3 - Endocrinology, Metabolism, and Diabetes
Editors: Schrier, Robert W.
Title: Internal Medicine Casebook, The: Real Patients, Real Answers, 3rd Edition
Copyright 2007 Lippincott Williams & Wilkins
> Table of Contents > Chapter 3 - Endocrinology, Metabolism, and Diabetes
function show_scrollbar() {}
Chapter 3
Endocrinology, Metabolism, and Diabetes
Irene E. Schauer
Jane E.B. Reusch
P.71
Adrenal Insufficiency
What are the general categories of adrenocortical insufficiency?
Can you explain why thyroid function tests should be evaluated in a patient with primary adrenal failure?
What are the characteristic signs and symptoms of acute and chronic adrenal insufficiency?
What criteria are used to make the diagnosis of adrenal insufficiency?
What are the considerations in deciding on long-term replacement therapy for Addison's disease?
What other metabolic abnormalities may occur in association with adrenal insufficiency?
What are the events that take place in the regulation of cortisol secretion by the hypothalamic pituitary adrenal axis?
What are the specific causes of primary and secondary adrenal failure?
Discussion
What are the general categories of adrenocortical insufficiency?
Adrenocortical insufficiency results primarily from deficient cortisol production and in some cases deficient aldosterone and androgen production by the adrenal gland. Because the adrenal cortex is normally stimulated by pituitary adrenocorticotropic hormone (ACTH; corticotropin), cortisol deficiency may result from adrenal disease (primary adrenal insufficiency or Addison's disease) or from pituitary or hypothalamic disease with ACTH deficiency (secondary adrenal insufficiency).
Can you explain why thyroid function tests should be evaluated in a patient with primary adrenal failure?
The association between autoimmune thyroiditis and autoimmune adrenal disease is well recognized. In general, patients with Addison's disease are afflicted more frequently with Hashimoto's thyroiditis than with Graves' disease. Approximately 50% or more of affected patients have high titers of thyroid antimicrosomal antibodies, although these patients often have no thyroid-related symptoms. Graves' hyperthyroidism can occur in association with primary adrenal failure. The association between thyroid failure and adrenal failure can also reflect hypopituitarism, with a consequent deficiency of both ACTH and thyroid-stimulating hormone (TSH). Therefore, abnormal results from thyroid function tests have been seen in the settings of both primary and secondary hypoadrenalism, making thyroid function tests an important component of the evaluation of a patient with primary or secondary adrenal failure.
What are the characteristic signs and symptoms of acute and chronic adrenal insufficiency?
Acute adrenal insufficiency is a potentially fatal medical emergency, and the clinical features include nausea, fever, and shock, progressing to
P.72
diarrhea, muscular weakness, increased and then decreased body temperature, hypoglycemia, hyponatremia, and hyperkalemia.The cardinal signs of chronic adrenal insufficiency are weakness, fatigue, and anorexia, along with gastrointestinal complaints of nausea, vomiting, diarrhea, and vague abdominal pain. Other symptoms include salt craving (20% of the patients) and muscle cramps. Physical findings may comprise weight loss, hyperpigmentation, hypotension, and vitiligo. The ear cartilage may calcify in patients with long-standing adrenal insufficiency.
What criteria are used to make the diagnosis of adrenal insufficiency?
The diagnosis of adrenocortical insufficiency is based primarily on the plasma cortisol determinations made during the rapid ACTH stimulation test (Cortrosyn test). Any screening tests for adrenal insufficiency must include determination of a basal level of cortisol and ACTH, together with a rapid ACTH stimulation test. This test is performed by administering 25 units (0.25 mg) of synthetic ACTH intravenously/intramuscularly (IV or IM) and measuring the response of cortisol and aldosterone. It is performed to assess initially whether the adrenals can respond to exogenous ACTH. A clearly normal response excludes the possibility of primary and chronic, but not acute, secondary adrenal failure. For the cortisol response to be normal, the cortisol level after ACTH administration should be at least 18 ng/dL and increased by at least 9 ng/dL above the basal levels. Normally, the aldosterone levels parallel the cortisol levels, with an increase of at least 14 ng/dL above the basal levels. Patients with Addison's disease exhibit very low cortisol levels and a clearly elevated ACTH level, whereas the levels of both tend to be low in patients with hypopituitarism. In the classic situation, the response of aldosterone to ACTH is absent in patients with primary adrenal failure, whereas it is preserved in patients with secondary adrenal failure. The measurement of aldosterone is not routine but can add diagnostic information for primary adrenal failure. A low-dose (1 g cortrosyn) cortrosyn stimulation test is also available and may be more sensitive when appropriate cutoff values are used. However, additional technical difficulties in cortrosyn administration and timing of blood tests have prevented this test from becoming routinely accepted.
What are the considerations in deciding on long-term replacement therapy for Addison's disease?
Long-term replacement therapy in patients with Addison's disease involves the oral administration of a cortisone preparation in physiologic replacement doses. Usually, two thirds of the total dose is given in the morning and the remainder is given in the evening to mimic the normal circadian secretion of cortisol. Cortisone acetate can be taken as a dose of 25 mg in the morning and 12.5 mg in the evening. Alternatively, hydrocortisone can be taken in a dosage of 30 to 40 mg per day. However, because cortisone must be converted to hydrocortisone in the body, hydrocortisone is considered the more physiologic agent. Despite this, prednisone (5 7.5 mg per day) is frequently prescribed for long-term replacement because it costs less than hydrocortisone. The side effects from the excessive administration of the above
P.73
glucocorticoids include increased appetite, weight gain, insomnia, edema, and hypertension.Mineralocorticoid replacement (fluorohydrocortisone therapy) is necessary in patients with primary Addison's disease, although the exact replacement dose must be titrated to the patient's response. Dramatic fluid retention may occur with the initial treatment, but this subsides once the dose is adjusted.
What other metabolic abnormalities may occur in association with adrenal insufficiency?
Hyperkalemia occurs frequently in patients with primary adrenal failure (approximately 64%). This is largely due to renal tubular absorption of potassium at the expense of sodium stemming from the mineralocorticoid deficiency. In addition, glucocorticoids help in maintaining the function of the sodium pump and the normal gradient between the intracellular and extracellular concentrations of sodium and potassium. Without cortisol, this gradient is not maintained, so that potassium moves out of the cell and sodium moves into the cell, thereby resulting in hyperkalemia. Of note, in patients with secondary (pituitary) adrenal insufficiency, the mineralocorticoid axis is intact and hyperkalemia, arising from the second mechanism only, is mild or absent.
Hypoglycemia occurs infrequently, and primarily in patients with Addison's disease who have fasted for any period. It is due to defective gluconeogenesis.
A mild acidosis may eventuate in patients with mineralocorticoid deficiency because of the decreased secretion of ammonia and hydrogen ions.
Circulating levels of antidiuretic hormone (ADH) may increase and contribute to the hyponatremia. The excessive loss of sodium by the renal tubules leads to an increased water loss. This is counterbalanced by an increase in the ADH levels, which tends to cause water retention. The low cardiac output and hypovolemia also serve as stimuli for ADH release.
The inability to excrete a water load was once used as a diagnostic test for Addison's disease. This phenomenon is primarily caused by glucocorticoid deficiency, even in the presence of euvolemia. A bolus of cortisol completely reverses the effect and a water diuresis ensues, but this also involves the interplay of other factors, such as an improvement in cardiac output, an increase in the effective circulating volume, an increase in the glomerular filtration rate, a reduction in ADH levels, and direct effects on the renal tubule.
Peripheral eosinophilia is a common finding in the setting of primary adrenal insufficiency.
What are the events that take place in the regulation of cortisol secretion by the hypothalamic pituitary adrenal axis?
Adrenocortical cell growth and steroid secretion are primarily controlled by the pituitary hormone ACTH. The secretory regulation of the hypothalamic pituitary adrenal axis involves the release of corticotropin-releasing hormone (CRH) by the hypothalamus into the hypophyseal portal system. This hormone causes the pituitary secretion of ACTH, which is transported by the peripheral circulation to the adrenal glands, where it is bound by
P.74
specific receptors and triggers steroid synthesis and secretion. Cortisol inhibits both CRH and ACTH release, whereas ACTH has a negative feedback effect on CRH release. Hormonal and neural input from higher brain centers stimulates or inhibits CRH synthesis and secretion in a 24-hour cycle, which causes both ACTH and cortisol secretion to exhibit a circadian rhythm. The circadian rhythm can be overcome by stress, however, leading to chronic cortisol synthesis. Cortisol circulates bound to cortisol-binding globulin (transcortin) and the free cortisol enters a cell and interacts with a specific receptor to exert its physiologic effects.What are the specific causes of primary and secondary adrenal failure?
Primary adrenal insufficiency (Addison's disease) is most commonly caused by idiopathic adrenal atrophy stemming from autoimmune destruction (68%), tuberculosis (17%), or some other etiology (15%). Ninety percent of the gland must have been destroyed before Addison's disease becomes apparent. Less common causes of adrenal insufficiency include other granulomatous diseases, such as histoplasmosis and sarcoidosis, or infiltrative diseases, such as amyloidosis, hemochromatosis, metastatic tumor, and adrenal leukodystrophy, as well as chronic anticoagulation and bilateral adrenal hemorrhage. Gram-negative septicemia, bilateral adrenalectomy, abdominal irradiation, adrenal vein thrombosis, adrenal artery embolus, and adrenolytic drugs are also rare causes of adrenal failure.
Adrenal insufficiency is found in some patients with acquired immunodeficiency syndrome (AIDS). The main presentation of adrenal insufficiency in AIDS is fatigue; electrolyte abnormalities are uncommon. Development of adrenal insufficiency in patients with at least one AIDS-defining disease is associated with poor prognosis.
Secondary adrenal insufficiency is commonly caused by iatrogenic corticosteroid therapy, which suppresses CRH and ACTH secretion and results in adrenal atrophy. Other, less common causes include pituitary and hypothalamic tumors, irradiation, trauma, pituitary necrosis, and lymphocytic hypophysitis surgical procedures.
Case
A 60-year-old man is hospitalized because of severe nausea, vomiting, and diarrhea of 4 days' duration. He admits to having experienced mild increasing fatigue and malaise for the last 6 months plus poor appetite, frequent abdominal cramps, and a 20-lb (9-kg) weight loss over the last 4 months. He feels dizzy in the morning and lightheaded after standing for more than an hour. He notes that he tends to take a nap in the late afternoon. Four days before presentation, abdominal cramps, vomiting, and diarrhea developed. He denies any skin changes and prolonged sun exposure. He admits to a decline in sexual desire. He has no history of hypertension, diabetes, asthma, or tuberculosis, and takes no medications.
Physical examination reveals a very tanned man, who appears acutely ill and somewhat dehydrated. He weighs 63 kg. His supine blood pressure (BP) is 106/68 mm Hg and his supine pulse is 90 beats per minute; his standing BP is 80/50 mm Hg and his standing pulse is 104 beats per minute.
P.75
His skin shows decreased turgor. His face, hands, extensor surfaces, chest, and back are notably tanned. The findings from the head, eyes, ear, nose, and throat examination are normal, except for the presence of hardened earlobes. No heart abnormalities are noted and his lungs are clear. Abdominal examination reveals the presence of diffuse tenderness, but no rebound or localized tenderness. The bowel sounds are hyperactive. There is decreased axillary hair. His testes are normal and central nervous system findings are unremarkable.
The following laboratory data are obtained: hemoglobin (Hgb), 10.6 g, normochromic normocytic anemia; white blood cell (WBC) count, 6,600 cells/mm3; sodium, 128 mEq/L; potassium, 5.9 mEq/L; creatinine, 2.0 mg/dL; bicarbonate (HCO3-), 20 mEq/L; chloride, 96 mEq/L; blood urea nitrogen (BUN), 39 mg/dL; and calcium, 11.1 mg/dL.
The chest radiographic study findings are normal and the abdominal radiographic study shows a normal gas pattern, but bilateral adrenal calcification. His electrocardiogram (ECG) is normal.
Seven months later, the patient becomes severely fatigued and weak and complains of cold intolerance, dry skin, somnolence, and constipation. Physical examination at that time reveals a pale patient, with a supine BP of 110/60 mm Hg and supine pulse of 64 per minute. He weighs 72 kg. His skin is dry and warm and exhibits decreased turgor. Periorbital freckling and vitiligo are present, as well as mild, diffuse thyromegaly. Neurologic examination reveals generalized muscle weakness and decreased deep tendon reflexes symmetrically.
Laboratory data are as follows: WBC, 6,900 cells/mm3 with normal differential; serum sodium, 135 mEq/L; potassium, 4.7 mEq/L; chloride, 99 mEq/L; HCO3-, 24.8 mEq/L; glucose, 78 mg/dL; creatinine, 1.0 mg/dL; and BUN, 18 mg/dL. Thyroid function tests reveal the following findings: serum thyroxine (T4), 3.2 g/dL (normal, 4 to 12 g/dL); triiodothyronine (T3) resin uptake, 20% (normal, 25% to 35%); and TSH, 16 U/mL (normal, 0.55.0 U/mL). The test result for antimicrosomal antibodies is positive, with a value of 1:50,000.
What is the most likely diagnosis in this patient?
What would be the first step in the diagnostic evaluation of this patient?
On the basis of the findings from the initial diagnostic evaluation, what is the diagnosis in this patient?
What would you recommend as an initial therapy?
How would you treat this patient's hypercalcemia?
What additional abnormalities may be seen in association with Addison's disease?
On the basis of the findings when the patient is seen 7 months later, what kind of thyroid disease does he have?
What is the most important advice to give this patient?
Case Discussion
What is the most likely diagnosis in this patient?
The most likely diagnosis in this patient is acute adrenal insufficiency resulting from either primary or secondary adrenal failure. This patient illustrates the
P.76
nonspecific nature of symptoms in the setting of chronic adrenal insufficiency, even though he exhibits the classic history and findings.This patient's hyperpigmentation and electrolyte changes suggest primary adrenal failure. The hyperkalemia and hyponatremia are due to mineralocorticoid deficiency, often seen in the setting of primary adrenal failure. Because ACTH is not the predominant regulator of aldosterone secretion, electrolyte abnormalities are less common in patients with secondary adrenal failure.
Adrenal crisis occurs when a stressful situation brings about decompensation. The nature of the stress may range from mild (e.g., the flu) to severe (e.g., trauma or surgery). Adrenal decompensation is marked by dehydration, hypovolemia, profound hypotension, hyponatremia, hyperkalemia, hypoglycemia, and hypothermia. Classic renal failure can mimic several aspects of chronic adrenal failure, including fatigue, malaise, anorexia, hyponatremia, and hyperkalemia. In this patient, the BUN and creatinine abnormalities are more indicative of prerenal azotemia than of acute renal failure.
Decreased libido, which is common in patients with hypopituitarism, can also be seen in patients with Addison's disease and is due to the debilitating nature of the illness, the associated primary gonadal failure, and possibly the decreased adrenal androgens.
Calcification of the auricular and costal cartilage is uncommon in patients with Addison's disease, but can occur incidentally. A lack of axillary hair is actually a more common finding in female patients. The amount of pubic hair may also be diminished.
What would be the first step in the diagnostic evaluation of this patient?
The ACTH stimulation test should be performed initially to assess whether the adrenal glands can respond to exogenous ACTH by increasing the levels of cortisol and aldosterone. Simultaneously, the plasma ACTH level should be measured because patients with Addison's disease have very low cortisol levels but elevated ACTH levels. It is critical to have the laboratory process the samples correctly (check with your laboratory to determine the appropriate process for blood collection). Adrenal autoantibody testing is now available and has a 70% sensitivity. In addition, because of the abdominal radiographic finding of adrenal calcification, a purified protein derivative (PPD) skin test should be performed to assess for tuberculosis.
An ACTH stimulation test reveals a basal cortisol level of 2.8 g/dL, which is then 2.8 g/dL at 30 minutes and 3.0 g/dL at 60 minutes. The aldosterone level is 2.5 ng/mL at 0 minutes, 2.5 ng/mL at 30 minutes, and 3.1 ng/mL at 60 minutes (normal values cortisol, 9 to 25 g/dL a.m. fasting, and 2 to 16 g/dL p.m. fasting; aldosterone, normal salt upright: men, 6 to 22 ng/dL; women, 4 to 31 ng/dL). The plasma ACTH level is found to be 779 pg/mL (normal, <580 pg/mL at 8:00 a.m. upright; 526 pg/mL at 8:00 a.m. supine; and <517 pg/mL at 4:00 p.m. supine). The PPD test result is negative.
On the basis of the findings from the initial diagnostic evaluation, what is the diagnosis in this patient?
The results of the ACTH stimulation test in this patient are clearly abnormal, showing subnormal responses to ACTH indicative of adrenal failure. This is the
P.77
classic situation in patients with primary adrenal failure, that is, the response of both cortisol and aldosterone to ACTH is absent; in secondary adrenal failure, the aldosterone response is preserved.The plasma ACTH level is markedly elevated in this patient, and such extreme elevations may be seen in the context of severe stress, such as that caused by surgery, anesthesia, and hypoglycemia. Calcification of the adrenal glands can occur in the setting of tuberculosis, histoplasmosis, and occasionally in autoimmune adrenal disease. Therefore, the cause of this patient's adrenal gland failure is primary adrenal failure, most likely secondary to the autoimmune destruction of the adrenals. The negative PPD result supports a nontuberculous etiology of the primary adrenal failure.
What would you recommend as an initial therapy?
Because the clinical presentation suggests adrenal crisis, therapy should be instituted immediately because adrenal crisis is a life-threatening emergency and any delay in treatment could prove fatal. Such therapy includes the immediate IV administration of a soluble corticosteroid preparation, such as hydrocortisone (100 mg), followed by rapid infusions of glucose and normal saline at a rate of 2 to 4 L per day. For true crisis, large volumes (2 to 3 L) of 0.9% saline solution or 5% dextrose in 0.9% saline should be infused intravenously as quickly as possible.
The glucocorticoids and volume repletion cause the serum potassium levels to decrease. Definitive diagnostic testing should be carried out after the acute therapy has been instituted. Mineralocorticoid therapy should be deferred until the patient can take medication orally.
How would you treat this patient's hypercalcemia?
Because this patient's hypercalcemia is mild, no special treatment other than hydration with normal saline is required. Both hypercalcemia and hypocalcemia have been reported to occur during an adrenal crisis. This may stem from dehydration, but may also be a consequence of the increased absorption of calcium from the gut, due to glucocorticoid deficiency. Occasionally, mild hypercalcemia and hyperparathyroidism may coexist with adrenal failure caused by a pituitary tumor that compromises the function of corticotrophs [multiple endocrine neoplasia type 1 (MEN 1)]. Hypocalcemia may occur in patients whose hypoadrenalism is a part of the autoimmune polyglandular syndrome type I (polyglandular failure).
What additional abnormalities may be seen in association with Addison's disease?
Other abnormalities that may arise in patients with Addison's disease include hypoglycemia, hyperkalemia, high ADH levels, metabolic acidosis, vitiligo, and high levels of antithyroid antibodies. All of these can be a frequent component of the clinical picture in patients with adrenal insufficiency.
On the basis of the findings when the patient is seen 7 months later, what kind of thyroid disease does he have?
The findings are consistent with those of Hashimoto's thyroiditis. Patients with idiopathic Addison's disease are prone to other autoimmune disorders, which may develop before or after adrenal failure is diagnosed. These disorders include Graves' hyperthyroidism, Hashimoto's thyroiditis, pernicious anemia, diabetes, hypoparathyroidism, primary hypogonadism, vitiligo, and moniliasis. Areas of vitiligo
P.78
form in 4% to 6% of the patients with Addison's disease, especially in those whose disease has an autoimmune cause.In this man who has a goiter, low T4 and high TSH levels, and strongly positive antithyroid antibody titers, levothyroxine therapy should be started, but only when adequate steroid replacement has been achieved and after the patient has been on steroid replacement therapy for at least 2 weeks. An adrenal crisis could be precipitated if levothyroxine is given to a patient who is in a hypoadrenal state because of the resulting increased metabolic demands that levothyroxine imposes on the body.
What is the most important advice to give this patient?
In any patient with adrenal insufficiency, it is critical to emphasize the need for increasing the dosage of glucocorticoids during periods of stress or illness, such as colds, flu, diarrhea, infections, trauma, or surgery. Failure to do so might precipitate the rapid development of an acute adrenal crisis. In addition, the patient must be instructed to wear an identification bracelet or carry a card at all times indicating that he has the disease and needs supplemental steroids during stress. This is a crucial life-preserving measure and cannot be overemphasized.
Suggested Readings
Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 2002;288(7):862 871.
Arafah BM. Medical management of hypopituitarism in patients with pituitary adenomas. Pituitary 2002;5:109 117.
Knapp PE, Arum SM, Melby JC. Relative adrenal insufficiency in critical illness: a review of the evidence. Curr Opin Endocrinol Diabetes 2004;11:147 152.
Lindsay JR, Nieman LK. The hypothalamic-pituitary-adrenal axis in pregnancy: challenges in disease detection and treatment. Endocr Rev 2005;26:775 799.
Nieman LK. Dynamic evaluation of adrenal hypofunction. J Endocrinol Invest 2003;26(7 Suppl):74 82.
Schrier RW. Current medical therapy, 2nd ed. New York: Raven Press, 1989.
Wilson JD, Foster DW, eds. Williams' textbook of endocrinology, 8th ed. Philadelphia: WB Saunders, 1992.
Cushing's Syndrome
What is the difference between Cushing's syndrome and Cushing's disease?
What is the most common cause of Cushing's syndrome?
What are the clinical features of Cushing's syndrome?
What are the biologic effects of glucocorticoids?
What are the screening tests used to diagnose Cushing's syndrome?
P.79
Discussion
What is the difference between Cushing's syndrome and Cushing's disease?
Cushing's syndrome refers to the phenotypic and clinical sequelae due to hypercortisolism resulting from any cause. Cushing's disease refers specifically to the hypercortisolism due to an ACTH-secreting pituitary corticotroph adenoma or pituitary corticotroph hyperplasia.
What is the most common cause of Cushing's syndrome?
The widespread use of potent corticosteroids in the practice of clinical medicine, particularly in the treatment of autoimmune, allergic, and pulmonary disorders, has made iatrogenic hypercortisolism the most common cause of Cushing's syndrome. However, once the iatrogenic causes are eliminated, pituitary adenoma (68%) becomes the most common cause.
What are the clinical features of Cushing's syndrome?
Cushing's syndrome is associated with many clinical features. Obesity, found in 94%, is the most common manifestation and weight gain is usually the earliest symptom of Cushing's syndrome. The obesity tends to be central, but fat can also be redistributed to the face (moon facies; 75%), as well as supraclavicular (80%) and dorsocervical areas ( buffalo hump ; 80%). The latter two areas, particularly the supraclavicular fat pad, are more specific findings for Cushing's syndrome.
Skin changes occur in 85% of the patients and arise because cortisol-induced atrophy of the epidermis leads to thinning and a transparent appearance of the skin, facial plethora, easy bruisability, and the formation of striae. The latter are purplish red areas that are depressed below the skin surface, but are wider than the pinkish white striae that appear after pregnancy or weight loss. Wounds heal slowly in these patients and may dehisce. Hyperpigmentation occurs in the setting of the ectopic ACTH syndrome, but is rare in patients with Cushing's disease and should not be found in those with primary adrenal Cushing's syndrome. Acne (40%) is also a symptom and is due to androgen excess; it may be more generalized than what the patient experienced before.
Hirsutism affects 80% of the patients and typically consists of a darkening and coarsening of the hair. Female patients complain of increased growth of hair over the face, upper thighs, abdomen, and breasts. Virilism occurs in approximately 20% of the cases of adrenal carcinoma. Hypertension is a problem in 75% of the patients. Elevated diastolic BP is a classic feature of spontaneous Cushing's syndrome, and it contributes greatly to the morbidity and mortality associated with the disorder. The increased sodium retention also leads to edema (18%). Congestive heart failure (22%) can be aggravated because of the increased BP and fluid load.
Gonadal dysfunction occurs in 75% of the patients. Elevated androgen levels can result in amenorrhea and infertility in 75% of affected premenopausal women. In men, the elevated cortisol level may cause a decrease in libido.
P.80
Muscle weakness arises in 60% of patients, in particular proximal weakness that most often occurs in the lower extremities. This weakness stems from the catabolic effects of glucocorticoids on muscle tissues, steroid-induced myopathy, and possibly electrolyte imbalances. Weakness can be assessed clinically by asking the patient to stand from the chair without assistance of arms. Radiographically detectable osteoporosis is present in most patients with Cushing's syndrome (60%), and back pain is the initial complaint in 58% of the cases. Pathologic fractures are found in the ribs and vertebrae in severe cases. It takes some time for the hypercortisolism to decalcify bone; therefore, Cushing's syndrome due to adrenal carcinoma and some ectopic ACTH cases is not present long enough to cause osteoporosis.
Psychological disturbances can arise in 40% of patients. These complaints range from mild symptoms, such as emotional lability, increased irritability, anxiety, insomnia, euphoria, poor concentration, poor memory, and mild depression, to severe symptoms, which include frank psychosis associated with delusions or hallucinations, paranoia, severe depression, and even suicidal behavior.
Renal calculi form in 15% of patients as a result of glucocorticoid-induced hypercalcemia. Renal colic may be the presenting symptom of Cushing's syndrome. Thirst and polyuria are seen in 10% of patients. The thirst is due to glucocorticoid-induced hyperglycemia [or worsening of existing diabetes mellitus (DM)] that causes an osmotic (glucose) diuresis. Diabetic ketoacidosis (DKA) and diabetic microvascular complications are rare in the diabetes seen with Cushing's syndrome.
What are the biologic effects of glucocorticoids?
From a molecular perspective, glucocorticoid hormones enter the cell by diffusion and activate specific gene transcription by binding to the nuclear glucocorticoid receptor. The glucocorticoid receptor is therefore a conditional transactivator that influences the rate of RNA polymerase II transcription initiation by binding to specific short DNA sequence elements (glucocorticoid response elements) in the promoter regulatory region of the various target genes. Although this is the best-established pathway of glucocorticoid action, other mechanisms that mediate the rapid effects of glucocorticoids, such as the fast-feedback inhibition of ACTH secretion and possibly modulation of the -aminobutyric acid receptor, must also exist.
In terms of their effects on metabolism, glucocorticoids accelerate hepatic gluconeogenesis by stimulating phosphoenolpyruvate carboxykinase and glucose-6-phosphatase activity, and induce a permissive effect in other gluconeogenic hormones (glucagon and catecholamines). Glucocorticoids also enhance hepatic glycogen synthesis and storage and inhibit glycogen breakdown. In muscles, glucocorticoids inhibit amino acid uptake and protein synthesis and stimulate protein breakdown as well as the release of amino acids, lactate, free fatty acids (FFAs), and glycerol. In adipose tissue, glucocorticoids primarily accelerate lipolysis, with a resultant release in the formation of glycerol and FFAs. Although glucocorticoids are lipolytic, an increased central fat deposition is a
P.81
classic feature of Cushing's syndrome. The steroid-induced increase in appetite and hyperinsulinemia may account for this, but the basis for this abnormal fat deposition in the setting of hypercortisolism remains unknown.What are the screening tests used to diagnose Cushing's syndrome?
A key aspect of the initial workup in a patient with suspected Cushing's syndrome is to distinguish true hypercortisolism from obesity, depression, or alcoholism, or a combination of these, because many clinical and laboratory features of these disorders display significant overlap. A key aid in establishing the clinical diagnosis of hypercortisolism is examining the patient's sequential photographs that span several years. Once Cushing's syndrome is suspected on clinical grounds, the overnight 1-mg dexamethasone suppression test (DST) and the 24-hour urinary free cortisol (UFC) determination are used as screening tests. If the results of the overnight 1-mg DST are normal (8 a.m. plasma cortisol <2 g/dL after the administration of 1 mg of dexamethasone at 11 p.m. the night before), the diagnosis is very unlikely. If the results of the UFC test are also normal (i.e., <90 to 100 g per day), Cushing's syndrome is effectively excluded. A third, recently available, but not yet widely accepted screening test is the late night salivary cortisol. This test takes advantage of the loss of normal circadian variation in cortisol level in Cushing's disease by measuring cortisol at a time when it is normally virtually absent. Several situations can cause false-positive results for the screening DST, including acute and chronic illness, obesity, high-estrogen states, certain drugs (phenytoin and phenobarbital), alcoholism, anorexia, renal failure, and depression. However, in the setting of obesity, high-estrogen states, and certain drugs, the results of a 24-hour UFC are normal. In the other situations, repeated testing is necessary to exclude the diagnosis. Rarely false-negative results can occur, such as in the event of prolonged dexamethasone clearance or episodic hypercortisolism.
Case
A 36-year-old white woman comes to you complaining of fatigue, irritability, depression, and a 30-lb (13.5-kg) weight gain over the last 2 years. She recounts that she has noticed a significant change in her energy level for at least the last 2 years. She states that she has always been a hard worker but 6 months before she had to quit her job as a waitress because of extreme muscle weakness and fatigue. She has also noted increased mood swings, manifested by increased irritability, spontaneous crying episodes, and depression. She reports that her face seems rounder than it was 2 years before. On further questioning, she admits that her menstrual periods have been irregular for the last 2 years. She also admits to drinking a six-pack of beer at least once a week, but denies smoking. She has also noted that she bruises easily. She denies any other medical problems, and states that she is not taking any medications. She specifically denies any glucocorticoid therapy. On asking about her family history, you find out that her mother has adult-onset DM.
Physical examination reveals an obese white woman who is crying while she sits on the examining table, but otherwise she does not appear to be very ill. Her weight is 193 lb (87 kg); height, 5 ft 7 in. (167.5 cm); BP, 165/100 mm Hg; and heart rate, 86 per minute and regular.
P.82
Her face is very round and plethoric compared with that in old photographs. Dorsocervical (buffalo hump) and supraclavicular fat pads are noted. She has mild facial hirsutism, some acne is noted over the face and chest, and wide purple striae are present on the lower abdomen. Her extremities are thin and she has proximal muscle weakness.
The following are the laboratory findings: fasting blood glucose, 180 mg/dL; potassium, 3 mEq/L; HCO3-, 34 mEq/L; liver function tests, all normal; 8 a.m. cortisol, 38 g/dL, which decreases to 32 g/dL after the administration of 1 mg of dexamethasone. The 24-hour UFC level is 876 g.
What is the most likely diagnosis in this patient, and why?
What studies would you perform to establish the anatomic cause of her hypercortisolism?
What is the role of magnetic resonance imaging (MRI) and computed tomographic (CT) scanning of the pituitary and adrenal glands, as well as inferior petrosal sinus sampling, in patients with Cushing's syndrome?
What is the optimal therapeutic approach for this patient?
Why is there a need for steroid therapy in the postoperative period, and sometimes beyond, in patients with Cushing's disease?
Case Discussion
What is the most likely diagnosis in this patient, and why?
Having excluded exogenous glucocorticoid medications in the history, the differential diagnosis list would include (a) pituitary corticotroph adenoma or hyperplasia (Cushing's disease), (b) ectopic ACTH or corticotropin-releasing factor (CRF) syndrome, (c) adrenal adenoma, (d) adrenal cancer, (e) obesity, (f) depression, and (g) alcoholism.
The most frequently encountered dilemma in the differential diagnosis of Cushing's syndrome is the clinical picture consisting of an obese, depressed patient who consumes excessive amounts of alcohol. These patients can display many of the phenotypic features and laboratory findings consistent with hypercortisolism, and yet not have Cushing's syndrome. Therefore, the patient's history of consuming a six-pack of beer per week is of concern because this could produce alcoholic pseudo-Cushing's syndrome. In this disorder, the effects of chronic alcoholism result in central obesity (ascites), a round plethoric face, easy bruising, and some abnormal results from the screening tests for Cushing's syndrome. However, this patient has no abnormal liver function findings and she has physical findings (a marked change in her facial appearance compared with that in old photographs, hypertension, dorsocervical and supraclavicular fat pads, purple abdominal striae, acne, and hirsutism) and laboratory data (hyperglycemia, hypokalemia, an elevated basal cortisol level that does not suppress in response to the 1-mg DST, and an elevated 24-hour UFC) that are all highly consistent with the clinical suspicion of hypercortisolism.
The lack of virilization and the relatively slow (>2 years) onset of the clinical symptoms argue against adrenal carcinoma. In addition, the lack of a smoking history and any hyperpigmentation, together with the slow onset, suggest that
P.83
ectopic ACTH arising from small cell lung carcinoma is unlikely to be the cause. This leaves pituitary adenoma (or hyperplasia), ectopic ACTH or CRF (from a carcinoid, pancreatic islet cell tumor, medullary thyroid carcinoma, or pheochromocytoma), and adrenal adenoma in the differential diagnosis. Given that pituitary adenomas constitute 68% of all noniatrogenic causes of hypercortisolism, this is the most likely diagnosis. However, further workup is required to document the precise source of the elevated cortisol levels in this patient.What studies would you perform to establish the anatomic cause of her hypercortisolism?
Once the diagnosis of hypercortisolism (Cushing's syndrome) has been confirmed by the findings of the clinical evaluation and screening laboratory tests, the combined use of the following diagnostic techniques can establish the diagnosis in almost all instances: determination of a basal plasma ACTH level, a high-dose (8 mg) DST, radiographic imaging, and inferior petrosal sampling (with or without CRF stimulation). By simultaneously measuring the plasma cortisol and ACTH levels the possibility of an adrenal adenoma can be assessed because the autonomous production of glucocorticoids by the adrenal adenoma suppresses ACTH to levels below 20 pg/mL. To differentiate between a pituitary adenoma and the ectopic tumor production of ACTH, several tests need to be performed because many of the laboratory and radiographic results can overlap for these two distinct causes of Cushing's syndrome. For example, the ACTH level can range between 40 and 200 pg/mL in the setting of Cushing's disease and between 100 and 10,000 pg/mL in the setting of ectopic ACTH. In the classic 2-day high-dose DST (2 mg of dexamethasone is given every 6 hours for 2 days, and 24-hour UFC samples are collected the day before and on the second day of dexamethasone administration), patients with pituitary tumors (Cushing's disease) typically exhibit a suppression to less than 50% of baseline values; those with ectopic ACTH or primary adrenal hypercortisolism display little or no reduction. However, some carcinoid tumors that produce ACTH ectopically maintain some degree of negative feedback through the influence of exogenous steroids, and the suppression observed may be equivalent to that seen in patients with pituitary tumors.
The abbreviated high-dose DST involves administering 8 mg of dexamethasone at 11 p.m. the night before and measuring the plasma cortisol level the next morning at 8 a.m. In this test, a suppression below 50% of basal plasma cortisol levels is seen in patients with pituitary tumor, but not in those with ectopic ACTH and primary adrenal cortisolism. This version of the high-dose DST is preferred because it appears to be more specific and does not require two 24-hour urine collections. For a more precise definition of the cause of the disorder, however, specific radiographic procedures must be performed.
What is the role of MRI and CT scanning of the pituitary and adrenal glands, as well as inferior petrosal sinus sampling, in patients with Cushing's syndrome?
The major problem with the CT and MRI evaluation of the pituitary and adrenal glands is that they can detect asymptomatic lesions in up to 15% and 8%, respectively, of the normal population. Because of this incidence of nonspecific radiographic lesions , the clinician must be cautious about basing the diagnosis of pituitary or adrenal Cushing's syndrome on the results of these imaging studies.
P.84
Pituitary adenomas causing Cushing's disease tend to be small (1 to 5 mm; rarely >10 mm), and are therefore detectable by contrast-enhanced CT scanning in as few as 30% to 35% of cases and by gadolinium DTPA-enhanced MRI in 55% of cases. Therefore, because of its better sensitivity, MRI has replaced CT in the assessment of these tumors. Patients whose imaging studies yield negative findings need to undergo inferior petrosal sampling to further document the pituitary anatomic location of the tumor. In addition, as already discussed, even if an abnormality is detected by these imaging methods, this does not constitute unequivocal evidence that the abnormality is responsible for the syndrome. As the resolution of CT and MRI improves, the ability to detect these incidental and clinically silent microadenomas will also increase and further confound the diagnostic workup. An ectopic CRF syndrome could also result in an enlarged pituitary due to corticotroph hyperplasia, and yet the primary disorder may actually be a carcinoid of the lung.
CT, MRI, ultrasonography, and isotope scanning with iodocholesterol can be used to define the nature of adrenal lesions. These procedures are not necessary in patients with ACTH hypersecretion, however. Nevertheless, some physicians use these tests to exclude the presence of a solitary adrenal adenoma or carcinoma, and thereby confirm the presence of bilateral adrenal hyperplasia or nodular adrenal hyperplasia in the setting of pituitary-based disease. These procedures are most useful for localizing adrenal tumors because these tumors must usually be larger than 1.5 cm to cause significant cortisol production and result in Cushing's syndrome. However, as noted previously, because of the 1% to 8% incidence of silent adrenal nodules biochemical testing must be performed with localization studies to ensure that the lesion identified is biologically significant.
To distinguish between the various causes of Cushing's syndrome when conflicting or overlapping data are obtained, bilateral simultaneous inferior petrosal venous sampling (with or without CRF stimulation) can successfully distinguish Cushing's disease from ectopic ACTH secretion and adrenal disease with greater accuracy than any other test. Because ACTH is rapidly metabolized (half-life, 7 to 12 minutes) and is secreted episodically, advantage can be taken of the concentration gradient between the pituitary venous drainage through the inferior petrosal sinus (central) and the peripheral venous values of ACTH to further determine whether an ACTH-producing corticotroph adenoma is present in the pituitary; the inclusion of CRF stimulation makes the test more sensitive. Bilateral and simultaneous inferior petrosal sinus samples are obtained to circumvent the problem of isolated secretory bursts or timing issues if catheters have to be repositioned. Therefore, ACTH samples are obtained from the inferior petrosal sinus, from the jugular bulb, and from other sites (e.g., superior or inferior vena cava), and the findings are compared with those from simultaneously obtained peripheral vein samples. In patients with Cushing's disease, the inferior petrosal sinus/peripheral (IPS : P) ratio of ACTH exceeds 2. In patients with ectopic ACTH, the ratio is less than 2 and selective venous sampling (e.g., of the pulmonary, pancreatic, or intestinal beds) may localize the ectopic tumor. The administration of CRF during bilateral inferior petrosal sinus sampling can increase the diagnostic accuracy of the test by eliciting
P.85
an ACTH response in the few patients with pituitary tumors who do not exhibit a diagnostic IPS : P gradient in the basal samples. Most patients with Cushing's disease have an IPS : P ratio greater than 3 after CRF stimulation, whereas patients with ectopic ACTH or adrenal disease have an IPS : P ratio of ACTH less than 3 after CRF stimulation. Inferior petrosal sinus sampling (with or without CRF stimulation) has not been extensively studied in the context of healthy people, however, and therefore the correct interpretation of the results requires that the patient must be hypercortisolemic at the time of the study so that the response of normal corticotrophs to CRF is suppressed.What is the optimal therapeutic approach for this patient, and why?
Once the tumor has been localized to the pituitary, the next goal is to surgically remove the corticotroph adenoma using the technique of selective transsphenoidal surgery. Because the tumors are small, it requires an experienced neurosurgeon to successfully identify and resect the adenoma. Meticulous exploration of the intrasellar contents is mandatory, and any identified adenoma is selectively removed, leaving the remaining normal pituitary intact. If the tumor cannot be identified, it is necessary to perform larger pituitary resections and, in some cases, a total hypophysectomy may be necessary. Transsphenoidal surgery is successful in approximately 85% of patients with microadenomas (tumor <10 mm), and surgical damage to the normal anterior pituitary is rare. The major side effects of the procedure include transient diabetes insipidus, cerebrospinal fluid leak, sinusitis, and, rarely, postoperative bleeding. All patients with Cushing's disease who are successfully treated with transsphenoidal surgery become adrenally insufficient for variable periods of time and must receive replacement doses of glucocorticoids (see question 5 which follows).
The success rates for transsphenoidal surgery drop drastically (15% to 25%) in the setting of large (>10 mm) tumors, locally invasive tumors, tumors not identified at surgery, and corticotroph hyperplasia. In these instances, adjunctive radiation therapy is usually administered. However, the major problem with radiation therapy is the lag time (6 to 12 months) for it to take effect and the 10% to 20% incidence of hypopituitarism and visual field deficits, even blindness, that may eventuate. A newer option is the more precise stereotactic radiosurgery using the gamma knife or photon knife. Risk of visual complications is largely eliminated, and the risk of pituitary deficiency is reduced.
Why is there a need for steroid therapy in the postoperative period and beyond, in patients with Cushing's disease?
The successful surgical removal of the ACTH-producing pituitary microadenoma eliminates the drive for adrenal glucocorticoid production and renders the patient dependent on the remaining normal corticotrophs. However, because these cells have been suppressed for years by the excess cortisol they are dormant. Therefore, those patients with Cushing's disease who have been successfully treated experience transient (1 to 18 months) adrenocortical insufficiency and require exogenous glucocorticoid support; in those patients not cured by the surgical procedure, the production of excessive amounts of glucocorticoids continues and they do not depend on an exogenous source of steroids.
P.86
Suggested Readings
Felig P, Baxter JD, Broadus AE, et al. eds. Diseases of the anterior pituitary. In: Endocrinology and metabolism, 2nd ed. New York: McGraw-Hill, 1987.
Findling JW, Raff H. Screening and diagnosis of Cushing's syndrome. Endocrinol Metab Clin North Am 2005;34:385 402.
Mansmann G, Lau J, Balk E, et al. The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev 2004;25(2):309 340.
Newell-Price J, Trainer P, Besser M, et al. The diagnosis and differential diagnosis of Cushing's syndrome and pseudo-Cushing's states. Endocr Rev 1998;19:657.
Schuff KG. Issues in the diagnosis of Cushing's syndrome for the primary care physician. Prim Care Office Pract 2003;30:791 799.
Tyrrell JB, Ron DC, Forsham PH. Glucocorticoids and adrenal androgens. In: Greenspan FS, ed. Basic and clinical endocrinology, 3rd ed. Norwalk, CT: Appleton & Lange, 1991.
Xiao XR, Ye LY, Shi LX, et al. Diagnosis and treatment of adrenal tumours: a review of 35 years' experience. Br J Urol 1998;82:199.
Diabetes Mellitus
What are the clinical manifestations of DM?
What are the major types of DM and what are their distinguishing features?
What are the major acute and chronic complications of the disease?
What aspects of the medical history require special emphasis?
What aspects of the physical examination require special attention?
What laboratory tests are essential in the evaluation of the patient with suspected diabetes?
What are the goals of diabetes therapy and what treatment modalities are available? How should these be individualized?
Discussion
What are the clinical manifestations of DM?
DM is a complex metabolic disorder characterized by abnormalities of carbohydrate, lipid, and protein metabolism resulting either from a deficiency of insulin or from target tissue resistance to its cellular metabolic effects. It is the most common endocrine-metabolic disorder and affects an estimated 22 million people in the United States, with the incidence of new cases increasing by more than 700,000 per year.
Diabetes is manifested by the finding of hyperglycemia and the time-dependent development of chronic complications (retinopathy, neuropathy, nephropathy, and accelerated atherosclerosis) resulting from the multiple metabolic derangements. Accordingly, the presenting clinical signs and symptoms can be due to hyperglycemia or the complications of the disease, or both. In general, the major classic symptoms of polydipsia, polyuria, weight loss,
P.87
and fatigue are found in the setting of new-onset diabetes in young patients whose disease is due to insulinopenia. On the other hand, older patients with diabetes may be relatively free of symptoms for a long time. In such patients, the diabetes is first detected either incidentally or because one of its chronic complications is discovered. It is estimated that approximately one third of all the adult cases of diabetes in the United States remain undiagnosed.What are the major types of DM and what are their distinguishing features?
The current classification (according to the National Diabetes Data Group) of DM and other categories of glucose intolerance consists of three clinical classes: (a) DM which includes type 1 diabetes mellitus (T1DM), [previously insulin-dependent diabetes mellitus (IDDM) or juvenile onset diabetes], and type 2 diabetes mellitus (T2DM), [previously non insulin-dependent diabetes mellitus (NIDDM)]; (b) impaired glucose tolerance/impaired fasting glucose; and (c) gestational DM. Of these, T1DM and T2DM represent the largest category and are discussed here in further detail. Impaired glucose tolerance and impaired fasting glucose are defined as an abnormality in glucose levels intermediate between normal and overt diabetes, whereas gestational DM is defined as carbohydrate intolerance with onset or first recognition during pregnancy.
T1DM constitutes approximately 5% to 10% of all cases of diabetes and is due to insulin deficiency resulting from the autoimmune destruction of insulin-producing pancreatic islet cells. Therefore, such patients are prone to ketoacidosis and are absolutely dependent on exogenous insulin to sustain life (hence the term insulin-dependent diabetes). The onset in these patients is relatively abrupt and occurs usually in youth (mean age, 12 years), although it may arise at any age and is often misdiagnosed in adults.
T2DM accounts for approximately 90% to 95% of all cases of diabetes. These patients have a dual impairment of insulin resistance (decreased target organ response to insulin, i.e., decreased glucose transport to muscle or ineffective suppression of hepatic glucose output) and inadequate insulin secretion to compensate for the insulin resistance. The recent obesity explosion, which is related to sedentary lifestyle and increased food intake, has exaggerated insulin resistance in susceptible people and contributed to the diabetes epidemic. Fig. 3-1 illustrates the natural history of the transition from impaired glucose tolerance to overt diabetes. T2DM is now affecting 3% to 6% of the population and occurring in younger people (even including children). There is usually a strong family history of DM in patients developing T2DM in youth.
What are the major acute and chronic complications of the disease?
DKA, hyperglycemic, hyperosmolar, nonketotic coma (HHNKC), and hypoglycemia are the major acute complications of DM. DKA is most commonly a complication of T1DM and is initiated by an absolute or relative insulin deficiency and an increase in counterregulatory hormones (glucagon, epinephrine), leading to the hepatic overproduction of glucose and ketone bodies. HHNKC is characterized by the insidious development of marked hyperglycemia, hyperosmolarity, dehydration, and prerenal azotemia in the
P.88
absence of significant hyperketonemia or acidosis. Finally, hypoglycemia can occur as an acute complication of the therapy of both T1DM and T2DM, and is the most common acute life-threatening complication of diabetes. It is most common with intensive insulin therapy, and recurrent hypoglycemia can induce a condition known as hypoglycemia unawareness, a blunting of the adrenergic and neuroglycopenic signs and symptoms of hypoglycemia. The risk of hypoglycemia unawareness can be minimized and existing unawareness treated by strict avoidance of hypoglycemia.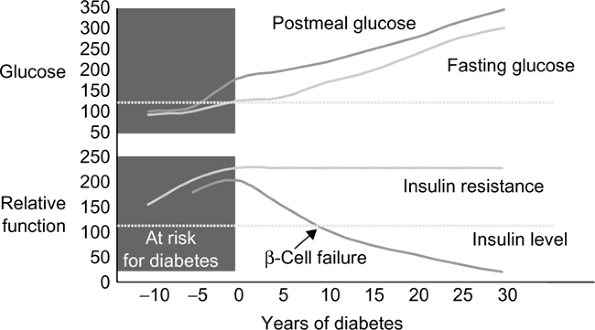
Figure 3-1 Natural history of type 2 diabetes mellitus. Adapted from
Bergenstal RM, Kendall DM, Franz MJ, et al. Management of type 2 diabetes: a systematic approach to meeting the standards of care. II: oral agents, insulin, and management of complications. In: Degroot LJ, Jameson JL, eds. Endocrinology. philadelphia: WB Saunders, 2001:821 835. with permission from Elsevier
.The most common chronic complication of diabetes and the leading cause of death for people with diabetes is cardiovascular (CV) disease. Seventy-seven percent of all hospitalizations and 80% of all mortality in diabetes is secondary to CV disease. Diabetes is an independent risk factor for CV disease. The incidence of CV events is so high in subjects with diabetes that diabetes is considered a CV risk equivalent. CV disease includes myocardial ischemia, stroke, and peripheral vascular disease. People with diabetes also have an increased incidence of heart failure, which will not be addressed in this section, as the pathophysiology is poorly understood. Outcomes after acute myocardial infarction (MI) in people with diabetes are worse than controls, but can be improved with intensive glycemic control in the hospital. Interventional studies demonstrate that lipid lowering significantly decreases mortality and CV events in people with diabetes. In fact, it appears that people with DM may benefit from statins regardless of initial low-density lipoprotein (LDL). Additional large prospective trials demonstrate decreased CV mortality with intensive BP control. There is no compelling data that improvement of glycemic control affects CV morbidity or mortality except in subjects with T1DM.
Microvascular complications (retinopathy, nephropathy and neuropathy) are specific to diabetes and are related directly to poor glycemic control with
P.89
a smaller contribution from hypertension and dyslipidemia. Diabetes is the leading cause of blindness in the United States. By 10 years' duration of diabetes, approximately 90% of individuals will have some degree of retinopathy. Retinopathy is largely a preventable complication of diabetes. Annual ophthalmologic examinations permit identification of individuals with progressive retinopathy. Two large multicenter studies have proved that early intervention at this stage with panretinal photocoagulation can prevent or decrease visual loss. The Diabetes Control and Complications Trial (DCCT) established that tight glycemic control also prevents or delays retinopathy. Macular edema, corneal ulceration, glaucoma, and cataracts are additional ocular complications of diabetes.Diabetes is the leading cause of renal failure/dialysis and transplantations nationwide. Forty percent to 60% of individuals with T1DM and 10% to 30% of individuals with T2DM will develop microalbuminuria, proteinuria, and end-stage renal disease secondary to diabetes. Hypertension and glycemic control are the primary factors that promote progression of nephropathy in people with diabetes. Normalization of BP and glucose dramatically slow the progression from incipient nephropathy (detectable microalbumin) to overt nephropathy. All people with DM should have a BP lower than 130/80 mm Hg, preferably much lower. Aggressive control of hyperglycemia (with intensive therapy) and BP [with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers] has been shown to retard the progression of nephropathy in patients with DM.
Neuropathy is a common complication of diabetes affecting more than 50% of patients with time. The most common form of nerve injury in diabetes is distal symmetric polyneuropathy, which occurs in a stocking-glove distribution; it can be painless or painful. This type of neuropathy increases the risk for traumatic foot injury and amputation. Other forms of neuropathy include: autonomic neuropathy (associated with an increased risk of CV death and hypoglycemia unawareness); mononeuritis multiplex (a vascular occlusion to a single nerve distribution that will typically recover with time); and diabetic amyotrophy (a profound, uncommon demyelinating neuromuscular wasting syndrome).
Diabetes is also associated with impaired blood flow and sensation to the extremities. This leads to a high incidence of mechanical trauma and infectious complications, leading to amputation and hospitalization. Diabetes is the most frequent cause of nontraumatic lower limb amputations. Each year, more than 56,200 amputations are performed among people with diabetes. This complication is largely preventable by appropriate footwear, regular foot examination, and education.
What aspects of the medical history require special emphasis?
A comprehensive medical history in a patient with suspected diabetes should be directed not only toward confirming the diagnosis but should also be used to review the nature of previous treatment programs and diabetes education, family history, the degree of past and recent glycemic control, and history of acute and chronic complications. Patients should also be queried
P.90
about their dietary, weight, and exercise history. Current medications for the management of diabetes, as well as other medications that may affect glycemic control, should be recorded. In addition, the presence, severity, and treatment of the acute and chronic complications of diabetes should be reviewed, including sexual function and dental care. All patients should have a careful history for diabetic health care maintenance documented at each visit. This includes glycemic control, lipid management (LDL <100, or <70 in high risk or known CV disease), BP management (<130/80 mm Hg), eye examination (annual), foot examination (each visit), diet, exercise, and self management (Fig. 3-2).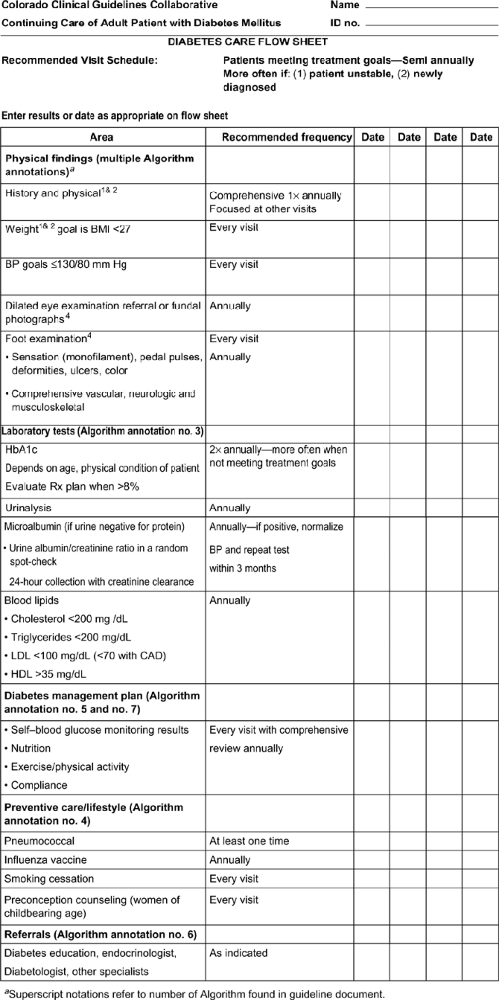
Figure 3-2 Diabetes care flow sheet. BMI, body mass index; BP, blood pressure; LDL, low-density lipoprotein; CAD, coronary artery disease; HDL, high-level lipoprotein.
What aspects of the physical examination require special attention?
The vital signs are critical for patients with diabetes. BP greater than 130/80 mm Hg increases the risk for all complications, resting tachycardia suggests autonomic nervous system dysfunction, and weight gain or loss provides valuable information on severity of illness and adherence to therapy. On physical examination, dentition is important as periodontal disease can impact glycemic control and is a risk factor for atherosclerosis (chronic inflammation). Complete CV examination [including bruits and ankle brachial index (ABI)] and evaluation for edema can detect CV disease and heart failure. Loss of respiratory variation in heart rate is an early warning of autonomic neuropathy. Foot examination, including pulses and monofilament testing, can identify high-risk feet and prevent amputations. The retinal examination [undilated by a primary care physician (PCP)] is not sensitive for detection of retinopathy and needs to be done by an
P.92
ophthalmologist or by using retinal photographs. It may be conducted by the PCP, but the needed formal annual evaluation should also be arranged.What laboratory tests are essential in the evaluation of the patient with suspected diabetes?
The American Diabetes Association (ADA) and regulatory agencies have established standards for laboratory evaluation of diabetes. The diagnosis of diabetes is formally made on the basis of one of the following criteria: (a) fasting glucose 126 mg/dL or more on two occasions, (b) random glucose 200 mg/dL or more on two occasions, or (c) one abnormal reading as above together with symptoms consistent with diabetes (polyuria, nocturia, polydipsia, weight loss, blurred vision). Hemoglobin AIc (HbAIc) is not yet recommended as a diagnostic test because of a lack of standardization of existing assays, but is increasingly being considered by the ADA and regulatory agencies as a potential diagnostic tool.
What are the goals of diabetes therapy and what treatment modalities are available? How should these be individualized?
In general, the goals of diabetes therapy are (a) to alleviate the signs and symptoms of the disease (e.g., polydipsia, polyuria, and nocturia); (b) to prevent the acute complications (i.e., hypoglycemia, DKA, and HHNKC); and (c) to prevent the long-term complications of the disease (i.e., retinopathy, nephropathy, neuropathy, and atherosclerotic CV disease). The DCCT first demonstrated that tight metabolic control of T1DM leads to definite beneficial effects on the rate of complications. This also held true for patients with T2DM in the recently completed United Kingdom Prospective Diabetes Study (UKPDS). Evidence documenting the importance of glycemic control for the prevention of microvascular complication is unequivocal. Therefore, intensive glycemic control is now routine with HbAIc goals of 6.5% to 7%. The limiting factor in such attempts is an increased frequency of hypoglycemic episodes. Recent data from the DCCT follow-up study Epidemiology of Diabetes Interventions and Complications (EDIC) now indicate that intensive control of blood glucose in T1DM also prevents macrovascular disease. In patients with T2DM, multitargeted therapy (lipid, BP, and glucose control) is the most effective strategy for CV disease prevention.
Insulin is required for glycemic control in T1DM whereas T2DM requires a multifaceted approach. Diet and exercise are the mainstays of T2DM therapy. They should be instituted first and patient adherence encouraged and maximized. Regardless of the ultimate regimen, diet and exercise remain important. Oral sulfonylurea agents that enhance -cell insulin secretion, metformin that decreases hepatic glucose output, or thiazolidinediones that enhance insulin action in the periphery are added to this treatment if diet and exercise alone fail to control hyperglycemia optimally. These agents can also be used in combination because they have different mechanisms and their actions are additive. Combination therapy with multiple classes of drugs is effective, but cost and monitoring for toxicity can be prohibitive. With increased duration of T2DM, -cell mass and function are diminished and lead to relative
P.93
insulin insufficiency. At some point, insulin therapy becomes necessary for optimal glycemic control. In fact, insulin therapy is the best way to normalize glucose in patients not responding well to oral agents and should be employed as soon as glucose rises and not as a last resort. New injectable agents that regulate glucagon, gastric emptying, satiety and insulin secretion: amylin and glucagon-like peptide-1 (GLP-1) agonists are recent additions to the list of anti-hyperglycemic agents. The most recent addition(s) are oral inhibitors of dipeptidyl peptidase 4 (DPP4), the enzyme that inactivates GLP-1. These agents are currently used by providers specializing in diabetes.
P.91
Case 1
A 14-year-old boy with an 8-year history of DM has been sick since yesterday when he began vomiting. His diabetes has been reasonably well controlled with a dosage of 20 units of glargine insulin taken daily. He uses a carbohydrate ratio of 1:20 and correction factor of 1:50 for mealtime bolus insulin. He has had several episodes of DKA in the past, but not for approximately 4 years. Yesterday, when he began vomiting, glucose concentration was 400 and his urine acetone was negative, so he took his usual dose of insulin. He has had intense polyuria and polydipsia for the last 24 hours. This morning, approximately 6 hours ago, his mother decided to withhold his insulin because of continued nausea and vomiting.
Physical examination reveals a drowsy young man who can respond to questioning. His BP is 90/70 mm Hg; pulse, 124 per minute; respirations, 30 per minute; and temperature, 38.3 C (100.9 F). His mucous membranes are dry and the ocular globes are soft and sunken, but the funduscopic findings are normal. Bowel sounds are absent and he has generalized abdominal tenderness without rebound. The deep tendon reflexes are hypoactive, but there are no localizing neurologic signs. The rest of the examination findings are normal.
Laboratory data consist of the following: Hgb, 16.4 g/dL; hematocrit (Hct), 53%; WBC, 16,942/ mm3 (93% polymorphonuclear leukocytes); BUN, 40 mg/dL; creatinine, 1.8 mg/dL; glucose, 847 mg/dL; serum ketones, strongly positive at 1:4 dilution; sodium, 126 mEq/L; potassium, 4.3 mEq/L; chloride, 100 mEq/L; and bicarbonate, 6 mEq/L. Urinalysis reveals a specific gravity of 1.030; glucose of 4+; acetone, strongly positive; and trace amounts of protein. Arterial blood gas analysis reveals a pH of 7.08, partial pressure of carbon dioxide (PCO2) of 12 mm Hg, and partial pressure of oxygen (PO2) of 80 mm Hg. An ECG shows sinus tachycardia with flat T waves. Chest radiographic study is normal. Abdominal radiographs show gastric distention, but otherwise the findings are normal.
What is the diagnosis and pathophysiologic process of this patient's disease?
How is the liver involved in the genesis of DKA?
What is the status of the patient's fluid and electrolyte levels?
What are the major goals of therapy?
What precipitated this episode of DKA?
Case Discussion
What is the diagnosis and pathophysiologic process of this patient's disease?
This patient has T1DM and is presenting with an episode of DKA. DKA is initiated by an absolute or relative insulin deficiency and an increase in the level
P.94
of counterregulatory catabolic hormones, leading to the hepatic overproduction of glucose and ketone bodies. Consistent with this, the patient's laboratory data show the presence of marked hyperglycemia, ketonemia, ketonuria, and severe metabolic acidosis. The patient's tachypnea is also consistent with his acidotic state.The destruction of pancreatic cells leading to T1DM is thought to be mediated by the activation of autoimmune processes in genetically predisposed people. The presence of antiislet and antiinsulin antibodies, the existence of inflammatory cells around the islet cells, and the temporary amelioration of new-onset T1DM by immunosuppressive therapy all provide strong evidence for an autoimmune basis of pancreatic -cell destruction.
How is the liver involved in the genesis of DKA?
Hepatic ketogenesis and the development of DKA depend on both the rate of substrate (FFA) supply to the liver and the activation of the hepatic ketogenic process, the latter being modulated by the relative increase in the glucagon-to-insulin ratio that prevails during DKA. The insulin deficiency leads to the activation of lipolysis and an increased supply of circulating FFA. In the liver these molecules undergo successive -oxidation to acetyl coenzyme A (CoA). During DKA the unrestrained FFA mobilization and oxidation trigger the production of excess amounts of acetyl CoA, which undergo condensation to acetoacetyl CoA, a precursor of the ketone bodies acetoacetate, acetone, and -hydroxybutyrate.
What is the status of the patient's fluid and electrolyte levels?
The patient's physical examination reveals signs of severe dehydration and intravascular hypovolemia (note his hypotension, tachycardia, and the dry mucous membranes). DKA, if not treated early, results in a severe total-body depletion of fluid (usually several liters) and electrolytes due to the following factors:
The hyperglycemia and hyperketonemia lead to osmotic diuresis and the urinary loss of fluid and electrolytes.
Because of acidosis, potassium is also shifted from the intracellular to extracellular fluid space and then lost during osmotic diuresis. Therefore, the serum potassium levels may not accurately reflect the total-body deficiency.
Vomiting, as in this patient, causes the further loss of fluid and electrolytes.
Muscle catabolism (proteolysis), which results from the insulin deficiency, leads to the loss of potassium, phosphate, magnesium, and nitrogen.
What are the major goals of therapy?
The immediate therapeutic goals are (a) to replenish the fluid (starting with isotonic saline) and electrolytes; and (b) to provide adequate insulin to inhibit lipolysis and ketogenesis and normalize carbohydrate metabolism, both in the liver (by inhibiting glucose production) and in the peripheral tissues (by enhancing disposal of glucose and ketone bodies). Insulin therapy is best administered in the form of a continuous IV infusion. During the fluid, electrolyte, and insulin therapy, the patient's blood glucose and electrolyte levels (especially potassium) should be monitored frequently and appropriate adjustments made. Additional therapeutic objectives include the identification and management of possible precipitating factors (e.g., infection, stress, and medication errors) and the implementation of measures to prevent the recurrence of DKA.
What precipitated this episode of DKA?
The immediate precipitating event of this patient's DKA is the withholding of insulin. An underlying stress or infection (e.g., gastroenteritis), which may also be present in this patient, should be evaluated and managed.
P.95
Case 2
A 63-year-old man is brought to the emergency room in an unconscious state. He was apparently in good health until 1 week before admission, when he experienced an insatiable thirst that he attempted to satisfy by drinking large quantities of beer and soda drinks. He had complained of having nocturia for several days, and had several bouts of diarrhea yesterday. He took to his bed yesterday and was found unconscious this morning. He takes no drugs, has not seen a physician for several years, and works regularly as a house painter. His health has been good previously. His mother had diabetes in her eighties and died of a stroke.
Physical examination reveals a deeply unconscious, acutely ill man who has several focal right-sided seizures during examination. His skin and mucous membranes are dry and his ocular globes are quite soft. His BP is 98/60 mm Hg, pulse is 120 per minute, and rectal temperature is 38 C (100.9 F), and he exhibits unlabored respirations at a rate of 13 per minute. Except for the findings of minimal hepatomegaly, absent knee jerks, and bilateral Babinski's reflexes, the examination findings are otherwise normal.
Laboratory data consist of the following: Hgb, 16.2 g/dL; Hct, 51%; and WBC, 21,340/ mm3 (92% polymorphonuclear leukocytes). Urinalysis reveals a specific gravity of 1.030; pH, 6.0; glucose, 4+; acetone, moderate amounts; and protein, trace amounts. Arterial blood gas analysis reveals a pH of 7.41, PCO2 of 35 mm Hg, and PO2 of 68 mm Hg. Both chest radiographic and head CT scan findings are normal. His ECG shows sinus tachycardia with nonspecific ST-T wave changes. Serum findings are BUN, 68 mg/dL; creatinine, 2.3 mg/dL; glucose, 1,420 mg/dL; ketones, trace amounts; sodium, 153 mEq/L; potassium, 4.6 mEq/L; chloride, 110 mEq/L; and bicarbonate, 26 mEq/L.
What is the diagnosis in this patient and how would you relate it to the major physical and laboratory findings?
What is the nature of this patient's endogenous insulin secretion, and is this type of diabetes hereditary?
Why did ketoacidosis not develop in this patient?
How is his liver involved in the pathogenesis of his hyperglycemia?
What are the major hormones that are counterregulatory to insulin action? Are they playing any role in this man's illness?
What would you predict about the state of his intravascular volume?
What are the major therapeutic goals in this patient?
Case Discussion
What is the diagnosis in this patient and how would you relate it to the major physical and laboratory findings?
This elderly patient presents in a comatose state preceded by several days of progressive symptoms of polyuria, polydipsia, and nocturia. His laboratory data show
P.96
the presence of marked hyperglycemia but no acidosis. In this setting, his moderate ketonemia and ketonuria are most likely secondary to starvation. Therefore, the diagnosis in this patient is HHNKC. His serum osmolality can be calculated using the formula: estimated osmolality = 2([Na] + [K]) + [glucose]/18 + [BUN]/2.8. For this patient, the estimated osmolality is calculated to be 418, which is consistent with a severe hyperosmolar state.What is the nature of this patient's endogenous insulin secretion, and is this type of diabetes hereditary?
This patient has T2DM. When T2DM is of short duration, such as in this patient, and when patients are obese, the endogenous insulin levels are typically normal or elevated. Such patients are still able to maintain sufficient endogenous insulin secretion to prevent ketoacidosis from developing under basal conditions. Only severe stress with elevated catecholamines plus glucagon and decreased insulin secretion will precipitate DKA in people with T2DM.
Heredity plays an important role in T2DM, although the mode of inheritance is largely unknown. T2DM is also a heterogeneous disorder, and different forms of genetic influences or defects may exist. Evidence for a genetic influence in the acquisition of T2DM include (a) a strong family history of the disease, (b) a very high prevalence of the disease in certain population groups (e.g., the Pima Indians and Micronesians of Nauru), (c) a concordance rate of 90% to 100% in monozygotic twins, and (d) an apparent autosomal dominant mode of transmission of maturity-onset diabetes of the young (an uncommon monogenic form of T2DM).
Why did ketoacidosis not develop in this patient?
This patient has sufficient endogenous insulin to prevent (a) lipolysis (FFA levels are lower in the setting of HHNKC than of DKA), and (b) full activation of the hepatic ketogenic system. In the presence of a reasonable level of endogenous insulin, the glucagon-to-insulin ratio is not high enough to lead to significant ketogenesis and ketoacidosis.
How is his liver involved in the pathogenesis of his hyperglycemia?
The hyperglycemia in this patient results from the increased hepatic production of glucose due to increased glycogenolysis and gluconeogenesis, and from the decreased uptake and utilization of glucose by the liver, muscle, and adipose tissue. All of these changes are due to the underlying insulin resistance of T2DM and the relative, but not absolute, insulin deficiency in the presence of acute stressful conditions. In addition, people with T1DM and underlying renal disease may present with HHNKC due to decreased clearance of insulin.
What are the major hormones that are counterregulatory to insulin action? Are they playing any role in this man's illness?
Glucagon, cortisol, catecholamines, and growth hormone (GH) are the chief insulin counterregulatory hormones that are elevated in major stressful conditions like HHNKC. Through the operation of several specific mechanisms, they counteract the effects of insulin, and this worsens the hyperglycemic state.
What would you predict about the state of his intravascular volume?
The intravascular volume is severely depleted in this patient (note the related findings revealed by the physical examination). The following sequence of events
P.97
may take place in patients with T2DM if they are not adequately treated: hyperglycemia osmotic diuresis loss of fluid and electrolytes dehydration worsening hyperosmolarity and osmotic diuresis elevated counterregulatory hormones hemoconcentration and hypovolemia prerenal azotemia circulatory insufficiency/shock/lactic acidosis irreversible coma death. Therefore, if this patient's condition is not rapidly treated, irreversible coma and death may ensue.What are the major therapeutic goals in this patient?
The major immediate therapeutic goals are (a) replacement of fluid and electrolytes, (b) correction of the hyperglycemia (relatively small doses of insulin are sufficient for patients in HHNKC compared with DKA), and (c) identification and management of the precipitating factors. HHNKC is a very serious medical emergency with a high risk of mortality unless an immediate, aggressive, and comprehensive management regimen is instituted. Once the acute situation is resolved, the diabetes may be managed in the long term either with diet and oral agents or with insulin.
Suggested Readings
American College of Endocrinology Task Force on Inpatient Diabetes and Metabolic Control. American College of Endocrinology position statement on inpatient diabetes and metabolic control. Endocr Pract 2004;10(1):77 82.
American Diabetes Association. Standards of medical care in diabetes 2006. Diabetes Care 2006;29(Suppl 1):S4 S42.
Bode BW, Braithwaite SS, Steed RD, et al. Intravenous insulin infusion therapy: indications, methods, and transition to subcutaneous insulin therapy. Endocr Pract 2004;10(Suppl 2):71 80.
Bretzel RG, Voigt K, Schatz H. The United Kingdom prospective diabetes study (UKPDS) implications for the pharmacotherapy of type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes 1998;106:369.
Cryer PE. Diverse causes of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med 2004;350(22):2272 2279.
Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin dependent diabetes mellitus. N Engl J Med 1993;329:977.
Eckel RH, Barouch WW, Ershow AG. Report of the National Heart, Lung, and Blood Institute-National Institute of Diabetes and Digestive and Kidney Diseases Working Group on the pathophysiology of obesity-associated cardiovascular disease. Circulation 2002;105(24):2923 2928.
Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005;365:1415 1428.
Eckel RH, Wassef M, Chait A, et al. Prevention conference VI: diabetes and cardiovascular disease: writing group II: pathogenesis of atherosclerosis in diabetes. Circulation 2002;105(18):e138 e143.
Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA 2001;285(19):2486 2497.
P.98
Grundy SM, Brewer HB, Cleeman JI, et al. For the conference participants. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation 2004;109:433 438.
Grundy SM, Cleeman JI, Merz CN, et al. National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. Circulation 2004;110(2):227 239.
Hill JO, Catenacci V, Wyatt HR. Obesity: overview of an epidemic. Psychiatr Clin North Am 2005;28:1 23.
Kushner RF, Roth JL. Assessment of the obese patient. Endocrinol Metab Clin North Am 2003;32:915 933.
Moghissi ES, Hirsch IB. Hospital management of diabetes. Endocrinol Metab Clin North Am 2005;34:99 116.
National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes 1979;28:1039.
Riddle MC. Glycemic management of type 2 diabetes: an emerging strategy with oral agents, insulins, and combinations. Endocrinol Metab Clin North Am 2005;34:77 98.
Turner RC. The U.K. prospective diabetes study: a review. Diabetes Care 1998;21:35.
UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34): UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352:854.
UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphoylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33): UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352:837.
Wyatt HR, Hill JO. What role for weight-loss medication? Weighing the pros and cons for obese patients. Postgrad Med 2004;115(1):38 40,43 45,58.
Disorders of the Thyroid
What are the key features in a patient's history that are important in assessing for a possible functional thyroid disorder?
What are the important physical examination findings?
What laboratory data are used to confirm or refute the existence of a functional thyroid abnormality?
Discussion
What are the key features in a patient's history that are important in assessing for a possible functional thyroid disorder?
When assessing a patient's history for clues to a functional thyroid disease, it is important to keep in mind that thyroid hormones in general control metabolism. Therefore, when questioning patients about their medical history, it is important to ask specifically about elements related to metabolism. For example, in the setting of hyperthyroidism, weight loss, anxiety, tremor, palpitations, heat intolerance, hyperdefecation, insomnia, restlessness, and changes in the hair or skin are important features. In contrast, in patients with
P.99
suspected hypothyroidism, look for clues that indicate decreased metabolic activity. These include weight gain; cold intolerance; constipation; dry, scaly skin; thick hair; depression; increased sleeping and fatigue; and generalized lethargy.What are the important physical examination findings?
Like the history, the physical examination should be performed to look for signs of hypermetabolism or hypometabolism. In the setting of hyperthyroidism, a fast pulse; tremor; sweating; thin, soft, and velvety hair; very brisk reflexes; and a hyperdynamic precordium are all features of increased metabolism. In addition, a very critical finding is an enlarged thyroid gland. If this is found in conjunction with a bruit, then the clinician can assume that the thyroid gland itself is overactive and overproducing thyroid hormone. In contrast, the findings characteristic of hypothyroidism include pale, sallow skin; thick hair; puffiness in the face and ankles; cool extremities; very delayed deep tendon relaxation; bradycardia; and a very quiet precordium. Again, an enlarged thyroid is an important physical examination finding. In this event, a firm, woody, or pebbly texture would indicate the presence of lymphocytic infiltration or Hashimoto's thyroiditis.
What laboratory data are used to confirm or refute the existence of a functional thyroid abnormality?
There is now a very sensitive and specific laboratory protocol to determine whether the patient has a functional thyroid disorder. The first diagnostic test should be measurement of the serum TSH level using the sensitive TSH assays. If this assay result proves to be within the normal range, then a functional abnormality of the thyroid has virtually been excluded. In contrast, an elevated TSH level means the thyroid gland is failing and the patient has primary thyroid gland failure, most commonly due to autoimmune thyroid disease. Conversely, if the serum TSH level is low and undetectable, this indicates hyperthyroidism due to Graves' disease, a multinodular goiter, a hot nodule, excessive thyroid hormone ingestion, subacute thyroiditis, postpartum thyroiditis, or silent thyroiditis. If the TSH value is abnormal, then thyroid hormone status should be assessed. This can be done either by obtaining a total T4 with a T3 resin uptake (to assess T4-binding globulin), or by simply ordering a free T4. Only in special circumstances is it necessary to test for total T3 or free T3 levels. Finally, in evaluating a patient with suspected hyperthyroidism, if both the TSH level is low and the free T4 level is high, the next step is to perform a radioactive iodine uptake test and scan. This test is very important in distinguishing causes of hyperthyroidism related to overproduction (i.e., Graves' disease, a multinodular goiter, or a hot nodule) from those related to excessive release but not production (i.e., subacute thyroiditis, postpartum thyroiditis, or silent thyroiditis), as well as excessive thyroid hormone ingestion. The scan also infers the therapeutic approach. In the setting of strong clinical evidence for hypothyroidism with a low or normal TSH, it is also important to consider the relatively rare possibility of central, or secondary, hypothyroidism (defective TSH production by the pituitary gland).
P.100
Case
A 31-year-old mother of two is seen because of complaints of headaches and amenorrhea, which have lasted for 3 months. She delivered her second child 10 months ago. The headaches developed after she was in a motor vehicle accident 4 months before. At that time, the patient experienced a temporary loss of consciousness but has since been normal; an extensive neurologic examination in the emergency room yielded negative findings. On further questioning, the patient also admits to a 15-lb (6.75-kg) weight loss despite a normal appetite, as well as mild heat intolerance and excessive sweating during the summer months. Recently, she has noted that her hands shake and her handwriting has become uneven. Of significance is her exercise history; she had been running 5 to 6 mi a day, 5 days a week, and has participated in marathon running competitions. However, over the last 3 months, her tolerance for exercise has decreased, and her running times have deteriorated. On questioning her about her family history, it is found that her mother takes levothyroxine for hypothyroidism, a maternal grandmother has T2DM, and her father has hyperlipidemia and coronary artery disease.
Physical examination reveals a well-developed, well-nourished, thin woman who appears somewhat anxious. Her BP is 130/50 mm Hg and her pulse is 120 beats per minute. Her hair is fine with streaks of gray. Her eyes exhibit no exophthalmos, but there is a stare and lid lag. The extraocular muscles are normal. The thyroid is diffusely enlarged at approximately 40 g. There is a high-intensity bruit audible over the right lobe of the thyroid. The cardiac examination reveals a normal first and second heart sound and a grade 1/6 systolic ejection murmur. The lungs are clear to auscultation and percussion. Abdominal examination findings are negative. Her hands exhibit an outstretched tremor and her skin is noted to be warm, smooth, and slightly moist. Her reflexes are symmetrically brisk. There is slight proximal muscle weakness detected in the thighs and shoulder girdle muscles.
What is the differential diagnosis in this patient, and should it include a normal pregnancy?
What is the most efficient approach to the laboratory evaluation in this patient?
To distinguish silent thyroiditis from Graves' hyperthyroidism, what is the most important diagnostic tool?
If the patient has silent thyroiditis, what is the appropriate therapy?
If the patient has Graves' disease and is treated with radioactive iodine, what is likely to occur?
If the patient is treated with antithyroid drugs, what is the likely short- and long-term prognosis?
Case Discussion
What is the differential diagnosis in this patient, and should it include a normal pregnancy?
A normal pregnancy can mimic many of the symptoms of hyperthyroidism, including increased energy, anxiety, heat intolerance, sweating, and, in areas of the world where iodine deficiency is common, a mild increase in the thyroid gland size. In addition, pregnancy can certainly decrease the tolerance for maximal exercise. Features that are not characteristic of pregnancy are the moderate (40 g) thyroid
P.101
enlargement, the lid lag and stare, and, most important, the bruit over the right side of the thyroid gland. A bruit in the thyroid gland reflects the presence of increased blood flow due to hyperplasia and excessive thyroid gland function. These features are absent in pregnancy, and therefore a bruit would not be heard. However, previously silent thyroid disease may become apparent during pregnancy. In addition, a 15-lb (6.75-kg) weight loss, despite a normal appetite, would be distinctly unusual in the pregnant state. Therefore, a normal pregnancy is an unlikely cause of this patient's symptoms. The differential diagnosis therefore consists of hyperthyroidism due to Graves' disease, a multinodular goiter, and silent thyroiditis. A multinodular goiter is unusual in a 31-year-old patient, and is usually seen in the older population. Furthermore, multinodular goiters are not associated with a bruit, even when producing hyperthyroidism. The diffuse enlargement of the thyroid gland at 40 g is also unusual in the setting of a multinodular goiter because, in this event, multiple nodules should be appreciated on the physical examination. A stare and lid lag can be found in patients with hyperthyroidism due to a multinodular goiter because these findings reflect the hyperthyroidism, not the autoimmune process.The important differential diagnostic exercise in this case should focus on whether the hyperthyroidism is due to Graves' disease or silent thyroiditis. Graves' disease is the most common cause of hyperthyroidism and is found more frequently in women than in men, with a ratio of 4:1. In addition, hyperthyroidism due to Graves' disease usually afflicts younger people between 20 and 50 years of age. The 15-lb (6.75-kg) weight loss, heat intolerance, excessive sweating, tremor, decreased exercise tolerance, moderate enlargement of the thyroid with a bruit, and the warm, smooth, and slightly moist skin are all characteristic of hyperthyroidism due to Graves' disease. In addition, the patient has a family history of autoimmune disease, in that the mother is being treated for hypothyroidism and the grandmother has adult-onset DM. In addition, the patient appears to have premature gray hair. The absence of exophthalmos does not conflict with this diagnosis because this finding may be clinically evident in only 10% to 20% of patients with Graves' hyperthyroidism. (However, when more sophisticated techniques for evaluating eye function are used, as many as 80% to 90% of the patients with Graves' hyperthyroidism prove to have discernible eye abnormalities.)
Hyperthyroidism due to silent thyroiditis is becoming an increasingly well-recognized diagnostic entity. The exact etiology of this disorder is obscure, but appears to be an autoimmune process. Silent thyroiditis is perhaps identical to postpartum thyroiditis, which arises in 5% to 8% of all pregnancies in the United States. This disorder appears usually between 2 and 6 months postpartum (10 months is slightly excessive). The patient exhibits the signs and symptoms of hyperthyroidism and an enlarged thyroid gland, but has no evidence of exophthalmos or pretibial
P.102
of the two disorders are quite different. Because amenorrhea can occur in all forms of hyperthyroidism, it is not a helpful clue for identifying the ultimate cause of the hyperthyroidism. Finally, the bruit over the right lobe of the thyroid is an important clue that points toward a diagnosis of Graves' disease in this patient. A bruit over the thyroid gland is usually not present in hyperthyroidism due to a multinodular goiter, silent thyroiditis, or subacute thyroiditis. Therefore, on the basis of the patient's history, physical examination findings, and statistical considerations the most likely diagnosis is hyperthyroidism due to Graves' disease. However, formal laboratory studies should be done first to determine whether hyperthyroidism is indeed present and then to identify the cause of the hyperthyroidism.Which is the most efficient approach to the laboratory evaluation in this patient?
The key issue in deciding on the nature of the laboratory evaluation is which test best determines if hyperthyroidism is indeed present. Total T4 and T3 resin uptake were traditionally regarded as the best tests for establishing the presence of hyperthyroidism and for distinguishing hyperthyroidism from a normal pregnancy. In all forms of hyperthyroidism, both total T4 and T3 resin uptake are elevated, whereas in pregnancy, the total T4 is elevated because of an increase in the T4-binding globulin level, and the T3 resin uptake is reduced, again because of the increased T4-binding globulin level. However, these same abnormalities in thyroid function can occur in the absence of pregnancy, such as in a patient taking birth control pills or replacement estrogen, or in a woman with congenital X-linked T4-binding globulin excess. Furthermore, confusion can arise when both hyperthyroidism and excessive estrogens coexist because the T4 concentration can be elevated and the T3 resin uptake may be variable (low, normal, or high values) in this setting. The total T3 is a good test for hyperthyroidism but because T3 is also bound to T4-binding globulin its levels are elevated in the context of a normal pregnancy or exogenous estrogen. The most efficient laboratory tests in this case are determinations of the free T4 and TSH levels. The free T4 level is elevated in hyperthyroidism but normal in pregnancy. Likewise, the TSH level is suppressed and undetectable in the setting of hyperthyroidism but normal in pregnancy. Therefore, this laboratory profile is ideal for discriminating between hyperthyroidism and pregnancy-related changes in thyroid levels. However, the free T4 and TSH levels cannot discriminate between hyperthyroidism due to Graves' disease, a multinodular goiter, or silent thyroiditis.
To distinguish silent thyroiditis from Graves' hyperthyroidism, what is the most important diagnostic tool?
The most important diagnostic tool for distinguishing silent thyroiditis from Graves' disease is a radioactive iodine uptake test. Graves' disease, which is a state of thyroid hormone overproduction, involves an excessive uptake of iodine into the thyroid gland and therefore a high radioactive iodine uptake. In contrast, silent thyroiditis is not an overproduction state but a state in which there is an excessive release of thyroid hormone into the circulation that suppresses TSH, stemming from autoimmune damage to thyroid follicular cells. These events render the thyroid gland incapable of taking up iodine. Therefore, in the setting of silent thyroiditis, the radioactive iodine uptake is very low, in striking contrast to the elevated values found in patients with Graves' hyperthyroidism.
If the patient has silent thyroiditis, what is the appropriate therapy?
Distinguishing silent thyroiditis from Graves' disease is important because the therapies for the two conditions are dramatically different. Because silent thyroiditis is a destructive process without overproduction, it does not respond to either antithyroid drugs or radioactive iodine. The low radioactive iodine uptake in silent thyroiditis renders radioactive iodine therapy ineffective, and the lack of overproduction of thyroid hormones negates the effectiveness of antithyroid drugs. Because silent thyroiditis is a self-limited process with a triphasic course, the recommended therapy is the judicious use of -blockers to control symptoms, particularly tremor and tachycardia, and observation of the patient during the spontaneous resolution of the process. Typically, the hyperthyroid phase lasts for 1 to 3 months, after which the process is dramatically reversed; in fact, hypothyroidism can occur transiently for another 1 to 3 months. During the hypothyroid phase, many patients benefit from a short course of thyroid hormone replacement. However, in most cases, both the hyperthyroidism and hypothyroidism resolve spontaneously and normal thyroid function is restored. In only 10% to 25% of cases does permanent hypothyroidism eventuate. Perhaps the most important clinical clue to the resolution of silent thyroiditis is normalization of the thyroid gland size. Because silent thyroiditis is a self-limited process that resolves spontaneously, thyroid surgery is usually unnecessary. However, this option can be reserved for particularly severe cases that have protracted or recurrent courses.
If the patient has Graves' disease and is treated with radioactive iodine, what is likely to occur?
The most common therapy for Graves' disease in the United States is radioactive iodine, which is given in the form of a small capsule containing 5 to 10 mCi of iodine-131 (131I). The radioactive iodine is quickly absorbed from the gastrointestinal tract into the bloodstream and then incorporated into the thyroid gland, where it induces radioactive damage and kills thyroid cells. Radioactive iodine that does not enter the thyroid gland is quickly excreted through the kidneys into the urine. Because radioactive iodine induces damage and eventual death of thyroid follicular cells, the chance of hypothyroidism developing is very high. In general, hypothyroidism develops in the first year in 50% to 60% of the patients treated; thereafter, the rate of development of hypothyroidism is 1% to 3% per year. Therefore, hypothyroidism will develop in most patients treated with radioactive iodine and they will require lifelong thyroid hormone replacement therapy.
If the patient is treated with antithyroid drugs, what is the likely short- and long-term prognosis?
Antithyroid drugs (propylthiouracil and methimazole) are derivatives of thiourea and their mechanism of action is to inhibit both thyroid hormone synthesis and, in the case of propylthiouracil, the peripheral conversion of T4 to T3. These drugs are an ideal choice of therapy for patients with hyperthyroidism due to Graves' disease. They are most commonly used in children and pregnant patients, and in relatively mild cases of hyperthyroidism in which the thyroid gland is only moderately enlarged. Propylthiouracil is given in a dose of approximately 100 mg three or four times a day. Methimazole (Tapazole; Eli Lilly, Indianapolis, IN) has a slightly longer half-life
P.104
than propylthiouracil and may be given in a single daily dose of 10 to 30 mg each day. Both agents exhibit a similar profile of side effects, which occur in 1% to 3% of the patients. The most common side effects are skin rash, urticaria, arthralgias, fever, and transient leukopenia. Minor gastrointestinal side effects and arthritis occur occasionally. The major rare side effect is agranulocytosis, which occurs in 0.2% to 0.5% of the patients. Other rare side effects include aplastic anemia, hepatitis, thrombocytopenia, vasculitis, and cholestatic jaundice. The side effects usually arise early in the course of therapy, and in the case of methimazole the reactions appear to be dose dependent.The antithyroid drugs are usually given for a 1- to 2-year period in the hope of inducing permanent remission. The frequency with which permanent remission takes place has been analyzed in many studies and has been found to occur in those patients who have mild disease and smaller thyroid glands. However, when all patients are considered, the chance for permanent remission is only 20% to 40%. Therefore, 60% to 80% of patients have a relapse of their hyperthyroidism, usually within 2 years of discontinuing the antithyroid drug. In cases of relapse, the antithyroid drug can either be reinstituted or a more definitive ablative form of therapy, such as radioactive iodine, can be administered.
P.103
Suggested Readings
Bartalena L, Marcocci C, Bogazzi F, et al. Relation between therapy for hyperthyroidism and the course of Graves' ophthalmopathy. N Engl J Med 1998;338:73.
Burguera B, Gharib H. Thyroid incidentalomas: prevalence, diagnosis, significance, and management. Endocrinol Metab Clin North Am 2000;29(1):187 203.
Castro MR, Gharib H. Continuing controversies in the management of thyroid nodules. Ann Intern Med 2005;142:926 931.
Cooper DS, Doherty GM, Haugen BR, et al. Management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2006;16(2):1 33.
Gharib H, Tuttle RM, Baskin HJ, et al. Subclinical thyroid dysfunction: a joint statement on management from the American Association of Clinical Endocrinologists, the American Thyroid Association, and the Endocrine Society. J Clin Endocrinol Metab 2005;90(1):581 585; discussion586 587.
Haugen BR. Initial treatment of differentiated thyroid carcinoma. Rev Endocr Metab Disord 2000;1(3):139 145.
Kahaly JG, Dillmann HW. Thyroid hormone action in the heart. Endocr Rev 2005;26:704 728.
Levy EG, Ridgway EC, Wartofsky L. Algorithms for diagnosis and management of thyroid disorders, 2003-2004. Available from: http://www.Thyroidtoday.com/ExpertOpinions/ThyroidDiseaseAlgorithms.pdf.
Sarlies NJ, Gourgiotis L. Thyroid emergencies. Rev Endocr Metab Disord 2003;4:129 136.
Stathatos N, Wartofsky L. The euthyroid sick syndrome: is there a physiologic rationale for thyroid hormone treatment? J Endocrinol Invest 2003;26(12):1174 1179.
Surks MI, Ortiz E, Daniels GH, et al. Subclinical thyroid disease: scientific review and guidelines for diagnosis and management. JAMA 2004;291(2):228 238.
P.105
Growth Hormone-Secreting Pituitary Tumors
What are the clinical symptoms of GH excess (i.e., acromegaly)?
What are the physical signs of acromegaly?
What is the best screening test to exclude the diagnosis of acromegaly?
What laboratory tests can confirm the diagnosis of acromegaly and assess other pituitary hormone functions?
What imaging studies are necessary?
What is the treatment of choice and what are the alternatives?
Discussion
What are the clinical symptoms of GH excess (i.e., acromegaly)?
Acromegaly is the clinical syndrome resulting from excessive GH production and is usually due to a pituitary tumor. The symptoms and signs are gradual in onset, which often cause diagnosis to be delayed for 6 to 8 years. The classic symptoms consist of headache, visual disturbances (due to an enlarging tumor compressing on the optic nerves), enlargement in glove and shoe size, excessive sweating, arthralgias, loss of libido, impotence in men, amenorrhea in women, muscle weakness, and problems with an underbite or spaces between the teeth.
What are the physical signs of acromegaly?
The physical signs of acromegaly include coarsening of the physical features (which can be determined by looking at old photographs); frontal bossing; thick, coarse skin; doughy, sweaty palms; prognathism (an enlarged mandible); widely spaced teeth and underbite; severe osteoarthritis; prominent lips, tongue, and nose; acanthosis nigricans; skin tags; enlargement of all organs; entrapment of peripheral nerves (i.e., carpal tunnel); hypertension with cardiomyopathy; obstructive sleep apnea visual field abnormalities; glucose intolerance; and diabetes.
What is the best screening test to exclude the diagnosis of acromegaly?
The best screening test for acromegaly is measurement of the insulin-like growth factor I (IGF-I; somatomedin C) level. This is a liver protein induced by GH and the test constitutes an integrated assessment of GH action. GH levels are pulsatile in nature fasting levels are usually less than 10 ng/mL in adults and the GH concentration increases at night during sleep. As the levels in patients with acromegaly can be as low as 5 10 ng/mL, the GH level is helpful if very high, but is neither sensitive nor specific if normal or mildly elevated.
What laboratory tests can confirm the diagnosis of acromegaly and assess other pituitary hormone functions?
To confirm the diagnosis and assess the pituitary function in a patient, the GH response to an oral glucose load is assessed. To perform this, 100 g oral glucose is given to the fasting patient and blood for GH determination is
P.106
obtained at 0, 30, 60, 90, and 120 minutes. In a normal response to a glucose load, the GH levels are suppressed to less than 2 ng/mL. In patients with acromegaly, the GH levels are either not suppressed or they paradoxically increase (approximately 30% of patients). This test, if done in conjunction with an IGF-1 test, is the best way of assessing a cure after surgery. In addition, approximately 30% of the patients show an increase in their GH levels after a thyrotropin-releasing hormone (500 g) IV push; healthy individuals do not. This test is more often used in a research setting or for the purpose of assessing tumor recurrence.Other blood tests for determining pituitary function include measurement of the prolactin level, which can be elevated because of the interruption of dopamine tone secondary to stalk compression or the cosecretion of prolactin by the tumor. The follicle-stimulating hormone (FSH), luteinizing hormone (LH), and testosterone or estrogen levels are used to assess the status of the reproductive axis. The thyroid status is checked by a free T4; measurement of the TSH level is not helpful because, if there is thyroid deficiency, it is secondary to TSH deficiency. The adrenal axis is assessed with a morning cortisol determination; if less than 5 mg/dL, a formal cosyntropin (Cortrosyn; Organon Teknika, Durham, NC) stimulation test of the ACTH reserve should be performed. Blood is drawn at 0, 30, and 60 minutes after one ampule (0.25 mg) of synthetic ACTH IV push or IM. In healthy subjects, the cortisol levels increase to 18 or more and are often double the baseline value. An -subunit level may be helpful as a tumor marker because some GH tumors cosecrete other pituitary hormones.
What imaging studies are necessary?
The imaging procedure of choice is an MRI scan of the pituitary. It provides the greatest detail of the tumor's extent and landmarks for the surgeon's use during surgical removal. A coronal CT scan with fine cuts can detect most large tumors, but a lateral skull film is not sensitive. Formal visual field testing should be done in all patients with macroadenomas to serve as a baseline for assessing postoperative improvement.
What is the treatment of choice and what are the alternatives?
The treatment of choice for these tumors, which are usually macroadenomas (>1 cm), is transsphenoidal resection of the tumor, although surgical cure is often difficult. If postoperative hormonal testing reveals continued abnormal GH production, radiation therapy is often necessary. Medical therapy with bromocriptine (if the tumor costains for prolactin) may also reduce the GH levels and tumor size, while the results of irradiation are awaited. Somatostatin analogs can inhibit GH production. They are now available in short-acting and long-acting forms. Long-acting octreotide normalizes IGF-I levels in 41% to 75% and reduces tumor size in 30% of subjects in published trials.
More recently a GH receptor antagonist, pegvisomant, has proved effective for reduction of IGF-I levels in acromegaly and has received U.S. Food and Drug Administration (FDA) approval. Studies demonstrate that pegvisomant normalizes IGF-I levels in more than 95% of patients who have
P.107
failed other therapies. Initial concerns that pituitary tumor growth would proceed unchecked with this treatment have not been borne out by experience to date.
Case
A 40-year-old man is seen because of headaches, muscle aches, and chronic low back and joint pain. As he enters the office, you notice his coarse facial features, frontal bossing, and large jaw. When you shake his hand, you find he has large, doughy, sweaty palms and, when he smiles, you note his teeth are widely spaced.
He has not seen a physician in 10 years and is taking no medications. His back and joint pain have been worsening for 6 years, but his headaches started 6 months ago.
His physical examination findings are significant for a BP of 150/100 mm Hg, pulse of 60 per minute, and respiratory rate of 12 per minute.
He returns in 2 weeks with old photographs that confirm a change in his physical appearance over time, and the laboratory test results confirm your clinical impression.
What is your initial diagnosis in this patient?
Besides the back and joint pain and the headaches, what other symptoms would you look for to confirm or refute your diagnosis?
Besides the physical features you observe initially, what other abnormalities would you look for on physical examination?
What laboratory tests should be performed initially?
What additional testing should be performed once the initial laboratory results are known?
What is the preferred treatment in this patient?
Case Discussion
What is your initial diagnosis in this patient?
Acromegaly should be your initial diagnosis.
Besides the back and joint pain and the headaches, what other symptoms would you look for to confirm or refute your diagnosis?
Other symptoms to look for in this patient include a change in glove, ring, and shoe sizes, spaces between the teeth and an underbite, decreased libido and impotence, sweating, new snoring, polyuria, polydipsia, and a change in vision.
Besides the physical features you observe initially, what other abnormalities would you look for on physical examination?
Other physical features to look for in this patient include thick coarse skin, skin tags, enlarged extremities and organs, entrapment neuropathies, visual field abnormalities, and decreased body hair and testicular size. Old pictures would confirm the clinical suspicion.
What laboratory tests should be performed initially?
Initial laboratory tests in this patient would consist of the measurement of IGF-1 (somatomedin C) and fasting GH levels. If the levels are elevated it would suggest the diagnosis of acromegaly, which would be confirmed if the GH levels did not
P.108
suppress to less than 2 ng/mL in response to a glucose load (some studies suggest 1 ng/mL, which may reflect the increased sensitivity of the assay).What additional testing should be performed once the initial laboratory results are known?
If the initial laboratory results indicate acromegaly, a fasting blood sugar test should be performed to rule out diabetes. Pituitary tests should include measurement of the prolactin, FSH, LH, testosterone, and -subunit levels. An MRI scan can show the extent of the tumor, and formal visual field testing should be performed.
What is the preferred treatment in this patient?
The preferred initial treatment is surgical removal of the tumor. Bromocriptine or a somatostatin (octreotide) analog may be useful as medical adjuncts. Pegvisomant may be considered for residual tumors that are refractory to other medical management. Radiation therapy may be indicated for the destruction of residual tumor if reoperation or surgical cure is not feasible. Postoperative hormonal testing is indicated to reassess pituitary function. Echocardiography and colonoscopy should be performed to evaluate for cardiomegaly and colon polyps.
Prolactin-Secreting Pituitary Tumors
What symptoms and signs are associated with an elevated prolactin level in women and in men?
What is the underlying pathophysiologic process responsible for the effects of elevated prolactin levels?
What are the causes of an elevated prolactin level other than a pituitary tumor?
What testing is necessary to confirm or refute a diagnosis of a prolactinoma?
What are the treatment options for a prolactinoma?
Discussion
What symptoms and signs are associated with an elevated prolactin level in women and in men?
In women, an elevated prolactin level is associated with disturbance of the menstrual cycle ranging from the occurrence of short cycles with an inadequate luteal phase, oligoovulation, and infertility, to amenorrhea. Galactorrhea, hirsutism, mood disturbances, and headaches are also frequent complaints. In men, symptoms include decreased libido, impotence, and infertility. Galactorrhea is a rare finding. Visual field defects are seen in the setting of large tumors. Osteopenia and fractures can occur in both sexes and are due to the secondary hypogonadism.
What is the underlying pathophysiologic process responsible for the effects of elevated prolactin levels?
Prolactin is under tonic inhibitory control from dopamine in the hypothalamus. Stalk compression, which causes dopamine tone to be inhibited, or prolactin secretion from a tumor inhibits the hypothalamic pituitary gonadal
P.109
axis at all three levels. The major effect, however, is termination of the gonadotropin-releasing hormone induced pulsatile release of the pituitary gonadotropins, LH and FSH. This disordered gonadotropin secretion then results in inadequate gametogenesis and steroidogenesis, and hence hypogonadism with or without infertility. For galactorrhea to occur, there must be estrogen priming of the breast in addition to an elevated prolactin level, which is why milk production does not develop in most men unless their prolactin level is chronically very elevated with suppression of testosterone release, elevation of the estradiol level, and gynecomastia.What are the causes of an elevated prolactin level other than a pituitary tumor?
Elevated prolactin levels may be due to physiologic causes such as pregnancy, stress, sleep, exercise, or frequent breast stimulation. Systemic disorders associated with elevated prolactin levels include hypothyroidism, hypoadrenalism, chronic renal failure (elevated production and decreased clearance), and liver failure. Drugs that elevate the prolactin level include phenothiazides, tricyclic antidepressants, opiates, metoclopramide, cimetidine, methyldopa, reserpine, and amphetamines, all of which interfere with dopamine inhibitory tone.
What testing is necessary to confirm or refute a diagnosis of a prolactinoma?
A prolactin level of more than 100 ng/mL suggests the presence of a tumor, although tumors or other causes can be associated with lower elevations. No stimulation or suppression test is needed. An MRI of the pituitary is necessary to detect a microadenoma and exclude a large pituitary or hypothalamic mass that is causing stalk compression.
What are the treatment options for a prolactinoma?
The treatment of choice for prolactinomas is the dopamine agonist, bromocriptine. It effectively lowers prolactin levels and reduces tumor size. Other dopamine agonists commonly used include cabergoline and pergolide. Surgical removal is reserved for noncompliant or bromocriptine-intolerant patients because of the high recurrence rate of 20% to 50% at 5 years. Major side effects of bromocriptine therapy include orthostatic dizziness, dry mouth, nausea, and vomiting, although these may be minimized by slow titration of the drug along with food intake at night. Lifelong therapy is probably necessary. Lack of treatment leads to prolonged gonadal steroid deficiency and the risk of osteopenia and fracture. The premature CV risk has not been assessed.
Case
A 28-year-old woman is seen because of irregular periods and infertility. Her menarche occurred at 12 years of age and she had regular periods with moliminal symptoms (breast tenderness, bloating, and cramping) until approximately 2 years ago. After that, her periods have become lighter and irregular without moliminal symptoms. She has decreased libido, occasional headaches, and is moody and irritable. She has noted a milky discharge from both nipples. She took birth control pills for 2 years, 5 years ago.
Her examination is significant for the following findings: normal visual fields, galactorrhea, and a decreased estrogen effect on the vaginal mucosa.
P.110
What is the most likely diagnosis in this patient?
What other historical facts are important to elicit in an effort to determine the cause of her symptoms?
What laboratory tests or studies would you have done?
What is the treatment of choice in this patient?
Case Discussion
What is the most likely diagnosis in this patient?
The most likely diagnosis in this patient is hyperprolactinemia.
What other historical facts are important to elicit in an effort to determine the cause of her symptoms?
It is important to find out whether she might be pregnant and whether she takes drugs that would inhibit dopamine tone. In addition, a history of hypothyroidism, hypoadrenalism, excessive breast stimulation, and renal or liver disease should be sought.
What laboratory tests or studies would you have done?
A serum prolactin level should be measured to determine the extent of the elevation. In addition, liver function studies and determination of the BUN and creatinine levels should be done to rule out liver or kidney disease. The human chorionic gonadotropin level should be measured to rule out pregnancy, as well as the TSH and cortisol levels, if there are symptoms or signs of hypothyroidism or hypoadrenalism. An MRI scan should be obtained to distinguish between a microadenoma and a macroadenoma.
What is the treatment of choice in this patient?
The treatment of choice is the dopamine agonist bromocriptine. Treatment is begun at night with the intake of food to decrease the side effects of postural hypotension, nausea, and dry mouth. The goal is to normalize the prolactin levels. Other long-acting dopamine agonists such as cabergoline, pergolide, and dihydroergotoxine are available if patients fail to tolerate bromocriptine.
Suggested Readings
Arafah BM. Medical management of hypopituitarism in patients with pituitary adenomas. Pituitary 2002;5:109 117.
Aron DC, Howlett TA. Pituitary incidentalomas. Endocrinol Metab Clin North Am 2000;29(1):205 221.
Melmed S, Casanueva FF, Cavagnini F, et al. Guidelines for acromegaly management. J Clin Endocrinol Metab 2002;87(9):4054 4058.
Molitch ME. Medical management of prolactin-secreting pituitary adenomas. Pituitary 2002;5:55 65.
Pickett CA. Diagnosis and management of pituitary tumors: recent advances. Prim Care Office Pract 2003;30:765 789.
Shimon I, Melmed S. Management of pituitary tumors. Ann Intern Med 1998;129:472.
P.111
Swearingen B, Barker FG II, Katznelson L, et al. Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab 1998;83:3419.
Hypercalcemia
What conditions can cause hypercalcemia?
What two medical conditions account for most cases of hypercalcemia?
In the hypercalcemic patient, what are the laboratory findings seen in the setting of hyperparathyroidism?
What is the treatment for hypercalcemia?
What are the indications for parathyroidectomy?
Discussion
What conditions can cause hypercalcemia?
The causes of hypercalcemia that need to be considered in any patient who exhibits a bona fide elevation in the serum calcium level as documented in at least three repeat determinations are listed in Table 3-1.
What two medical conditions account for most cases of hypercalcemia?
Of the many causes of hypercalcemia listed in Table 3-1, the most common are malignancy (45%) and hyperparathyroidism (45%). The lengthy differential diagnosis (see Table 3-1) includes the other 10% of the causes. Hence, from a practical standpoint, hypercalcemic disorders can be broken down into two categories: parathyroid hormone (PTH)-mediated hypercalcemia and non PTH-mediated hypercalcemia.
In the hypercalcemic patient, what are the laboratory findings seen in the setting of hyperparathyroidism?
For the sake of simplicity, the many causes of hypercalcemia can be separated into two categories according to the PTH level and laboratory findings result from the presence or absence of the action of PTH. (Tables 3-2 and 3-3).
What is the treatment of hypercalcemia?
A hypercalcemic emergency is diagnosed when the calcium level exceeds 14 mg/dL or the patient exhibits symptoms of hypercalcemia, consisting of profound weakness, impaired mental function, nausea and vomiting, and central nervous system depression leading to stupor, lethargy, or coma. Urgent treatment of the hypercalcemia is mandatory in these situations (Table 3-4).
What are the indications for parathyroidectomy?
The following indications for parathyroidectomy in hyperparathyroid patients have been proposed by a National Institutes of Health (NIH) consensus conference:
Patient younger than 50 years
Elevated serum calcium to a concentration of 1.0 to 1.6 mg/dL above normal laboratory values
History of a life-threatening hypercalcemic episode
Reduced creatinine clearance
Presence of kidney stones
Urine calcium excretion of greater than 400 mg per 24 hours
Bone mass reduced by more than 2 standard deviations below normal
P.112
Table 3-1 Causes of Hypercalcemia | |
|---|---|
|
Case
A 47-year-old white male computer consultant is seen in the walk-in clinic complaining of severe right hip pain and difficulty in walking. He has been taking ibuprofen for pain relief. The pain in his right hip and right proximal lower extremity has been present for approximately 4 months, and has progressed to become a sharp, localized right hip joint pain during the past month. The patient has noted a 10-lb (4.5-kg) weight loss over the preceding 2 months, but ascribes this to a self-enforced diet. He has nocturia with two to
P.113
five micturitions per night, and complains of excessive thirst. In addition, he is aware of a decrease in his ability to concentrate over the preceding several months.
Table 3-2 Parathyroid Hormone Action on Kidney | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
His past medical history is significant for nephrolithiasis requiring hospitalization 10 years earlier, and an upper gastrointestinal hemorrhage 5 years ago, secondary to peptic ulcer disease. His family history is noncontributory.
Radiographic studies of the patient's pelvis and right hip show a lytic lesion in the right sacrum and femoral acetabulum. The patient is admitted for further evaluation.
Physical examination reveals a pleasant, middle-aged man who is experiencing considerable pain in his right hip. His BP is 160/98 mm Hg; pulse, 96 beats per minute; respiratory rate, 20 per minute; and temperature, 97.8 F (36.8 C). No significant skin
P.114
P.115
lesions are found. On examination of the oral pharynx, a white mass on the hard palate is noted. The patient has no cervical, axillary, or inguinal adenopathy. Thyroid examination reveals a fullness in the left lower lobe. Range-of-motion exercises of the right lower extremity elicit severe right hip tenderness. A neurologic examination reveals diminished strength in the right hip flexors and extenders with normal deep tendon reflexes throughout. The patient's mental status is appropriate.
Table 3-3 Causes of Hypercalcemia | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Table 3-4 Therapy for Hypercalcemia | ||
|---|---|---|
|
Initial laboratory data reveal the following: WBC, 5,800; Hgb, 13.3 g/dL; Hct, 39.7%; and platelet count, 274 103mm3. The following electrolyte and serum chemistry values are reported: sodium, 138 mEq/L; potassium, 3.9 mEq/L; chloride, 108 mEq/L; CO2, 21.5 mEq/L; BUN, 18 mg/dL; creatinine, 1.0 mg/dL; and fasting glucose, 94 mg/dL. Other significant laboratory values include the following: calcium, 11.5 mg/dL; phosphate, 2.0 mg/dL; total protein, 6.8 g/dL; albumin, 2.8 g/dL; and magnesium, 1.7 mEq/L. Urinalysis findings are normal. The erythrocyte sedimentation rate is 9 mm per hour and the alkaline phosphatase level is 396 IU/L.
Chest film findings are normal. In a review of the pelvis and hip radiographic studies, lytic changes with bony destruction are found in both hemipelves, but these are greater on the right. Right femoral head involvement is also noted.
A pelvic CT scan shows the existence of multiple destructive soft tissue lesions in the bone of the pelvis; the largest of the lesions measures 8 cm. A radionuclide bone scan reveals increased uptake in the pelvic lesions and in several ribs, as well. A large-bore needle biopsy specimen from the gingivopalatal mass and the right ilium shows the appearance of a giant cell tumor mixed with fibroblasts.
Special endocrine studies reveal an ionized calcium level of 2.7 mmol/L (normal, 1.15 to 1.35 mmol/L). The 24-hour urine calcium and phosphate excretions are 290 and 856 mg, respectively.
Given the patient's hypoalbuminemia of 2.8 g/dL, what is the corrected calcium level?
What is the explanation for the patient's polyuria and polydipsia?
Based solely on the patient's admission electrolyte levels, what is the likely diagnosis?
What is the most likely explanation for the multiple bone lesions in this patient?
What is the special laboratory test that needs to be performed in this patient?
What is the best localizing procedure in patients such as this one?
Case Discussion
Given the patient's hypoalbuminemia of 2.8 g/dL, what is the corrected calcium level?
As a rule, approximately 45% of the measured serum calcium is protein bound; 55% is diffusible. The protein-bound fraction is greater for albumin than for globulin. For a serum calcium level of 10 mg/dL, approximately 0.8 mg/dL is bound to globulin and 3.7 mg/dL is bound to albumin. In the setting of a low albumin state, approximately 1 g of albumin binds 0.8 mg of calcium. For example, this patient has
P.116
a serum calcium level of 11.5 mg/dL and a serum albumin level of 2.8 g/dL. The corrected calcium level is calculated as follows:
What is the explanation for the patient's polyuria and polydipsia?
Hypercalcemia causes a vasopressin-resistant nephrogenic diabetes insipidus. This can promote dehydration in hypercalcemic patients, thereby aggravating the symptoms and worsening the hypercalcemia.
Based solely on the patient's admission electrolyte levels, what is the likely diagnosis?
The electrolyte levels in this patient strongly support a diagnosis of primary hyperparathyroidism. Hypophosphatemia is seen in nearly 40% to 60% of patients with hyperparathyroidism, and its presence depends on the dietary phosphate intake. In addition, the chloride concentration greater than 104 mmol/L and the serum bicarbonate value in the mildly acidotic range suggest hyperparathyroidism. A chloride-to-phosphate ratio of greater than 33 is seen in the setting of hyperparathyroidism. In this patient, this ratio is 54, which indicates PTH-mediated hypercalcemia. An elevated 1,25-dihydroxyvitamin D level may be seen in patients with primary hyperparathyroidism, but, if there is magnesium deficiency, these levels may be normal or low.
What is the most likely explanation for the multiple bone lesions in this patient?
The turnover state of bone formation and resorption is high in patients with hyperparathyroidism. The classic histologic picture found in bone biopsy specimens is an increased number of osteoclasts, together with increased tetracycline labeling and increased rates of bone formation. The marrow in these patients may show focal areas of fibrosis. In extremely advanced cases of hyperparathyroidism, osteoclastomas or giant cell tumors of bone may be seen. This patient had multiple such tumors.
What is the special laboratory test that needs to be performed in this patient?
The special laboratory test that needs to be done in this patient is measurement of his PTH level, which proves to be markedly elevated to a value of 811 pg/mL (normal, 10 to 65 pg/mL).
What is the best localizing procedure in patients such as this one?
Approximately 80% to 90% of patients with primary hyperparathyroidism have a single parathyroid adenoma, 10% to 15% have parathyroid hyperplasia, and less than 1% have parathyroid carcinoma. The preoperative localizing procedures such as a Sestamibi scan add to the management of this disease as they permit a directed minimally invasive approach. However, the most reliable approach to localization is an experienced surgeon who can, in almost all cases, remove the adenoma or identify the parathyroid hyperplasia and remove three and one-half glands. This patient proved to have a large, 16-g parathyroid adenoma, which was identified easily and removed.
P.117
Suggested Readings
Bilezikian JB. Management of acute hypercalcemia. N Engl J Med 1992;326:1196.
Bilezikian JP, Brandi ML, Rubin M, et al. Primary hyperparathyroidism: new concepts in clinical, densitometric and biochemical features. J Intern Med 2005;257(1):6 17.
Broadus AE, Mangin M, Ikeda K, et al. Humoral hypercalcemia of cancer: identification of a novel parathyroid hormone-like peptide. N Engl J Med 1988;319:556.
Colao A, Di Sarno A, Landi ML, et al. Long-term and low-dose treatment with cabergoline induces macroprolactin shrinkage. J Clin Endocrinol Metab 1997;82:3574.
Consensus Development Conference. Diagnosis and management of asymptomatic primary hyperparathyroidism. Ann Intern Med 1991;114:593.
Davies PH, Stewart SE, Lancranjan L, et al. Long-term therapy with long-acting octreotide (Sandostatin-LAR) for the management of acromegaly. Clin Endocrinol 1998;48:311.
De Luca F, Baron J. Molecular biology and clinical importance of the Ca(2+)-sensing receptor. Curr Opin Pediatr 1998;10:435.
Henderson JE, Shustik C, Kremer R, et al. Circulating concentrations of parathyroid hormone-like peptide in malignancy and in hyperparathyroidism. J Bone Miner Res 1990;5:105.
Inzucchi SE. Management of hypercalcemia. Diagnostic workup, therapeutic options for hyperparathyroidism and other common causes. Postgrad Med 2004;115(5):27 36.
Inzucchi SE. Understanding hypercalcemia: its metabolic basis, signs and symptoms. Postgrad Med 2004;115(4):69 76.
Lufkin EG, Kao PC, Heath H. Parathyroid hormone radioimmunoassays in the differential diagnosis of hypercalcemia due to primary hyperparathyroidism or malignancy. Ann Intern Med 1987;160:559.
Muratori M, Arosio M, Gambino G, et al. Use of cabergoline in the long-term treatment of hyperprolactinemic and acromegalic patients. J Endocrinol Invest 1997;20:537.
Ralston SH, Gallacher SJ, Patel U, et al. Cancer-associated hypercalcemia: morbidity and mortality. Ann Intern Med 1990;112:499.
Yeh PJ, Chen JW. Pituitary tumors: surgical and medical management. Surg Oncol 1997;6:67.
Hypoglycemia
What constitutes medically significant hypoglycemia?
What are the common symptoms of hypoglycemia?
What is the best first step in classifying hypoglycemia?
What are the causes of medically significant hypoglycemia?
In people with diabetes, what factors are associated with an increased risk of hypoglycemia?
What is reactive hypoglycemia and how should it be evaluated?
Discussion
What constitutes medically significant hypoglycemia?
Medically significant hypoglycemia is diagnosed on the basis of only three findings (Whipple's triad): (a) blood glucose level of less than 50 mg/dL; (b) the
P.118
presence of symptoms consistent with hypoglycemia; and (c) the resolution of symptoms after the ingestion of carbohydrates. The lower limit of normal for glucose is 70 mg/dL, but this is the lower limit for healthy people after a 12-hour fast. During a 72-hour fast, up to 40% of healthy women may have blood glucose values below 45 mg/dL and some as low as between 20 and 30 mg/dL. These low values may also be seen in apparently healthy women 3 to 4 hours after the administration of 75 g of glucose orally (the oral glucose tolerance test), but almost none have symptoms of hypoglycemia and, therefore, medically significant hypoglycemia. Conversely, many people who exhibit symptoms consistent with hypoglycemia 3 to 4 hours after eating, which respond to the ingestion of carbohydrate, also do not have true hypoglycemia. The blood glucose levels in these individuals are rarely less than 50 mg/dL at the time they experience symptoms. These people have a condition that has been called postprandial syndrome or functional hypoglycemia.What are the common symptoms of hypoglycemia?
The symptoms of hypoglycemia can be divided into two categories: adrenergic and neuroglycopenic (Table 3-5). A substantial reduction in the blood glucose level stimulates the release of cortisol, GH, glucagon, and catecholamines. The attendant rise in sympathetic nervous system activity is experienced as nervousness, sweating, and palpitations. Because the brain is
P.119
critically dependent on glucose for normal neuronal functioning, inadequate delivery of glucose to the brain rapidly results in alterations in mentation, which can take many forms. The signs and symptoms of neuroglycopenia can even mimic those associated with structural brain lesions or psychiatric conditions.Table 3-5 Symptoms of Hypoglycemia
Adrenergic Neuroglycopenic Anxiety Headache Nervousness Blurred vision Tremulousness Paresthesias Sweating Weakness Hunger Tiredness Palpitations Confusion Irritability Dizziness Pallor Amnesia Nausea Incoordination Flushing Abnormal mentation Angina Behavioral change Feeling cold Difficulty waking in the morning Senile dementia Organic personality syndrome Transient hemiplegia Transient aphasia Seizures Coma What is the best first step in classifying hypoglycemia?
There are a variety of methods for categorizing the conditions that cause hypoglycemia, but none of these schemes is completely satisfactory. One approach is to divide the causes into those involving increased insulin levels, those involving increased glucose consumption, or those involving decreased glucose production. In reality, however, most of the causes of hypoglycemia embrace a combination of these mechanisms. An alternative and more useful scheme is based on the history and physical examination findings. The key features of this approach are to assess whether the hypoglycemia occurs with fasting or postprandially, and whether the affected person appears healthy. In general, the hypoglycemia that occurs with fasting or that is found in people who appear generally ill is a more ominous form of the disorder.
What are the causes of medically significant hypoglycemia?
The specific causes of hypoglycemia are numerous (Table 3-6). The history, physical examination, and initial laboratory tests are performed in an effort to rule out the common causes.
The most common cause of hypoglycemia overall is the administration of a hypoglycemic agent, either insulin or an oral hypoglycemic agent. These medications may have been prescribed for the control of diabetes or may be ingested in error. If this cause is not obvious from the patient's history, the diagnosis can be made by performing an oral hypoglycemic screen on a sample of plasma, or by measuring the insulin and C peptide level at the time of hypoglycemia. C peptide is a by-product of endogenous insulin production. If the insulin producing hypoglycemia is exogenous, the insulin level is high and the C peptide level is suppressed.
In one series consisting of hospitalized patients with hypoglycemia, the second most common cause of hypoglycemia was renal failure. Renal failure causes hypoglycemia for several reasons. First, because the kidneys play an important role in insulin clearance, insulin clearance may be decreased and insulin levels inappropriately high in the presence of renal failure. Second, during prolonged fasting, the kidneys may be responsible for as much as 30% of the net gluconeogenesis that takes place, and this would be compromised in the setting of renal failure. Finally, it appears that uremic toxins may suppress hepatic glucose output. As with other forms of hypoglycemia, inadequate caloric intake during a medical illness often contributes to the development of hypoglycemia.
Hypoglycemia may occur in association with a number of tumors including islet cell tumors and non islet cell tumors (Table 3-7). The latter are usually large tumors located in the mediastinum or retroperitoneum. The mechanism by which these tumors cause hypoglycemia remains somewhat obscure. One
P.120
explanation may be high levels of glucose extraction and utilization by the tumor mass. A second contributing feature is poor nutrition in these patients. An increased activity of IGF-II has been shown in some patients with non islet cell tumors. IGF-II can interact with the insulin receptor, although with less affinity than insulin itself. Normally, IGF-II cleaves to a smaller protein with minimal insulin-like activity. It has been shown that although the IGF-II levels are not increased in these patients with hypoglycemia associated with cancer, there are increased levels of big IGF-II. This is the uncleaved form of the hormone that has more insulin-like activity.Table 3-6 Etiologic Classification of Hypoglycemia
- Hypoglycemia associated predominantly with fasting
- Hypersecretion of insulin due to islet cell adenoma, carcinoma, hyperplasia, or nesidioblastosis
- Hepatic disease
- Generalized hypofunction
- Ethanol hypoglycemia associated with prior poor nutrition and decreased glycogen stores
- Sepsis
- Endocrine deficiencies
- Anterior pituitary insufficiency growth hormone, adrenocorticotropic hormone
- Adrenocortical insufficiency
- Hypothyroidism
- Large nonislet cell tumors
- Renal disease
- Deficient carbohydrate stores or intake
- Severe inanition
- Severe exercise
- Autoimmune with insulin antibodies or antibodies to the insulin receptor
- Drug induced
- Reactive or stimulative hypoglycemia
- Idiopathic functional hypoglycemia
- Alimentary hyperinsulinism
- Prediabetic functional hypoglycemia
- Endocrine deficiencies
- Factitious and artifactual hypoglycemia
- Surreptitious insulin administration
- Surreptitious sulfonylurea ingestion
- Elevated leukocyte count leukemia or polycythemia
- Hypoglycemia of infancy
- Abnormalities in hormone secretion
- Abnormalities of production and utilization of metabolic fuels
- Abnormalities in substrate availability
Another common cause of hypoglycemia is the ingestion of a drug that stimulates peripheral glucose utilization, inhibits hepatic glucose production,
P.121
or stimulates insulin release, and there are a large number of such drugs. The drugs most often implicated are in part a function of the age of the patient (Table 3-8). Alcohol may actually be the most common drug associated with hypoglycemia because it causes an increase in the ratio of nicotinamide adenine dinucleotide hydrogenase (NADH) to NAD+, which decreases the gluconeogenic capacity of the liver. The antiparasitic drug pentamidine is now widely used in the treatment of Pneumocystis carinii pneumonia in patients with AIDS. It can produce hypoglycemia by injuring the pancreatic islet cells, thereby causing insulin release and inappropriate hyperinsulinemia. As with all forms of hypoglycemia, inadequate caloric intake often contributes to the development of symptomatic hypoglycemia.Table 3-7 Non Islet Cell Tumors Associated with Hypoglycemia
- Mesenchymal
- Mesothelioma
- Fibrosarcoma
- Rhabdomyosarcoma
- Leiomyosarcoma
- Liposarcoma
- Hemangiopericytoma
- Carcinomas
- Hepatic: hepatoma, biliary carcinoma
- Adrenocortical carcinoma
- Genitourinary: hypernephroma, Wilms' tumor, prostate carcinoma
- Reproductive: cervical carcinoma, breast carcinoma
- Neurologic and neuroendocrine
- Pheochromocytoma
- Carcinoid tumor
- Neurofibroma
- Hematologic
- Leukemias
- Lymphoma
- Myeloma
Leukemia and polycythemia vera can cause pseudohypoglycemia because of the high WBC or Hct value in these settings, which can result in continued glucose consumption in the test tube after the blood sample has been obtained. In this situation, the blood glucose level is extremely low but the patient is without symptoms. To determine the actual blood glucose level in such patients, blood should be drawn into a tube that contains a substance that poisons the blood elements and prevents glycolysis from occurring after collection.
Postprandial (reactive) hypoglycemia can occur in as many as 20% of patients after gastric surgical procedures. This condition is also called
P.122
alimentary hypoglycemia and can occur after a variety of procedures, including gastric bypass, gastrectomy, gastroenterostomy, pyloroplasty, and vagotomy. Although biochemical hypoglycemia is not rare in these patients during a long oral glucose tolerance test, symptomatic hypoglycemia is uncommon.Table 3-8 Drugs Associated with Hypoglycemia in a Variety of Age-groups
Age Range (yr) No. of Patients Drugs Most Frequently Used (No. of Cases) Newborn 47 Sulfonylurea (mother) (14); propranolol (19); ritodrine, etc. (14) 0-2 26 Salicylate (17); propranolol (9) 2-10 48 Alcohol (28); quinine (15); propranolol (3); sulfonylurea (2) 11-30 79 Sulfonylurea (34); insulin (factitious) (20); quinine (10); alcohol
(8); insulin + druga(3); insulin + alcohol (2); propranolol (2)31-40 78 Alcohol (50); sulfonylurea (14); quinine (4); insulin + alcohol
(3) or drug (3); insulin (factitious) (2); propranolol (2)41-50 71 Alcohol (33); sulfonylurea (19); insulin + alcohol (5); pro pranolol
(3); alcohol + drug (2); quinine (2); disopyramide (1)51-60 177 Sulfonylurea (86); alcohol (72); propranolol (4);
sulfonylurea + insulin (3) or alcohol (3) or drug (3);
disopyramide (3); quinine (1)61-70 242 Sulfonylurea (173); alcohol (35); sulfonylurea + drug (10) or
phenylbutazone (8) or insulin (4); disopyramide (5); pro
pranolol (3)Older than 71 273 Sulfonylurea (219); alcohol (23); sulfonylurea + drug (12) or
phenformin (6); disopyramide (5); propranolol (3)Total 1,041 (69%)b aAn agent without intrinsic hypoglycemic activity. bPercentage of 1,418 patients for whom data were available. Other causes of hypoglycemia are much less common. Fasting by itself is a rare cause. However, extremely long periods of inadequate nutrition are required for hypoglycemia to occur in the absence of other metabolic defects. This is seen in the setting of anorexia nervosa and starvation. Likewise, liver disease produces hypoglycemia only in its most severe forms or in conjunction with inadequate caloric intake. Hypoglycemia is occasionally produced by the presence of autoantibodies either to insulin itself or to the insulin receptor, but these conditions usually occur in the presence of a known autoimmune syndrome. Finally, age plays an important role in the susceptibility to hypoglycemia. Elderly people lose counterregulatory hormone responses to hypoglycemia, are frequently on multiple medications, and may have mild organ dysfunction (renal failure, liver dysfunction, or congestive heart failure), all of which can increase the likelihood of multifactorial hypoglycemia. In addition, elderly patients may have dementia that can interfere with insight and normal food-seeking behavior.
- Hypoglycemia associated predominantly with fasting
In people with diabetes, what factors are associated with an increased risk of hypoglycemia?
Hypoglycemia occurs all too frequently in treated diabetic patients, and is either directly or indirectly the cause of death in 3% to 5% of all patients with T1DM. It results from excessive insulin administration, inadequate caloric intake, or excessive exercise. In nondiabetic people, if hypoglycemia develops, a number of hormones respond to increased glucose production and maintain a normal blood sugar level. In addition, the person notices symptoms of hypoglycemia and ingests carbohydrate to counteract these. In people with long-standing diabetes, however, there may be hypoglycemic unawareness and autonomic neuropathy, both of which blunt the normal response to hypoglycemia. Another factor that plays a role in the hypoglycemia that occurs in diabetic patients has to do with the introduction of recombinant human insulin, such that very few patients now remain on purified pork insulin.
During the development of T1DM, there may be a period when islet cells are damaged but still retain their capacity to synthesize and store insulin. During this period, insulin may be released in a dysfunctional manner in response to nonphysiologic stimuli, or in inappropriate quantities. This may result in episodes of symptomatic hypoglycemia, but, later, as the islet cells are completely destroyed, insulinopenia and hyperglycemia predominate.
Historically, there has been a strong desire to normalize the blood glucose levels in patients with diabetes in an effort to prevent long-term complications. With the advent of home glucose monitoring, multiple daily injections of short- and long-acting insulins, insulin pumps, and glycosylated Hgb determinations, tight control is attainable. What has been learned, however, is that there is a trade-off, in that tight control can be achieved only by accepting a substantial increase in the risk of symptomatic and life-threatening hypoglycemia. The DCCT has demonstrated that tight control of blood glucose in patients with T1DM prevents or delays the development of diabetic complications. It is prudent to strive toward tight control while avoiding frequent hypoglycemic episodes.
What is reactive hypoglycemia and how should it be evaluated?
The term hypoglycemia is recognized by much of the lay public as a common problem that occurs at 10:30 a.m. in women whose breakfast consisted of a cup of coffee and a strawberry Danish. Some physicians have evaluated these reactive hypoglycemic symptoms with oral glucose tolerance tests. However, this approach is problematic because most of the women with these symptoms do not have blood glucose levels that are less than 50 mg/dL at the time of their symptoms and, in fact, most of these symptoms resolve spontaneously without the ingestion of carbohydrate. In addition, some healthy women can have blood glucose values that are less than 50 mg/dL 3 to 4 hours after a 100-g oral glucose load, and yet not have symptoms. In general, these people do not have a serious illness and virtually never have an insulinoma in the absence of more typical episodes that occur with fasting. Instead, they need reassurance and a practical approach to their symptoms. Diets that are low in carbohydrate, high
P.124
in protein, and high in fiber have not been conclusively shown to be of benefit, and extreme diets should be avoided. The regular ingestion of a balanced diet in perhaps four to five meals over the course of the day instead of the traditional three may be of benefit in these people. In some cases, reactive hypoglycemia may be a result of insulin resistance, hyperinsulinemia in response to a high carbohydrate meal, and mismatch of insulin and glucose clearance after the meal. In these cases, hypoglycemia may respond to insulin sensitizers as well as the above measures.
P.123
Case
A 52-year-old white woman has an 18-month history of episodic confusion and poor work performance but neurologic evaluation, including CT scan of the head and an electroencephalogram, is unrevealing. Dilantin and then phenobarbital are prescribed but do not alter the frequency of the attacks, and are eventually discontinued. On the day of admission, she has a generalized seizure at work. The paramedics are called, find her unconscious, and administer naloxone hydrochloride [Narcan (Du Pont Merck Pharmaceutical, Wilmington, DE)] and 1 ampule of 50% dextrose IV. She then regains consciousness. Her blood glucose level before receiving the 50% dextrose is 28 mg/dL. She denies consuming alcohol or taking any prescription medications. Her family history is unremarkable and she has no history of gastric surgery. On physical examination, she is found to be a thin woman who appears to be in good health. Her examination findings are normal, as are her initial laboratory results.
What is the likely diagnosis in this patient?
If she had a family history of this problem, what other endocrine tumors would you look for?
What diagnostic test, or tests, are useful in making this diagnosis?
If the results of the biochemical studies indicate she has an insulinoma, what should the next test be?
What is the proper therapy for an insulinoma?
Case Discussion
What is the likely diagnosis in this patient?
The patient's history suggests the presence of an insulinoma because the hypoglycemia is severe, recurrent, progressive, symptomatic, and reversed by the administration of IV glucose. The symptomatic episodes of hypoglycemia associated with an insulinoma may occur in the postprandial state, but almost never exclusively in this state (Table 3-9). Most people with adrenal insufficiency, tumor-associated hypoglycemia, or alimentary hypoglycemia have other signs or symptoms, appear ill, or have a known surgical history.
If she had a family history of this problem, what other endocrine tumors would you look for?
There are three generally recognized syndromes of MEN [or multiple endocrine adenomatosis (MEA)]. People with MEN 1 can have tumors of the pituitary (e.g.,
P.125
prolactinomas or Cushing's disease), the pancreas (insulinoma and gastrinoma most commonly), or the parathyroid glands. Usually, hypercalcemia due to hyperparathyroidism develops first. Those affected with MEN 2A are at risk for medullary carcinoma of the thyroid, pheochromocytoma, and, less commonly, hyperparathyroidism. All these features can be found in people with MEN 2B, together with mucosal neuromas. These syndromes can occur either in families or sporadically. In all patients with insulinomas, the serum calcium and prolactin levels should be checked and a complete history and physical examination performed to look for evidence of the other potentially associated conditions. Among the cases of sporadic nonfamilial insulinomas, 80% are solitary and benign, 11% are multiple and benign, and 6% are single and malignant. The remaining 3% of the patients have multiple malignant tumors or islet hyperplasia. Ten percent of the insulinomas occur in association with MEN 1, and are multifocal 80% of the time.Table 3-9 Association of Hypoglycemia Symptoms with Eating in People with Insulinoma
Timing of Symptoms No. of Patients Percentage of Total 1. Symptoms during or after overnight fast
only (before breakfast)20 26 2. Fasting and daytime postprandial (before
lunch or dinner) symptoms21 27 3. Symptoms after missed meal only 6 8 4. Postprandial (before lunch and dinner)
symptoms only23 29 5. Uncertain about timing of symptoms 7 9 6. No symptoms experienced 1/78 1/100 Symptoms exacerbated by exercise 24 31 What diagnostic test, or tests, are useful in making this diagnosis?
The traditional diagnostic approach in patients with a suspected insulinoma is a supervised 72-hour fast. If symptomatic hypoglycemia develops and the blood glucose level is less than 50 mg/dL, then insulin and C peptide levels should be determined. In one series of patients with insulinomas, hypoglycemia occurred in the first 12 hours of fasting in 29%, within 24 hours in 71%, within 48 hours in 92%, within 60 hours in 92%, and within 72 hours in 98%. In this series, the blood glucose level at the time symptoms appeared was less than 46 mg/dL in 100%, less than 39 mg/dL in 70%, less than 35 mg/dL in 50%, and less than 28 mg/dL in 25%. Because the insulin secretion from an insulinoma is often sporadic, not all insulinomas can be diagnosed on the basis of a single fast. It is important to determine the C peptide level to demonstrate that the insulin is produced endogenously. A proinsulin level can also be helpful in diagnosing insulinomas. Proinsulin is the prohormone from which insulin and C peptide are cleaved, and accounts for only 15% to 20% of the circulating immunoreactive insulin in healthy people. In those
P.126
with an insulinoma, however, it constitutes 30% to 90% of the insulin mass. There are other specialized tests for evaluating a patient with a suspected insulinoma but, although advocated by some, they are usually not necessary. A serum drug screen to rule out a drug-induced hypoglycemia and an ACTH stimulation test to rule out adrenal insufficiency are useful in the evaluation of hypoglycemia of unknown cause, but are not helpful in establishing the diagnosis of hyperinsulinism.If the results of the biochemical studies indicate she has an insulinoma, what should the next test be?
Once the biochemical diagnosis of insulinoma has been established, an anatomic study is usually done. Although no single study is completely satisfactory, abdominal CT scanning and ultrasonography possess a relatively high sensitivity, pose no risk to the patient, and are relatively inexpensive, making them a good first step. Abdominal ultrasonography is advocated by some as the superior study, but its utility varies from institution to institution. Angiography is more sensitive but carries some risk and is quite expensive. Some groups have advocated transhepatic venous sampling. In this method, by measuring insulin levels in the venous blood draining a particular region of the pancreas, the tumor can be localized, although not visualized. The newest preoperative localizing technique is endoscopic ultrasonography. In this technique, the ultrasound transducer is endoscopically placed in the duodenum. From this site, the head of the pancreas can be well visualized, yielding a better sensitivity than that of traditional abdominal ultrasonography. However, this technology is not yet widely available. The main problem with all these approaches is that most insulinomas are small (average, 1.5 cm, 2 g), and the diagnosis hinges on the clinical presentation and the results of biochemical studies. If the anatomic studies are unrevealing and the biochemical results are convincing, the patient should undergo exploratory surgery performed by an experienced pancreatic surgeon. For this reason, extensive preoperative anatomic studies are not advocated.
What is the proper therapy for an insulinoma?
Surgical removal performed by an experienced surgeon is the primary form of therapy for insulinomas. Intraoperative direct ultrasonographic examination of the pancreas combined with manual palpation by an experienced surgeon successfully localizes the tumor 80% to 90% of the time. Once the tumors are resected, most patients are cured. For those who are not cured by surgical means, long-acting somatostatin analogs can be used to decrease the frequency and severity of the hypoglycemic episodes. Diazoxide, verapamil, phenytoin, and propranolol have been used successfully in a few cases. In these patients, frequent scheduled meals are an important component of therapy.
Suggested Readings
Adrogue HJ. Glucose homeostasis and the kidney. Kidney Int 1992;42:1266.
Field JB. Hypoglycemia: definition, clinical presentations, classification, and laboratory tests. Endocrinol Metab Clin North Am 1989;18:27.
Fischer KF, Lees JA, Newman JH. Hypoglycemia in hospitalized patients: causes and outcomes. N Engl J Med 1986;315:1245.
P.127
Kurlan R. Postprandial (reactive) hypoglycemia and restless leg syndrome: related neurologic disorders? Mov Disord 1998;13:619.
Leonetti F, Foniciello M, Iozzo P, et al. Increased nonoxidative glucose metabolism in idiopathic reactive hypoglycemia. Metabolism 1996;45:606.
Piedrola G, Cassado JL, Lopez E, et al. Clinical features of adrenal insufficiency in patients with acquired immunodeficiency syndrome. Clin Endocrinol 1996;45:97.
Ross RJ, Trainer PJ. Endocrine investigation: Cushing's syndrome. Clin Endocrinol 1998;49:153.
Seltzer HS. Drug-induced hypoglycemia: a review of 1418 cases. Endocrinol Metab Clin North Am 1989;18:163.
Service FJ. Hypoglycemias. West J Med 1991;154:442.
Service FJ. Hypoglycemia. Endocrinol Metab Clin North Am 1997;26:937.
Soderbergh A, Winqvist O, Norheim I, et al. Adrenal autoantibodies and organ-specific autoimmunity in patients with Addison's disease. Clin Endocrinol 1996;45:453.
Metabolic Bone Disease
Which diseases of bone are considered to be metabolic in origin?
What is osteopenia?
What conditions may cause osteopenia?
What are the risk factors for osteoporosis?
What simple laboratory tests can help assess the patient with osteopenia?
When are bone density measurements indicated?
Discussion
Which diseases of bone are considered to be metabolic in origin?
Metabolic bone diseases are those conditions in which all the metabolic bone units throughout the skeleton are equally affected by the disease process. These diseases include osteoporosis, osteomalacia, osteitis fibrosa cystica, and osteogenesis imperfecta. Diseases that affect either a single area or multiple areas in bone are considered localized bone diseases, and include Paget's disease, fibrous dysplasia, bone cysts, healing fractures, Sudeck's atrophy, and injury disuse osteoporosis.
What is osteopenia?
Osteopenia constitutes a diagnosis based on radiographic findings, in that the mineral content of the bones is seen to be reduced on radiography. Usually, before these studies can show bone loss, however, approximately 30% to 40% of the skeleton must have demineralized. In addition, osteopenia is now defined as a bone density that is 1 to 2.5 standard deviations below the mean for young women, on dual energy x-ray absorptiometry (DEXA) imaging. Any of the metabolic bone conditions listed can cause osteopenia.
What conditions may cause osteopenia?
There are many disease processes to be considered in the osteopenic patient. The condition most often encountered in such patients is age-related,
P.128
idiopathic osteoporosis. Type I osteoporosis is postmenopausal osteoporosis and is usually manifested clinically by vertebral fractures; type II osteoporosis has been termed senile osteoporosis and is characterized by hip fracture.There are many secondary causes of osteopenia seen in the setting of nutritional deficiency; renal, liver, gastrointestinal, and endocrine and metabolic disease; drug usage; and certain lifestyles (Table 3-10). In many of these conditions, alterations in the calcium level or vitamin D metabolism, secondary hyperparathyroidism, osteomalacia, acidosis, or a combination of these conditions is the underlying mechanism responsible for the osteopenia.
What are the risk factors for osteoporosis?
Smoking, poor calcium intake, immobilization, malnutrition, a hypogonadal state, and a family history are all risk factors for osteoporosis. Smoking is a risk factor because it induces hepatic enzymes to inactivate circulating sex hormones, such as estrogen. A hypogonadal state can occur in either men or women, but in women it may result from a total hysterectomy and oophorectomy or from the spontaneous menopausal state, both of which lead to lowered estrogen levels. Other factors include the ingestion of soft drinks, most of which contain phosphoric acid. This substance increases the ingested phosphate load, which in turn depresses the serum calcium level and stimulates PTH release. Coffee is a calciuretic substance, and, as such, excessive consumption contributes to osteoporosis. The fat cell can act as an endocrine organ; therefore, in lean people whose fat cell mass is decreased, the conversion of adrenal androgens to estrogens is decreased, and this can lead to osteoporosis. Some of the lifestyle risk factors can be modified to prevent osteoporosis.
What simple laboratory tests can help assess the patient with osteopenia?
A complete blood count with erythrocyte sedimentation rate and a standard serum chemistry profile that includes electrolyte, calcium, phosphate, alkaline phosphatase, creatinine, BUN, calcium, and phosphate measurements plus liver function tests are the simple blood tests needed. A 24-hour urine specimen is obtained for determination of the calcium, phosphate, and creatinine content. Bone densitometry establishes the severity of bone loss. All these laboratory tests can be used to quickly assess the patient with osteopenia. If the patient has anemia and an elevated sedimentation rate, the clinician should consider the possibility of a multiple myeloma and have either a serum protein or urine protein electrophoresis, or both, performed. Abnormalities in calcium balance can be assessed by identifying hypocalcemic or hypercalcemic disorders. Abnormalities of liver and kidney function represent secondary causes of osteoporosis. The electrolyte levels help suggest the presence of renal tubular acidosis syndromes. Alkaline phosphatase is a marker of bone osteoblast function and its measurement helps identify those patients with high-turnover osteoporosis or osteomalacia. A 24-hour urine calcium determination can identify patients who have idiopathic hypercalciuria or low urine calcium losses, suggesting a calcium-deficient state. An extremely low urine phosphate value may reflect the consumption of a vegetarian diet. A 25-hydroxy vitamin
P.129
P.130
D level assesses for vitamin D deficiency, a condition now recognized to be more common than previously appreciated. Diagnosis is important, as repletion is essential before bisphosphonate therapy. Other laboratory tests, including measurement of the PTH level, serum osteocalcin level, and urine hydroxyproline or hydroxypyridinium, are reserved for those patients in whom these are specifically indicated. A bone density measurement establishes the presence or absence of significant osteoporosis.Table 3-10 Causes of Osteopenia
- Idiopathic age-related
- Juvenile
- Young adults
- Postmenopausal (type I)
- Senile (type II)
- Secondary to disease states
- Metabolic conditions
- Calcium deficiency
- Vitamin D deficiency states
- Malnutrition
- Idiopathic hypercalciuria
- Renal tubular acidosis and other systemic acidosis
- Diabetes mellitus
- Scurvy
- Endocrine conditions
- Thyrotoxicosis
- Cushing's syndrome
- Male and female hypogonadal state
- Hypoamenorrheic female runners
- Prolactinoma
- Hyperparathyroidism
- Renal disease
- Gastrointestinal-liver disease
- InheriTable connective tissue disease
- Osteogenesis imperfecta
- Homocystinuria
- Ehlers-Danlos syndrome
- Marfan's syndrome
- Bone marrow infiltration
- Multiple myeloma
- Lymphoma
- Leukemia
- Drugs
- Dilantin
- Phenobarbital
- Thyroid hormone
- Corticosteroids
- Prolonged heparin therapy
- Lifestyle
- Nutrition
- Alcohol
- Smoking
- Inactivity
- Immobilization
- Excessive coffee and soft drinks
- Miscellaneous
- Rheumatoid arthritis
- Systemic mastocytosis
- Metabolic conditions
- Idiopathic age-related
When are bone density measurements indicated?
The National Osteoporosis Foundation has recommended that bone mineral density be measured in all postmenopausal women younger than 65 years with one or more risk factors for osteoporosis other than menopause, and in all women older than 65. Formal recommendations for screening in men do not exist at this time, but screening should be considered in men with risk factors (especially hypogonadism, steroid use, or untreated hyperparathyroidism) or after a fracture.
Case
A thin, 55-year-old, white, postmenopausal woman is seen in her primary care clinic because of muscle aches and weakness. She has been seen by numerous physicians for evaluation of this condition, and has been referred to the psychiatry department for treatment of a stress reaction. The patient's past medical history is significant for a gastrectomy approximately 15 years earlier for the treatment of peptic ulcer disease. She has noticed loose stools since that time. The patient admits to a poor calcium intake, but otherwise consumes a nonvegetarian diet. She suffers hot flashes and insomnia, but has never been evaluated for estrogen therapy. During her evaluation, osteopenic changes are noted on the chest film. The patient's laboratory evaluation reveals the following findings: calcium, 8.4 mg/dL (normal, 8.7 to 10.3 mg/dL); phosphate, 2.0 mg/dL (normal, 2.7 to 4.5 mg/dL); chloride, 108 mEq/L; sodium, 145 mEq/L; potassium, 4.5 mEq/L; CO2, 23 mEq/L; and albumin, 4.1 g/dL. Her kidney and liver function test results are normal. The alkaline phosphatase level is elevated to 380 IU/L (normal, 39 to 117 IU/L). Her 24-hour urine excretion of calcium is 40 mg (normal, 100 to 300 mg); creatinine, 1.1 g; total hydroxyproline, 86 mg (normal, 25 to 77 mg); and phosphate, 780 mg (normal, 400 to 800 mg). The osteocalcin level is 7.1 ng/mL (normal, 1.8 to 6.6 ng/mL).
What are the risk factors for osteoporosis in this patient?
On the basis of the patient's history and laboratory findings, what is the differential diagnosis?
What additional laboratory tests should be obtained in this patient?
On the basis of the laboratory findings, what would you anticipate the bone biopsy specimen to show?
What should the treatment be in this patient?
What would you advise this patient regarding the advantages and disadvantages of estrogen replacement therapy?
P.131
Case Discussion
What are the risk factors for osteoporosis in this patient?
This thin, white, postmenopausal woman with poor calcium intake is at risk for osteoporosis.
On the basis of the patient's history and laboratory findings, what is the differential diagnosis?
This patient's history suggests that, at her age of 55 years, she is entering a postmenopausal state, as indicated by the hot flashes and insomnia. In addition, poor calcium balance may be likely because of her lifelong history of poor calcium intake and the gastrectomy for peptic ulcer disease, which could lead to poor vitamin D absorption. Confirming a state of negative calcium balance is the patient's hypocalcemia, low urine calcium excretion, and electrolyte levels, all of which suggest the presence of secondary hyperparathyroidism with hyperchloremia and low serum phosphate levels.
What additional laboratory tests should be obtained in this patient?
The patient may be deficient in vitamin D. Measuring the 25-hydroxyvitamin D level, which is the major circulating form of vitamin D, can establish the diagnosis of simple vitamin D deficiency. Some patients may also have a deficiency of 1,25-dihydroxyvitamin D, particularly older patients and those with renal disease. A PTH value can establish the diagnosis of secondary hyperparathyroidism due to a calcium-deficient state stemming from the vitamin D deficiency. Once treatment is initiated, a PTH value that returns to normal confirms a state of normal calcium balance.
In this patient, the 25-vitamin D level is 10 ng/mL (normal, 16 to 74 ng/mL) and the PTH value is 120 pg/mL (normal, 10 to 65 pg/mL).
On the basis of the laboratory findings, what would you anticipate the bone biopsy specimen to show?
A tetracycline-labeled bone biopsy is performed by having the patient ingest 250 mg of tetracycline four times a day for 3 days, then withhold the tetracycline for 10 days, and then take tetracycline for another 3 days. These two tetracycline labels determine the rate of bone formation. Osteoclast counts can be determined from bone histomorphologic analysis, and the amount of tetracycline that has surfaced can be measured as an indication of active bone formation. This patient proved to have a high-turnover osteoporosis with an increased tetracycline surface and an increased osteoclast count, as borne out by the high PTH level. In addition, the high osteocalcin, alkaline phosphatase, and urinary hydroxyproline or pyridinium levels indicate a state of high bone turnover. Early in vitamin D deficiency (hypovitaminosis-D I), secondary hyperparathyroidism predominates, leading to a high-turnover osteoporosis. In the setting of severe vitamin D deficiency, especially childhood rickets (hypovitaminosis-D II and III), a low bone-turnover state exists in which there is little tetracycline uptake.
What should the treatment be in this patient?
This patient has a combined disorder of both estrogen and vitamin D deficiency contributing to her presumed osteopenia. There is no doubt that she will benefit
P.132
from vitamin D repletion and this should be initiated immediately. She will most likely not respond to small doses of vitamin D, but may require 50,000 units of vitamin D (ergocalciferol), given once or twice weekly, or calcifediol (Calderol), 20 to 50 g daily, because of the poor gastrointestinal absorption of vitamin D stemming from her gastrectomy. Before the release of the results of the Women's Health Initiative (WHI), this patient would also have been treated with hormone replacement therapy of daily estrogen with either daily or cyclic progesterone. Since the WHI, this has become a decision that requires careful consideration of the risks and benefits of hormone therapy described further below. Another effective means of treating osteoporosis is with bisphosphonates. The initial experience with increased bone density using bisphosphonates was with etidronate (Didronel). However, etidronate was never formally approved for osteoporosis and has been superceded by the newer oral bisphosphonates, alendronate (Fosamax), risedronate (Actonel), and recently ibandronate (Boniva). All three are available as oral formulations and have been shown to increase bone mineral density and decrease vertebral fracture risk. Alendronate and risedronate can be administered daily or weekly, ibandronate daily or monthly. Other bisphosphonates (pamidronate, etidronate, and zoledronate) are approved for bone preservation and serum calcium reduction in malignancy, but not for osteoporosis. Bisphosphonates should not be administered to patients with vitamin D or calcium deficiency before at least partial repletion.What would you advise this patient regarding the advantages and disadvantages of estrogen replacement therapy?
Estrogens are effective agents for treating osteoporosis by stabilizing bone density and preventing fractures. However, estrogen therapy alone in a patient with an intact uterus is associated in a dose-dependent manner with an increased incidence of endometrial cancer; that can be abolished by the addition of 10 to 14 days of a progestin at least three to four times annually. On the basis of observational studies, hormone replacement therapy was previously thought to have additional cardioprotective benefits. However, such benefits were not found in the large randomized portion of the WHI. This study, in fact, demonstrated that hormone replacement therapy is not without risk and may increase the risk for CV disease and stroke, as well as for breast cancer. A caveat to this conclusion is that a large portion of the women in the study were many years postmenopausal and may have responded very differently to hormone therapy than women who were recently estrogen sufficient. However, as the only large randomized controlled study to date, the WHI must be carefully considered when the decision to start estrogen for osteoporosis is made.
As a result of the WHI, estrogen therapy should now be considered as treatment for osteoporosis only in women who are symptomatically postmenopausal. Even in these women it is contraindicated in patients who have: (a) a personal history of estrogen-related neoplasia of the breast, (b) a personal or strong family history of breast carcinoma, (c) a personal history of thromboembolic disease or known vascular disease, or (d) significant CV risk factors, especially tobacco use, obesity, or hypertension. Estrogen therapy is relatively contraindicated in patients
P.133
with estrogen-related headaches. The use of estrogen therapy may also be associated with an increased incidence of gallstones. A marked triglyceride elevation may develop in some patients when estrogen therapy is initiated; hence, lipid levels need to be checked within 4 to 8 weeks of starting therapy. Patients who develop adverse lipid abnormalities to oral estrogens may do better with transdermal estradiol therapy.
Suggested Readings
Armanento-Villereal R, Villereal DT, Avioli LV, et al. Estrogen status and heredity are major determinants of premenopausal bone mass. J Clin Invest 1992;90:2464.
Berenson JR, Lipton A. Pharmacology and clinical efficacy of bisphosphonates. Curr Opin Oncol 1998;10:566.
Fulfaro F, Casuccio A, Ticozzi C, et al. The role of bisphosphonates in the treatment of painful metastatic bone disease: a review of phase III trials. Pain 1998;78:157.
Hodsman BA, Bauer DC, Dempster DW, et al. Parathyroid hormone and teriparatide for the treatment of osteoporosis: a review of the evidence and suggested guidelines for its use. Endocr Rev 2005;26:688 703.
Hofeldt FD. Proximal femoral fractures. Clin Orthop 1987;218:12.
Holick MF. Vitamin D: the underappreciated D-lightful hormone that is important for skeletal and cellular health. Curr Opin Endocrinol Diabetes 2002;9:87 98.
Jackson RD, Wactawski-Wende J, Lacroix AZ, et al. For the Women's Health Initiative Investigators. Effects of conjugated equine estrogen on risk of fractures and BMD in postmenopausal women with hysterectomy: results from the women's health initiative randomized trial. J Bone Miner Res 2006;21(6):817 828.
McClung M, Clemmesen B, Daifotis A, et al. Alendronate prevents postmenopausal bone loss in women without osteoporosis: a double-blind, randomized, controlled trial: Alendronate Osteoporosis Prevention Study Group. Ann Intern Med 1998;128:253.
Olszynski WP, Shawn DK, Adachi JD, et al. Osteoporosis in men: epidemiology, diagnosis, prevention, and treatment. Clin Ther 2004;26(1):15 28.
Parfitt AM, Rao DS, Stanciu AR, et al. Irreversible bone loss in osteomalacia: comparison of radial photon absorptiometry with iliac bone histomorphometry during treatment. J Clin Invest 1985;76:2403.
Riggs BL, Melton U. The prevention and treatment of osteoporosis. N Engl J Med 1992;327:620.
Rosen CJ. Postmenopausal osteoporosis. N Engl J Med 2005;353(6):595 603.
Rubin MR, Bilezikian JP. New anabolic therapies in osteoporosis. Endocrinol Metab Clin N Am 2003;32(1):285 307.
Srivastava AK, Vliet EL, Lewiecki EM, et al. Clinical use of serum and urine bone markers in the management of osteoporosis. Curr Med Res Opin 2005;21(7):1015 1026.
Stein E, Shane E. Secondary osteoporosis. Endocrinol Metab Clin N Am 2003;32:115 134.
Udell JA, Fischer MA, Brookhart MA, et al. Effect of the women's health initiative on osteoporosis therapy and expenditure in medicaid. J Bone Miner Res 2006;21(5):765 771.
Wimalawansa SJ. A four-year randomized controlled trial of hormone replacement and bisphosphonate, alone or in combination, in women with postmenopausal osteoporosis. Am J Med 1998;104:219.
P.134
Erectile Dysfunction
Case 1
A 65-year-old man presented with erectile dysfunction he had noted gradual onset of difficulty in achieving and maintaining an erection during the last 4 years. He had had hypertension for 10 years and has recently been told that his blood cholesterol level was high. His family history was positive for coronary artery disease, hypertension, and hypercholesterolemia.
The patient's medications included atenolol, 50 mg twice a day; hydrochlorothiazide, 50 mg per day; and aspirin, 325 mg per day. He had smoked a pack of cigarettes a day for 30 years, but quit 2 years earlier. He drank three beers each night.
Physical examination showed a BP of 160/90 mm Hg, the presence of arcus corneae, and an S4 heart sound. The liver and testicular examinations were normal, as were reflexes. The pedal pulses were diminished.
Laboratory test results were testosterone, 450 ng/dL (normal, 300 to 1,000 ng/dL); liver enzymes, normal; total cholesterol, 350 mg/dL; triglycerides, 300 mg/dL; and high-density lipoprotein (HDL), 25 mg/dL.
Case Discussion
From the history alone, it would be expected that this patient's erectile dysfunction had a vascular cause and perhaps iatrogenic exacerbation. Coronary artery disease is a risk factor for erectile dysfunction, and recent studies have suggested that merely having a history of hypercholesterolemia points to an underlying vascular etiology. His long-standing hypertension also suggests vascular disease.
This patient is taking two medications that have been associated with erectile dysfunction. Among the classes of currently used antihypertensive agents, -blockers and diuretics are most often at fault. Of the diuretics, hydrochlorothiazide is more of a problem than furosemide.
Smoking, of course, increases the risk of vascular disease. Excessive alcohol intake is directly toxic to the testicles and can result in decreased testosterone production. Alcohol is also directly toxic to the liver, and the resulting liver dysfunction can cause imbalance in testosterone and estradiol metabolism, which is often associated with gynecomastia.
The patient's BP reading indicates that his hypertension is inadequately controlled, and the S4 heart sound indicates that the hypertension is long standing and has affected his heart. The presence of arcus corneae signifies prolonged hypercholesterolemia. Diminished pedal pulses offer further evidence for vascular disease.
Hypogonadism cannot be reliably detected by clinical assessment alone; hence, measurement of the testosterone level was indicated. Liver function testing was performed in light of the history of significant alcohol intake. The lipid panel confirmed hypercholesterolemia.
There have been many studies on how to distinguish between psychogenic and vascular erectile dysfunction for example, by monitoring for nocturnal erections. No controlled study has shown that the methods change the management strategy; however,
P.135
the workup can be limited to the history, physical examination, and some laboratory testing to exclude other treatable causes of erectile dysfunction.
In this patient, atenolol and hydrochlorothiazide were replaced with enalapril. The patient was counseled on dietary changes that would help lower his cholesterol level. A vacuum pump device was prescribed for the erectile dysfunction.
Two months later, the patient's BP was normal. He reported successful resumption of sexual intercourse using the vacuum pump.
Evaluation of this patient's erectile dysfunction provided the opportunity to address the underlying hypertension and hypercholesterolemia. Otherwise, he might not have presented until a stroke or heart attack occurred.
Changing antihypertensive medication is especially important if the initiation of treatment and onset of erectile dysfunction coincide. In this case, a medication change was further justified because of inadequate BP control. ACE inhibitors do not appear to cause erectile dysfunction and calcium channel blockers rarely do, so these are the drugs that may be prescribed if medication is interfering with sexual functioning. Unfortunately, a change in antihypertensive medication alone is unlikely to restore erectile function.
Correction of this patient's blood lipids is long overdue. If dietary changes do not sufficiently improve his lipid profile within a few months, he will be a candidate for therapy with an HMG-CoA reductase inhibitor.
The vacuum pump device can treat erectile dysfunction in a case like this. The vacuum pump device consists of a Lucite tube and pump; the suction pulls blood into the penis. Once an erection has been produced, a rubber ring is placed at the base of the penis to maintain the erection. The vacuum pump has no major side effects, it can be used as often as the patient wishes, it can be used in all types of erectile dysfunction, and it has the highest success rate it is effective in 90% to 95% of cases. Obviously, it is not meant for a man who is not in a stable relationship, largely because of poor patient acceptance. There has been some speculation that vacuum pump devices might be contraindicated in patients taking warfarin because of the potential for ecchymosis from the ring, but studies have eliminated that concern.
Case 2
A 52-year-old man with diabetes presented with erectile dysfunction. His pubertal development had been normal. The diabetes had been diagnosed 15 years earlier. At the time of diagnosis, he had had problems with impotence that resolved as the hyperglycemia was brought under control. Erectile dysfunction had returned gradually during the last 2 years. He rarely had morning erections. The erectile dysfunction has created stress in his relationship with his wife.
The patient had taken an oral hypoglycemic agent for 5 years after diagnosis of diabetes and had been on insulin for the last 10 years. He had diabetic complications, including mild retinopathy, proteinuria, and mild peripheral neuropathy. Symptoms of gastroparesis had developed during the last 6 months.
His current insulin regimen consisted of 30 units of NPH (neutral protamine Hagedorn) and 15 units of regular insulin in the morning, and 10 units of NPH and 8 units of regular insulin in the evening.
Other medications included lisinopril (15 mg per day) and simvastatin (10 mg per day). He did not smoke or drink excessive amounts of alcohol.
P.136
Noteworthy findings on the physical examination included a BP reading of 120/80 mm Hg without significant orthostasis, retinopathy, absence of an S4 heart sound, and slightly soft testes. Sensation to pinprick on the calf was decreased.
Laboratory test results were serum testosterone, 200 ng/dL; total cholesterol, 150 mg/dL; triglycerides, 250 mg/dL; and HDL, 35 mg/dL. Glycosylated Hgb was 10% (normal, <6.5%).
Case Discussion
Diabetes is one of the most common causes of erectile dysfunction. A combination of vascular and neurologic disease is usually at fault, although hormone deficiency, medications, and psychogenic aspects also may be involved. All five components may be present in a single patient.
Men with T2DM often have acute erectile dysfunction at the onset of the disease, simply as a result of severe hyperglycemia. The mechanism of erectile dysfunction may include hypogonadotropic hypogonadism as well as metabolic and neurologic dysfunction (caused by glucose toxicity) in the testes. Vascular factors may also be involved because the hyperglycemia is usually associated with severe hyperlipidemia. The erectile dysfunction associated with new-onset diabetes may improve when hyperglycemia is brought under control.
In a patient with long-standing diabetes, the presence of other end-organ complications makes it more likely that erectile dysfunction is due to diabetes. In this patient, clinical assessment suggests a strong neurogenic component; the diminished sensation denotes peripheral neuropathy and the gastroparesis indicates autonomic neuropathy (although the lack of orthostasis suggests that the neuropathy is not severe). The proteinuria suggests a vascular component and, even though the absence of an S4 argues against that, it should be remembered that an S4 is not always present in diabetic patients with coronary artery disease.
Drug-induced erectile dysfunction does not appear to be an issue in this patient because neither the ACE inhibitor nor the HMG-CoA reductase inhibitor causes erectile dysfunction.
The testicular softness suggests a minor hormonal component, and indeed the testosterone level is slightly decreased. A low-normal or slightly low testosterone level is a typical finding in diabetic patients with erectile dysfunction. Although the reading confirms that hormone deficiency is one of his problems, a testosterone level of 200 ng/dL would not by itself cause significant hypogonadism and symptoms.
In addition, this patient's BP needs to be monitored; if it increases, he will need an additional antihypertensive medication because he is already taking a maximal dose of lisinopril. Increasing data suggest that tight control of BP with ACE inhibitors helps prevent both the renal and the vascular complications of diabetes. Consequently, aggressive antihypertensive therapy to lower BP to less than 130/85 mm Hg is indicated.
The patient's LDL level is low, but the triglyceride level is not optimal. If changes in his diet do not reduce the triglyceride level, he will be a candidate for treatment with gemfibrozil or a statin.
P.137
The patient was managed with intracavernosal injection of alprostadil and androgen replacement with a low-dose testosterone patch. He reported improved erectile function, increased energy, and a sense of well-being. In addition, the patient received dietary counseling and the insulin regimen was adjusted. The glycosylated Hgb decreased to 8%.
In diabetic patients with erectile dysfunction, injection of alprostadil into the corpora cavernosa of the penis can be effective. The treatment is particularly suited to diabetic patients because they often have neurologic complications, making the injections less painful than in other patients. In addition, those who are taking insulin are already familiar with needles and syringes and are less likely to be squeamish about injecting the penis. Intracavernosal injection is effective in approximately 65% of cases. The vacuum pump is held in reserve as second-line treatment.
Implantable penile prostheses were commonly used to treat erectile dysfunction in the 1970s and 1980s. They are used much less frequently today because they are expensive and may have many complications. Infection and poor wound healing are particular problems in diabetic patients, often necessitating removal of the implant at which point the option for injection therapy has been eliminated. However, some of the newer implants may be appropriate for young men with severe erectile dysfunction that does not respond to other therapy.
Intraurethral placement of vasoactive medication was introduced as an alternative to intracavernosal injections. However, several studies have shown it to be less effective, with a success rate as low as 30% in diabetic patients.
Side effects of intracavernosal injection include priapism and penile fibrosis. Patients with neurogenic or psychogenic erectile dysfunction should use a low dose of alprostadil. If the dose is too high, the risk of priapism is significant. When priapism occurs, the patient has to go to an emergency room, where he is treated with IV epinephrine or an 18-gauge needle that is inserted into the corpora cavernosa to withdraw blood. There have been only rare reports of more severe consequences, such as loss of the penis due to infarction.
The rate of priapism as a complication varies according to the agent used. Alprostadil has a much lower risk of priapism and fibrosis than do papaverine and phentolamine. However, alprostadil is more likely to cause a burning sensation. For that reason, it used to be mixed with papaverine, but papaverine has been withdrawn as a treatment for outpatients. Because of neuropathy, diabetic patients may not experience a burning sensation with alprostadil.
It is not clear whether better control of this patient's diabetes during the previous 10 years would have prevented erectile dysfunction. It seems logical that tight control of blood glucose levels will forestall erectile dysfunction, just as it can prevent retinopathy, renal failure, and macrovascular disease. Nevertheless, there are no prospective, double-blinded, placebo-controlled studies to confirm that long-term tight blood glucose control reduces the incidence of erectile dysfunction.
Case 3
A 48-year-old man had experienced acute onset of erectile dysfunction 6 months earlier. He had no other medical problems. Pubertal development had been normal. He was the father of three children.
P.138
On further questioning, the patient said that he had lost his job 4 months ago. He was having problems in his relationship with his wife, and had increased his alcohol consumption from two beers a week to four beers a day.
The physical examination was normal. The serum testosterone level was 450 ng/dL.
The patient was advised that his drinking was probably contributing to his erectile dysfunction and that he should reduce his intake. Referral for psychological counseling was offered, but he refused because of the cost. Instead, the physician discussed the patient's circumstances with him. A 6-week trial of yohimbine, 5.4 mg thrice a day, was prescribed.
The patient returned 8 weeks later and reported some improvement in erectile function.
He felt that yohimbine had been helpful; however, he had also found a job, was experiencing less psychological stress, and had reduced his alcohol consumption.
What was the major factor in this patient's erectile dysfunction?
How do you approach psychological erectile dysfunction?
What are the pharmacologic options for treatment?
Case Discussion
What was the major factor in this patient's erectile dysfunction?
Although the history in this case indicated that psychological stress was the major trigger for the erectile dysfunction, it was important to consider the possibility of other components. As noted, erectile dysfunction rarely results from an isolated cause. In this case, further questioning was needed to reveal that alcohol was almost certainly a major contributor.
Obtaining an accurate history of alcohol intake is notoriously difficult. Instead of asking the patient, Do you drink? ask, When you drink, do you drink beer, whiskey, or wine? After identifying the drink of choice, pick a large amount and let patients come down from there; with beer, for example, ask if they drink a six-pack at a time. Determining the true amount of alcohol intake often requires several discussions.
Also ask patients when they drink, because they may not understand that intermittent drinking can have persistent effects. Some patients who drank heavily on the weekend and nothing at all during the week may present with erectile dysfunction and painful right-sided gynecomastia (which was worse on Mondays). Their liver enzymes were not severely elevated, but the drinking had nevertheless caused a symptomatic imbalance of testosterone and estradiol.
In patients with a history of chronic alcohol abuse, liver function tests should be ordered. Their erectile function may not return even if they reduce their alcohol intake. Because this patient's increase in alcohol intake was fairly acute, his erectile function improved as soon as he began to drink less.
How do you approach psychological erectile dysfunction?
Despite the ubiquity of the psychological component in erectile dysfunction, there have been no controlled studies to show whether psychotherapy or counseling actually helps. Even assuming that such intervention would be helpful, there are no
P.139
data on the best approach. Should patients receive behavioral therapy? Counseling? Is simply talking with the primary care physician sufficient? considerations such as these can help in approaching psychological erectile dysfunction, but there are no clear answers.The primary care physician should at least acknowledge psychological stress as a component of erectile dysfunction. Sometimes acknowledging the problem is enough; the patient just needs to talk about it. Sometimes further intervention is required. Whether this is provided by the primary care physician depends on his or her level of comfort with that aspect of treatment. Insurance coverage is often an important factor as well.
What are the pharmacologic options for treatment?
The options for treatment of erectile dysfunction have radically changed with the introduction of sildenafil (Viagra), the first truly effective oral medication for this condition, and more recently approved related medications, vardenafil (Levitra) and tadalafil (Cialis).
Advances in our knowledge of the physiology of erection have facilitated understanding of the pharmacodynamics of sildenafil. Erection is initiated by dilation of the arterial bed, which increases blood flow and pressure; it is maintained by restriction of venous outflow. Previously it was believed that the parasympathetic system was critical in maintaining erection. Now, we know that the major player is the nonadrenergic, noncholinergic (NA-NC) system, which was identified 50 years ago but never studied in detail until relatively recently. The NA-NC system uses nitric oxide as a neurotransmitter. Through its second messenger, cyclic guanine monophosphate (cGMP), nitric oxide triggers relaxation of penile endothelium and smooth muscle, allowing expansion of the lacunar spaces within the corpora and the trapping of blood by compression of peripheral draining venules.
Sildenafil, a type 5 phosphodiesterase inhibitor, prevents the breakdown of cGMP, thereby prolonging erection. It has no effect on libido and does not cause erection without stimulation, but it maintains an erection once it has been achieved. Although the NA-NC system is particularly prominent in the penis, it is also found in the heart, the brain, and other organs. Its presence in the eye explains the blue visual hue that some patients experience after taking sildenafil.
The most common side effects of sildenafil are headache, flushing, and dyspepsia. It can also decrease BP. Because the decrease in BP may be synergistic with the hypotensive action of nitrates, sildenafil is contraindicated in patients taking a medication that contains nitrates, such as nitroglycerin.
In addition, sildenafil alters the half-life of many other medications, and many medications change the half-life of sildenafil. The list of agents that can interact with sildenafil includes such common medications as nonselective -blockers, erythromycin, itraconazole, potassium-sparing diuretics, and cimetidine. It is not known whether those interactions affect the side effects of sildenafil, particularly the incidence or severity of hypotension. In initial clinical trials, hypotension was reported in approximately 3% of patients, but those trials included a large percentage of young men with psychogenic impotence. Obviously, patients with vascular disease or diabetes have more problems with BP regulation and theoretically with orthostatic
P.140
hypotension. Deaths have been reported among patients taking sildenafil since it became available. The FDA is investigating those deaths.The first study of data on different patient populations taking sildenafil was published several months after the drug became available for clinical use. Although the package insert indicated an overall efficacy of 82% (vs. 24% for placebo), analysis since has found more modest efficacy of 68% in patients with hypertension, 57% in diabetes, and 61% after transurethral prostatectomy. Moreover, published results are frequently obtained in a selected patient population, not from general clinical use.
EAN: 2147483647
Pages: 14