are malignancy, tuberculosis, and recent abdominal surgery or trauma.
Ascites in patients with cirrhosis results from portal hypertension (increased hydrostatic pressure); hypoalbuminemia (decreased oncotic pressure); peripheral vasodilation, perhaps mediated by endotoxin-induced release of nitric oxide from splanchnic and systemic vasculature, with resulting increases in renin and angiotensin levels and sodium retention by the kidneys; impaired liver inactivation of aldosterone; and increased aldosterone secretion secondary to increased renin production. Free water excretion is also impaired in cirrhosis, and hyponatremia may develop.
In all patients with cirrhotic ascites, dietary sodium intake may initially be restricted to 2000 mg/d; the intake of sodium may be liberalized slightly after diuresis ensues. In some patients, there is a rapid diminution of ascites on bed rest and dietary sodium restriction alone. In individuals with ascites, the urinary excretion of sodium is usually less than 10 mEq/L. Restriction of fluid intake (800–1000 mL/d) is required for patients with hyponatremia (serum sodium < 125 mEq/L). Treatment of severe hyponatremia with vasopressin receptor antagonists is under study.
a. Diuretics
Spironolactone, generally in combination with furosemide, should be used in patients who do not respond to salt restriction. An initial trial of furosemide 80 mg intravenously demonstrating a rise in urine sodium to 750 mmol in 8 hours may predict response to diuretic therapy. The initial dose of spironolactone is 100 mg daily and may be increased by 100 mg every 3–5 days (up to a maximal conventional daily dose of 400 mg/d, although higher doses have been used) until diuresis is achieved, typically preceded by a rise in the urinary sodium concentration. Monitoring for hyperkalemia is important. In patients who cannot tolerate spironolactone because of side effects, such as painful gynecomastia, amiloride (another potassium-sparing diuretic) may be used in a dose of 5–10 mg daily. Diuresis is augmented by the addition of a loop diuretic such as furosemide. This potent diuretic, however, will maintain its effect even with a falling glomerular filtration rate, with resultant prerenal azotemia. The dose of furosemide ranges from 40 mg/d to 160 mg/d, and the drug should be administered while monitoring blood pressure, urinary output, mental status, and serum electrolytes, especially potassium.
The goal of weight loss in the ascitic patient without associated peripheral edema should be no more than 1–1.5 lb/d (0.5–0.7 kg/d).
b. Large-volume paracentesis
In patients with massive ascites and respiratory compromise, ascites re-
P.671
fractory to diuretics, or intolerable diuretic side effects, large-volume paracentesis (4–6 L) is effective. Intravenous albumin concomitantly at a dosage of 10 g/L of ascites fluid removed protects the intravascular volume, although the usefulness of this practice is debated. Moreover, use of albumin at approximately $15 per gram adds considerable expense to the procedure. Large-volume paracentesis can be repeated daily until ascites is largely resolved and may decrease the need for hospitalization. If possible, diuretics should be continued in the hope of preventing recurrent ascites.
c. Transjugular intrahepatic portosystemic shunt (TIPS)
TIPS is an effective treatment of variceal bleeding refractory to standard therapy (eg, endoscopic band ligation or sclerotherapy) and has shown benefit in the treatment of severe refractory ascites. The technique involves insertion of an expandable metal stent between a branch of the hepatic vein and portal vein over a catheter inserted via the internal jugular vein. Increased renal sodium excretion and control of ascites refractory to diuretics can be achieved in about 75% of selected cases. The success rate is lower in patients with underlying renal insufficiency. TIPS appears to be the treatment of choice for refractory hepatic hydrothorax (translocation of ascites across the diaphragm to the pleural space); video-assisted thoracoscopy with pleurodesis using talc may be effective when TIPS is contraindicated. Complications of TIPS include hepatic encephalopathy in 20–30% of cases, infection, shunt stenosis in up to 60%, and shunt occlusion in up to 30% of cases. Long-term patency usually requires periodic shunt revisions. In most cases, patency can be maintained by balloon dilation, local thrombolysis, or placement of an additional stent. Because of the complications associated with TIPS and uncertainty about its long-term efficacy (reduced hepatic perfusion as a result of TIPS may conceivably shorten a patient's survival), it is currently preferred in patients who require short-term control of variceal bleeding or ascites until liver transplantation can be performed—as opposed to patients in need of definitive control of bleeding or ascites but in whom liver transplantation is not a consideration. In patients with refractory ascites, TIPS results in lower rates of ascites recurrence and hepatorenal syndrome but a higher rate of hepatic encephalopathy than occur with repeated large-volume paracentesis; a benefit in survival has been demonstrated in one study but not in two others. Renal insufficiency, refractory encephalopathy, and hyperbilirubinemia are associated with mortality after TIPS.
d. Peritoneovenous shunts
In the past, peritoneovenous shunts were advocated for use in patients with refractory ascites. These shunts may be effective but carry a considerable complication rate: disseminated intravascular coagulation in 65% of patients (25% symptomatic; 5% severe), bacterial infections in 4–8%, congestive heart failure in 2–4%, and variceal bleeding from sudden expansion of intravascular volume. TIPS is now preferred for refractory ascites.
2. Spontaneous bacterial peritonitis
Spontaneous bacterial peritonitis is heralded by abdominal pain, increasing ascites, fever, and progressive encephalopathy in a patient with cirrhotic ascites; symptoms are typically mild. Paracentesis reveals an ascitic fluid with, most commonly, a total white cell count of up to 500 cells/mcL with a high percentage of PMNs (> 250/mcL) and a protein concentration of 1 g/dL or less, corresponding to decreased ascitic opsonic activity. Rapid diagnosis of bacterial peritonitis can be made with a high degree of accuracy with rapid reagent strips (“dipsticks”) that detect leukocyte esterase in ascitic fluid. Cultures of ascites give the highest yield—80–90% positive—using blood culture bottles inoculated at the bedside. Common isolates are Escherichia coli and pneumococci. (Gram-positive cocci are the most common isolates in patients who have undergone invasive procedures such as central venous line placement.) Anaerobes are rare. Pending culture results, if there are 250 or more PMN/mcL, intravenous antibiotic therapy should be initiated with cefotaxime, 2 g intravenously every 8–12 hours for at least 5 days. Ceftriaxone and amoxicillin-clavulanic acid are alternatives. Oral ofloxacin, 400 mg twice daily, or a 2-day course of intravenous ciprofloxacin, 200 mg twice daily, followed by oral ciprofloxacin, 500 mg twice daily for 5 days, may be effective in selected patients. Supplemental administration of intravenous albumin may reduce mortality. Response to therapy can be documented, if necessary, by a decrease in the PMN count of at least 50% on repeat paracentesis 48 hours after initiation of therapy. The overall mortality rate is high—up to 30% during hospitalization and up to 70% by 1 year. In survivors, the risk of recurrent peritonitis may be decreased by long-term norfloxacin, 400 mg orally daily (although in recurrence the causative organism is often resistant to quinolones). In high-risk cirrhotic patients (eg, those with ascitic protein < 1 g/dL, serum bilirubin > 2.5 mg/dL, acute variceal bleeding), first episodes of peritonitis may be prevented by prophylactic norfloxacin, ciprofloxacin (500 mg twice a day), or trimethoprim-sulfamethoxazole (one double-strength tablet five times a week).
3. Hepatorenal syndrome
Hepatorenal syndrome occurs in up to 10% of patients with advanced cirrhosis and ascites and is characterized by azotemia in the absence of shock or significant proteinuria and by failure of renal function to improve following intravenous infusion of 1.5 L of isotonic saline. Oliguria, hyponatremia, and low urinary sodium are typical features. Hepatorenal syndrome is diagnosed only when other causes of renal failure (including prerenal azotemia and acute tubular necrosis) have been excluded. Type I hepatorenal syndrome is characterized by doubling of the serum creatinine to a level greater than 2.5 mg/ dL or by halving of the creatinine clearance to less than 20 mL/min in less than 2 weeks. Type II hepatorenal syndrome is more slowly progressive and chronic. The cause is unknown, but the pathogenesis
P.672
involves intense renal vasoconstriction, possibly because of impaired synthesis of renal vasodilators such as prostaglandin E2 and decreased total renal blood flow; histologically, the kidneys are normal. Treatment is often ineffective. Improvement may follow intravenous infusion of the long-acting vasoconstrictor ornipressin and albumin (but with a high rate of ischemic side effects), ornipressin and dopamine, terlipressin (a long-acting vasopressin analog not available in the United States) and albumin, norepinephrine and albumin, or administration of the somatostatin analog octreotide subcutaneously, midodrine, an aadrenergic drug, orally, and albumin intravenously. Prolongation of survival has been associated with use of the molecular adsorbent recirculating system (MARS), a modified dialysis method that selectively removes albumin-bound substances. Improvement and sometimes normalization of renal function may also follow placement of a TIPS. Mortality is high without liver transplantation, death being due to complicating infection or hemorrhage.
4. Hepatic encephalopathy
Hepatic encephalopathy is a state of disordered central nervous system function resulting from failure of the liver to detoxify noxious agents of gut origin because of hepatocellular dysfunction and portosystemic shunting. The clinical spectrum ranges from day-night reversal and mild intellectual impairment to coma. Patients with minimal hepatic encephalopathy have no recognizable clinical symptoms but demonstrate mild cognitive and psychomotor deficits on standardized tests. Ammonia is the most readily identified and measurable toxin but is not solely responsible for the disturbed mental status. Pathogenic factors may include production of false neurotransmitters, increased sensitivity of central nervous system neurons to the inhibitory neurotransmitter γ-aminobutyric acid (GABA), an increase in circulating levels of endogenous benzodiazepines, decreased activity of urea-cycle enzymes due to zinc deficiency, decreased brain levels of myoinositol, deposition of manganese in the basal ganglia, and swelling of astrocytes in the brain. Bleeding into the intestinal tract may significantly increase the amount of protein in the bowel and precipitate rapid development of encephalopathy. Other precipitants include constipation, alkalosis, and potassium deficiency induced by diuretics, opioids, hypnotics, and sedatives; medications containing ammonium or amino compounds; paracentesis with attendant hypovolemia; hepatic or systemic infection; and portosystemic shunts (including TIPS).
Dietary protein should be withheld during acute episodes if the patient cannot eat. When the patient resumes oral intake, protein intake should be 60–80 g/d as tolerated; vegetable protein is better tolerated than meat protein. Gastrointestinal bleeding should be controlled and blood purged from the gastrointestinal tract. This can be accomplished with 120 mL of magnesium citrate by mouth or nasogastric tube every 3–4 hours until the stool is free of gross blood, or by administration of lactulose. The value of treating patients with minimal hepatic encephalopathy is uncertain.
Lactulose, a nonabsorbable synthetic disaccharide syrup, is digested by bacteria in the colon to shortchain fatty acids, resulting in acidification of colon contents. This acidification favors the formation of ammonium ion in the NH4+ NH3 + H+ equation; NH4+ is not absorbable, whereas NH3 is absorbable and thought to be neurotoxic. Lactulose also leads to a change in bowel flora so that fewer ammonia-forming organisms are present. When given orally, the initial dose of lactulose for acute hepatic encephalopathy is 30 mL three or four times daily. The dose should then be titrated so that two or three soft stools per day are produced. When rectal use is indicated because of the patient's inability to take medicines orally, the dose is 300 mL of lactulose in 700 mL of saline or sorbitol as a retention enema for 30–60 minutes; it may be repeated every 4–6 hours. Lactilol is a less sweet disaccharide alternative available as a powder in some countries.
The ammonia-producing intestinal flora may also be controlled with neomycin sulfate, 0.5–1 g orally every 6 or 12 hours for 7 days. Side effects of neomycin include diarrhea, malabsorption, superinfection, ototoxicity, and nephrotoxicity, usually only after prolonged use. Alternative antibiotics are vancomycin, 1 g orally twice daily, or metronidazole, 250 mg orally three times daily. Patients who do not respond to lactulose alone may improve with a 1-week course of an antibiotic in addition to lactulose.
Opioids and sedatives metabolized or excreted by the liver are avoided. If agitation is marked, oxazepam, 10–30 mg, which is not metabolized by the liver, may be given cautiously by mouth or by nasogastric tube. Zinc deficiency should be corrected, if present, with oral zinc sulfate, 600 mg/d in divided doses. There is limited evidence that eradication of Helicobacter pylori, which generates ammonia in the stomach, may improve encephalopathy. Sodium benzoate, 10 g daily, and ornithine aspartate, 9 g three times daily, may lower blood ammonia levels, but there is less experience with these drugs than with lactulose. The benzodiazepine competitive antagonist flumazenil is effective in about 30% of patients with severe hepatic encephalopathy, but the drug is short-acting and intravenous administration is required. Use of special dietary supplements enriched with branched-chain amino acids is usually unnecessary except in occasional patients who are intolerant of standard protein supplements. Treatment by modulating the gut flora with prebiotic and probiotic agents is under study.
5. Anemia
For iron deficiency anemia, ferrous sulfate, 0.3-g enteric-coated tablets, one tablet three times daily after meals, is effective. Folic acid, 1 mg/d orally, is indicated in the treatment of macrocytic anemia associated with alcoholism. Transfusions with packed red blood cells may be necessary to replace blood loss.
P.673
6. Hemorrhagic tendency
Severe hypoprothrombinemia may be treated with vitamin K (eg, phytonadione, 5 mg orally or subcutaneously daily). This treatment is ineffective when synthesis of coagulation factors is impaired because of severe hepatic disease. In such cases, correcting the prolonged prothrombin time requires large volumes of fresh frozen plasma. Because the effect is transient, plasma infusions are not indicated except for active bleeding or before an invasive procedure. Use of recombinant factor VII may be an alternative.
7. Hemorrhage from esophageal varices
See Chapter 14.
8. Hepatopulmonary syndrome
Shortness of breath in patients with cirrhosis may result from pulmonary restriction and atelectasis caused by massive ascites. The hepatopulmonary syndrome is the triad of chronic liver disease, an increased alveolar-arterial gradient while the patient is breathing room air, and intrapulmonary vascular dilations or arteriovenous communications that result in a right-to-left intrapulmonary shunt and occurs in 4–29% of patients with cirrhosis. The syndrome is presumed to result from failure of the diseased liver to clear circulating pulmonary vasodilators. Patients often have dyspnea and arterial deoxygenation in the upright position (orthodeoxia) and relieved by recumbency. The diagnosis should be suspected in a cirrhotic patient with a pulse oximetry level ≤ 97%. Contrast-enhanced echocardiography is a sensitive screening test for detecting pulmonary vascular dilations, whereas macroaggregated albumin lung perfusion scanning is more specific and is used to confirm the diagnosis. High-resolution CT may be useful for detecting dilated pulmonary vessels that may be amenable to embolization in patients who respond poorly to supplemental oxygen and is under study. Medical therapy has been disappointing; however, intravenous methylene blue may improve oxygenation in patients by inhibiting nitric oxide-induced vasodilation. The syndrome may reverse with liver transplantation, although postoperative mortality is increased in patients with a preoperative arterial oxygen tension < 60 mm Hg or with substantial intrapulmonary shunting. TIPS may provide palliation in patients with hepatopulmonary syndrome awaiting transplantation. Liver transplantation is contraindicated in patients with moderate to severe pulmonary hypertension (mean pulmonary pressure > 35 mm Hg). Pulmonary hypertension occurs in 0.7% of patients with cirrhosis and is thought to result from an excess of circulating vasoconstrictors, particularly endothelin-1. In some cases, treatment with epoprostenol or bosentan may reduce pulmonary hypertension and thereby facilitate liver transplantation.
C. LIVER TRANSPLANTATION
Liver transplantation is indicated in selected cases of irreversible, progressive chronic liver disease, acute hepatic failure, and certain metabolic diseases in which the metabolic defect is in the liver. Absolute contraindications include malignancy (except small hepatocellular carcinomas in a cirrhotic liver), advanced cardiopulmonary disease (except pulmonary arteriovenous shunting due to portal hypertension and cirrhosis), and sepsis. Relative contraindications include age over 70, portal and mesenteric vein thrombosis, active alcohol or drug abuse, HIV infection, severe malnutrition, and lack of patient understanding. With the emergence of effective antiretroviral therapy for HIV disease, a major cause of mortality in these patients has shifted to liver disease caused by HCV and HBV infection; preliminary experience suggests that the outcome of liver transplantation is comparable to that for non–HIV-infected liver transplant recipients. Alcoholics should be abstinent for 6 months. Liver transplantation should be considered in patients with worsening functional status, rising bilirubin, decreasing albumin, worsening coagulopathy, refractory ascites, recurrent variceal bleeding, or worsening encephalopathy. The major impediment to more widespread use of liver transplantation is a shortage of donor organs. Increasingly, adult living donor liver transplantation is an option for some patients. Five-year survival rates as high as 80% are now reported. Hepatocellular carcinoma, hepatitis B and C, and some cases of Budd-Chiari syndrome and autoimmune liver disease may recur in the transplanted liver. The incidence of recurrence of hepatitis B can be reduced by preoperative and postoperative treatment with lamivudine or adefovir dipivoxil and perioperative administration of HBIG. Immunosuppression is achieved with a combination of cyclosporine or tacrolimus, corticosteroids, azathioprine, and mycophenolate mofetil and may be complicated by infections, renal failure, neurologic disorders, and drug toxicity as well as graft rejection, vascular occlusion, or bile leaks. Patients are at risk for obesity, diabetes, and hyperlipidemia.
Prognosis
Factors determining survival include the patient's ability to stop the intake of alcohol as well as the Child-Turcotte-Pugh class (Table 15-5). The MELD score, which incorporates the serum bilirubin and creatinine levels and the international normalized ratio (INR), is also a measure of mortality risk in patients with end-stage liver disease and is particularly useful for predicting shortand intermediate-term survival and determining allocation priorities for donor livers. Hematemesis, jaundice, and ascites are unfavorable signs. In patients with a relatively low MELD score (< 21) and a low priority for liver transplantation, a low serum sodium concentration (< 135 mEq/L) and persistent ascites appear to be independent predictors of mortality (rate of 40% in 180 days when both are present). In established cases with severe hepatic dysfunction (serum albumin < 3 g/dL, bilirubin > 3 mg/dL, ascites, encephalopathy,
P.674
cachexia, and upper gastrointestinal bleeding), only 50% survive 6 months. The risk of death in this subgroup of patients with advanced cirrhosis is associated with renal insufficiency, cognitive dysfunction, ventilatory insufficiency, age ≥ 65 years, and prothrombin time ≥ 16 seconds. Obesity appears to be a risk factor for cirrhosis-related death or hospitalization in nonalcoholic patients. Patients with cirrhosis are at risk for the development of hepatocellular carcinoma, with rates of 3–5% per year for alcoholic and viral hepatitis-related cirrhosis (see later). Liver transplantation has markedly improved the outlook for patients who are acceptable candidates and are referred for evaluation early. In-hospital mortality from variceal bleeding in patients with cirrhosis has declined from over 40% in 1980 to 15% in 2000. Medical treatments to reverse hepatic fibrosis are under investigation.
Table 15-5. Modified Child-Turcotte-Pugh classification for cirrhosis. | |||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||
Caldwell SH et al: Recombinant factor VII (rVIIa) as a hemostatic agent in liver disease: a break from convention in need of controlled trials. Hepatology 2004;39:592.
Cardenas A et al: Review article: hepatic hydrothorax. Aliment Pharmacol Ther 2004;20:271.
C rdoba J et al: Normal protein diet for episodic hepatic encephalopathy: results of a randomized study. J Hepatol 2004;41: 38.
Gin s P et al: Management of cirrhosis and ascites. N Engl J Med 2004;350:1646.
Heuman DM et al: Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death. Hepatology 2004;40:802.
Hoeper MM et al: Portopulmonary hypertension and hepatopulmonary syndrome. Lancet 2004;363:1461.
Neff GW et al: Review article: current status of liver transplantation in HIV-infected patients. Aliment Pharmacol Ther 2004;20:993.
Runyon BA: Management of adult patients with ascites due to cirrhosis. Hepatology 2004;39:841.
PRIMARY BILIARY CIRRHOSIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Occurs in middle-aged women.
Often asymptomatic.
Elevation of alkaline phosphatase, positive antimitochondrial antibody, elevated IgM, increased cholesterol.
Characteristic liver biopsy.
In later stages, can present with fatigue, jaundice, features of cirrhosis, xanthelasma, xanthoma, steatorrhea.
General Considerations
Primary biliary cirrhosis is a chronic disease of the liver characterized by autoimmune destruction of intrahepatic bile ducts and cholestasis. It is insidious in onset, occurs usually in women aged 40–60, and is often detected by the chance finding of elevated alkaline phosphatase levels. Estimated incidence and prevalence rates in the United States are 4.5 and 65.4 per 100,000, respectively, in women, and 0.7 and 12.1 per 100,000, respectively, in men. The frequency of the disease among first-degree relatives of affected persons is 1.3–6%, and the concordance rate in identical twins is high. The disease is progressive and may be complicated by steatorrhea, xanthomas, xanthelasma, osteoporosis, osteomalacia, and portal hypertension. It may be associated with Sj gren's syndrome, autoimmune thyroid disease, Raynaud's syndrome, scleroderma, hypothyroidism, and celiac disease. Infection with Novosphingobium aromaticivorans or Chlamydia pneumoniae may be triggering or causative in primary
P.675
biliary cirrhosis; other triggers, including viruses, such as human betaretrovirus, and xenobiotics, are also suspected. X-chromosome monosomy may be a predisposing factor.
Clinical Findings
A. SYMPTOMS AND SIGNS
Many patients are asymptomatic for years. The onset of clinical illness is insidious and is heralded by fatigue and pruritus. With progression, physical examination reveals hepatosplenomegaly. Xanthomatous lesions may occur in the skin and tendons and around the eyelids. Jaundice, steatorrhea, and signs of portal hypertension are late findings. The risk of osteopenia and osteoporosis is increased in patients with primary biliary cirrhosis (who tend to be older women) possibly due in part to polymorphisms of the vitamin D receptor.
B. LABORATORY FINDINGS
Blood counts are normal early in the disease. Liver biochemical tests reflect cholestasis with elevation of alkaline phosphatase, cholesterol (especially high-density lipoproteins), and, in later stages, bilirubin. Antimitochondrial antibodies (directed against the dihydrolipoamide acetyltransferase component of pyruvate dehydrogenase or other 2-oxo-acid enzymes in mitochondria) are present in 95% of patients, and serum IgM levels are elevated. ANAs with distinctive specificities (eg, against gp210 in the nuclear envelope or nucleoporin p62) may be detected in specialized laboratories.
Diagnosis
The diagnosis of primary biliary cirrhosis is based on the detection of cholestatic liver chemistries (often initially an isolated elevation of the alkaline phosphatase) and antimitochondrial antibodies in serum. Liver biopsy is not essential for diagnosis but permits histologic staging: I, portal inflammation with granulomas; II, bile duct proliferation, periportal inflammation; III, interlobular fibrous septa; and IV, cirrhosis.
Differential Diagnosis
The disease must be differentiated from chronic biliary tract obstruction (stone or stricture), carcinoma of the bile ducts, primary sclerosing cholangitis, sarcoidosis, cholestatic drug toxicity (eg, chlorpromazine), and in some cases chronic hepatitis. Patients with a clinical and histologic picture of primary biliary cirrhosis but no antimitochondrial antibodies are said to have “autoimmune cholangitis,” which has been associated with lower serum IgM levels and a greater frequency of smooth muscle and ANAs. Many such patients are found to have antimitochondrial antibodies by immunoblot against recombinant proteins (rather than standard immunofluorescence). Some patients have overlapping features of primary biliary cirrhosis and autoimmune hepatitis.
Treatment
Treatment is primarily symptomatic. Cholestyramine (4 g) or colestipol (5 g) in water or juice three times daily may be beneficial for the pruritus. Rifampin, 150–300 mg orally twice daily, is inconsistently beneficial. Opioid antagonists (eg, naloxone, 0.2 mcg/kg/ min by intravenous infusion, or naltrexone, 50 mg/d by mouth) show promise in the treatment of pruritus. The 5-HT3 serotonin receptor antagonist ondansetron may also provide some benefit. For refractory pruritus, plasmapheresis or extracorporeal albumin dialysis may be needed. Deficiencies of vitamins A, K, and D may occur if steatorrhea is present and is aggravated when cholestyramine or colestipol is administered. Calcium supplementation (500 mg three times daily) may help prevent osteomalacia but is of uncertain benefit in osteoporosis. For patients with established osteoporosis, treatment options include hormone replacement therapy in older women, bisphosphonates, and calcitonin.
Because of its lack of toxicity, ursodeoxycholic acid (10–15 mg/kg/d in one or two doses) is the preferred medical treatment (and only treatment approved by the US Food and Drug Administration) for primary biliary cirrhosis and has been shown to slow the progression of disease (particularly in early-stage disease), improve longterm survival, reduce the risk of developing esophageal varices, and delay the need for liver transplantation. Ursodeoxycholic acid therapy has also been reported to reduce the risk of recurrent colorectal adenomas in patients with primary biliary cirrhosis. Colchicine (0.6 mg twice daily) and methotrexate (15 mg/wk) have had some reported benefit in improving symptoms and serum levels of alkaline phosphatase and bilirubin. Methotrexate may also improve liver histology in some patients. Penicillamine, corticosteroids, and azathioprine have proved to be of no benefit. Mycophenolate mofetil is under study. For patients with advanced disease, liver transplantation is the treatment of choice.
Prognosis
Without liver transplantation, survival averages 7–10 years once symptoms develop. Progression to liver failure is associated with the presence of anti-centromere antibodies. In advanced disease, adverse prognostic markers are older age, high serum bilirubin, edema, low serum albumin, prolonged prothrombin time, and variceal hemorrhage. Among asymptomatic patients, at least one-third will become symptomatic within 15 years. The risk of hepatobiliary malignancies appears to be increased in patients with primary biliary cirrhosis. Liver transplantation for advanced primary biliary cirrhosis is associated with a 1-year survival rate of 85–90%. The disease recurs in the graft in 20% of patients by 3 years, but this does not seem to affect survival.
Bergasa NV et al: Primary biliary cirrhosis: report of a focus study group. Hepatology 2004;40:1013.
Invernizzi P et al: Frequency of monosomy X in women with primary biliary cirrhosis. Lancet 2004;363:533.
P.676
Prince MI et al: Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut 2004;53:865.
Selmi C et al: Epidemiology and pathogenesis of primary biliary cirrhosis. J Clin Gastroenterol 2004;38:264.
Talwalkar JA et al: Primary biliary cirrhosis. Lancet 2003;362:53.
HEMOCHROMATOSIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Usually diagnosed because of elevated iron saturation or serum ferritin or a family history.
Most patients are asymptomatic; the disease is rarely recognized clinically before the fifth decade.
Hepatic abnormalities and cirrhosis, congestive heart failure, hypogonadism, and arthritis.
HFE gene mutation (usually C282Y/C282Y) is found in most cases.
General Considerations
Hemochromatosis is an autosomal recessive disease caused in many cases by a mutation in the HFE gene on chromosome 6. The alteration leads to substitution of tyrosine for cysteine at position 282 (C282Y) in a region of the gene product involved in interaction with β2-microglobulin. The HFE protein is thought to play an important role in the process by which duodenal crypt cells sense body iron stores. The presence of the mutation apparently reduces surface expression of an HFE-β2 microglobulin complex on duodenal crypt cells, decreasing the affinity of the transferrin receptor for transferrin. This impairs transferrin-mediated uptake of iron from the circulation into crypt cells and results in up-regulation of duodenal metal-transporter-1 and ferroportin-1 expression on the luminal side of villous cells, leading in turn to increased iron absorption from the intestine. About 85% of persons with well-established hemochromatosis are homozygous for the C282Y mutation. The frequency of the gene mutation averages 7% in Northern European and North American white populations, resulting in a 0.5% frequency of homozygotes (of whom 40–70% will develop iron overload and even fewer will develop clinical symptoms). By contrast, the gene mutation and hemochromatosis are uncommon in African-American and Asian-American populations. A second genetic mutation leading to substitution of aspartic acid for histidine at position 63 (H63D) of the same protein may contribute to the development of hemochromatosis in a small percentage (1.5%) of persons who are compound heterozygotes for C282Y and H63D. A third HFE mutation, S65C, appears to lead to mild to moderate hepatic iron overload without fibrosis in some cases. Rare instances of hemochromatosis result from mutations in the transferrin receptors 1 and 2, ferroportin, and other modifier genes.
The disorder is characterized by increased accumulation of iron as hemosiderin in the liver, pancreas, heart, adrenals, testes, pituitary, and kidneys. Cirrhosis is nine times more likely to develop in affected persons who drink alcohol excessively than in those who do not. Eventually, hepatic and pancreatic insufficiency, congestive heart failure, and hypogonadism may develop. The disease is rarely recognized clinically before the fifth decade. Heterozygotes do not develop cirrhosis in the absence of associated disorders such as viral hepatitis or NAFLD. A juvenile-onset variant is characterized by severe iron overload, cardiac dysfunction, hypogonadotropic hypogonadism, and a high mortality rate and is not associated with the C282Y mutation but is linked to a mutation of a gene on chromosome 1q designated HJV that produces a protein called hemojuvelin or rarely to a mutation in the HAMP gene that encodes a protein called hepcidin.
Clinical Findings
A. SYMPTOMS AND SIGNS
The onset is usually after age 50—earlier in men than in women; however, because of widespread liver biochemical testing and iron screening, the diagnosis can be made long before symptoms develop. Early symptoms are nonspecific (eg, fatigue, arthralgias). Later clinical manifestations include arthropathy, hepatomegaly and evidence of hepatic insufficiency (late finding), skin pigmentation (combination of slate-gray due to iron and brown due to melanin, sometimes resulting in bronze color), cardiac enlargement with or without heart failure or conduction defects, diabetes mellitus with its complications, and impotence in men. Bleeding from esophageal varices may occur, and in patients in whom cirrhosis develops, there is a 15–20% incidence of hepatocellular carcinoma. Affected patients are at increased risk of infection with Vibrio vulnificus, Listeria monocytogenes, Yersinia enterocolitica, and other siderophilic organisms.
B. LABORATORY FINDINGS
Laboratory findings include mildly abnormal liver tests (AST, alkaline phosphatase), an elevated plasma iron with greater than 50% saturation of the transferrin (after an overnight fast), and an elevated serum ferritin (although a normal iron saturation and normal ferritin does not exclude the diagnosis).
C. IMAGING
CT and MRI may show changes consistent with iron overload of the liver, but these techniques are not sensitive enough for screening. Testing for HFE mutations is indicated in any patient with evidence of iron overload and in siblings of patients with confirmed hemochromatosis.
P.677
D. LIVER BIOPSY
In patients who are homozygous for C282Y, liver biopsy is often indicated to determine whether cirrhosis is present. Biopsy can be deferred, however, in patients under age 40 in whom the serum ferritin level is < 1000 mcg/L, serum AST level is normal, and no hepatomegaly is present; the likelihood of cirrhosis is low in these persons. Liver biopsy is also indicated when iron overload is suspected even though the patient is not homozygous for C282Y. In patients with hemochromatosis, the liver biopsy characteristically shows extensive iron deposition in hepatocytes and in bile ducts and the hepatic iron index—hepatic iron content per gram of liver converted to micromoles and divided by the patient's age—is generally greater than 1.9.
Treatment
Early diagnosis and treatment in the precirrhotic phase of hemochromatosis is of great importance. Affected patients should avoid foods rich in iron (such as red meat), alcohol, vitamin C, raw shellfish, and supplemental iron. Weekly phlebotomies of 1 or 2 units (250–500 mL) of blood (each containing about 250 mg of iron) is indicated in all symptomatic patients, those with a serum ferritin level of at least 1000 mcg/ L, and those with an increased fasting iron saturation and should be continued for up to 2–3 years to achieve depletion of iron stores. This process is monitored by hematocrit and serum iron determinations. When iron store depletion is achieved (iron saturation < 50% and serum ferritin level < 50 mcg/L), maintenance phlebotomies (every 2–4 months) are continued (although compliance has been reported to decrease with time). The chelating agent deferoxamine is indicated for patients with hemochromatosis and anemia or in those with secondary iron overload due to thalassemia who cannot tolerate phlebotomies. The drug is administered intravenously or subcutaneously in a dose of 20–40 mg/kg/d infused over 24 hours and can mobilize 30 mg of iron per day. However, treatment is painful and time-consuming. Complications of hemochromatosis—arthropathy, diabetes, heart disease, portal hypertension, and hypopituitarism—also require treatment.
The course of the disease is favorably altered by phlebotomy therapy. In precirrhotic patients, cirrhosis may be prevented. Cardiac conduction defects and insulin requirements improve with treatment. In patients with cirrhosis, varices may reverse, and the risk of variceal bleeding declines. However, cirrhotic patients must be monitored for the development of hepatocellular carcinoma. Liver transplantation for advanced cirrhosis associated with severe iron overload, including hemochromatosis, has been reported to lead to survival rates that are lower than those for other types of liver disease because of cardiac complications and an increased risk of infections.
Screening
Genetic testing is recommended for all first-degree family members of the proband; children of an affected person (C282Y homozygote) need to be screened only if the patient's spouse carries the C282Y or H63D mutation. Screening all white men over age 30 or all adults over age 20 by measurement of the transferrin saturation or perhaps the unbound ironbinding capacity has been recommended by some, but the value of screening has been questioned on the basis of recent observations that morbidity and mortality from hemochromatosis are lower than expected. Patients with otherwise unexplained chronic liver disease, arthritis, impotence, and late-onset type 1 (and possibly type 2) diabetes mellitus should be screened for iron overload.
Beutler E: Natural history of hemochromatosis. Mayo Clin Proc 2004;79:305.
Dubois S et al: Review article: targeted screening for hereditary hemochromatosis in high-risk groups. Aliment Pharmacol Ther 2004;20:1.
Limdi JK et al: Hereditary hemochromatosis. QJM 2004;97:315.
Morrison ED et al: Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann Intern Med 2003;138:627.
Pietrangelo A: Hereditary hemochromatosis—a new look at an old disease. N Engl J Med 2004;350:2383.
WILSON'S DISEASE
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Characterized by excessive deposition of copper in the liver and brain.
Rare autosomal recessive disorder that usually occurs between the first and third decades.
Serum ceruloplasmin, the plasma copper-carrying protein, is low.
Urinary excretion of copper and hepatic copper concentration are high.
General Considerations
Wilson's disease (hepatolenticular degeneration) is a rare autosomal recessive disorder that usually occurs between the first and third decades. The worldwide prevalence is about 30 per million population. The condition is characterized by excessive deposition of copper in the liver and brain. The genetic defect, localized to chromosome 13, has been shown to affect a copper-transporting adenosine triphosphatase (ATP7B) in the liver and leads to copper accumulation in the liver and oxidative damage of hepatic mitochondria. Most patients are compound heterozygotes (ie, carry
P.678
two different mutations). Over 200 different mutations in the Wilson disease gene have been identified, making genetic diagnosis impractical. The H1069Q mutation accounts for 40% of disease alleles in populations of Northern European descent and may be associated with a neurologic presentation.
The major physiologic aberration in Wilson's disease is excessive absorption of copper from the small intestine and decreased excretion of copper by the liver, resulting in increased tissue deposition, especially in the liver, brain, cornea, and kidney. Serum ceruloplasmin, the plasma copper-carrying protein, is low. Urinary excretion of copper is high.
Clinical Findings
Wilson's disease tends to present as liver disease in adolescents and neuropsychiatric disease in young adults, but there is great variability. The diagnosis should always be considered in any child or young adult with hepatitis, splenomegaly with hypersplenism, Coombsnegative hemolytic anemia, portal hypertension, and neurologic or psychiatric abnormalities. Wilson's disease should also be considered in persons under 40 years of age with chronic or fulminant hepatitis.
Hepatic involvement may range from elevated liver tests (although the alkaline phosphatase may be low) to cirrhosis and portal hypertension. The neurologic manifestations are related to basal ganglia dysfunction and include a resting, postural, or kinetic tremor and dystonia of the bulbar musculature with resulting dysarthria and dysphagia. Psychiatric features include behavioral and personality changes and emotional lability. The pathognomonic sign of the condition is the brownish or gray-green Kayser-Fleischer ring, which represents fine pigmented granular deposits in Descemet's membrane in the cornea close to the endothelial surface. The ring is usually most marked at the superior and inferior poles of the cornea. It can frequently be seen with the naked eye and almost invariably by slit-lamp examination. It may be absent in patients with hepatic manifestations only but is usually present in those with neuropsychiatric disease. Renal calculi, the Fanconi defect, renal tubular acidosis, hypoparathyroidism, and hemolytic anemia may occur in patients with Wilson's disease.
Diagnosis
The diagnosis can be challenging and is based on demonstration of increased urinary copper excretion (> 100 mcg/24 h) or low serum ceruloplasmin levels (< 20 mcg/dL), and elevated hepatic copper concentration (> 250 mcg/g of dry liver). However, increased urinary copper and low serum ceruloplasmin levels are not specific for Wilson's disease. In equivocal cases (when the serum ceruloplasmin level is normal), the diagnosis may require demonstration of low radiolabeled copper incorporation into ceruloplasmin or urinary copper determination after a penicillamine challenge. Liver biopsy may show acute or chronic hepatitis or cirrhosis. MRI of the brain may show increased basal ganglia copper even early in the course of the disease.
Treatment
Early treatment to remove excess copper is essential before it can produce neurologic or hepatic damage. Early in the treatment phase, restriction of dietary copper (shellfish, organ foods, and legumes) may be of value. Oral penicillamine (0.75–2 g/d in divided doses) is the drug of choice and enhances urinary excretion of chelated copper. Pyridoxine, 50 mg per week, is added, since penicillamine is an antimetabolite of this vitamin. If penicillamine treatment cannot be tolerated because of gastrointestinal, hypersensitivity, or autoimmune reactions, consider the use of trientine, 250–500 mg three times a day. Oral zinc acetate, 50 mg three times a day, interferes with intestinal absorbtion of copper, promotes fecal copper excretion, and may be used as maintenance therapy after decoppering with a chelating agent or as first-line therapy in presymptomatic or pregnant patients. Ammonium tetrathiomolybdate, which complexes copper in the intestinal tract, has shown promise as initial therapy for neurologic Wilson's disease.
Treatment should continue indefinitely. Supplemental vitamin E, an antioxidant, has been recommended but not rigorously studied. Once the serum nonceruloplasmin copper level is within the normal range, the dose of chelating agent can be reduced to the minimum necessary for maintaining that level. The prognosis is good in patients who are effectively treated before liver or brain damage has occurred. Liver transplantation is indicated for fulminant hepatitis (often after plasma exchange as a stabilizing measure), end-stage cirrhosis, and, in selected cases, intractable neurologic disease. Family members, especially siblings, require screening with serum ceruloplasmin, liver function tests, and slit-lamp examination.
Ferenci P: Review article: diagnosis and current therapy of Wilson's disease. Aliment Pharmacol Ther 2004;19:157.
Stapelbroek JM et al: The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson's disease: results of a meta-analysis. J Hepatol 2004;41:758.
HEPATIC VEIN OBSTRUCTION (Budd-Chiari Syndrome)
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Right upper quadrant pain and tenderness.
Ascites.
Imaging studies show occlusion/absence of flow in the hepatic vein(s) or inferior vena cava.
Clinical picture is similar in veno-occlusive disease but major hepatic veins are patent.
P.679
General Considerations
Factors that predispose patients to Budd-Chiari syndrome, including hereditary and acquired hypercoagulable states, can be identified in 75% of affected patients. Occlusion of the hepatic veins may occur from a variety of causes. Many cases are associated with polycythemia vera or other myeloproliferative diseases, which may be subclinical. In some cases, an underlying predisposition to thrombosis (eg, hyperprothrombinemia [factor II G20210A mutation], activated protein C resistance [factor V Leiden mutation], protein C or S or antithrombin deficiency, the methylenetetrahydrofolate reductase mutation, antiphospholipid antibodies) can be identified. Hepatovenous obstruction may be associated with caval webs, right-sided heart failure or constrictive pericarditis, neoplasms that cause hepatic vein occlusion, paroxysmal nocturnal hemoglobinuria, Beh et's syndrome, blunt abdominal trauma, use of birth control pills, and pregnancy. Some cytotoxic agents and pyrrolizidine alkaloids (“bush teas”) may cause hepatic veno-occlusive disease (occlusion of terminal venules, also known as sinusoidal obstruction syndrome), which mimics Budd-Chiari syndrome clinically. Veno-occlusive disease is common in patients who have undergone bone marrow transplantation, particularly those with pretransplant aminotransferase elevations or fever during cytoreductive therapy with cyclophosphamide, azathioprine, carmustine, busulfan, or etoposide or those receiving high-dose cytoreductive therapy or high-dose total body irradiation. In India, China, and South Africa, Budd-Chiari syndrome is often the result of occlusion of the hepatic portion of the inferior vena cava, presumably due to prior thrombosis, and the clinical presentation is mild but the course is frequently complicated by hepatocellular carcinoma.
Clinical Findings
A. SYMPTOMS AND SIGNS
The presentation may be fulminant, acute, subacute, or chronic. An insidious (subacute) onset is most common. Clinical manifestations generally include tender, painful hepatic enlargement, jaundice, splenomegaly, and ascites. With chronic disease, bleeding varices and hepatic coma may be evident; hepatopulmonary syndrome may occur.
B. IMAGING
Hepatic imaging studies may show a prominent caudate lobe, since its venous drainage may not be occluded. The screening test of choice is duplex Doppler ultrasonography, which has a sensitivity of 85% for detecting evidence of hepatic venous or inferior vena caval thrombosis. MRI with spin-echo and gradient-echo sequences and intravenous gadolinium injection allows visualization of the obstructed veins and collateral vessels. Direct venography can delineate caval webs and occluded hepatic veins (“spider-web” pattern) most precisely.
C. LIVER BIOPSY
Percutaneous or transjugular liver biopsy frequently shows a characteristic centrilobular congestion and fibrosis and often multiple large regenerating nodules. Liver biopsy is often contraindicated in veno-occlusive disease because of thrombocytopenia, and the diagnosis is based on clinical findings.
Treatment
Ascites should be treated with fluid and salt restriction and diuretics. Treatable causes of Budd-Chiari syndrome should be sought. Prompt recognition and treatment of an underlying hematologic disorder may avoid the need for surgery, but the optimal anticoagulation regimen is uncertain. Infusion of a thrombolytic agent into recently occluded veins has been attempted with success. In patients with veno-occlusive disease, defibrotide, an adenosine receptor agonist that increases endogenous tissue plasminogen activator levels, has shown promise. TIPS placement may be attempted in patients with persistent hepatic congestion or failed thrombolytic therapy, although late TIPS dysfunction is common. Surgical decompression (side-to-side portacaval, mesocaval, or mesoatrial shunt) of the congested liver may be required to relieve persistent hepatic congestion but has not been proven to improve long-term survival. Balloon angioplasty, in some cases with placement of an intravascular metallic stent, is preferred in patients with an inferior vena caval web and may be feasible in patients with a short segment of thrombosis in the hepatic vein. Liver transplantation is considered in patients with fulminant hepatic failure, cirrhosis and hepatocellular dysfunction, and failure of a portosystemic shunt. Patients often require lifelong anticoagulation and treatment of the underlying myeloproliferative disease; antiplatelet therapy with aspirin and hydroxyurea has been suggested as an alternative to warfarin in patients with a myeloproliferative disorder. The overall 5-year survival rate is 50–80%. Adverse prognostic factors in patients with Budd-Chiari syndrome are older age, advanced Child-Turcotte-Pugh stage, ascites, encephalopathy, elevated total bilirubin, prolonged prothrombin time, elevated serum creatinine, and histologic features of acute liver disease superimposed on chronic liver injury.
Hern ndez-Guerra M et al: PTFE-covered stents improve TIPS patency in Budd-Chiari syndrome. Hepatology 2004;40:1197.
Menon KVN et al: The Budd-Chiari syndrome. N Engl J Med 2004;350:578.
Murad SD et al: Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004;39:500.
P.680
R ssle M et al: The Budd-Chiari syndrome: outcome after treatment with the transjugular intrahepatic portosystemic shunt. Surgery 2004;135:394.
Sharma S et al: Pharmacological thrombolysis in Budd-Chiari syndrome: a single centre experience and review of the literature. J Hepatol 2004;40:172.
THE LIVER IN HEART FAILURE
Shock liver, or ischemic hepatopathy, results from an acute fall in cardiac output due, for example, to acute myocardial infarction or arrhythmia, usually in a patient with passive congestion of the liver. Clinical hypotension may be absent (or unwitnessed). In some cases, the precipitating event is arterial hypoxemia due to respiratory failure. The hallmark is a rapid and striking elevation of serum aminotransferase levels (often > 5000 units/L); an early rapid rise in the serum lactate dehydrogenase level is also typical, but elevations of serum alkaline phosphatase and bilirubin are usually mild. The prothrombin time may be prolonged, and encephalopathy may develop. The mortality rate due to the underlying disease is high, but in patients who recover, the aminotransferase levels return to normal quickly, usually within 1 week—in contrast to viral hepatitis.
In patients with passive congestion of the liver due to right-sided heart failure, the serum bilirubin level may be elevated, occasionally as high as 40 mg/ dL, due in part to hypoxia of perivenular hepatocytes. Serum alkaline phosphatase levels are normal or slightly elevated. Hepatojugular reflux is present, and with tricuspid regurgitation the liver may be pulsatile. Ascites may be out of proportion to peripheral edema, with a high serum ascites-albumin gradient (> 1.1) and a protein content of more than 2.5 g/dL. In severe cases, signs of encephalopathy may develop.
Denis C et al: Acute hypoxic hepatitis (“liver shock”): still a frequently overlooked cardiological diagnosis. Eur J Heart Fail 2004;6:561.
Myers RP et al: Cardiac hepatopathy: clinical, hemodynamic, and histologic characteristics and correlations. Hepatology 2003;37:393.
Rosenberg PM, Friedman LS: The liver in circulatory failure. In: Schiff's Diseases of the Liver, 9th ed. Schiff ER et al (editors). Lippincott Williams & Wilkins, 2003.
NONCIRRHOTIC PORTAL HYPERTENSION
Noncirrhotic portal hypertension must be considered in the differential diagnosis of splenomegaly or upper gastrointestinal bleeding due to esophageal or gastric varices in patients with little if any liver dysfunction. This syndrome may be due to portal vein thrombosis (with so-called cavernous transformation), splenic vein obstruction (presenting as gastric varices without esophageal varices), schistosomiasis, noncirrhotic intrahepatic portal sclerosis, nodular regenerative hyperplasia, or arterial-portal vein fistula. Aside from splenomegaly, the physical findings are not remarkable, although hepatic decompensation can follow severe gastrointestinal bleeding or a concurrent hepatic disorder, and intestinal infarction may occur when portal vein thrombosis is associated with mesenteric venous thrombosis. Endoscopy shows esophageal or gastric varices. The liver tests are usually normal, but there may be findings of hypersplenism. Magnetic resonance angiography (MRA) of the portal system is generally confirmatory. Magnetic resonance cholangiography may demonstrate compression of the bile duct by a large portal cavernoma. Needle biopsy of the liver may be indicated to diagnose schistosomiasis and noncirrhotic intrahepatic portal sclerosis and may demonstrate sinusoidal dilatation. An underlying hypercoagulable state is found in many patients with portal vein thrombosis; these include mutation G20210A of prothrombin, factor V Leiden mutation, protein C and S deficiency, antiphospholipid syndrome, mutation TT677 of methylenetetrahydrofolate reductase, elevated factor VIII levels, and myeloproliferative disorders. It is possible, however, that deficiency of protein C and S—as well as of antithrombin—is a secondary phenomenon due to portosystemic shunting and reduced hepatic blood flow. Portal vein thrombosis may occur in up to 15% of patients with cirrhosis and may be associated with hepatocellular carcinoma. Splenic vein thrombosis may complicate pancreatitis or pancreatic cancer.
If splenic vein thrombosis is the cause of variceal bleeding, splenectomy is curative. For other causes, band ligation or sclerotherapy is initiated for variceal bleeding and portosystemic shunting is reserved for failures of endoscopic therapy. Anticoagulation may be indicated for isolated acute portal vein thrombosis if a hypercoagulable disorder is identified.
Amitrano L et al: Risk factors and clinical presentation of portal vein thrombosis in patients with liver cirrhosis. J Hepatol 2004;40:736.
Condat B et al: Portal cavernoma-associated cholangiopathy: a clinical and MR cholangiography coupled with MR portography imaging study. Hepatology 2003;37:1302.
Saadoun D et al: Association of idiopathic hepatic sinusoidal dilatation with the immunological features of the antiphospholipid syndrome. Gut 2004;53:1516.
PYOGENIC HEPATIC ABSCESS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Fever, right upper quadrant pain, jaundice.
Often in setting of biliary disease but up to 40% are “cryptogenic” in origin.
Detected by imaging studies.
P.681
General Considerations
The liver can be invaded by bacteria via (1) the portal vein (pylephlebitis); (2) the common duct (ascending cholangitis); (3) the hepatic artery, secondary to bacteremia; (4) direct extension from an infectious process; and (5) traumatic implantation of bacteria through the abdominal wall. Risk factors for liver abscess include older age and male gender. Predisposing conditions include malignancy, diabetes, inflammatory bowel disease, cirrhosis, and liver transplantation.
Ascending cholangitis resulting from biliary obstruction due to a stone, stricture, or neoplasm is the most common identifiable cause of hepatic abscess in the United States. In 10% of cases, liver abscess is secondary to appendicitis or diverticulitis. At least 40% of abscesses have no demonstrable cause and are classified as cryptogenic. A dental source is identified in some such cases. The most frequently encountered organisms are E coli, Klebsiella pneumoniae, Proteus vulgaris, Enterobacter aerogenes, and multiple microaerophilic and anaerobic species (eg, Streptococcus milleri). Staphylococcus aureus is usually the causative organism in patients with chronic granulomatous disease. Unusual causative organisms include Salmonella, Haemophilus, and Yersinia. Hepatic candidiasis, tuberculosis, and actinomycosis are seen in immunocompromised patients and those with hematologic malignancies. Rarely, hepatocellular carcinoma can present as a pyogenic abscess because of tumor necrosis, biliary obstruction, and superimposed bacterial infection. The possibility of an amebic liver abscess must always be considered (see Chapter 35).
Clinical Findings
A. SYMPTOMS AND SIGNS
The presentation is often insidious. Fever is almost always present and may antedate other symptoms or signs. Pain may be a prominent complaint and is localized to the right hypochondrium or epigastric area. Jaundice, tenderness in the right upper abdomen, and either steady or swinging fever are the chief physical findings.
B. LABORATORY FINDINGS
Laboratory examination reveals leukocytosis with a shift to the left. Liver function studies are nonspecifically abnormal. Blood cultures are positive in 50–100% of cases.
C. IMAGING
Chest roentgenograms usually reveal elevation of the diaphragm if the abscess is on the right side. Ultrasound, CT, or MRI may reveal the presence of intrahepatic defects. On MRI, characteristic findings include high signal intensity on T2-weighted images and rim enhancement. Hepatic candidiasis is seen usually in the setting of systemic candidiasis, and on CT scan the characteristic appearance is that of multiple “bullseyes,” but imaging studies may be negative in neutropenic patients.
Treatment
Treatment should consist of antimicrobial agents (a third-generation cephalosporin and metronidazole) that are effective against coliform organisms and anaerobes. Antibiotics are administered for 2–3 weeks, and sometimes up to 6 weeks. If the abscess is at least 5 cm in diameter or the response to antibiotic therapy is not rapid, intermittent needle aspiration or catheter or surgical (eg, laparoscopic) drainage should be done. The mortality rate is still substantial (≥ 8% in most studies) and is highest in patients with underlying biliary malignancy or severe multiorgan dysfunction. Hepatic candidiasis often responds to intravenous amphotericin B (total dose of 2–9 g). Fungal abscesses are associated with mortality rates of up to 50% and are treated with intravenous amphotericin B and drainage.
Kaplan GG et al: Population-based study of the epidemiology of and the risk factors for pyogenic liver abscess. Clin Gastroenterol Hepatol 2004;2:1032.
Rahimian J et al: Pyogenic liver abscess: recent trends in etiology and mortality. Clin Infect Dis 2004;39:1654.
Yu SC et al: Treatment of pyogenic liver abscess: prospective randomized comparison of catheter drainage and needle aspiration. Hepatology 2004;39:932.
NEOPLASMS OF THE LIVER
1. Hepatocellular Carcinoma
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
In Western countries, usually a complication of cirrhosis.
Characteristic CT and MRI features and elevated serum alpha-fetoprotein may obviate the need for a confirmatory biopsy.
General Considerations
Malignant neoplasms of the liver that arise from parenchymal cells are called hepatocellular carcinomas; those that originate in the ductular cells are called cholangiocarcinomas.
Hepatocellular carcinomas are associated with cirrhosis in 80% of cases. Incidence rates are rising rapidly (twofold since 1978) in the United States and other Western countries, presumably because of the increasing prevalence of cirrhosis caused by chronic hepatitis C infection over the past 2–3 decades. In Western countries, risk factors for hepatocellular carcinoma in patients known to have cirrhosis are male
P.682
gender, age > 55 years (although there has been an increase in the number of younger cases), nonwhite ethnicity, anti-HCV positivity, prothrombin time < 75% of control, and platelet count < 75,000/mcL. In Africa and most of Asia, hepatitis B is of major etiologic significance, whereas in Western countries and Japan, hepatitis C (sometimes in combination with “occult” HBV infection) and alcoholic cirrhosis are the most common risk factors. Other associations include hemochromatosis (and possibly the C282Y carrier state), aflatoxin exposure (associated with mutation of the P53 gene), α1-antiprotease (α1-antitrypsin) deficiency, and tyrosinemia. Increasingly, cirrhosis resulting from NAFLD and insulin resistance syndrome has been associated with hepatocellular carcinomas. The fibrolamellar variant of hepatocellular carcinoma occurs in young women and is characterized by a distinctive histologic picture, absence of risk factors, and indolent course.
Histologically, hepatocellular carcinoma is made up of cords or sheets of cells that roughly resemble the hepatic parenchyma. Blood vessels such as portal or hepatic veins are commonly involved by tumor.
Clinical Findings
A. SYMPTOMS AND SIGNS
The presence of a hepatocellular carcinoma may be unsuspected until there is deterioration in the condition of a cirrhotic patient who was formerly stable. Cachexia, weakness, and weight loss are associated symptoms. The sudden appearance of ascites, which may be bloody, suggests portal or hepatic vein thrombosis by tumor or bleeding from the necrotic tumor.
Physical examination may show tender enlargement of the liver, with an occasionally palpable mass. In Africa, the typical presentation in young patients is a rapidly expanding abdominal mass. Auscultation may reveal a bruit over the tumor or a friction rub when the process has extended to the surface of the liver.
B. LABORATORY FINDINGS
Laboratory tests may reveal leukocytosis, as opposed to the leukopenia that is frequently encountered in cirrhotic patients. Anemia is common, but a normal or elevated hematocrit may be found in up to one-third of patients owing to elaboration of erythropoietin by the tumor. Sudden and sustained elevation of the serum alkaline phosphatase in a patient who was formerly stable is a common finding. Hepatitis B surface antigen is present in a majority of cases in endemic areas, whereas in the United States anti-HCV is found in up to 40% of cases. Alpha-fetoprotein levels are elevated in up to 70% of patients with hepatocellular carcinoma in Western countries (although the sensitivity is lower in blacks); however, mild elevations are also often seen in patients with chronic hepatitis. Serum levels of des-gamma-carboxy prothrombin are elevated in up to 90% of patients with hepatocellular carcinoma, but they may also be elevated in patients with vitamin K deficiency, chronic hepatitis, and metastatic cancer. Cytologic study of ascitic fluid rarely reveals malignant cells.
C. IMAGING
Arterial phase helical CT scanning and MRI with contrast enhancement are the preferred imaging studies for determining the location and vascularity of the tumor. Lesions smaller than 2 cm may be difficult to characterize. Ultrasound is less sensitive and operator dependent but is used to screen for hepatic nodules in high-risk patients. Contrast-enhanced ultrasound has sensitivity and specificity approaching those of arterial phase helical CT. Positron emission tomography is under study.
D. LIVER BIOPSY AND STAGING
Liver biopsy is diagnostic, although seeding of the needle tract by tumor is a potential risk (1–3%), and biopsy can be deferred if imaging studies and alpha-fetoprotein levels are diagnostic (eg, serum alpha-fetoprotein > 400 ng/mL and a mass lesion on imaging of a cirrhotic liver) or if surgical resection is planned. Staging in the TNM classification includes the following definitions: T0: no evidence of primary tumor; T1: solitary tumor without vascular invasion; T2: solitary tumor with vascular invasion or multiple tumors none more than 5 cm; T3: multiple tumors more than 5 cm or tumor involving a major branch of the portal or hepatic vein; and T4: tumors with direct invasion of adjacent organs other than the gallbladder or with perforation of the visceral peritoneum. Alternative staging systems also incorporate liver function, tumor aggressiveness and growth rate, general health of the patient, and treatment but require refinement and validation. For example, the CLIP (Cancer of the Liver Italian Program) classification incorporates the Child-Turcotte-Pugh stage, tumor morphology (uninodular, multinodular, extensive), alpha-fetoprotein (less than or greater than 400 ng/mL), and vascular invasion.
Treatment
Surgical resection of solitary hepatocellular carcinomas may result in cure if liver function is preserved (Child class A or possibly B). Laparoscopic liver resection has been performed in selected cases. Treatment of underlying chronic viral hepatitis, adjuvant chemotherapy, and adaptive immunotherapy may lower postsurgical recurrence rates. Liver transplantation may be appropriate for small unresectable tumors in a patient with advanced cirrhosis, with reported 5-year survival rates of up to 75%. The recurrence-free survival may be better for liver transplantation than for resection in patients with well-compensated cirrhosis and small tumors (one tumor < 5 cm or three or fewer tumors each < 3 cm in diameter). However, liver transplantation is often impractical because of the donor organ shortage and living donor liver transplantation may be considered
P.683
in these cases. Chemotherapy, hormonal therapy with tamoxifen, and long-acting octreotide have not been shown to prolong life, but chemoembolization via the hepatic artery is palliative and may prolong survival. Injection of absolute ethanol into, radiofrequency ablation of, or cryotherapy of small tumors (< 4 cm) may prolong survival in patients who are not candidates for resection; these interventions may provide a “bridge” to liver transplantation. New radiation therapy techniques and novel biologic approaches (eg, antiangiogenesis agents and inhibitors of growthfactor signaling) are under study. For patients whose disease progresses despite treatment or who present with advanced tumors, vascular invasion, or extrahepatic spread, meticulous efforts at palliative care are essential (see Chapter 5). Severe pain may develop in such patients due to expansion of the liver capsule by the tumor and require concerted efforts at pain management, including the use of opioids (see Chapter 5).
Prognosis
In the United States, overall 1and 5-year survival rates for patients with hepatocellular carcinoma are 23% and 5%, respectively. Five-year survival rates rise to 56% for patients with localized resectable disease (T1, T2, T3, selected T4; N0; M0) but are virtually nil for those with localized unresectable or advanced disease. The fibrolamellar variant has a better prognosis than conventional hepatocellular carcinoma, with a 5-year survival rate of 32%.
Screening & Prevention
In the patient with chronic hepatitis B or cirrhosis caused by HCV or alcohol, surveillance for the development of hepatocellular carcinoma should be considered with regular (eg, every 6 months) alpha-fetoprotein testing and ultrasonography. The risk of hepatocellular carcinoma in a patient with cirrhosis is 3–5% a year. The diagnosis of hepatocellular carcinoma is established (without the need for biopsy) for lesions > 2 cm when characteristic arterial hypervascularity is demonstrated on both helical CT and MRI (or on either CT or MRI if the serum alpha-fetoprotein level is higher than 400 mg/L). In a population of patients with cirrhosis, over 60% of nodules < 2 cm in diameter detected on a screening ultrasound proved to be hepatocellular carcinoma. Mass vaccination programs against HBV in developing countries are leading to reduced rates of hepatocellular carcinoma, whereas in the United States and western Europe the incidence of hepatocellular carcinoma is rising. Successful treatment of hepatitis C in patients with cirrhosis reduces the subsequent risk of hepatocellular carcinoma.
Caturelli E et al: Ultrasound guided fine needle biopsy of early hepatocellular carcinoma complicating liver cirrhosis: a multicentre study. Gut 2004;53:1356.
El-Serag HB et al: Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004;126:460.
El-Serag HB et al: Is fibrolamellar carcinoma different from hepatocellular carcinoma? A US population-based study. Hepatology 2004;39:798.
Lin SM et al: Radiofrequency ablation improves prognosis compared with ethanol injection for hepatocellular carcinoma ≤ 4 cm. Gastroenterology 2004;127:1714.
Seeff LB et al: Introduction: The burden of hepatocellular carcinoma. Gastroenterology 2004;127(5 Suppl 1):S1.
Sherman M et al: AASLD single-topic research conference on hepatocellular carcinoma: conference proceedings. Hepatology 2004;40:1465.
Shiratori Y et al: Antiviral therapy for cirrhotic hepatitis C: association with reduced hepatocellular carcinoma development and improved survival. Ann Intern Med 2005;142:105.
2. Benign Liver Neoplasms
The most common benign neoplasm of the liver is the cavernous hemangioma, often an incidental finding on ultrasound or CT scan. This lesion may enlarge in women who take hormonal therapy and must be differentiated from other space-occupying intrahepatic lesions, usually by MRI. Rarely, fine-needle biopsy is necessary to differentiate these lesions and does not appear to carry an increased risk of bleeding. Surgical resection of cavernous hemangiomas is rarely necessary but may be required for abdominal pain or to exclude malignancy.
In addition to rare instances of sinusoidal dilatation and peliosis hepatis, two distinct benign lesions with characteristic clinical, radiologic, and histopathologic features have been described in women taking oral contraceptives—focal nodular hyperplasia and hepatic adenoma. Focal nodular hyperplasia occurs at all ages but is probably not caused by oral contraceptives. It is often asymptomatic and appears as a hypervascular mass, occasionally with a central hypodense “stellate” scar on CT scan or MRI. Microscopically, focal nodular hyperplasia consists of hyperplastic units of hepatocytes with a central stellate scar containing proliferating bile ducts. It is not a true neoplasm but a nonspecific reaction to vascular abnormalities. The prevalence of hepatic hemangiomas is increased in patients with focal nodular hyperplasia. A variant termed “telangiectatic focal nodular hyperplasia” is associated with overexpression of the gene that encodes angiopoietin-1, a protein involved in blood vessel maturation, and appears to be a true neoplasm, like hepatic adenoma.
Hepatic adenoma occurs most commonly in the third and fourth decades of life and is usually caused by oral contraceptives; the clinical presentation is often one of acute abdominal pain due to necrosis of the tumor with hemorrhage. Hepatic adenomas occur in patients with glycogen storage disease. Rare instances of multiple hepatic adenomas in association with maturity-onset diabetes of the young occur in
P.684
families with a germline mutation in hepatocyte nuclear factor 1α. The tumor is hypovascular and reveals a cold defect on liver scan. Grossly, the cut surface appears structureless. As seen microscopically, the hepatic adenoma consists of sheets of hepatocytes without portal tracts or central veins. The only physical finding in focal nodular hyperplasia or hepatic adenoma is a palpable abdominal mass in a minority of cases. Liver function is usually normal. Arterial phase helical CT and MRI with contrast can distinguish an adenoma from focal nodular hyperplasia in 80–90% of cases.
Treatment of focal nodular hyperplasia is resection only in the symptomatic patient. The prognosis is excellent. Hepatic adenoma often undergoes necrosis and rupture, and resection is advised even in asymptomatic persons. In selected cases, laparoscopic resection may be feasible. Regression of benign hepatic tumors may follow cessation of oral contraceptives.
Glinkova V et al: Hepatic hemangiomas: possible association with female sex hormones. Gut 2004;53:1352.
Paradis V et al: Telangiectatic focal nodular hyperplasia: a variant of hepatocellular adenoma. Gastroenterology 2004;126:1323.
DISEASES OF THE BILIARY TRACT
CHOLELITHIASIS (GALLSTONES)
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Often asymptomatic.
Classic biliary pain characterized by infrequent episodes of steady severe pain in epigastrium or right upper quadrant with radiation to right scapula.
Detected on ultrasound.
General Considerations
Gallstones are more common in women than in men and increase in incidence in both sexes and all races with aging. In the United States, over 10% of men and 20% of women have gallstones by age 65; the total exceeds 20 million people. Although cholesterol gallstones are less common in black people, cholelithiasis attributable to hemolysis occurs in over a third of individuals with sickle cell anemia. Native Americans of both the Northern and Southern Hemispheres have a high rate of cholesterol cholelithiasis, probably because of a predisposition resulting from “thrifty” (LITH) genes that promote efficient calorie utilization and fat storage. As many as 75% of Pima and other American Indian women over the age of 25 years have cholelithiasis. Other genetic mutations that predispose persons to gallstones have been identified. Obesity is a risk factor for gallstones, especially in women. Rapid weight loss also increases the risk of symptomatic gallstone formation. There is evidence that glucose intolerance and elevated serum insulin levels (insulin resistance syndrome) are risk factors for gallstones. A low-carbohydrate diet and physical activity may help prevent gallstones. Consumption of caffeinated coffee appears to protect against gallstones in women, and a high intake of polyunsaturated and monounsaturated fats reduces the risk of gallstones in men on an energy-balanced diet. A high-fiber diet reduces the risk of cholecystectomy in women. Hypertriglyceridemia may promote gallstone formation by impairing gallbladder motility. The incidence of gallstones is high in individuals with Crohn's disease; approximately one-third of those with inflammatory involvement of the terminal ileum have gallstones due to disruption of bile salt resorption that results in decreased solubility of the bile. The incidence of cholelithiasis is also increased in patients with diabetes mellitus and in those with cirrhosis. Drugs such as clofibrate, octreotide, and ceftriaxone can cause gallstones. In contrast, aspirin and other nonsteroidal anti-inflammatory drugs may protect against gallstones. Prolonged fasting (over 5–10 days) can lead to formation of biliary “sludge” (microlithiasis), which usually resolves with refeeding but can lead to gallstones or biliary symptoms. Pregnancy, particularly in obese women, is associated with an increased risk of gallstones and of symptomatic gallbladder disease. Hormone replacement therapy appears to increase the risk of gallbladder disease and need for cholecystectomy.
Pathogenesis of Gallstones
Gallstones are classified according to their predominant chemical composition as cholesterol or calcium bilirubinate stones. The latter comprise less than 20% of the stones found in Europe or the United States but 30–40% of stones found in Japan.
Three compounds comprise 80–95% of the total solids dissolved in bile: conjugated bile salts, lecithin, and cholesterol. Cholesterol is a neutral sterol, and lecithin is a phospholipid; both are almost completely insoluble in water. However, bile salts in combination with lecithin are able to form multimolecular aggregates (and micelles) or vesicles that solubilize cholesterol in an aqueous solution. Precipitation of cholesterol microcrystals may come about because of increased biliary secretion of cholesterol, defective formation of vesicles, an excess of factors promoting the nucleation of cholesterol crystals (or deficiency of antinucleating factors), or delayed emptying of the gallbladder.
Table 15-6. Diseases of the biliary tract. | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
P.685
Clinical Findings
Table 15-6 lists the clinical and laboratory features of several diseases of the biliary tract as well as their treatment. Cholelithiasis is frequently asymptomatic and is discovered in the course of routine radiographic study, operation, or autopsy. There is generally no need for prophylactic cholecystectomy in an asymptomatic person unless the gallbladder is calcified, gallstones are over 3 cm in diameter, or the patient is a candidate for cardiac transplantation. Ultimately, symptoms (biliary colic) develop in 10–25% of patients, and complications (such as acute cholecystitis) develop in 3% by 10 years. “Symptomatic” cholelithiasis usually means characteristic right upper quadrant or epigastric discomfort or pain (biliary pain). Occasionally, small intestinal obstruction due to “gallstone ileus” presents as the initial manifestation of cholelithiasis.
Treatment
Laparoscopic cholecystectomy is the treatment of choice for symptomatic gallbladder disease. The minimal trauma to the abdominal wall makes it possible for patients to go home within 1 day of the procedure and to return to work within 7 days (instead of weeks for those undergoing standard open cholecystectomy). In many cases, the procedure may be performed on an outpatient basis. This procedure is suitable in most patients, including those with acute cholecystitis. If problems are encountered, the surgery can be converted to a conventional open cholecystectomy. Bile duct injuries occur in 0.1% of cases done by experienced surgeons. Cholecystectomy may increase the risk of esophageal and proximal small intestinal adenocarcinoma because of increased duodenogastric reflux and changes in intestinal exposure to bile, respectively. A conservative approach to biliary colic is advised in pregnant patients, but for patients with repeated attacks of biliary colic or acute cholecystitis, cholecystectomy can be performed—even by the laparoscopic route—preferably in the second trimester. Enterolithotomy alone is considered adequate treatment in most patients with gallstone ileus.
Persistence of symptoms after removal of the gallbladder (postcholecystectomy syndrome) implies either mistaken diagnosis, functional bowel disorder, technical error, retained or recurrent common bile duct stone, or spasm of the sphincter of Oddi (see below).
P.686
Cheno- and ursodeoxycholic acids are bile salts that when given orally for up to 2 years dissolve some cholesterol stones and may be considered in selected patients who refuse cholecystectomy. The dose is 7 mg/ kg/d of each or 8–13 mg/kg of ursodeoxycholic acid in divided doses daily. They are most effective in patients with a functioning gallbladder, as determined by gallbladder visualization on oral cholecystography, and multiple small “floating” gallstones (representing not more than 15% of patients with gallstones). In half of patients, gallstones recur within 5 years after treatment is stopped. Ursodeoxycholic acid, 500–600 mg daily, reduces the risk of gallstone formation with rapid weight loss, as occurs after bariatric surgery.
Lithotripsy in combination with bile salt therapy for single radiolucent stones less than 20 mm in diameter was an option in the past but is no longer generally used in the United States.
Cirillo DJ et al: Effect of estrogen therapy on gallbladder disease. JAMA 2005;293:330.
Ko CW et al: Incidence, natural history, and risk factors for biliary sludge and stones during pregnancy. Hepatology 2005;41:359.
Lublin M et al: Symptoms before and after laparoscopic cholecystectomy for gallstones. Am Surg 2004;70:836.
Tsai CJ et al: Long-term intake of dietary fiber and decreased risk of cholecystectomy in women. Am J Gastroenterol 2004;99:1364.
Tasi CJ et al: The effect of long-term intake of cis unsaturated fats on the risk for gallstone disease in men: a prospective cohort study. Ann Intern Med 2004;141:514.
ACUTE CHOLECYSTITIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Steady, severe pain and tenderness in the right hypochondrium or epigastrium.
Nausea and vomiting.
Fever and leukocytosis.
General Considerations
Cholecystitis is associated with gallstones in over 90% of cases. It occurs when a stone becomes impacted in the cystic duct and inflammation develops behind the obstruction. Acalculous cholecystitis should be considered when unexplained fever or right upper quadrant pain occurs within 2–4 weeks of major surgery or in a critically ill patient who has had no oral intake for a prolonged period; multiorgan failure is often present. Primarily as a result of ischemic changes secondary to splanchnic vasoconstriction or intravascular coagulation, gangrene may develop, resulting in perforation. Although generalized peritonitis is possible, the leak usually remains localized and forms a chronic, wellcircumscribed abscess cavity. Acute cholecystitis caused by infectious agents (eg, cytomegalovirus, cryptosporidiosis, or microsporidiosis) may occur in patients with AIDS.
Clinical Findings
A. SYMPTOMS AND SIGNS
The acute attack is often precipitated by a large or fatty meal and is characterized by the sudden appearance of steady pain localized to the epigastrium or right hypochondrium, which may gradually subside over a period of 12–18 hours. Vomiting occurs in about 75% of patients and in half of instances affords variable relief. Right upper quadrant abdominal tenderness (often with Murphy's sign, or inhibition of inspiration by pain on palpation of the right upper quadrant) is almost always present and is usually associated with muscle guarding and rebound pain. A palpable gallbladder is present in about 15% of cases. Jaundice is present in about 25% of cases and, when persistent or severe, suggests the possibility of choledocholithiasis. Fever is typical.
B. LABORATORY FINDINGS
The white blood cell count is usually high (12,000–15,000/mcL). Total serum bilirubin values of 1–4 mg/dL may be seen even in the absence of common duct obstruction. Serum aminotransferase and alkaline phosphatase are often elevated—the former as high as 300 units/ mL, or even higher when associated with ascending cholangitis. Serum amylase may also be moderately elevated.
C. IMAGING
Plain films of the abdomen may show radiopaque gallstones in 15% of cases. 99mTc hepatobiliary imaging (using iminodiacetic acid compounds), also known as the HIDA scan, is useful in demonstrating an obstructed cystic duct, which is the cause of acute cholecystitis in most patients. This test is reliable if the bilirubin is under 5 mg/dL (98% sensitivity and 81% specificity for acute cholecystitis). False-positive results can occur with prolonged fasting, liver disease, and chronic cholecystitis. Right upper quadrant abdominal ultrasound may show the presence of gallstones but is not sensitive for acute cholecystitis (67% sensitivity, 82% specificity); findings suggestive of acute cholecystitis are gallbladder wall thickening, pericholecystic fluid, and sonographic Murphy's sign.
Differential Diagnosis
The disorders most likely to be confused with acute cholecystitis are perforated peptic ulcer, acute pancreatitis, appendicitis in a high-lying appendix, perforated colonic carcinoma or diverticulum of the hepatic flexure, liver abscess, hepatitis, pneumonia with pleurisy on the right side, and even myocardial ischemia. Definite localization of pain and tenderness in the right hypochondrium, with radiation around to the infrascapular
P.687
area, strongly favors the diagnosis of acute cholecystitis. True cholecystitis without stones suggests the rare possibility of polyarteritis nodosa affecting the cystic artery.
Complications
A. GANGRENE OF THE GALLBLADDER
Continuation or progression of right upper quadrant abdominal pain, tenderness, muscle guarding, fever, and leukocytosis after 24–48 hours suggests severe inflammation and possible gangrene of the gallbladder. Necrosis may occasionally develop without definite signs in the obese, diabetic, elderly, or immunosuppressed patient. Other serious acute complications include gallbladder perforation (usually with formation of a pericholecystic abscess), emphysematous cholecystitis (secondary infection with a gas-forming organism), and empyema.
B. CHOLANGITIS
Cholangitis classically presents with Charcot's triad, namely, fever and chills, right upper quadrant pain, and jaundice. Although 95% of patients who present with this picture will have common duct stones, only a minority of patients with acute cholecystitis have common duct stones that will present in this manner.
C. CHRONIC CHOLECYSTITIS AND OTHER COMPLICATIONS
Chronic cholecystitis results from repeated episodes of acute cholecystitis or chronic irritation of the gallbladder wall by stones and is characterized pathologically by varying degrees of chronic inflammation of the gallbladder. Calculi are usually present. In about 4–5% of cases, the villi of the gallbladder undergo polypoid enlargement due to deposition of cholesterol that may be visible to the naked eye (“strawberry gallbladder,” cholesterolosis). In other instances, adenomatous hyperplasia of all or part of the gallbladder wall may be so marked as to give the appearance of a myoma (pseudotumor). Hydrops of the gallbladder results when acute cholecystitis subsides but cystic duct obstruction persists, producing distention of the gallbladder with a clear mucoid fluid. Occasionally, a stone in the neck of the gallbladder may compress the bile duct and cause jaundice (Mirizzi's syndrome). Xanthogranulomatous cholecystitis is a rare variant of chronic cholecystitis characterized by grayish-yellow nodules or streaks, representing lipid-laden macrophages, in the wall of the gallbladder.
Cholelithiasis with chronic cholecystitis may be associated with acute exacerbations of gallbladder inflammation, common duct stone, fistulization to the bowel, pancreatitis and, rarely, carcinoma of the gallbladder. Calcified (porcelain) gallbladder has generally been thought to have a high association with gallbladder carcinoma and to be an indication for cholecystectomy, although the risk of gallbladder cancer may be higher when calcification is mucosal rather than intramural.
Treatment
Acute cholecystitis will usually subside on a conservative regimen (withholding of oral feedings, intravenous alimentation, analgesics, and intravenous antibiotics—generally a third-generation cephalosporin with the addition of metronidazole in severe cases). Meperidine may be preferable to morphine for pain because of less spasm of the sphincter of Oddi. Because of the high risk of recurrent attacks (up to 10% by 1 month and over 30% by 1 year), cholecystectomy—generally laparoscopically— should generally be performed within 2–3 days after hospitalization (and immediately if perforation or gangrene is suspected). If nonsurgical treatment has been elected, the patient (especially if diabetic or elderly) should be watched carefully for recurrent symptoms, evidence of gangrene of the gallbladder, or cholangitis. In high-risk patients, ultrasound-guided aspiration of the gallbladder, if feasible, or percutaneous cholecystostomy may postpone or even avoid the need for surgery. Cholecystectomy is mandatory when there is evidence of gangrene or perforation.
Surgical treatment of chronic cholecystitis is the same as for acute cholecystitis. If indicated, cholangiography can be performed during laparoscopic cholecystectomy. Choledocholithiasis can also be excluded by either preoperative or postoperative ERCP or MRCP.
Prognosis
The overall mortality rate of cholecystectomy is less than 1%, but hepatobiliary tract surgery is a more formidable procedure in the elderly, in whom the mortality rate is 5–10%. A technically successful surgical procedure in an appropriately selected patient is generally followed by complete resolution of symptoms.
Alobaidi M et al: Current trends in imaging evaluation of acute cholecystitis. Emerg Radiol 2004;10:256.
Ito K et al: Percutaneous cholecystostomy versus gallbladder aspiration for acute cholecystitis: a prospective randomized controlled trial. AJR Am J Roentgenol 2004;183:193.
Tambyraja AL et al: Outcome of laparoscopic cholecystectomy in patients 80 years and older. World J Surg 2004;28:745.
Tsumura H et al: An evaluation of laparoscopic cholecystectomy after selective percutaneous transhepatic gallbladder drainage for acute cholecystitis. Gastrointest Endosc 2004;59:839.
Yusoff IF et al: Diagnosis and management of cholecystitis and cholangitis. Gastroenterol Clin North Am 2003;32:1145.
PRE& POSTCHOLECYSTECTOMY SYNDROMES
Precholecystectomy
In a small group of patients (mostly women) with biliary pain, conventional radiographic studies of the upper gastrointestinal tract and gallbladder—including
P.688
cholangiography—are unremarkable. In some such cases, emptying of the gallbladder is markedly reduced on gallbladder scintigraphy following injection of cholecystokinin; cholecystectomy may be curative. In some cases, histologic examination of the resected gallbladder shows chronic cholecystitis or microlithiasis. An additional diagnostic consideration is sphincter of Oddi dysfunction (see below).
Postcholecystectomy
Following cholecystectomy, some patients complain of continuing symptoms, ie, right upper quadrant pain, flatulence, and fatty food intolerance. The persistence of symptoms in this group of patients suggests the possibility of an incorrect diagnosis prior to cholecystectomy, eg, esophagitis, pancreatitis, radiculopathy, or functional bowel disease. It is important to rule out the possibility of choledocholithiasis or common duct stricture as a cause of persistent symptoms. Pain may also be associated with dilation of the cystic duct remnant, neuroma formation in the ductal wall, foreign body granuloma, or traction on the common duct by a long cystic duct.
The clinical presentation of right upper quadrant pain, chills, fever, or jaundice should suggest biliary tract disease. Endoscopic ultrasonography or retrograde cholangiography may be necessary to demonstrate or exclude biliary tract disease. Biliary pain associated with elevated liver tests and a dilated bile duct suggests sphincter of Oddi dysfunction or stenosis. Biliary manometry may be useful for documenting elevated baseline sphincter of Oddi pressures typical of sphincter dysfunction when biliary pain is associated with elevated liver tests (twofold) or a dilated bile duct (> 12 mm) but is not necessary when both are present. (Analogous criteria have been developed for pancreatic sphincter dysfunction.) Biliary scintigraphy and MRCP following intravenous administration of secretin show promise as screening tests for sphincter dysfunction. Endoscopic sphincterotomy is most likely to relieve symptoms when they are associated with elevated liver chemistry tests, a dilated common duct, or an elevated sphincter of Oddi pressure, although many patients continue to have some pain. In some cases, treatment with calcium channel blockers, longacting nitrates, or possibly injection of the sphincter with botulinum toxin may be beneficial. In refractory cases, surgical sphincteroplasty or removal of the cystic duct remnant may be considered.
Petersen BT: Amn evidence-based review of sphincter of Oddi dysfunction: part 1, presentations with “objective” biliary findings (types I and II). Gastrointest Endosc 2004;59:525.
Petersen BT: Sphincter of Oddi dysfunction: part 2: Evidencebased review of the presentations, with “objective” pancreatic findings (types I and II) and of presumptive type III. Gastrointest Endosc 2004;59:670.
Piccinni G et al: Diagnosis and treating sphincter of Oddi dysfunction: a critical literature review and reevaluation. J Clin Gastroenterol 2004;38:350.
CHOLEDOCHOLITHIASIS & CHOLANGITIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Often a history of biliary pain or jaundice.
Sudden onset of severe right upper quadrant or epigastric pain, which may radiate to the right scapula or shoulder.
Occasional patients present with painless jaundice.
Nausea and vomiting.
Fever, which may be followed by hypothermia and gram-negative shock, jaundice, and leukocytosis.
Stones in bile duct most reliably detected by ERCP or endoscopic ultrasound.
General Considerations
About 15% of patients with gallstones have choledocholithiasis (common bile duct stones). The percentage rises with age, and the frequency in elderly people with gallstones may be as high as 50%. Common duct stones usually originate in the gallbladder but may also form spontaneously in the common duct after cholecystectomy. The risk is increased twofold in persons with a juxtapapillary duodenal diverticulum. The stones are frequently “silent” as no symptoms result unless there is obstruction.
Clinical Findings
A. SYMPTOMS AND SIGNS
A history of biliary pain or prior jaundice may be obtained. Biliary pain results from rapid increases in common bile duct pressure due to obstructed bile flow. The features that suggest the presence of a common duct stone are (1) frequently recurring attacks of right upper abdominal pain that is severe and persists for hours, (2) chills and fever associated with severe pain, and (3) a history of jaundice associated with episodes of abdominal pain. The combination of pain, fever (and chills), and jaundice represents Charcot's triad and denotes the classic picture of cholangitis. The addition of altered sensorium and hypotension (Reynold's pentad) connotes acute suppurative cholangitis and represents an endoscopic emergency.
Hepatomegaly may be present in calculous biliary obstruction, and tenderness is usually present in the right upper quadrant and epigastrium. Common duct obstruction lasting longer than 30 days results in liver damage leading to cirrhosis. Hepatic failure with portal hypertension occurs in untreated cases.
P.689
B. LABORATORY FINDINGS
Acute obstruction of the bile duct typically produces a transient albeit striking increase in serum aminotransferase levels (> 1000 units/L). Bilirubinuria and elevation of serum bilirubin are present if the common duct remains obstructed; levels commonly fluctuate. Serum alkaline phosphatase levels rise more slowly and are suggestive of obstructive jaundice. Not uncommonly, serum amylase elevations are present because of secondary pancreatitis. When extrahepatic obstruction persists for more than a few weeks, differentiation of obstruction from chronic cholestatic liver disease becomes more difficult. Leukocytosis is present in patients with cholangitis. Prolongation of the prothrombin time can result from the obstructed flow of bile to the intestine. In contrast to hepatocellular dysfunction, hypoprothrombinemia due to obstructive jaundice will respond to 10 mg of parenteral vitamin K or water-soluble oral vitamin K (phytonadione, 5 mg) within 24–36 hours.
C. IMAGING
Ultrasonography, CT scan, and radionuclide imaging may demonstrate dilated bile ducts and impaired bile flow. Endoscopic ultrasonography, helical CT, and magnetic resonance cholangiography have been found to be accurate in demonstrating common duct stones and, if available, may be used in patients thought to be at low or intermediate risk for choledocholithiasis. ERCP (occasionally with intraductal ultrasound) or percutaneous transhepatic cholangiography provides the most direct and accurate means of determining the cause, location, and extent of obstruction. If the likelihood that obstruction is caused by a stone or that cholangitis is present is high, ERCP is the procedure of choice because it permits sphincterotomy with stone extraction or stent placement. Endoscopic balloon dilation of the sphincter of Oddi is associated with a higher rate of pancreatitis than endoscopic sphincterotomy and is generally reserved for patients with coagulopathy, in whom the risk of bleeding is lower with balloon dilation than with sphincterotomy.
Differential Diagnosis
The most common cause of obstructive jaundice is common duct stone. Next in frequency is carcinoma of the pancreas, ampulla of Vater, or common duct. Extrinsic compression of the common duct may result from metastatic carcinoma (usually from the gastrointestinal tract or breast) involving porta hepatis lymph nodes or, rarely, from a large duodenal diverticulum. Gallbladder cancer extending into the common duct often presents as obstructive jaundice. Chronic cholestatic liver diseases (primarily biliary cirrhosis, sclerosing cholangitis, drug-induced) must be considered. Hepatocellular jaundice can usually be differentiated by the history, clinical findings, and liver tests, but liver biopsy is necessary on occasion. Recurrent pyogenic cholangitis should be considered in persons from Asia (and occasionally elsewhere) with intrahepatic biliary stones (particularly in the left ductal system) and with recurrent cholangitis.
Treatment
Common duct stone in a patient with cholelithiasis or cholecystitis is usually treated by endoscopic sphincterotomy and stone extraction followed by laparoscopic cholecystectomy. For the poor-risk patient with cholelithiasis and choledocholithiasis, however, cholecystectomy may be deferred after endoscopic sphincterotomy because the risk of subsequent cholecystitis is low. ERCP with sphincterotomy should be performed before cholecystectomy in patients with gallstones and jaundice (serum total bilirubin > 5 mg/dL), a dilated common bile duct (> 7 mm), or stones in the bile duct seen on ultrasound or CT scans. (Stones may ultimately recur in up to 12% of patients, particularly when the bile duct diameter is ≥ 15 mm or there are brown pigment stones at time of the initial sphincterotomy.) In patients with biliary pancreatitis that resolves rapidly, the stone usually passes into the intestine, and ERCP prior to cholecystectomy is not necessary if an intraoperative cholangiogram is done.
Choledocholithiasis discovered at laparoscopic cholecystectomy may be managed via laparoscopic common duct exploration or, if necessary, conversion to open surgery or by postoperative endoscopic sphincterotomy. Operative findings of choledocholithiasis are palpable stones in the common duct, dilation or thickening of the wall of the common duct, or stones in the gallbladder small enough to pass through the cystic duct. Laparoscopic intraoperative cholangiography should be done at the time of cholecystectomy in patients with liver enzyme elevations but a common duct diameter of less than 5 mm; if a ductal stone is found, the duct is explored. In the postcholecystectomy patient with choledocholithiasis, endoscopic sphincterotomy with stone extraction is preferable to transabdominal surgery. Lithotripsy (endoscopic or external), direct cholangioscopy, or biliary stenting may be a therapeutic consideration for large stones. For the patient with a T tube and common duct stone, the stone may be extracted via the T tube. A properly placed T tube should drain bile at the operating table and continuously thereafter; otherwise, it should be considered blocked or dislocated. The volume of bile drainage varies from 100 mL to 1000 mL daily (average, 200–400 mL). Above-average drainage may be due to obstruction at the ampulla (usually by edema).
Postoperative antibiotics are not administered routinely after biliary tract surgery. Cultures of the bile are always taken at operation. If biliary tract infection was present preoperatively or is apparent at operation, ampicillin (500 mg every 6 hours intravenously) with gentamicin (1.5 mg/kg every 8 hours) and metronidazole (500 mg every 6 hours) or ciprofloxacin (250 mg intravenously every 12 hours) or a third-generation cephalosporin (eg, cefoperazone, 1–2 g intravenous every 12 hours) is administered postoperatively until the results of sensitivity
P.690
tests on culture specimens are available. A T-tube cholangiogram should be done before the tube is removed, usually about 3 weeks after surgery. A small amount of bile frequently leaks from the tube site for a few days.
Urgent ERCP, sphincterotomy, and stone extraction are generally indicated for choledocholithiasis complicated by ascending cholangitis and are preferred to surgery. Before ERCP, liver function should be evaluated thoroughly. Prothrombin time should be restored to normal by parenteral administration of vitamin K (see above). Ciprofloxacin, 250 mg intravenously every 12 hours, penetrates well into bile and is effective treatment for cholangitis. An alternative regimen in severely ill patients is mezlocillin, 3 g intravenously every 4 hours, plus either metronidazole or gentamicin (or both). The dose of metronidazole is 500 mg intravenously every 6 hours (if there has been no prior manipulation of the duct); the dose of gentamicin is 2 mg/kg intravenously as loading dose, plus 1.5 mg/kg every 8 hours adjusted for renal function. Aminoglycosides should not be given for more than a few days because the risk of aminoglycoside nephrotoxicity is increased in patients with cholestasis. Emergent decompression of the bile duct, generally by ERCP, is required for patients who are septic or fail to improve on antibiotics within 12–24 hours. Medical therapy alone is most likely to fail in patients with tachycardia, serum albumin < 3 g/dL, marked hyperbilirubinemia, and prothrombin time > 14 seconds on admission. If sphincterotomy cannot be performed, decompression by a biliary stent or nasobiliary catheter can be done. Once decompression is achieved, antibiotics are generally continued for at least another three days. Elective cholecystectomy can be undertaken after resolution of cholangitis, unless the patient remains unfit for surgery.
Baron TH et al: Endoscopic balloon dilation of the biliary sphincter compared to endoscopic biliary sphincterotomy for removal of common bile duct stones during ERCP: a metaanalysis of randomized, controlled trials. Am J Gastroenterol 2004;99:1455.
DiSario JA et al: Endoscopic balloon dilation compared with sphincterotomy for extraction of bile duct stones. Gastroenterology 2004;127:1291.
Hui CK et al: Outcome of emergency ERCP for acute cholangitis in patients 90 years of age or older. Aliment Pharmacol Ther 2004;19:1153.
Tse F et al: The elective evaluation of patients with suspected choledocholithiasis undergoing laparoscopic cholecystectomy. Gastrointest Endosc 2004;60:437.
BILIARY STRICTURE
Benign biliary strictures are the result of surgical anastomosis or injury in about 95% of cases. The remainder of cases are caused by blunt external injury to the abdomen, pancreatitis, erosion of the duct by a gallstone, or prior endoscopic sphincterotomy.
Signs of injury to the duct may or may not be recognized in the immediate postoperative period. If complete occlusion has occurred, jaundice will develop rapidly; more often, however, a tear has been accidentally made in the duct, and the earliest manifestation of injury may be excessive or prolonged loss of bile from the surgical drains. Bile leakage may predispose to localized infection, which in turn accentuates scar formation and the ultimate development of a fibrous stricture.
Cholangitis is the most common complication of stricture. Typically, the patient experiences episodes of pain, fever, chills, and jaundice within a few weeks to months after cholecystectomy. Physical findings may include jaundice during an attack of cholangitis and right upper quadrant abdominal tenderness.
Serum alkaline phosphatase is usually elevated. Hyperbilirubinemia is variable, fluctuating during exacerbations and usually remaining in the range of 5–10 mg/dL. Blood cultures may be positive during an episode of cholangitis. ERCP or PTC can be valuable in demonstrating the stricture, permitting biopsy and cytologic specimens, and allowing dilation and stent placement, thereby avoiding surgical repair in some cases. Placement of multiple plastic stents may be more effective than placement of a single stent. Metal stents, which cannot be removed endoscopically, are generally avoided in benign strictures. When malignancy cannot be excluded with certainty, additional endoscopic diagnostic approaches may be considered—if available—including endoscopic ultrasonography, intraductal ultrasound, and choledochoscopy (cholangioscopy).
Differentiation from cholangiocarcinoma may ultimately require surgical exploration. Significant hepatocellular disease due to secondary biliary cirrhosis will inevitably occur if a biliary stricture is not treated. Operative treatment of a stricture frequently necessitates performance of an end-to-end ductal repair, choledochojejunostomy, or hepaticojejunostomy to reestablish bile flow into the intestine.
Kim JH et al: Percutaneous transhepatic cholangioscopic treatment of patients with benign bilio-enteric anastomotic strictures. Gastrointest Endosc 2003;58:733.
Lee JH et al: Endoscopic ultrasound and fine-needle aspiration of unexplained bile duct strictures. Am J Gastroenterol 2004; 99:1069.
Morelli J et al: Long-term outcomes for patients with post-liver transplant anastomotic biliary strictures treated by endoscopic stent placement. Gastrointest Endosc 2003;58:374.
PRIMARY SCLEROSING CHOLANGITIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Most common in men aged 20–50 years.
Often associated with ulcerative colitis.
Progressive jaundice, itching, and other features of cholestasis.
Diagnosis based on characteristic cholangiographic findings.
10% risk of cholangiocarcinoma.
P.691
General Considerations
Primary sclerosing cholangitis is an uncommon disease characterized by a diffuse inflammation of the biliary tract leading to fibrosis and strictures of the biliary system. The disease is most common in men aged 20–40, with a prevalence rate in the United States of 21 per 100,000 men and 6 per 100,000 women, and is closely associated with ulcerative colitis (and occasionally Crohn's colitis), which is present in approximately twothirds of patients with primary sclerosing cholangitis; however, clinically significant sclerosing cholangitis develops in only 1–4% of patients with ulcerative colitis. As in ulcerative colitis, smoking is associated with a decreased risk of primary sclerosing cholangitis. Primary sclerosing cholangitis is associated with the histocompatible antigens HLA-B8 and -DR3 or -DR4, suggesting that genetic factors may play an etiologic role. ANCA, with fluorescent staining characteristics and target antigens distinct from those found in patients with Wegener's granulomatosis or vasculitis, are found in 70% of patients. In patients with AIDS, sclerosing cholangitis may result from infections caused by cytomegalovirus, cryptosporidium, or microsporidium. Rarely, a clinical and radiologic picture of sclerosing cholangitis can follow an episode of septic shock.
Clinical Findings
A. SYMPTOMS AND SIGNS
Primary sclerosing cholangitis presents as progressive obstructive jaundice, frequently associated with malaise, pruritus, anorexia, and indigestion. Patients may be diagnosed in the presymptomatic phase because of an elevated alkaline phosphatase level. Complications of chronic cholestasis, such as osteoporosis and malabsorption of fat-soluble vitamins, may occur late in the course. Esophageal varices are most likely in patients with a platelet count below 150,000/mcL, low serum albumin, and advanced histologic stage. In patients with primary sclerosing cholangitis, ulcerative colitis is frequently characterized by rectal sparing and backwash ileitis.
B. DIAGNOSTIC FINDINGS
The diagnosis of primary sclerosing cholangitis has generally been made by endoscopic retrograde cholangiography; increasingly, magnetic resonance cholangiography is being used as a noninvasive diagnostic approach, but it is less sensitive than ERCP for visualizing the intrahepatic ducts. Biliary obstruction by a stone or tumor should be excluded. The disease may be confined to small intrahepatic bile ducts, in which case ERCP is normal and the diagnosis is suggested by liver biopsy. These patients have a longer survival and lower rate of cholangiocarcinoma than patients with involvement of the large ducts. Liver biopsy also allows staging, which is based on the degree of fibrosis. In addition to ANCA, patients may have serum antinuclear, anticardiolipin, and antithyroperoxidase antibodies and rheumatoid factor. Occasional patients have clinical and histologic features of both sclerosing cholangitis and autoimmune hepatitis. Even more rarely, an association with chronic pancreatitis (sclerosing pancreaticocholangitis) is seen, and this entity is often responsive to corticosteroids. In general, the diagnosis of primary sclerosing cholangitis is difficult to make after biliary surgery or intrahepatic artery chemotherapy, which may result in bile duct injury. Primary sclerosing cholangitis must be distinguished from idiopathic adulthood ductopenia, a rare disorder affecting young to middle-aged adults who manifest cholestasis resulting from loss of interlobular and septal bile ducts yet who have a normal cholangiogram.
Complications
Cholangiocarcinoma may complicate the course of primary sclerosing cholangitis in at least 10% of cases and may be difficult to diagnose by cytologic examination or biopsy because of false-negative results. A serum CA 19-9 level > 100 units/mL is suggestive but not diagnostic of cholangiocarcinoma. Patients with ulcerative colitis and primary sclerosing cholangitis are at high risk for colorectal neoplasia.
Treatment
Treatment with corticosteroids and broad-spectrum antimicrobial agents has been used with inconsistent and unpredictable results. Episodes of acute bacterial cholangitis may be treated with ciprofloxacin. Ursodeoxycholic acid in standard doses (10–15 mg/kg/d) may improve liver function test results but does not appear to alter the natural history. However, recent experience suggests that high-dose ursodeoxycholic acid (20 mg/ kg/d) may reduce cholangiographic progression and liver fibrosis. Careful endoscopic evaluation of the biliary tree may permit balloon dilation of localized strictures. If there is a major stricture, short-term placement of a stent may relieve symptoms and improve biochemical abnormalities with sustained improvement after the stent is removed. Repeated balloon dilation of a recurrent dominant bile duct stricture may improve survival. However, long-term stenting may increase the rate of complications such as cholangitis. In patients without cirrhosis, surgical resection of a dominant bile duct stricture may lead to longer survival than endoscopic therapy by decreasing the subsequent risk of cholangiocarcinoma. In patients with ulcerative colitis, primary sclerosing cholangitis is an independent risk factor for the development of colorectal dysplasia and cancer, and strict adherence to a colonoscopic surveillance program
P.692
is advisable. Treatment with ursodeoxycholic acid has been reported to reduce the risk of colorectal dysplasia and carcinoma in patients with ulcerative colitis and primary sclerosing cholangitis. For patients with cirrhosis and clinical decompensation, liver transplantation is the procedure of choice.
Prognosis
Survival of patients with primary sclerosing cholangitis averages 10 years once symptoms appear. Adverse prognostic markers are older age, higher serum bilirubin and AST levels, lower albumin levels, and a history of variceal bleeding. Variceal bleeding is also a risk factor for cholangiocarinoma. Actuarial survival rates with liver transplantation are as high as 85% at 3 years, but rates are much lower once cholangiocarcinoma has developed. Following transplantation, patients have an increased risk of nonanastomotic biliary strictures and—in those with ulcerative colitis—colon cancer. The retransplantation rate is higher than that for primary biliary cirrhosis. Those patients who are unable to undergo liver transplantation will ultimately require high-quality palliative care (see Chapter 5).
Bj ro K et al: Liver transplantation for primary sclerosing cholangitis. J Hepatol 2004;40:570.
Burak K et al: Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol 2004;99:523.
Talwalkar JA et al: Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology 2004;40:39.
Loftus EV Jr et al: PSC-IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 2005;54:91.
Zein CO et al: Prevalence and predictors of esophageal varices in patients with primary sclerosing cholangitis. Hepatology 2004;39:204.
CARCINOMA OF THE BILIARY TRACT
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Presents with obstructive jaundice, usually painless, often with dilated biliary tree.
Pain is more common in gallbladder carcinoma than cholangiocarcinoma.
A Courvoisier (dilated) gallbladder may be detected.
Diagnosis by cholangiography with biopsy and brushings for cytology.
General Considerations
Carcinoma of the gallbladder occurs in approximately 2% of all people operated on for biliary tract disease. It is notoriously insidious, and the diagnosis is often made unexpectedly at surgery. Cholelithiasis (often large, symptomatic stones) is usually present. Other risk factors are chronic infection of the gallbladder with Salmonella typhi, gallbladder polyps over 1 cm in diameter, mucosal calcification of the gallbladder (porcelain gallbladder), and anomalous pancreaticobiliary ductal junction. Genetic factors include K-ras and p53 mutations. Spread of the cancer—by direct extension into the liver or to the peritoneal surface—may be the initial manifestation. The TNM classification includes the following stages: Tis, carcinoma in situ; T1a, tumor invades lamina propria, and T1b, tumor invades muscle layer; T2, tumor invades perimuscular connective tissue, no extension beyond serosa (visceral peritoneum) or into liver; T3, tumor perforates the serosa or directly invades the liver or adjacent organ or structure; T4, tumor invades main portal vein or hepatic artery or invades multiple extrahepatic organs or structures; N1, regional lymph node metastasis; and M1, distant metastasis.
Carcinoma of the bile ducts (cholangiocarcinoma) accounts for 3% of all cancer deaths in the United States, and the incidence and mortality rates have increased dramatically in the past 2 decades. It affects both sexes equally but is more prevalent in persons aged 50–70. Two-thirds arise at the confluence of the hepatic ducts (Klatskin tumors), and one-fourth arise in the distal extrahepatic bile duct; the remainder are intrahepatic. Staging is similar to that for carcinoma of the gallbladder. The frequency of carcinoma in persons with choledochal cysts has been reported to be over 14% at 20 years, and surgical excision is recommended. There is an increased incidence in patients with Caroli's disease, chronic intrahepatic lithiasis, a biliary-enteric anastomosis, and ulcerative colitis, especially those with primary sclerosing cholangitis. In southeast Asia, infection of the bile ducts with helminths (Clonorchis sinensis, Opisthorchis viverrini, Fasciola hepatica) is associated with chronic cholangitis and an increased risk of cholangiocarcinoma.
Clinical Findings
A. SYMPTOMS AND SIGNS
Progressive jaundice is the most common and usually the first sign of obstruction of the extrahepatic biliary system. Pain in the right upper abdomen with radiation into the back is usually present early in the course of gallbladder carcinoma, but this occurs later in the course of bile duct carcinoma. Anorexia and weight loss are common and often associated with fever and chills due to cholangitis. Rarely, hematemesis or melena results from erosion of tumor into a blood vessel (hemobilia). Fistula formation between the biliary system and adjacent organs may also occur. The course is usually one of rapid deterioration, with death occurring within a few months.
Physical examination reveals profound jaundice. A palpable gallbladder with obstructive jaundice usually
P.693
is said to signify malignant disease (Courvoisier's law); however, this clinical generalization has been proved to be accurate only about 50% of the time. Hepatomegaly is usually present and is associated with liver tenderness. Ascites may occur with peritoneal implants. Pruritus and skin excoriations are common.
B. LABORATORY FINDINGS
Laboratory examination reveals predominantly conjugated hyperbilirubinemia, with total serum bilirubin values ranging from 5 to 30 mg/dL. There is usually concomitant elevation of the alkaline phosphatase and serum cholesterol. AST is normal or minimally elevated. An elevated CA 19-9 level may help distinguish cholangiocarcinoma from a benign biliary stricture (in the absence of cholangitis).
C. IMAGING
Ultrasonography and CT may show a gallbladder mass in gallbladder carcinoma and intrahepatic mass or biliary dilation in carcinoma of the bile ducts. CT may also show involved regional lymph nodes and atrophy of a hepatic lobe because of vascular encasement with compensatory hypertrophy of the unaffected lobe. MRI with MRCP and gadolinium enhancement permits visualization of the biliary tree and detection of vascular invasion and obviates the need for angiography and, in some cases, direct cholangiography. Preliminary observations suggest that positron emission tomography can detect cholangiocarcinomas as small as 1 cm, but false-positive results occur. The most helpful diagnostic studies before surgery are either percutaneous transhepatic or endoscopic retrograde cholangiography with biopsy and cytologic specimens, although false-negative biopsy and cytology results are common. Endoscopic ultrasound with fine-needle aspiration of tumors, choledochoscopy, and intraductal ultrasonography also have roles in the diagnosis of cholangiocarcinoma.
Treatment
In young and fit patients, curative surgery may be attempted if the tumor is well localized. The 5-year survival rate for localized carcinoma of the gallbladder (stage 1, T1a, N0, M0) is as high as 80% with laparoscopic cholecystectomy but drops to 15%, even with a more extended open resection, if there is muscular invasion (T1b). If the tumor is unresectable at laparotomy, biliary-enteric bypass (eg, Roux-en-Y hepaticojejunostomy) can be performed. Carcinoma of the bile ducts is curable by surgery in less than 10% of cases. Palliation can be achieved by placement of a selfexpandable metal stent via an endoscopic or percutaneous transhepatic route. Plastic stents are less expensive but more prone to occlude than metal ones; they are suitable for patients expected to survive only a few months. Preliminary experience suggests that photodynamic therapy in combination with stent placement prolongs survival when compared with stent placement alone in patients with nonresectable cholangiocarcinoma. Radiotherapy may relieve pain and contribute to biliary decompression. There is limited response to chemotherapy such as with gemcitabine. In general, the prognosis is poor, with few patients surviving for more than 12 months after surgery. Although cholangiocarcinoma is generally considered to be a contraindication to liver transplantation because of rapid tumor recurrence, an 80% 5-year survival rate has been reported in patients with stage I and II cholangiocarcinoma undergoing chemoradiation and exploratory laparotomy followed by liver transplantation.
For those patients whose disease progresses despite treatment, meticulous efforts at palliative care are essential (see Chapter 5).
Eloubeidi MA et al: Endoscopic ultrasound-guided fine needle aspiration biopsy of suspected cholangiocarcinoma. Clin Gastroenterol Hepatol 2004;2:209.
Gores GJ: Cholangiocarcinoma: pathogenesis, diagnosis, and management. Semin Liver Dis 2004;24:113.
Levy MJ et al: Palliation of malignant extrahepatic biliary obstruction with plastic versus expandable metal stents: an evidence-based approach. Clin Gastroenterol Hepatol 2004;2: 273.
Shaib YH et al: Rising incidence of intrahepatic cholangiocarcinoma in the United States: a true increase? J Hepatol 2004;40:472.
Rajagopalan V et al: Gallbladder and biliary tract carcinoma: a comprehensive review, Parts I and II. Oncology (Huntingt) 2004;18:889 and 1049.
DISEASES OF THE PANCREAS
ACUTE PANCREATITIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Abrupt onset of deep epigastric pain, often with radiation to the back.
History of previous episodes, often related to alcohol intake.
Nausea, vomiting, sweating, weakness.
Abdominal tenderness and distention and fever.
Leukocytosis, elevated serum amylase, elevated serum lipase.
General Considerations
Acute pancreatitis is thought to result from “escape” of activated pancreatic enzymes from acinar cells into surrounding tissues. Most cases are related to biliary tract disease (a passed gallstone, usually < 5 mm in diameter) or heavy alcohol intake. The exact pathogenesis
P.694
is not known but may include edema or obstruction of the ampulla of Vater, resulting in reflux of bile into pancreatic ducts, or direct injury to the acinar cells. Among the numerous other causes or associations are hypercalcemia, hyperlipidemias (chylomicronemia, hypertriglyceridemia, or both), abdominal trauma (including surgery), drugs (including azathioprine, mercaptopurine, asparaginase, pentamidine, didanosine, valproic acid, tetracyclines, estrogen and tamoxifen [by raising serum triglycerides] sulfonamides, thiazides, and possibly glucocorticoids), vasculitis, infections (eg, mumps, cytomegalovirus, Mycobacterium avium complex), peritoneal dialysis, cardiopulmonary bypass, ERCP, and genetic mutations that also predispose to chronic pancreatitis (see below). In patients with pancreas divisum, a congenital anomaly in which the dorsal and ventral pancreatic ducts fail to fuse, acute pancreatitis may result from stenosis of the minor papilla with obstruction to flow from the accessory pancreatic duct, although concomitant mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene have also been reported to account for acute pancreatitis in some patients with pancreas divisum. Acute pancreatitis may also result from anomalous union of the pancreaticobiliary duct. Rarely, acute pancreatitis may be the presenting manifestation of a pancreatic or ampullary neoplasm. Apparently “idiopathic” acute pancreatitis is often caused by occult biliary microlithiasis and may be caused by sphincter of Oddi dysfunction involving the pancreatic duct.
Smoking may increase the risk of alcoholic and idiopathic pancreatitis. Pathologic changes vary from acute edema and cellular infiltration to necrosis of the acinar cells, hemorrhage from necrotic blood vessels, and intraand extrapancreatic fat necrosis. All or part of the pancreas may be involved.
Clinical Findings
A. SYMPTOMS AND SIGNS
Epigastric abdominal pain, generally abrupt in onset, is steady, boring, and severe and often made worse by walking and lying supine and better by sitting and leaning forward. The pain usually radiates into the back but may radiate to the right or left. Nausea and vomiting are usually present. Weakness, sweating, and anxiety are noted in severe attacks. There may be a history of alcohol intake or a heavy meal immediately preceding the attack, or a history of milder similar episodes or biliary colic in the past.
The abdomen is tender mainly in the upper abdomen, most often without guarding, rigidity, or rebound. The abdomen may be distended, and bowel sounds may be absent with associated ileus. Fever of 38.4–39 C, tachycardia, hypotension (even true shock), pallor, and cool clammy skin are often present. Mild jaundice is common. Occasionally, an upper abdominal mass due to the inflamed pancreas or a pseudocyst may be palpated. Acute renal failure (usually prerenal) may occur early in the course of acute pancreatitis.
B. ASSESSMENT OF SEVERITY
Ranson's criteria are generally used in assessing the severity of acute alcoholic pancreatitis on presentation (pancreatitis due to other causes is assessed by similar criteria).
When three or more of the following are present on admission, a severe course complicated by pancreatic necrosis can be predicted with a sensitivity of 60–80%:
Age over 55 years.
White blood cell count over 16,000/mcL.
Blood glucose over 200 mg/dL.
Serum LDH over 350 units/L.
AST over 250 units/L.
Development of the following in the first 48 hours indicates a worsening prognosis:
Hematocrit drop of more than ten percentage points.
BUN rise greater than 5 mg/dL.
Arterial Po2 of less than 60 mm Hg.
Serum calcium of less than 8 mg/dL.
Base deficit over 4 mEq/L.
Estimated fluid sequestration of more than 6 L.
Mortality rates correlate with the number of criteria present:
Number of criteria Mortality rate 0–2 1% 3–4 16% 5–6 40% 7–8 100%
An APACHE II score ≥ 8 also correlates with mortality.
C. LABORATORY FINDINGS
Serum amylase and lipase are elevated—usually more than three times the upper limit of normal—within 24 hours in 90% of cases; their return to normal is variable depending on the severity of disease; lipase remains elevated longer than amylase. Leukocytosis (10,000–30,000/mcL), proteinuria, granular casts, glycosuria (10–20% of cases), hyperglycemia, and elevated serum bilirubin may be present. Blood urea nitrogen and serum alkaline phosphatase may be elevated and coagulation tests abnormal. In patients with clear evidence of acute pancreatitis, a serum ALT level of more than 80 units/L suggests biliary pancreatitis. Decrease in serum calcium may reflect saponification and correlates well with severity of disease. Levels lower than 7 mg/dL (when serum albumin is normal) are associated with tetany and an unfavorable prognosis.
P.695
Patients with acute pancreatitis caused by hypertriglyceridemia generally have fasting triglyceride levels above 1000 mg/dL. An early rise in the hematocrit value above 47% suggests hemoconcentration and is thought to predict severe disease. An elevated C-reactive protein concentration after 48 hours also suggests the development of pancreatic necrosis.
Table 15-7. Severity index for acute pancreatitis. | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
Other tests that offer the possibility of simplicity, rapidity, ease of use, and low cost—including urinary trypsinogen-2, trypsinogen activation peptide, and carboxypeptidase B—are under study, and a urinary dipstick for trypsinogen activation peptide is nearing commercial release. In patients in whom ascites or left pleural effusions develop, fluid amylase content is high. Electrocardiography may show ST–T wave changes.
D. IMAGING
Plain radiographs of the abdomen may show gallstones, a “sentinel loop” (a segment of air-filled small intestine most commonly in the left upper quadrant), the “colon cutoff sign”—a gas-filled segment of transverse colon abruptly ending at the area of pancreatic inflammation—or linear focal atelectasis of the lower lobe of the lungs with or without pleural effusion. CT scan is useful in demonstrating an enlarged pancreas when the diagnosis of pancreatitis is uncertain, in detecting pseudocysts, and in differentiating pancreatitis from other possible intra-abdominal catastrophes. Intravenous contrast-enhanced CT following aggressive volume resuscitation is of particular value after the first 3 days of severe acute pancreatitis to identify areas of necrotizing pancreatitis and to assess prognosis (Table 15-7), although the use of intravenous contrast may increase the risk of complications of pancreatitis and of renal failure and should be avoided when the serum creatinine level is greater than 1.5 mg/dL. MRI appears to be a suitable alternative to CT. The presence of a fluid collection in the pancreas correlates with an increased mortality rate. CT-guided needle aspiration of areas of necrotizing pancreatitis after the third day may disclose infection, usually by enteric organisms, which invariably leads to death unless surgical debridement is performed. The presence of gas bubbles on CT scan implies that infection by gasforming organisms is present. Ultrasonography is less reliable, because the echoes are deflected by the gasdistended small intestine frequently associated with pancreatitis but is the initial imaging study required when pancreatitis is thought to be caused by gallstones. Endoscopic ultrasonography is useful in identifying occult biliary disease (eg, small stones, sludge), which, including microlithiasis, is present in a majority of patients with apparently idiopathic acute pancreatitis. ERCP is generally not indicated after a first attack of acute pancreatitis unless there is associated cholangitis or jaundice, but MRCP or endoscopic ultrasonography can be considered. After repeated attacks of idiopathic acute pancreatitis, aspiration of bile for crystal analysis may confirm the suspicion of microlithiasis, and manometry of the pancreatic duct sphincter may detect sphincter of Oddi dysfunction as a cause of pancreatitis.
Differential Diagnosis
Acute pancreatitis must be differentiated from an acutely perforated duodenal ulcer, acute cholecystitis, acute intestinal obstruction, leaking aortic aneurysm, renal colic, and acute mesenteric vascular insufficiency or thrombosis. Serum amylase may also be elevated in high intestinal obstruction, in gastroenteritis, in mumps not involving the pancreas (salivary amylase), in ectopic pregnancy, after administration of opioids, and after abdominal surgery. Serum lipase may also be elevated in many of these conditions.
Complications
Intravascular volume depletion secondary to leakage of fluids in the pancreatic bed and ileus with fluid-filled loops of bowel may result in prerenal azotemia and even acute tubular necrosis without overt shock. This usually occurs within 24 hours of the onset of acute pancreatitis and lasts 8–9 days. Some patients require peritoneal dialysis or hemodialysis.
As mentioned above, sterile or infected necrotizing pancreatitis may complicate the course of 5–10% of cases and accounts for most of the deaths. The risk of infection does not correlate with the extent of necrosis. Pancreatic necrosis is often associated with fever, leukocytosis and, in some cases, shock and is associated
P.696
with organ failure (eg, pulmonary, renal, gastrointestinal bleeding) in 50% of cases. Because infected pancreatic necrosis is almost always an indication for operative treatment, fine-needle aspiration of necrotic tissue under CT guidance should be performed (if necessary, repeatedly) for Gram stain and culture.
A serious complication of acute pancreatitis is acute respiratory distress syndrome (ARDS); cardiac dysfunction may be superimposed. It usually occurs 3–7 days after the onset of pancreatitis in patients who have required large volumes of fluid and colloid to maintain blood pressure and urinary output. Most patients with ARDS require assisted respiration with positive end-expiratory pressure.
Pancreatic abscess is a suppurative process characterized by rising fever, leukocytosis, and localized tenderness and epigastric mass usually 6 or more weeks into the course of acute pancreatitis. This may be associated with a left-sided pleural effusion or an enlarging spleen secondary to splenic vein thrombosis. In contrast to infected necrosis, the mortality rate is low following drainage.
Pseudocysts, encapsulated fluid collections with high enzyme content, commonly appear in pancreatitis when CT scans are used to monitor the evolution of an acute attack. Pseudocysts that are smaller than 6 cm in diameter often resolve spontaneously. They most commonly are within or adjacent to the pancreas but can present anywhere (eg, mediastinal, retrorectal), by extension along anatomic planes. Pseudocysts are multiple in 14% of cases. Pseudocysts may become secondarily infected, necessitating drainage as for an abscess. Erosion of the inflammatory process into a blood vessel can result in a major hemorrhage into the cyst.
Pancreatic ascites may present after recovery from acute pancreatitis as a gradual increase in abdominal girth and persistent elevation of the serum amylase level in the absence of frank abdominal pain. Marked elevations in the ascitic protein (> 3 g/dL) and amylase (> 1000 units/L) concentrations are typical. The condition results from rupture of the pancreatic duct or drainage of a pseudocyst into the peritoneal cavity.
Rare complications of acute pancreatitis include hemorrhage caused by erosion of a blood vessel to form a pseudoaneurysm and colonic necrosis. Chronic pancreatitis develops in about 10% of cases. Permanent diabetes mellitus and exocrine pancreatic insufficiency occur uncommonly after a single acute episode.
Treatment
A. MANAGEMENT OF ACUTE DISEASE
In most patients, acute pancreatitis is a mild disease that subsides spontaneously within several days. The pancreas is “rested” by a regimen of withholding food and liquids by mouth, bed rest and, in patients with moderately severe pain or ileus and abdominal distention or vomiting, nasogastric suction. Pain is controlled with meperidine, up to 100–150 mg intramuscularly every 3–4 hours as necessary. In those with severe hepatic or renal dysfunction, the dose may need to be reduced. (Morphine has been thought to cause sphincter of Oddi spasm but is probably an acceptable alternative.) Oral intake of fluid and foods can be resumed when the patient is largely free of pain and has bowel sounds (even if the serum amylase is still elevated). Clear liquids are then given first, and gradual advancement to a low-fat diet is prescribed, guided by the patient's tolerance and by the absence of pain. Following recovery from acute biliary pancreatitis, laparoscopic cholecystectomy is generally performed, although in selected cases endoscopic sphincterotomy alone may be done. In patients with recurrent pancreatitis associated with pancreas divisum, insertion of a stent in the minor papilla (or minor papilla sphincterotomy) may reduce the frequency of subsequent attacks, although complications of such therapy are frequent.
In more severe pancreatitis—particularly necrotizing pancreatitis—there may be considerable leakage of fluids, necessitating large amounts of intravenous fluids to maintain intravascular volume. The importance of aggressive intravenous hydration cannot be overemphasized. Calcium gluconate must be given intravenously if there is evidence of hypocalcemia with tetany. Infusions of fresh frozen plasma or serum albumin may be necessary in patients with coagulopathy or hypoalbuminemia. With colloid solutions, there may be an increased risk of developing ARDS. If shock persists after adequate volume replacement (including packed red cells), pressors may be required. For the patient requiring a large volume of parenteral fluids, central venous pressure and blood gases should be monitored at regular intervals. Total parenteral nutrition (including lipids) should be considered in patients who have severe pancreatitis and ileus and will be without oral nutrition for at least 7–10 days. Enteral nutrition via a jejunal feeding tube is preferable but may not be tolerated in some patients with an ileus. Imipenem (500 mg every 8 hours intravenously) and possibly cefuroxime (1.5 g intravenously three times daily, then 250 mg orally twice daily) administered to patients with sterile pancreatic necrosis appear to reduce the risk of pancreatic infection and mortality, but the routine use of antibiotics in the absence of pancreatic necrosis is not indicated. The role of intravenous somatostatin in severe acute pancreatitis is uncertain, but octreotide is thought to have no benefit. There is conflicting evidence as to whether the risk of pancreatitis after ERCP can be reduced by the administration of somatostatin or gabexate mesilate, a protease inhibitor. Lexipafant, an antagonist of platelet-activating factor, appears to be of no benefit.
The patient with severe pancreatitis requires attention in an intensive care unit. Close follow-up of white blood count, hematocrit, serum electrolytes, serum calcium, serum creatinine, blood urea nitrogen, serum AST and lactate dehydrogenase, and arterial blood gases is mandatory. Cultures of blood, urine, sputum, and pleural effusion (if present) and needle aspirations of areas of pancreatic necrosis (with CT guidance) should be obtained.
P.697
B. TREATMENT OF COMPLICATIONS AND FOLLOW-UP
A surgeon should be consulted in all cases of severe acute pancreatitis. If the diagnosis is in doubt and investigations indicate a strong possibility of a serious surgically correctable lesion (eg, perforated peptic ulcer), exploration is indicated. When acute pancreatitis is unexpectedly found on exploratory laparotomy, it is usually wise to close without intervention. If the pancreatitis appears mild and cholelithiasis is present, cholecystectomy or cholecystostomy may be justified. When severe pancreatitis results from choledocholithiasis—particularly if jaundice (serum total bilirubin > 5 mg/dL) or cholangitis is present—ERCP with endoscopic sphincterotomy and stone extraction is indicated. MRCP may be useful in selecting patients for therapeutic ERCP.
Operation may improve survival in patients with necrotizing pancreatitis and clinical deterioration with multiorgan failure or lack of resolution by 4–6 weeks. Surgery is nearly always indicated for infected necrosis. The goal of surgery is to debride necrotic pancreas and surrounding tissue and establish adequate drainage. Outcomes are best if surgery is delayed until the necrosis has organized, usually about 4 weeks after disease onset. In selected cases, nonsurgical drainage of necrotizing pancreatitis under radiologic or endoscopic guidance may be feasible depending on local expertise. Peritoneal lavage has not been shown to improve survival in severe acute pancreatitis, in part because the risk of late septic complications is not reduced.
The development of a pancreatic abscess is an indication for prompt percutaneous or surgical drainage. Chronic pseudocysts require endoscopic, percutaneous catheter, or surgical drainage when infected or associated with persisting pain, pancreatitis, or common duct obstruction. For pancreatic infections, imipenem, 500 mg every 8 hours intravenously, is a good antibiotic because it achieves bactericidal levels in pancreatic tissue for most causative organisms.
Prognosis
Mortality rates for acute pancreatitis have declined from at least 10% to around 5% in the past 20 years, but the mortality rate for severe acute pancreatitis (more than three Ranson criteria) remains at least 20%. Half of deaths occur within the first 2 weeks, usually from multiorgan failure. Later deaths occur because of complications of infected necrosis. Recurrences are common in alcoholic pancreatitis.
Arvanitakis M et al: Computed tomography and magnetic resonance imaging in the assessment of acute pancreatitis. Gastroenterology 2004;126:175.
Isenmann R et al: Prophylactic antibiotic treatment in patients with predicted severe acute pancreatitis: a placebo-controlled, double-blind trial. Gastroenterology 2004;126:997.
Klein SD et al: Pancreas divisum, and evidence-based review: part I, pathophysiology. Gastrointest Endosc 2004;60:419.
Klein SD et al: Pancreas divisum, and evidence-based review: part II, patient selection and treatment. Gastrointest Endosc 2004;60:585.
Mortele KJ et al: A modified CT severity index for evaluating acute pancreatitis: improved correlation with patient outcome. AJR 2004;183:1261.
Swaroop VS et al: Severe acute pancreatitis. JAMA 2004;291: 2865.
Tenner S: Initial management of acute pancreatitis: critical issues during the first 72 hours. Am J Gastroenterol 2004;99:2489.
Whitcomb DC et al: Pancreatic diseases: novel mechanisms and management. Gastroenterol Clin North Am 2004;33:717.
CHRONIC PANCREATITIS
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Chronic or intermittent epigastric pain, steatorrhea, weight loss, abnormal pancreatic imaging.
A mnemonic for the predisposing factors of chronic pancreatitis is TIGAR-O: toxic-metabolic, idiopathic, genetic; autoimmune, recurrent and severe acute pancreatitis, or obstructive.
General Considerations
Chronic pancreatitis occurs most often in patients with alcoholism (70–80% of all cases). The risk of chronic pancreatitis increases with the duration and amount of alcohol consumed, but pancreatitis develops in only 5–10% of heavy drinkers. Ethanol is thought to cause secretion of insoluble pancreatic proteins that calcify and occlude the pancreatic duct. Progressive fibrosis and destruction of functioning glandular tissue then occur, perhaps as a result of repeated episodes of necroinflammation and activation of pancreatic stellate cells. About 2% of patients with hyperparathyroidism develop pancreatitis. In tropical Africa and Asia, tropical pancreatitis, related in part to malnutrition, is the most common cause of chronic pancreatitis. A stricture, stone, or tumor obstructing the pancreas can lead to obstructive chronic pancreatitis. Autoimmune chronic pancreatitis is associated with hypergammaglobulinemia (IgG4 in particular) and often with ANAs and antibodies to carbonic anhydrase IV and is responsive to corticosteroids. About 10–20% of cases are idiopathic. Genetic factors may predispose to chronic pancreatitis in some of these cases. For example, a mutant trypsinogen gene (PRSS1) for hereditary pancreatitis, transmitted as an autosomal dominant trait with variable penetrance, has been identified on chromosome 7. Furthermore, mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene have been identified in as many as 50% of patients with idiopathic chronic
P.698
pancreatitis and no other clinical features of cystic fibrosis. Mutations of the pancreatic secretory trypsin inhibitory gene (PSTI, serine protease inhibitor, SPINK1) and possibly the gene for uridine 5′diphosphate glucuronosyltransferase have also been associated with idiopathic chronic pancreatitis. A useful mnemonic for the predisposing factors to chronic pancreatitis is TIGAR-O: toxic-metabolic, idiopathic, genetic; autoimmune, recurrent and severe acute pancreatitis, or obstructive.
The pathogenesis of chronic pancreatitis may be explained by the SAPE (sentinel acute pancreatitis event) hypothesis by which the first (sentinel) acute pancreatitis event initiates an inflammatory process that results in both injury and later fibrosis. In many cases, chronic pancreatitis is a self-perpetuating disease characterized by chronic pain or recurrent episodes of acute pancreatitis and ultimately by pancreatic exocrine or endocrine insufficiency. After many years, chronic pain may resolve spontaneously or as a result of surgery tailored to the cause of pain. Over 80% of adults develop diabetes 25 years after the clinical onset of chronic pancreatitis.
Clinical Findings
A. SYMPTOMS AND SIGNS
Persistent or recurrent episodes of epigastric and left upper quadrant pain with referral to the upper left lumbar region are typical. Anorexia, nausea, vomiting, constipation, flatulence, and weight loss are common. Abdominal signs during attacks consist chiefly of tenderness over the pancreas, mild muscle guarding, and ileus. Attacks may last only a few hours or as long as 2 weeks; pain may eventually be almost continuous. Steatorrhea (as indicated by bulky, foul, fatty stools) may occur late in the course.
B. LABORATORY FINDINGS
Serum amylase and lipase may be elevated during acute attacks; however, a normal amylase does not exclude the diagnosis. Serum alkaline phosphatase and bilirubin may be elevated owing to compression of the common duct. Glycosuria may be present. Excess fecal fat may be demonstrated on chemical analysis of the stool; pancreatic insufficiency generally is confirmed by response to therapy with pancreatic enzyme supplements; the secretin stimulation test can be used if available, as can detection of decreased fecal chymotrypsin or elastase levels, although the latter tests lack sensitivity and specificity. Vitamin B12 malabsorption is detectable in about 40% of patients, but clinical deficiency of vitamin B12 and fat-soluble vitamins is rare. Accurate diagnostic tests are available for the major trypsinogen gene mutations, but because of uncertainty about the mechanisms linking heterozygous CFTR and PSTI mutations with pancreatitis, genetic testing for mutations in these two genes is not currently recommended.
C. IMAGING
Plain films show calcifications due to pancreaticolithiasis in 30% of affected patients. CT may show calcifications not seen on plain films as well as ductal dilation and heterogeneity or atrophy of the gland. Occasionally, the findings raise suspicion of pancreatic cancer (“tumefactive chronic pancreatitis”). ERCP is the most sensitive imaging study for chronic pancreatitis and may show dilated ducts, intraductal stones, strictures, or pseudocyst, but the results may be normal in patients with so-called minimal change pancreatitis. MRCP and endoscopic ultrasonography (with pancreatic tissue sampling) are promising alternatives to ERCP. Characteristic imaging features of autoimmune chronic pancreatitis include diffuse enlargement of the pancreas and irregular narrowing of the main pancreatic duct.
Complications
Opioid addiction is common. Other frequent complications include often brittle diabetes mellitus, pancreatic pseudocyst or abscess, cholestatic liver enzymes with or without jaundice, common bile duct stricture, steatorrhea, malnutrition, and peptic ulcer. Pancreatic cancer develops in 4% of patients after 20 years; the risk may relate to tobacco and alcohol use. In patients with hereditary pancreatitis, the risk of pancreatic cancer rises after age 50 and reaches 19% by age 70.
Treatment
Correctable coexistent biliary tract disease should be treated surgically.
A. MEDICAL MEASURES
A low-fat diet should be prescribed. Alcohol is forbidden because it frequently precipitates attacks. Narcotics should be avoided if possible. Steatorrhea is treated with pancreatic supplements that are selected on the basis of their high lipase activity. A total dose of 30,000 units of lipase in capsules is given with meals (Table 15-8). Higher doses may be required in some cases. The tablets should be taken at the start of, during, and at the end of a meal. Concurrent administration of H2-receptor antagonists (eg, ranitidine, 150 mg twice daily), a proton pump inhibitor (eg, omeprazole, 20–60 mg daily), or sodium bicarbonate, 650 mg before and after meals, decreases the inactivation of lipase by acid and may thereby further decrease steatorrhea. In selected cases of alcoholic pancreatitis and in cystic fibrosis, entericcoated microencapsulated preparations may offer an advantage. However, in patients with cystic fibrosis, highdose pancreatic enzyme therapy has been associated with strictures of the ascending colon. Pain secondary to idiopathic chronic pancreatitis may be alleviated in some cases by the use of pancreatic enzymes (not enteric-coated) or octreotide, 200 mcg subcutaneously three times daily. Associated diabetes should be treated
P.699
(see Chapter 27). Autoimmune chronic pancreatitis is treated with prednisone 40 mg/d for 1–2 months, followed by a taper of 5 mg every 2–4 weeks.
Table 15-8. Selected pancreatic enzyme preparations. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B. SURGICAL AND ENDOSCOPIC TREATMENT
Endoscopic therapy or surgery may be indicated in chronic pancreatitis to drain persistent pseudocysts, relieve biliary obstruction, treat other complications, attempt to relieve pain, or exclude pancreatic cancer. The objectives of such interventions are to eradicate biliary tract disease, ensure a free flow of bile into the duodenum, and eliminate obstruction of the pancreatic duct. Liver fibrosis may regress after biliary drainage. Distal common bile duct obstruction may be relieved by endoscopic placement of multiple bile duct stents. When obstruction of the duodenal end of the pancreatic duct can be demonstrated by ERCP, dilation or placement of a stent in the duct or resection of the tail of the pancreas with implantation of the distal end of the duct by pancreaticojejunostomy may be successful. When the pancreatic duct is diffusely dilated, anastomosis between the duct after it is split longitudinally and a defunctionalized limb of jejunum (modified Puestow procedure), in some cases combined with resection of the head of the pancreas, is associated with relief of pain in 80% of cases. In advanced cases, subtotal or total pancreatectomy may be considered as a last resort but has variable efficacy and is associated with a high rate of pancreatic insufficiency and diabetes. Perioperative administration of somatostatin or octreotide may reduce the risk of postoperative pancreatic fistulas. Endoscopic or surgical drainage is indicated for symptomatic pseudocysts and, in many cases, those over 6 cm in diameter. Endoscopic ultrasound may facilitate selection of an optimal site for endoscopic drainage. Pancreatic ascites or pancreaticopleural fistulas due to a disrupted pancreatic duct can be managed by endoscopic placement of a stent across the disrupted duct. Fragmentation of stones in the pancreatic duct by lithotripsy and endoscopic removal of stones from the duct, pancreatic sphincterotomy, or pseudocyst drainage may relieve pain in selected patients. For patients with chronic pain and nondilated ducts, a percutaneous celiac plexus nerve block may be considered under either CT or endoscopic ultrasound guidance, with pain relief in approximately 50% of patients.
Prognosis
Chronic pancreatitis is a serious disease that often leads to chronic disability. The prognosis is best in patients with recurrent acute pancreatitis caused by a remediable condition such as cholelithiasis, choledocholithiasis, stenosis of the sphincter of Oddi, or hyperparathyroidism. Medical management of the hyperlipidemia frequently associated with the condition may also prevent recurrent attacks of pancreatitis. In alcoholic pancreatitis, pain relief is most likely when a dilated pancreatic duct can be decompressed. In patients with disease not amenable to decompressive surgery, addiction to narcotics is a frequent outcome of treatment.
Baillie J: Pancreatic pseudocysts (Part I). Gastrointest Endosc 2004;59:873.
Baillie J: Pancreatic pseudocysts (Part II). Gastrointest Endosc 2004;60:105.
Catalano MF et al: Treatment of symptomatic distal common bile duct stenosis secondary to chronic pancreatitis: comparison of single vs. multiple simultaneous stents. Gastrointest Endosc 2004;60:945.
Howes N et al: Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol 2004;2:252.
Kim K et al: Autoimmune chronic pancreatitis. Am J Gastroenterol 2004;99:1605.
Stevens T et al: Pathogenesis of chronic pancreatitis: an evidencebased review of past theories and recent developments. Am J Gastroenterol 2004;99:2256.
Whitcomb DC: Value of genetic testing in the management of pancreatitis. Gut 2004;53:1710.
CARCINOMA OF THE PANCREAS & THE PERIAMPULLARY AREA
![]() ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Obstructive jaundice (may be painless).
Enlarged gallbladder (may be painful).
Upper abdominal pain with radiation to back, weight loss, and thrombophlebitis are usually late manifestations.
P.700
General Considerations
Carcinoma is the most common neoplasm of the pancreas. About 75% are in the head and 25% in the body and tail of the organ. Carcinomas involving the head of the pancreas, the ampulla of Vater, the distal common bile duct, and the duodenum are considered together, because they are usually indistinguishable clinically; of these, carcinomas of the pancreas constitute over 90%. They comprise 2% of all cancers and 5% of cancer deaths. Risk factors include age, obesity, tobacco use, chronic pancreatitis, prior abdominal radiation, and family history. About 7–8% of patients with pancreatic cancer have a family history of pancreatic cancer in a first-degree relative, compared with 0.6% of control subjects. Pancreatic cancer can also occur as part of several hereditary syndromes, including hereditary pancreatitis, familial atypical multiple mole melanoma, Peutz-Jeghers syndrome, ataxiatelangiectasia, familial breast cancer (BRCA-2), and hereditary nonpolyposis colorectal cancer. Neuroendocrine tumors account for 2–5% of pancreatic neoplasms. Cystic neoplasms account for only 1% of pancreatic cancers, but they are important because they are often mistaken for pseudocysts. A cystic neoplasm should be suspected when a cystic lesion in the pancreas is found in the absence of a history of pancreatitis. Whereas serous cystadenomas (which account for 32–39% of cystic pancreatic neoplasms) are benign, mucinous cystic neoplasms (defined by the presence of ovarian stroma) (10–45%), intraductal papillary mucinous neoplasms (21–33%), and solid pseudopapillary neoplasms (< 10%) are premalignant, although their prognoses are better than the prognosis of adenocarcinoma of the pancreas, unless the neoplasm is locally advanced.
Clinical Findings
A. SYMPTOMS AND SIGNS
Pain is present in over 70% of cases and is often vague, diffuse, and located in the epigastrium or left upper quadrant when the lesion is in the tail. Radiation of pain into the back is common and sometimes predominates. Sitting up and leaning forward may afford some relief, and this usually indicates that the lesion has spread beyond the pancreas and is inoperable. Diarrhea, perhaps due to maldigestion, is an occasional early symptom. Migratory thrombophlebitis is a rare sign. Weight loss is a common but late finding and may be associated with depression. Occasionally a patient presents with acute pancreatitis in the absence of an alternative cause. Jaundice is usually due to biliary obstruction by a cancer in the pancreatic head. A palpable gallbladder is also indicative of obstruction by neoplasm (Courvoisier's law), but there are frequent exceptions. A hard, fixed, occasionally tender mass may be present. In advanced cases, a hard periumbilical (Sister Joseph's) nodule may be palpable.
B. LABORATORY FINDINGS
There may be mild anemia. Glycosuria, hyperglycemia, and impaired glucose tolerance or true diabetes mellitus are found in 10–20% of cases. The serum amylase or lipase level is occasionally elevated. Liver function tests may suggest obstructive jaundice. Steatorrhea in the absence of jaundice is uncommon. Occult blood in the stool is suggestive of carcinoma of the ampulla of Vater (the combination of biliary obstruction and bleeding may give the stools a distinctive silver appearance). CA 19-9, with a sensitivity of 70% and a specificity of 87%, has not proved sensitive enough for early detection of pancreatic cancer; increased values are also found in acute and chronic pancreatitis and cholangitis. Point mutations in codon 12 of the K-ras oncogene are found in 70–100%, and inactivation of the tumor suppressor genes P16 on chromosome 9, P53 on chromosome 17, and MADH4 on chromosome 18 are found in 95%, 75%, and 55% of pancreatic cancers, respectively.
C. IMAGING
With carcinoma of the head of the pancreas, the upper gastrointestinal series may show a widening of the duodenal loop, mucosal abnormalities in the duodenum ranging from edema to invasion or ulceration, spasm, or compression. Ultrasound is not reliable because of interference by intestinal gas. Multiphase thin-cut spiral CT scanning is generally the initial diagnostic procedure and detects a mass in over 80% of cases. CT scanning identifies metastases, delineates the extent of the tumor, and allows for percutaneous fineneedle aspiration for cytologic studies and tumor markers. MRI is an alternative to CT scanning. Preliminary experience suggests that positron emission tomography is a sensitive technique for detecting pancreatic cancer and metastases. Selective celiac and superior mesenteric arteriography may demonstrate vessel invasion by tumor, a finding that would interdict attempts at surgical resection, but it is less widely used since the advent of multiphase spiral CT. Endoscopic ultrasonography, if available, is more sensitive than CT scanning for detecting pancreatic cancer and equivalent to CT scanning for determining nodal involvement and resectability. A normal endoscopic ultrasound excludes pancreatic cancer. Endoscopic ultrasonography may also be used to guide fine-needle aspiration for tissue diagnosis and tumor markers. ERCP may clarify an ambiguous CT scan or MRI study by delineating the pancreatic duct system or confirming an ampullary or biliary neoplasm. In patients with bile duct obstruction, preoperative endoscopic placement of a biliary stent does not appear to reduce operative mortality or morbidity. MRCP appears to be at least as sensitive as ERCP in diagnosing pancreatic cancer. In some centers, pancreatoscopy or intraductal ultrasonography can be used to evaluate filling defects in the pancreatic duct and assess resectability of intraductal papillary mucinous tumors. With
P.701
obstruction of the splenic vein, splenomegaly or gastric varices are present, the latter delineated by endoscopy, endoscopic ultrasonography, or angiography.
Cystic neoplasms can be distinguished by their appearance on CT, endoscopic ultrasonography, and ERCP and features of the cyst fluid on gross and cytologic analysis. For example, serious cystadenomas may have a central scar or honeycomb appearance; mucinous cystadenomas are unilocular or multilocular and contain mucin-rich fluid with high carcinoembryonic antigen levels; and intraductal papillary mucinous neoplasms are associated with a dilated pancreatic duct and extrusion of gelatinous material from the ampulla. Staging by the TNM classification includes the following definitions: Tis: carcinoma in situ; T1: tumor limited to the pancreas, 2 cm or less in greatest dimension; T2: tumor limited to the pancreas, more than 2 cm in greatest dimension; T3: tumor extends beyond the pancreas but without involvement of the celiac axis or the superior mesenteric artery; T4, tumor involves the celiac axis or the superior mesenteric artery (unresectable primary tumor).
Treatment
Abdominal exploration is usually necessary when cytologic diagnosis cannot be made or if resection is to be attempted, which includes about 30% of patients. In a patient with a localized mass in the head of the pancreas and without jaundice, laparoscopy may detect tiny peritoneal or liver metastases and thereby avoid resection in 4–13% of patients. Radical pancreaticoduodenal (Whipple) resection is indicated for lesions strictly limited to the head of the pancreas, periampullary zone, and duodenum (T1, N0, M0). Five-year survival rates are 20–25% in this group and as high as 40% in those with negative resection margins and without lymph node involvement. The best results are achieved at centers that specialize in the multidisciplinary treatment of pancreatic cancer. Adjuvant fluorouracil-based chemotherapy or gemcitabine appear to be of benefit, but the value of adjuvant radiation therapy is uncertain. When resection is not feasible, endoscopic stenting of the bile duct or cholecystojejunostomy is performed to relieve jaundice. A gastrojejunostomy is also done if duodenal obstruction is expected to develop later; alternatively, endoscopic placement of a self-expandable duodenal stent may be feasible. Combined irradiation and chemotherapy may be used for palliation of unresectable cancer confined to the pancreas. Chemotherapy has been disappointing in metastatic pancreatic cancer, although improved response rates have been reported with gemcitabine. Novel small molecules, including fluoropyrimidines, nucleoside cytidine analogues, and topoisomerase inhibitors, are under study. Celiac plexus nerve block or thoracoscopic splanchnicectomy may improve pain control. Photodynamic therapy is under study.
Surgical resection is indicated for all mucinous cystic neoplasms, symptomatic serous cystadenomas, and cystic tumors that remain undefined after spiral CT, endoscopic ultrasound, and diagnostic aspiration. In the absence of locally advanced disease, survival is higher than for adenocarcinoma. Endoscopic resection or ablation may be feasible for ampullary adenomas.
Prognosis
Carcinoma of the pancreas, especially in the body or tail, has a poor prognosis. Reported 5-year survival rates range from 2% to 5%. Lesions of the ampulla have a better prognosis, with reported 5-year survival rates of 20–40% after resection; jaundice and lymph node involvement are adverse prognostic factors. In carefully selected patients, resection of cancer of the pancreatic head is feasible and results in reasonable survival. In persons with a family history of pancreatic cancer, screening with spiral CT and endoscopic ultrasonography should be considered beginning 10 years before the age at which pancreatic cancer was diagnosed in a family member.
For those patients whose disease progresses despite treatment, meticulous efforts at palliative care are essential (see Chapter 5).
Bettschart V et al: Presentation, treatment and outcome in patients with ampullary tumours. Br J Surg 2004;91:1600.
Brugge WR et al: Cystic neoplasms of the pancreas. N Engl J Med 2004;351:1218.
DeWitt J et al: Comparison of endoscopic ultrasonography and multidetector computed tomography for detecting and staging pancreatic cancer. Ann Intern Med 2004;141:753.
Levy MJ et al: Evaluation and management of cystic pancreatic tumors: emphasis on the role of EUS FNA. Clin Gastroenterol Hepatol 2004;2:639.
Li D et al: Pancreatic cancer. Lancet 2004;363:1049.
Reddy RP et al: Pancreatic cystic neoplasm defined by ovarian stroma: demographics, clinical features, and prevalence of cancer. Clin Gastroenterol Hepatol 2004;2:1026.
Takhar AS et al: Recent developments in diagnosis of pancreatic cancer. BMJ 2004;329:668.
EAN: 2147483647
Pages: 71
- ERP Systems Impact on Organizations
- Challenging the Unpredictable: Changeable Order Management Systems
- ERP System Acquisition: A Process Model and Results From an Austrian Survey
- Enterprise Application Integration: New Solutions for a Solved Problem or a Challenging Research Field?
- The Effects of an Enterprise Resource Planning System (ERP) Implementation on Job Characteristics – A Study using the Hackman and Oldham Job Characteristics Model