4 - Vitreoretinal Disorders
Editors: Tasman, William; Jaeger, Edward A.
Title: Wills Eye Hospital Atlas of Clinical Ophthalmology , The, 2nd Edition
Copyright 2001 Lippincott Williams & Wilkins
> Table of Contents > Chapter 4 - Vitreoretinal Disorders
function show_scrollbar() {}
Chapter 4
Vitreoretinal Disorders
Carl D. Regillo
William E. Benson
Neal H. Atebara
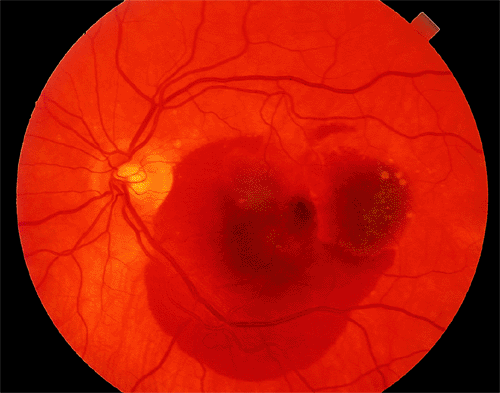 |
| A 67-year-old woman with age-related macular degeneration and sudden visual acuity loss in her left eye. There is subretinal pigment epithelial hemorrhage in the macular from underlying choroidal neovascularization. |
P.168
Anatomic Considerations
Layers of the Retina
The retinal pigment epithelium (RPE) is a single layer of melanin-containing cells that extends from the optic nerve to the ora serrata and continues anteriorly as the pigment epithelium of the pars plana (Fig. 4.1A). The RPE is separated from the choriocapillaris by Bruch's membrane. The basement membrane of the RPE forms the inner layer of Bruch's membrane. The layer of rods and cones is formed by the outer and inner segments of the photoreceptor cells (i.e., rods and cones). The outer segments of these cells contain visual pigment molecules that absorb light energy and propagate a nerve impulse.
The external limiting membrane is not a true membrane; it is composed of a series of intercellular connections called zonulae adherentes that unite the plasma membranes of adjacent photoreceptors and M ller's cells. This fenestrated membrane separates the outer segments of the rods and cones from their cell bodies and nuclei that form the outer nuclear layer. The axons of the rods and cones and the dendrites of the bipolar cells form, and synapse within, the outer plexiform layer, which also includes processes of the horizontal cells and M ller's cells. The bipolar cells receive the visual impulse from the rods and cones and are considered the first-order neurons. The inner nuclear layer consists of the cell bodies and nuclei of bipolar, amacrine, M ller's, and horizontal cells.
The inner plexiform layer is the zone of synapse between the axons of the bipolar cells and the dendrites of the second-order neurons, the ganglion cells. Processes of amacrine, ganglion, M ller's, and bipolar cells are also contained in this layer. The ganglion cell layer contains the cell bodies and nuclei of the ganglion cells. The axons of these ganglion cells form the nerve fiber layer. These axons course through the retina and gather to form the optic nerve. The visual impulse initiated in the photoreceptor cells is transferred to the bipolar cells, then to the ganglion cells, and does not synapse again until the lateral geniculate body. The internal limiting membrane separates the retina from the vitreous. A true basement membrane, the internal limiting
P.169
membrane is derived chiefly from the footplates of the giant glial cells of M ller that extend between the internal and external limiting membranes.
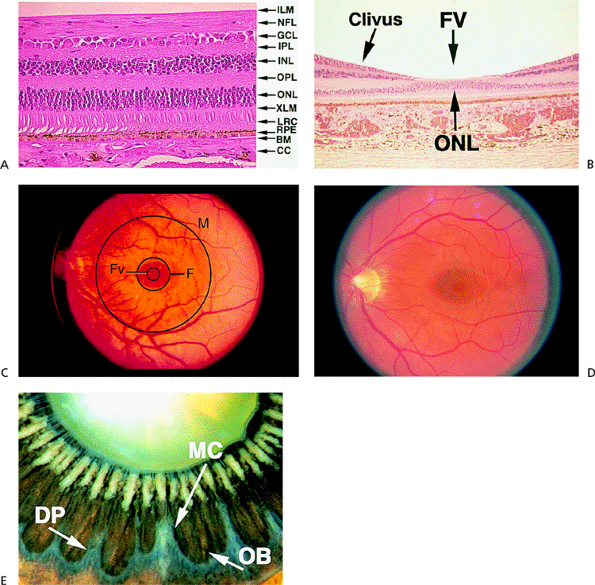 |
Figure 4.1. A: Histologic section of the retina, showing the normal layers. (BM, Bruch's membrane; CC, choriocapillaris; GCL, ganglion cell layer; ILM, internal limiting membrane; INL, inner nuclear layer; IPL, inner plexiform layer; LRC, layer of rods and cones; NFL, nerve fiber layer; ONL, outer nuclear layer; OPL, outer plexiform layer; RPE, retinal pigment epithelium; XLM, external limiting membrane.) B: Histologic section of the fovea. (FV, foveola.) C: The posterior pole, illustrating the borders of the macula (M), fovea (F), and foveola (FV). D: Clinical view of the normal macula. E: Posterior view of the ora serrata in an enucleated eye of an 87-year-old patient. Notice the mature cataract, the pseudoexfoliation on the zonules, and the hyalinization of the ciliary processes. (DP, dentate processes; MC, meridional complex; OB, oral bay; A, B, and E courtesy of Ralph C. Eagle Jr, M.D., Philadelphia, PA.) |
Macula
The Anatomic Macula
There are important anatomic modifications in the macula that subserve the highest level of visual efficiency and color perception. Histologically, the macula is the region of the posterior retina in which the ganglion cell layer is more than one cell layer thick (Fig. 4.1B). This area is about 6 mm in diameter. The central portion of the macula is the fovea or pit. It is 1.5 mm wide (i.e., 1 disc diameter) and consists of a sloping area of clivus and the foveola, which is the floor of the concavity. The foveola is 0.33 mm wide. The RPE cells in the fovea are tall and narrow. In the foveola, the layer of rods and cones and the outer nuclear layer consist entirely of specialized cone cells. These cone cells are tall and narrow, densely packed, and resemble rod cells. Their nuclei are multilayered in the outer nuclear layer. Rod cells and the inner retinal layers are added progressively along the clivus of the fovea. The internal limiting membrane remains intact but thins over the foveola. The outer plexiform layer, also called Henle's fiber layer, is obliquely oriented in the clivus. It is estimated that 10% of the total number of retinal cones are in the foveal area. The center of the fovea is devoid of blood vessels and receives its nutrients from the choriocapillaris. The outer nuclear layer and ganglion cell layer are thickest in the immediate parafoveal area.
P.170
The Clinical Macula
The macula is a somewhat ill-defined area of the posterior pole that is about 4 disc diameters wide (6 mm) and extends from the axial center of the retina to a point near the disc margin on the nasal side and to an equal radius temporally (Fig. 4.1C). The central portion of the macula is the fovea, which is 1 disc diameter wide. The center or floor of the foveal concavity is the foveola. The foveola lies 0.5 mm inferior to a line drawn horizontally through the center of the disc.
Ophthalmoscopically, the fovea appears darker than the surrounding retina because of increased concentration of pigment in the RPE (Fig. 4.1D). A circular light reflex can be seen reflected from the thickened peripheral edge of the fovea in younger individuals, and a central light spot is reflected from the pit of the foveola. The fovea sometimes appears yellowish because of a carotenoid pigment called xanthophyll in the retina. The fine arterioles and venules derived from the central retinal artery form an arcade around the fovea, but the inner portion of the fovea itself is devoid of blood vessels.
Ora Serrata
The ora serrata marks the anterior extension of the neural retina and the junction between the neurosensory retina and the pars plana of the ciliary body (Fig. 4.1E). Because the nasal retina extends farther anteriorly than the temporal retina, the ora serrata lies 6 mm posterior to the limbus on the nasal aspect of the globe and 7 mm temporally. The peripheral retina gradually thins, with a loss of distinct anatomic layers as it approaches the ora. At this point, the multilayered sensory retina becomes continuous with the monolayer of nonpigmented cells that forms the inner half of the ciliary epithelium. The RPE continues as the pigment epithelium of the ciliary body. The internal limiting membrane thins and eventually interweaves with the collagenous filaments of the vitreous base.
The ora is not uniform but is made of alternating dentate processes and oral bays, which are more prominent nasally. A dentate process is an extension of the hypoplastic peripheral retina into the ciliary body. An oral bay is a posterior extension of the pars plana into the peripheral retina. If a dentate process undergoes hypertrophy, particularly of its glial elements, in a linear configuration, the resulting entity is called a meridional fold. If this fold is contiguous with a ciliary process, the resultant structure is a meridional complex.
The vitreous base spans an area of 6 mm, 2 mm anterior and 4 mm posterior to the ora. In this area, vitreous fibers are tightly bound to the internal limiting membrane. This process is particularly prominent in the area of meridional folds. Disruption of the vitreous base from its attachment to the peripheral retina may produce a tear and subsequent retinal detachment.
Myelinated Nerve Fibers
Normally, the myelin covering of the optic nerve fibers stops at the lamina cribrosa (i.e., level of the sclera). Sometimes the myelin continues onto the retina, which can be seen with the ophthalmoscope. Myelinated nerve fibers appear pearly white with feathery edges (Fig. 4.2A) and assume a nerve fiber bundle configuration. They are usually attached to the disc but may appear anywhere in the retina. Scotomatous visual field defects of variable density may be demonstrated.
Coloboma
Clinical Features
Coloboma is a congenital condition that results from a failure of the embryonic fissure to close, and it may involve the optic nerve, retina, and choroid. Anterior segment structures also may be involved. The inner and outer layers of the optic cup are abnormal in this area of faulty closure. The outer layer (i.e., RPE) is usually absent, and the choroid fails to develop. The inner layer, which gives rise to the sensory retina, is often present but attenuated. However, blood vessels often can be seen coursing through the defect.
Retinochoroidal colobomas are located inferior or inferonasal to the disc. They are glistening white and sharply demarcated, and some have patchy pigmentation. They may be isolated or may extend to the inferior periphery (Fig. 4.2B, C). Breaks may occur within or at the edge of the attenuated sensory retina over the coloboma and can result in rhegmatogenous retinal detachment.
Retinochoroidal colobomas are frequently associated with chromosomal abnormalities or multisystem diseases such as trisomy 13 and Goldenhar's syndrome.
Management
No treatment is available for this congenital anomaly or prevention of retinal breaks and detachments. Rhegmatogenous retinal detachments that occasionally develop are repaired with vitrectomy techniques.
Asteroid Hyalosis
Asteroid hyalosis is a degenerative process within the vitreous, characterized by large numbers of whitish, suspended, spherical opacities (Fig. 4.2D and E).
Clinical Features
Viewed through the ophthalmoscope, these vitreous opacities are more numerous and more uniform than those associated with the age-related vitreous liquefaction process. The condition is unilateral in 75% of patients and is more common among those older than 60 years of age. The composition is thought to be consistent with calcium-containing lipid. There is no known cause and no association with systemic or other ocular disease.
Patients occasionally complain of a film in front of their vision but are amazingly asymptomatic considering the debris seen by the ophthalmologist. Asteroid hyalosis almost never causes a significant loss of vision.
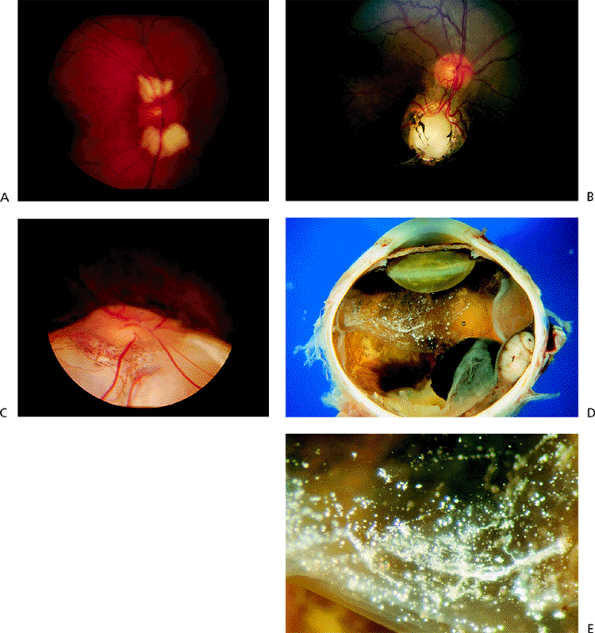 |
Figure 4.2. A: Myelinated nerve fibers contiguous with the disc. B: Sharply demarcated, isolated retinochoroidal coloboma inferior to the disc. C: More extensive retinochoroidal coloboma extending to the inferior periphery. D: Gross specimen demonstrates asteroid hyalosis within the detached vitreous in an eye that was removed because of a malignant melanoma. E: Magnified view of the opacities. (Courtesy of Ralph C. Eagle Jr, M.D., Philadelphia, PA.) |
P.171
Management
No treatment needed as asteroid hyalosis is asymptomatic.
Hereditary Macular Dystrophies
Stargardt's Disease
Stargardt's disease is an inherited disease in which the quantity of lipofuscin pigment in all RPE cells is markedly increased. The disease is also referred to as fundus flavimaculatus, especially when the yellow flecks dominate the clinical picture (see below). The vast majority of Stargardt's cases are autosomal recessive, and the genetic defect has been traced to mutations in the ABCR gene.
Clinical Features
The onset of the disease occurs between 6 and 20 years of age. The initial symptom is decreased visual acuity. In advanced cases, there may be decreased night vision. When visual acuity is first diminished, the macula may appear normal, and the diagnosis may be extremely difficult to make. Later, mild periforeal pigment epithelial atrophy is detected. Still later, the atrophic changes may progress to a bull's-eye appearance. In some cases, the pigment epithelium is diffusely atrophic.
Focal hyperaccumulations of lipofuscin account for the characteristic pisciform (i.e., fish-like) yellow flecks (Fig. 4.3). These may be localized to the macula, scattered throughout the postequatorial area, or absent. Over time, some disappear, and new ones appear. Fluorescein angiography shows areas of hyperfluorescence that do not necessarily
P.172
correspond to the flecks seen on clinical examination. Another important finding is that the diffuse pattern of hyperconcentrated lipofuscin in pigment epithelial cells partially blocks the background choroidal fluorescence normally seen on fluorescein angiography. This dark or silent choroid is seen in 85% of cases. Some cases are complicated by secondary choroidal neovascularization (CNV).
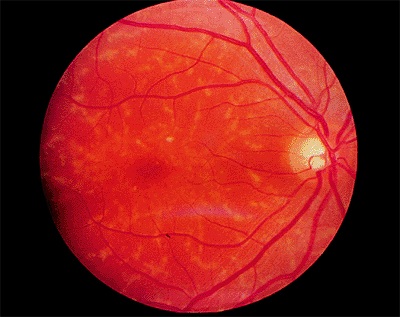 |
Figure 4.3. Pisciform lesions and macular bull's-eye atrophy in patient with Stargardt's disease. |
Electrophysiologic testing typically shows a normal electroretinogram (ERG) and electrooculogram (EOG), especially in earlier stages of the condition. Later, however, the ERG may be mildly to moderately decreased. The visual prognosis is poor. Most patients have a visual acuity of 20/200 or worse by 40 years of age.
Young patients without the flecks are often misdiagnosed as malingerers or hysterics. Cone dystrophy, which is discussed later in the chapter, must be ruled out.
Management
Periodic monitoring, genetic counseling, and support for low vision constitute management.
Best's Vitelliform Dystrophy
Best's vitelliform dystrophy is an autosomal dominantly inherited condition characterized by the accumulation of lipofuscin in RPE cells throughout the fundus and especially in the macula. The genetic defect has recently been shown to be a mutation in a novel retina-specific gene (VMD2) located on chromosome 12. Electrophysiologically, the condition is characterized by diffuse RPE dysfunction (see below).
Clinical Features
The most common fundus finding is the egg yolk macular lesion (Fig. 4.4), which is hypofluorescent on fluorescein angiography. It has been observed in patients as young as 1 week of age, but the fundus may be normal until middle age. Some patients have multiple, yellow lesions (i.e., multifocal Best's dystrophy). Most patients' eyes are hyperopic, and many are symptom free, even if the egg yolk appearance is present. With time, the lesion may progress to a pseudohypopyon stage. The most common late finding is the scrambled egg stage, which may or may not be associated with a CNV.
Unless CNV occurs, the visual prognosis is good (i.e., 20/50 to 20/100). Best's vitelliform dystrophy is one of a very few conditions in which the ERG is normal but the EOG is abnormal. This condition should be differentiated from adult-onset foveomacular vitelliform dystrophy.
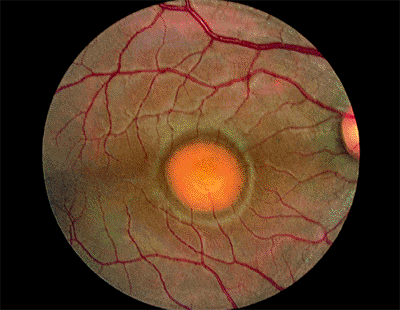 |
Figure 4.4. Egg yolk lesion in a patient with Best's vitelliform dystrophy. |
Management
Periodic monitoring, genetic counseling, and support of low vision constitute management of Best's vitelliform dystrophy.
Pattern Dystrophies
The pattern dystrophies include adult-onset foveomacular vitelliform dystrophy, butterfly dystrophy, reticular dystrophy of the pigment epithelium, and coarse pigment mottling in the macula (i.e., fundus pulverulentus). Most forms are inherited as an autosomal dominant trait. A few patients have different patterns in opposite eyes, and some progress from one pattern to another. In some pedigrees, family members express different patterns. For these reasons, it is thought that they all are probably expressions of the same disorder. Mutations in the peripherin/RDS gene have been found in some patients but not others.
Adult-Onset Foveomacular Vitelliform Dystrophy
Adult-onset foveomacular vitelliform dystrophy (AOFVD) is characterized by a slightly elevated, round, yellow lesion at the level of the RPE. The lesion often will have a small, pigmented dot in its center (Fig. 4.5).
Clinical Features
The lesions are usually bilateral and are about one third to one half of the disc diameter. Sometimes, there are adjacent small, yellow flecks. The lesions are hypofluorescent on fluorescein angiography but often have a hyperfluorescent ring. Symptoms such as metamorphopsia or slightly decreased vision usually do not appear until middle age, but many patients remain asymptomatic for life. The prognosis is excellent, except for the occasional case in which CNV develops.
Best's vitelliform dystrophy may closely resemble AOFVD, especially when the AOFVD lesions are large, but the EOG results in AOFVD are normal or only slightly reduced.
P.173
Many patients are misdiagnosed as having a pigment epithelial detachment (PED) caused by age-related macular degeneration (AMD). A fluorescein angiogram can distinguish AOFVD from AMD-related PED as the latter lesion would be expected to be hyperfluorescent.
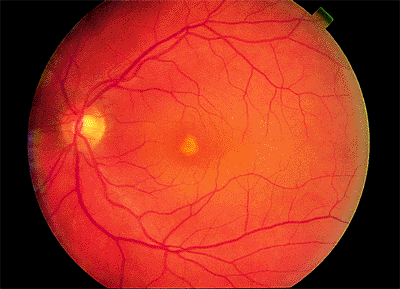 |
Figure 4.5. Central yellow deposit in a patient with pattern dystrophy. |
Butterfly Dystrophy
Butterfly dystrophy is a pattern dystrophy in which there is irregularly branching yellow-gray pigment figure in the macula (Fig. 4.6). Like AOFVD, it is usually bilateral and symmetric and located at the level of the RPE. The visual acuity is normal or only slightly decreased. The ERG is normal, but the EOG is often moderately abnormal, indicating a widespread abnormality in the RPE. Onset may occur between the teens and middle age. As with AOFVD, the fluorescein angiogram shows the pigment figure itself to be hypofluorescent and have a surrounding hyperfluorescent halo.
Reticular Dystrophy of the Retinal Pigment Epithelium
Reticular dystrophy of the RPE is a pattern dystrophy in which the pigment pattern looks like chicken wire, and pigment sometimes extends out of the macular area.
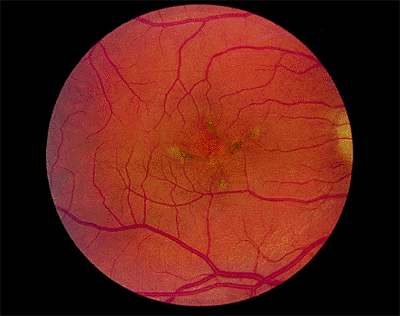 |
Figure 4.6. Deposits at the level of the pigment epithelium in a patient with butterfly dystrophy. |
Cone Dystrophy
Cone dystrophy, which is inherited as an autosomal dominant trait or sporadically, is characterized by progressive deterioration of cones.
Clinical Features
The onset of symptoms has a somewhat bimodal distribution; it usually occurs between 4 and 8 years of age or between the late teens and 30 years of age. The chief presenting symptom is decreased visual acuity, but color vision also is abnormal. Photophobia (or hemeralopia) is common. Because rod function is normal or nearly normal, patients see well in dim light, but their vision deteriorates in bright light, in which rod function is suppressed and normal cone function is essential. The prognosis is poor, with most patients deteriorating to 20/200 or worse. In some cases, the condition may progress to cone-rod dystrophy with night blindness in addition to the poor central vision.
Early in the course of the disease, the macula may be normal (Fig. 4.7A) or have only mild RPE depigmentation and be indistinguishable from early cases of Stargardt's disease without pisciform lesions. ERG shows decreased cone function before any decrease in visual acuity or color vision is detected. A sensitive indicator of cone dysfunction is a decreased or flat tracing in response to a 30 cycle per second stimulus (i.e., flicker ERG). Dark adaptation shows a rod phase only. Fluorescein angiography may show small RPE
P.174
window-type transmission defects early in the course of the disease and establish the existence of central pigment epithelial degeneration (Fig. 4.7B). Later, there may be a bull's-eye macula, diffuse granular-appearing pigment epithelial atrophy, or geographic atrophy of the macula (Fig. 4.8).
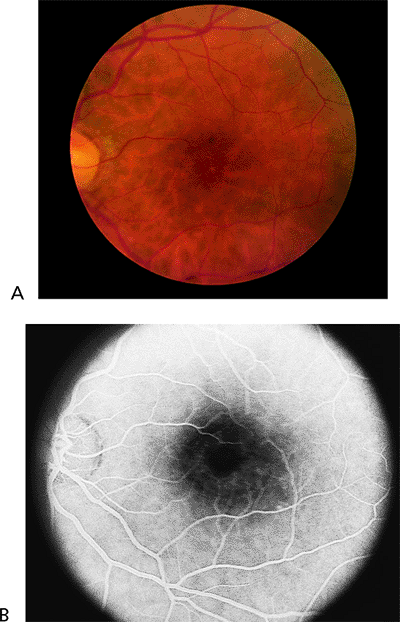 |
Figure 4.7. A: The fundus appears almost normal in a patient with cone dystrophy. B: Fluorescein angiography reveals a bull's-eye area of depigmentation of the retinal pigment epithelium. |
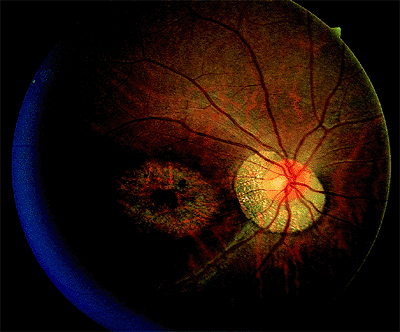 |
Figure 4.8. Severe central macular atrophy in a patient with cone dystrophy. |
This condition should be differentiated from Stargardt's disease, malingering, and hysteria.
Management
There is no effective treatment. Genetic counseling and low vision support should be suggested.
Exudative Maculopathies
Central Serous Chorioretinopathy
Idiopathic central serous chorioretinopathy (CSCR) is a condition in which there is a small collection of relatively clear subretinal fluid in the macula (Fig. 4.9).
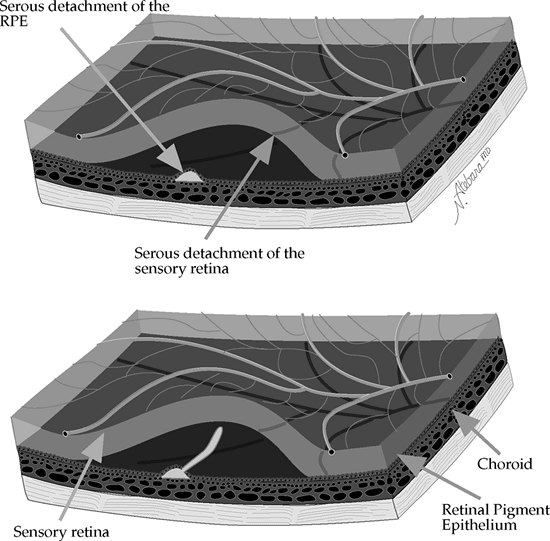 |
Figure 4.9. In idiopathic central serous chorioretinopathy, a small serous detachment of the retinal pigment epithelium (RPE) in the macular or paramacular area may be followed by serous detachment of the overlying and surrounding sensory retina (top). Because the fluorescein leaking through the defect in the RPE is lighter than the subretinal fluid, it rises, resulting in the classic smokestack appearance (bottom). (Courtesy of Neil Atebara, M.D., Philadelphia, PA.) |
P.175
Clinical Features
Symptoms include metamorphopsia, micropsia, decreased color vision, and decreased visual acuity. The visual acuity occasionally is normal. If decreased, it typically improves with a pinhole or with plus lenses.
Eighty percent of cases are men, and most patients are between 30 and 50 years of age. It is rare in persons of African descent. There is no known cause, although some researchers have associated the condition with an aggressive, hard-driving, type A personality. Corticosteroids can exacerbate the condition.
On fundus examination, absence of the foveal reflex and blurring of the choroidal pattern are excellent clues to the presence of subtle subretinal fluid. When the fluid collection is more prominent, a blister-like elevation is readily appreciated with indirect ophthalmoscopy (Fig. 4.10A). By carefully positioning the slit lamp beam through a 90 or 60 diopter lens or through a Goldmann lens, the retinal elevation can be seen, and the clear nature of the subretinal fluid can be appreciated. Outer retinal yellow-white precipitates are common. Granular RPE mottling caused by past attacks may also be noticed.
On fluorescein angiography, 80% of patients show a small dot of hyperfluorescence that slowly expands (Fig. 4.10B, C). Rapid leakage of fluorescein, which rises upward in the characteristic smokestack pattern, is seen in 20% of these patients.
The most common and important differential diagnosis is CNV, which can occur in conditions such as ocular histoplasmosis and AMD. Indirect signs of CNV include hemorrhage and lipid exudate, both of which would not be expected with CSCR. Furthermore, a focal, gray-green pigmented membrane under the retina representing the CNV itself may be directly visible ophthalmoscopically. On fluorescein angiography, the neovascular vessels may be seen in the early phases, and there is usually more leakage than there is in idiopathic CSCR. Less commonly, conditions that cause serous macular detachment, such as eclampsia or Harada's disease, must be ruled out.
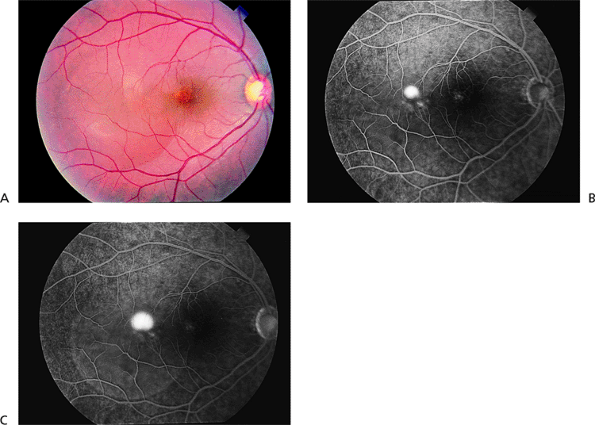 |
Figure 4.10. A: The serous elevation is mostly temporal to but includes the center of the macula in a patient with central serous chorioretinopathy. B: The middle phase of the fluorescein angiogram shows a small dot of hyperfluorescence. C: The late phase shows additional leakage, with some leakage into the serous elevation of the retina. |
Management
Idiopathic CSCR usually is a benign condition. Resolution of the subretinal fluid is expected in most cases, usually within 3 to 4 months. Two thirds of patients recover 20/20 vision, although mild color vision or contrast sensitivity abnormalities may persist. Between 25% and 50% of patients have recurrent attacks. Laser photocoagulation of the leaking spot accelerates resolution of the subretinal fluid, but it does not result in better final visual acuity or better final color vision when performed early in the course of typical CSCR. Also, it does not reduce the rate of recurrence. Furthermore, in rare cases, laser therapy may result in secondary CNV. In general, laser treatment should be reserved for patients who have an occupational need for normal vision, patients with recurrences who have had permanent visual loss from previous episodes, and patients who have had attacks persisting more than 5 months.
Age-Related Macular Degeneration
AMD is a progressive deterioration of Bruch's membrane, the RPE, and the choriocapillaris. It is the leading cause of
P.176
irreversible, severe central vision loss in people over the age of 50 years in the United States. The clinical hallmark of the disease is drusen (singular: druse) in the macula. Drusen observed ophthalmoscopically represent focal accumulation of undigested products of RPE cells under the basal lamina of Bruch's membrane. Histopathologically, however, AMD is characterized by diffuse thickening of the inner aspect of Bruch's membrane.
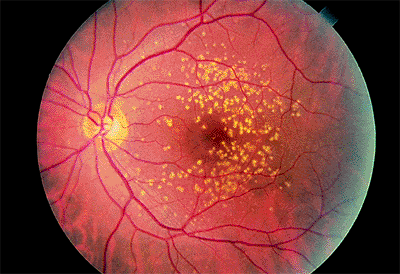 |
Figure 4.11. Multiple, hard drusen in early-stage, age-related macular degeneration. |
There are two basic clinical variants of AMD: the dry, atrophic type and the wet, exudative or neovascular type. The dry type is much more common, accounting for 90% of cases, but it results in only 10% of AMD eyes that suffer severe (20/200 or worse) visual loss.
Clinical Features
Hard drusen are small (<64 m) and well defined (Fig. 4.11). Soft drusen are larger and often have mottled overlying pigment epithelium (Fig. 4.12). On fluorescein angiography, both types are hyperfluorescent, representing either a window defect in the RPE or a staining defect. They do not show leakage and, therefore, would not be expected to exhibit increasing brightness or size into the middle and late phases of the angiographic study.
The dry type produces progressive atrophy of the pigment epithelium and the choriocapillaris of varying degrees over time. Large areas of macular RPE-choriocapillaris loss can result in severe vision loss (if the fovea is involved) and are often referred to as geographic atrophy (Fig. 4.13). Fluorescein angiography may reveal the large choroidal vessels. Again, there is no leakage of dye.
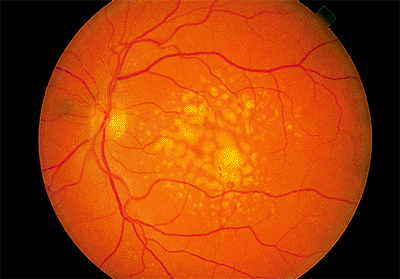 |
Figure 4.12. Multiple, soft, confluent drusen. |
In the wet type, an exudative macular detachment occurs as a result of CNV formation and secondary leakage of fluid, lipid exudate, or blood into the subretinal or sub-RPE space (Fig. 4.14A). Typically, as the CNV grows and leaks over weeks to months, there is progressive visual loss. In most cases, subretinal fibrosis eventually sets in to some degree, and the exudative changes ultimately organize into a so-called disciform scar, the end stage of wet AMD (Fig. 4.14B). Larger amounts of exudation and bigger disciform scars involving the fovea usually correlate with poorer visual outcomes.
There are two basic categories of CNV growth patterns on fluorescein angiography, classic and occult. Classic CNV shows up as well-defined, lacy hyperfluorescence in the early phases and demonstrates profuse late leakage (Fig. 4.15). Occult CNV can vary in its appearance but, in general, is characterized by more late-appearing and less intense fluorescein leakage. The borders of occult CNV may be well delineated or poorly delineated and can also be obscured by thick blood or rapid leakage of dye when there is an associated serous pigment epithelial detachment (Fig. 4.16). Indocyanine green angiography can sometimes be helpful as an adjunct to fluorescein angiography in detecting or better delineating the location of occult CNV (Fig. 4.17). Classic CNV tends to grow faster and result in more severe vision loss over a shorter time frame than occult CNV.
The differential diagnosis of dry AMD includes pattern macular dystrophy and old CSCR. For wet AMD, simulating conditions include active CSCR, retinal arterial macroaneurysm, and idiopathic polypoidal choroidal vasculopathy.
Management
There is no treatment for visual loss from dry AMD. Furthermore, there is no proven treatment to slow AMD progression and prevent the dry to wet transformation. There are, however, several different treatment options for wet AMD that can help reduce the risk of severe vision
P.177
loss. Because the best results with these treatments typically come about when CNV is detected early in its development, patients with dry AMD should be monitored regularly. This is especially important for those who have eyes with high-risk features such as numerous soft drusen, focal RPE clumps, and fellow eyes with wet AMD. Patients with these features should self-monitor their central vision with an Amsler grid and be reevaluated should there be any new central visual disturbance.
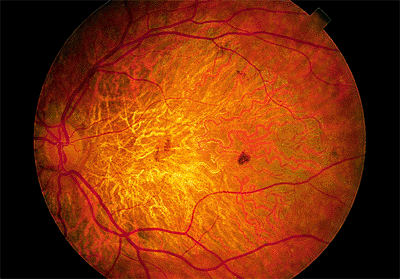 |
Figure 4.13. Dry, age-related macular degeneration with severe atrophy of the pigment epithelium and choriocapillaris. |
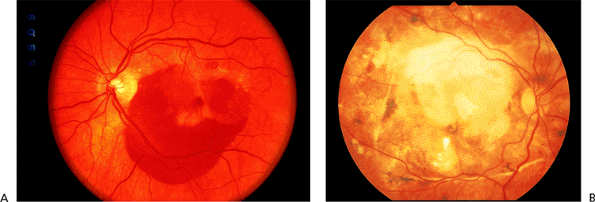 |
Figure 4.14. A: Acute exudative age-related macular degeneration with subretinal hemorrhage. B: End-stage exudative macular degeneration with inactive subretinal fibrosis ( disciform scar ). |
Treatment of wet AMD depends on the location and type of CNV (and may also be influenced by the associated exudative features, such as blood). In general, effective forms of treatment currently exist for CNV that is mostly classic and well delineated (with minimal associated blood). If the CNV is nonsubfoveal in location, then standard, ablative laser photocoagulation is the treatment of choice (Fig. 4.15). For similar-appearing CNV that has extended through the foveal center, photodynamic therapy is employed. Currently, there is no treatment proven to effectively reduce vision loss in eyes that have wet AMD with
P.178
CNV that is mostly occult. Indocyanine green angiography may identify laser-treatable areas in some eyes with occult CNV (Fig. 4.17).
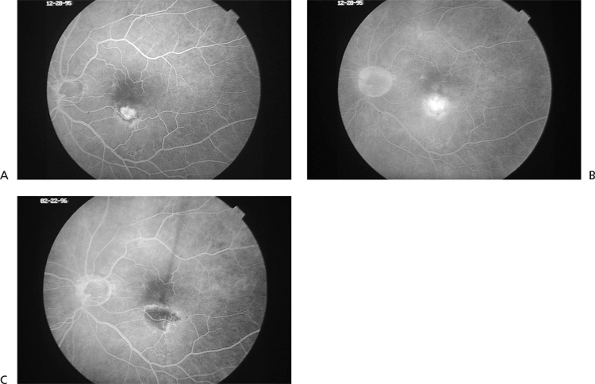 |
Figure 4.15. Early (A) and late (B) frame fluorescein angiogram photographs of classic, extrafoveal choroidal neovascularization in age-related macular degeneration. C: Fluorescein angiogram photograph taken 3 weeks after successful laser photocoagulation treatment. There is no evidence of persistent choroidal neovascularization. |
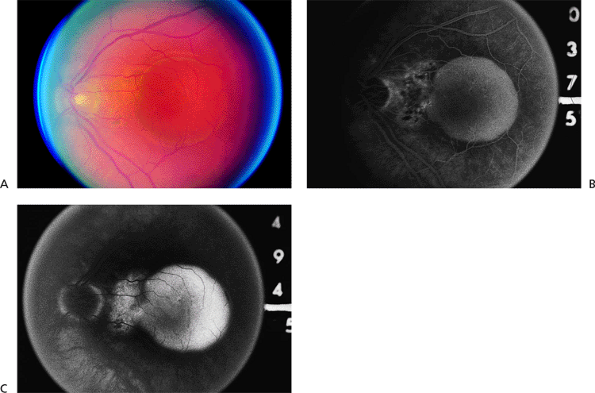 |
Figure 4.16. A: Serous retinal pigment epithelial detachment (SPED) in a patient with age-related macular degeneration. B: The middle phase of the fluorescein angiogram demonstrates total filling of the pigment epithelial detachment and stippled hyperfluorescence nasal to the detachment. C: At 494 seconds after fluorescein administration, leakage of dye in the area nasal to the SPED is consistent with occult choroidal neovascularization. |
The Macular Photocoagulation Study (MPS) showed that eyes with classic CNV located more than 200 m from the center of the foveal avascular zone ( extrafoveal ) and, to a lesser degree, CNV positioned 1 to 199 m from the center of the foveal avascular zone ( juxtafoveal ) have about a 50% reduction in severe vision loss with laser photocoagulation treatment compared to untreated control eyes (Fig. 4.18). The treatment results are best in patients whose CNV is adequately initially treated (i.e., confluent laser application over the entire lesion extending 100 m beyond its border). Despite good treatment, however, CNV recurrence
P.179
rates remain high (over 50%), and eyes with recurrent CNV will typically have worse vision outcomes. There is no evidence that any available wavelength (e.g., green vs. red) provides the best treatment results.
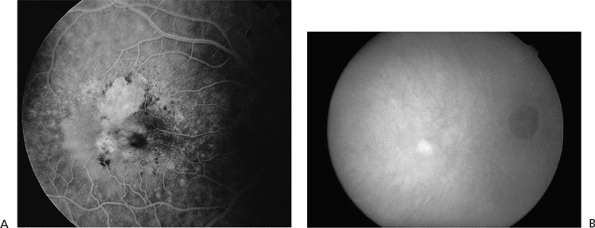 |
Figure 4.17. A: Fluorescein angiogram showing occult choroidal neovascularization adjacent to geographic atrophy. B: Corresponding indocyanine green angiogram showing a well-defined spot of hyperfluorescence representing the neovascular focus. (From Regillo CD. The present role of indocyanine green angiography in ophthalmology. Curr Opin Ophthalmol 1999;10:189, with permission.) |
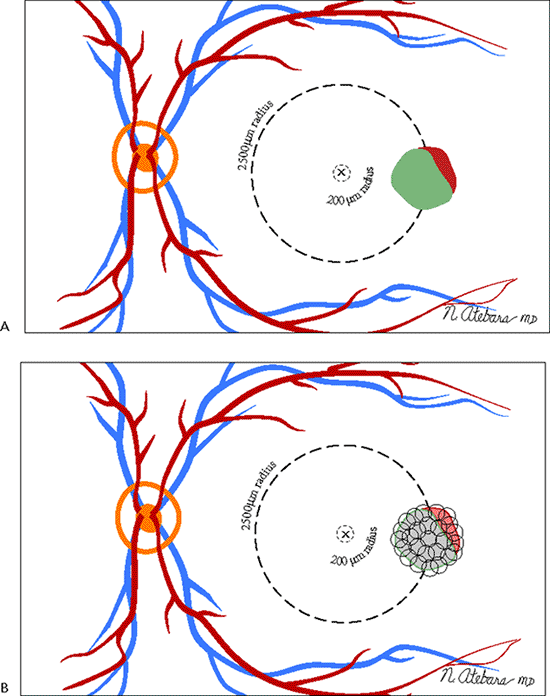 |
Figure 4.18. A: Extrafoveal choroidal neovascularization lies between 200 and 2,500 m from the center of the foveal avascular zone. The Macular Photocoagulation Study (MPS) found that 41% of untreated eyes suffered six or more lines of visual loss at 1 year, compared with only 24% of the treated eyes. After 5 years, 64% of the untreated eyes suffered severe visual loss, compared with 46% of the treated eyes. B: In the MPS, extrafoveal choroidal neovascularization is treated with confluent laser burns that extend 100 m beyond the choroidal neovascular membrane and adjacent blood or blocked fluorescence. |
For subfoveal CNV that is mostly the classic type, photodynamic therapy (PDT) was recently proven to statistically reduce the risk of vision loss. PDT involves the intravenous infusion of a photosensitizing dye followed shortly thereafter by the application of a long duration, nonthermal laser light with a wavelength specific for the particular dye in use. Unlike standard photocoagulation, PDT does not destroy the overlying sensory retina and, therefore, is a more selective CNV treatment technique. It is thought to work by promoting temporary thrombosis of CNV. Because there is
P.180
often reperfusion of CNV 6 to 12 weeks after treatment, PDT may require retreatment every 3 months for 1 to 2 years to ultimately achieve permanent involution of CNV and complete resolution of exudation (Fig. 4.19).
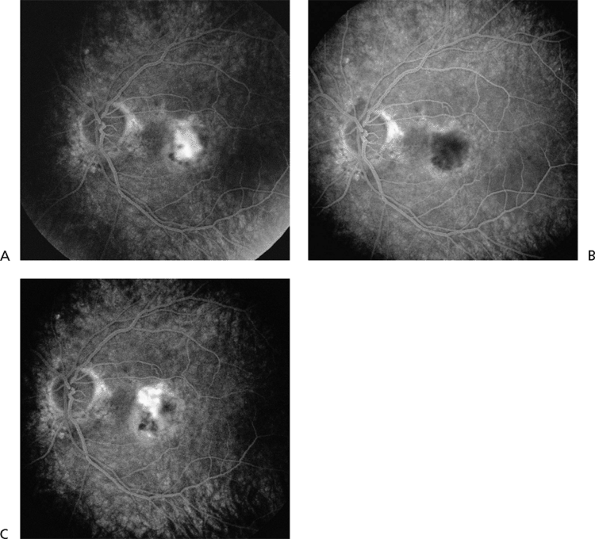 |
Figure 4.19. Fluorescein angiogram photographs before and after photodynamic therapy. A: Before treatment, there is classic, subfoveal choroidal neovascularization (CNV). B: One week after treatment, the CNV is hypofluorescent or nonperfused. C: Six weeks after treatment, there is partial reperfusion of the original CNV lesion. (From Regillo CD. Update on photodynamic therapy. Curr Opin Ophthalmol 2000;11:166, with permission.) |
Other forms of treatment for exudative CNV that are occasionally utilized include macular translocation to treat small, subfoveal CNV; pneumatic displacement or surgical evacuation for large, submacular hemorrhage; and transpupillary thermotherapy to manage occult, subfoveal CNV. Antiangiogenesis drugs are currently in early stages of investigation.
Presumed Ocular Histoplasmosis Syndrome
Presumed ocular histoplasmosis syndrome (POHS) is an acquired condition that appears to develop in certain individuals exposed to the histoplasmosis organism, which, in turn, results in bilateral, focal chorioretinal lesions. There is a high prevalence of POHS in the Ohio/Mississippi River valleys, where histoplasmosis is endemic. The condition is typically detected in young and middle-aged white adults, male or female.
Clinical Features
The classic clinical triad of POHS includes the following signs (Fig. 4.20):
Punched-out chorioretinal scars ( histo spots )
Peripapillary chorioretinal atrophy
Maculopathy: subretinal fibrosis from CNV
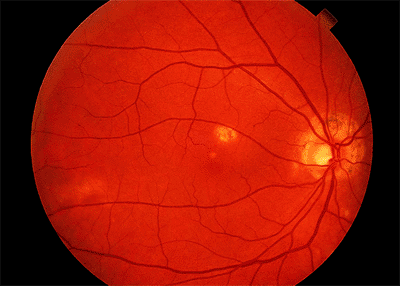 |
Figure 4.20. Presumed ocular histoplasmosis syndrome. Note the peripapillary atrophy and macular punched-out chorioretinal scar ( histo spot ). |
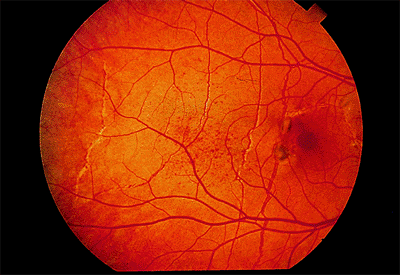 |
Figure 4.21. Peau d'orange in an eye with multiple angioid streaks and two small foci of choroidal neovascularization. |
P.181
Because POHS is not an active infectious or inflammatory process, anterior chamber or vitreous cells are not present. Over time, one may observe peripapillary atrophy enlargement, histo spots can enlarge, new spots develop (5% to 10% cases), and CNV form in the macula (de novo or from a small macular histo spot).
Vision loss in POHS is almost always the result of macular CNV. The risk of CNV in the eye of a patient whose fellow eye developed CNV in the past varies depending on the following features:
If disk and macula are normal appearing: 1% over 4 years
If there are peripapillary changes only: 4% over 4 years
If there is a macular scar (i.e., macular histo spots) present: 25% over 4 years
Management
The only proven treatment for POHS-related nonsubfoveal CNV is laser photocoagulation. As with wet AMD, the MPS demonstrated the efficacy of laser (compared to no treatment) with both extrafoveal and juxtafoveal CNV. Persistent or recurrent CNV rates after laser treatment are also relatively high but lower than for laser treatment of AMD-related CNV. For subfoveal CNV, surgical removal of CNV may be beneficial. The role of surgery is currently being investigated in the Submacular Surgery Trial (SST). PDT is another potential option for subfoveal CNV management.
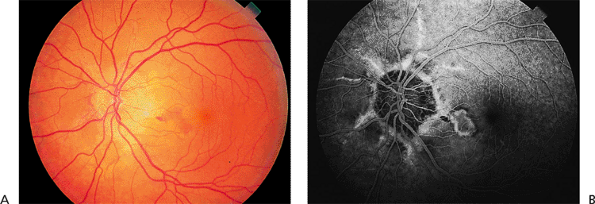 |
Figure 4.22. A: Subretinal hemorrhage and fluid in a patient with pseudoxanthoma elasticum and angioid streaks. B: The fluorescein angiogram shows classic choroidal neovascularization along one of the streaks in the macula. |
Angioid Streaks
Angioid streaks are cracks in a thickened, calcified, and brittle Bruch's membrane.
Clinical Features
Angioid streaks can be straight or jagged and may intersect. They are mostly radial but can also be concentric to the optic disc. Fine mottling of the pigment epithelium, called peau d'orange (i.e., orange skin pattern), temporal to the macula is often present (Fig. 4.21). Other associated peripheral findings are focal atrophic spots that resemble histo spots and small subretinal crystalline deposits. There is a high risk of subretinal hemorrhage from mild blunt trauma and a high risk of CNV emanating from the streaks (Fig. 4.22).
The most commonly associated systemic condition is pseudoxanthoma elasticum, which can be diagnosed clinically (60% of all patients with angioid streaks) or by scar biopsy (85%). Angioid streaks plus disc drusen are virtually diagnostic of pseudoxanthoma elasticum. Other associated conditions include Paget's disease, sickle cell hemoglobinopathy, and Ehlers-Danlos syndrome. The prevalence of angioid streaks in patients with Paget's disease is somewhat controversial. Paget's disease was found in 10% of a series of patients with angioid streaks, but angioid streaks were found in only one of a series of 70 patients with Paget's disease.
Other causes of CNV must be ruled out. In some patients, the angioid streaks are not prominent and may be overlooked. Fluorescein angiography often will show the streaks more readily appearing as hyperfluorescent lines, and it will be necessary to identify CNV when suspected.
Management
The long-term visual prognosis is relatively poor. In some patients, nonsubfoveal CNV can be treated successfully with laser photocoagulation, but the recurrence rate is high.
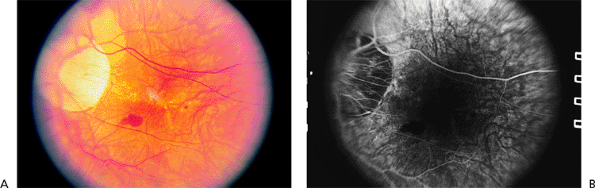 |
Figure 4.23. A: Peripapillary atrophy and a small subretinal hemorrhage in a patient with high myopia. There are a few small lacquer cracks. B: Fluorescein angiography shows blockage in the area of submacular hemorrhage. There is no choroidal neovascular membrane. |
P.182
Myopic Degeneration
In very long eyes, the retina and choriocapillaris are thin, causing myopic macular degeneration.
Clinical Features
The risk of complications increases with increasing axial length of the globe. Early findings include thinning of the macular pigment epithelium, peripapillary atrophy, and tilting of the optic disc. Later complications include breaks in Bruch's membrane called lacquer cracks (Fig. 4.23), posterior staphyloma, small macular hemorrhages (Fig. 4.23), CNV (Fig. 4.24), and severe macular atrophy. Foerster-Fuchs spots are localized areas of pigment epithelial proliferation. Macular holes may lead to retinal detachment.
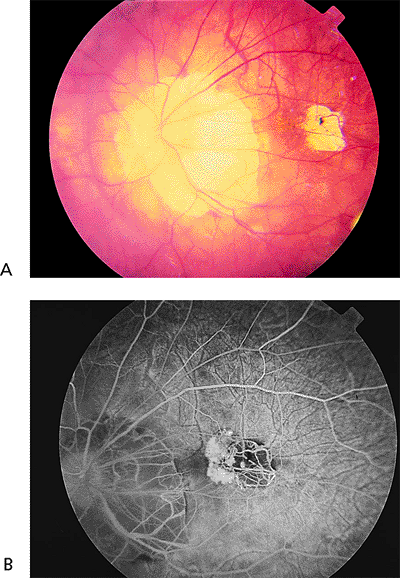 |
Figure 4.24. A: Peripapillary atrophy and a focal area of pigment epithelial atrophy in a patient with high myopia who complained of distorted vision. B: Fluorescein angiography reveals classic choroidal neovascularization nasal to the area of pigment epithelial atrophy. |
Management
Laser photocoagulation of CNV is initially effective, but later enlargement of the laser scar may cause decreased vision. Therefore, laser treatment is recommended only for CNV located 100 m from the foveal center. For subfoveal CNV, PDT appears to be beneficial to decrease the risk of further vision loss. Retinal detachment caused by myopic macular holes is best treated by vitrectomy with gas-liquid exchange or by injection of gas alone. The hole usually does not require laser treatment.
Macular Hole and Pucker
Idiopathic Macular Hole
Focal contraction of the posterior hyaloid face can tear the thin central macula, leading to a macular hole. Most occur in elderly women and are spontaneous in nature. Rarely, they result from severe blunt ocular trauma.
Clinical Features
In Gass's classification, a stage I impending macular hole is characterized by a localized foveolar detachment, with loss of the foveolar depression and the presence of a yellow macular spot (stage IA) or ring (stage IB) (Fig. 4.25). Visual acuity is minimally affected and usually better than 20/50. Approximately half of stage I eyes progress to stage II, in which the hole begins to develop and, by definition, is small (<400 m). Typically, vision is in the 20/50 to 20/80 range. Stage III, a completed hole, is larger than a stage II hole ( 400 m) and often has a cuff (i.e., halo) of subretinal fluid (Fig. 4.26). There also may be an overlying pseudooperculum. Commonly, there are some drusen-like deposits at the base of the hole. The vitreous is
P.183
still attached posteriorly at this stage. The visual acuity usually drops to 20/200. In stage IV, there is complete posterior vitreous detachment. The risk of developing a macular hole in a fellow eye is as high as 15%.
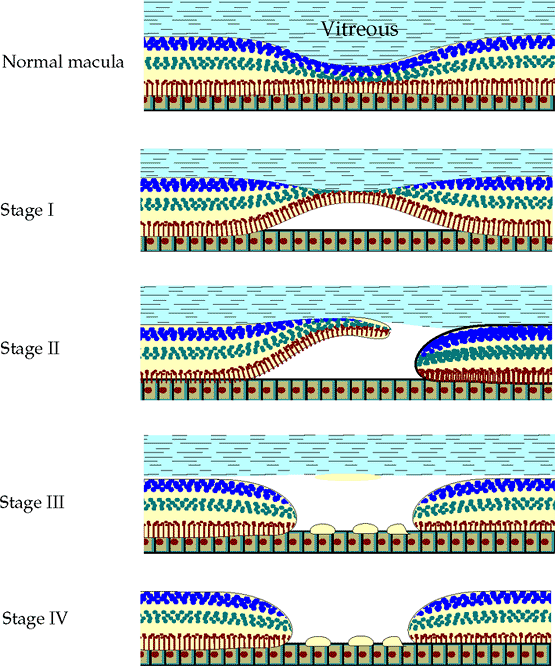 |
Figure 4.25. Stages of macular hole development. In stage I, localized contraction of the perifoveal vitreous causes a traction detachment of the fovea, characterized clinically as a small round yellow spot or ring in the fovea. Stage II is marked by a small full-thickness opening (<400 m in diameter). The hole gradually enlarges into a fully developed stage III macular hole that is usually approximately one-third disc diameter in size ( 400 m in diameter). It often has a small rim of subretinal fluid and yellow deposits at the retinal pigment epithelium. A pseudooperculum may or may not be present. A fully developed macular hole with a posterior vitreous detachment is classified as stage IV. |
Macular pucker with pseudohole and a lamellar macular hole are the entities most commonly confused with macular hole. A lamellar hole does not have a cuff of subretinal fluid, nor is there significant pucker. Clinically, a Watske Allen test is useful to differentiate a true full-thickness macular hole from a pseudohole. Ocular coherence tomography (OCT) can also be helpful.
Management
Kelly and Wendel were the pioneers in the surgical repair of macular holes. The surgery consists of a standard pars plana vitrectomy, posterior vitreous separation, possible membrane peeling, and injection of a long-acting gas. The patient then remains in a face-down position for 1 to 2 weeks after the operation. Over 90% of the holes can be anatomically closed, and two thirds or more of these eyes will have some degree of improved vision. Both anatomic and functional results are better with holes of less than 2 years in duration and of smaller size (i.e., stage II holes).
After the vitrectomy, most patients develop a cataract within 1 to 2 years that is dense enough to necessitate extraction to ultimately achieve the best potential visual acuity. Retinal detachment and other complications of vitrectomy also occur.
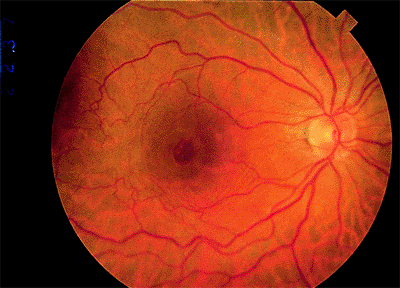 |
Figure 4.26. Photograph of a full-thickness macular hole with a small cuff of subretinal fluid. |
P.184
Macular Pucker
Macular pucker, also called surface wrinkling retinopathy and cellophane retinopathy, is caused by growth and contraction of an epiretinal membrane over the macular area. In most cases, a posterior vitreous detachment has preceded the membrane formation.
Clinical Features
The characteristic findings are wrinkling of the retina and straightening or zigzagging of retinal vessels (Fig. 4.27). In many cases, fluorescein angiography reveals macular edema leakage. In some cases, a central defect in the membrane (i.e., pseudohole) resembles a full-thickness macular hole. In most cases the visual acuity and fundus appearance remain stable after the epiretinal membrane contracts.
Macular pucker with pseudohole may resemble a full-thickness or a lamellar macular hole. In contrast to a full-thickness macular hole, the vision may be only slightly reduced, there is no cuff of subretinal fluid, and there are no drusen-like central deposits.
Management
In patients with a visual acuity of about 20/50 or less and symptoms of distortion or blur, vitrectomy with peeling of the membrane can be offered. Although some patients recover 20/20 vision, the median postoperative visual acuity improvement is approximately 2 to 3 Snellen lines. Vision outcomes are probably better with macular pucker of relatively recent (<1 year) onset.
Traumatic Maculopathies
Commotio Retinae
Commotio retinae is caused by the contrecoup mechanism. Shock waves caused by the impact traverse the fluid-filled eye and strike the retina. When commotio retinae is located in the macula, it is called Berlin's edema.
Clinical Features
The opacification of the outer retinal layers (Fig. 4.28) is caused by disruption of the outer segments of the photoreceptors and not by edema. In mild cases, fluorescein angiography shows no leakage (Fig. 4.29), indicating minimal damage to the pigment epithelium and an excellent prognosis for full return of vision. However, if there is subretinal hemorrhage or leakage on fluorescein angiography, the RPE is badly damaged, and the visual prognosis is poor.
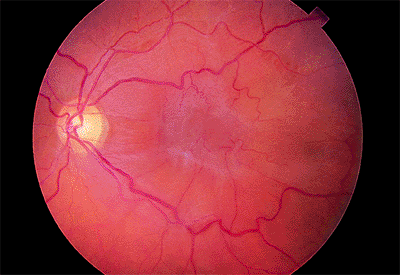 |
Figure 4.27. Severe macular pucker. Note the gray-appearing central epiretinal membrane and tortuosity of the perifoveal vessels. |
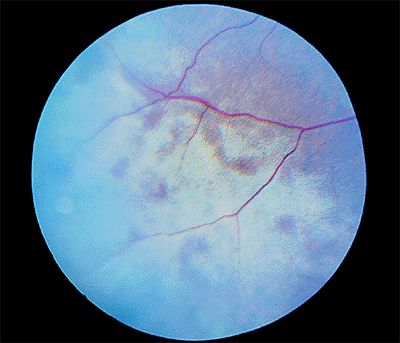 |
Figure 4.28. Peripheral commotio retinae. |
Management
There is no treatment for commotio retinae.
Choroidal Rupture
As blunt trauma compresses the eye on its anteroposterior axis and expands it on its horizontal axis, it may cause a
P.185
choroidal rupture, because Bruch's membrane is relatively rigid and breaks more easily. With it, the RPE and choriocapillaris break as well.
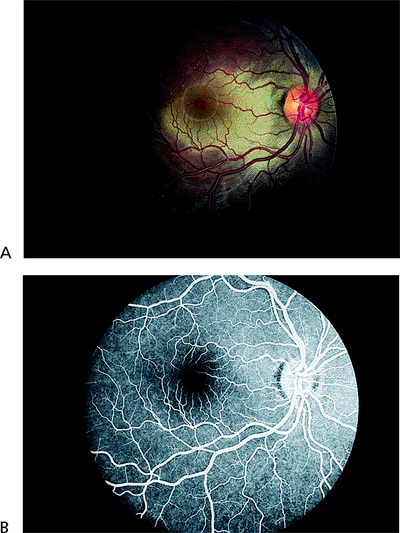 |
Figure 4.29. A: Berlin's edema. B: Normal fluorescein angiography in a case of Berlin's edema. |
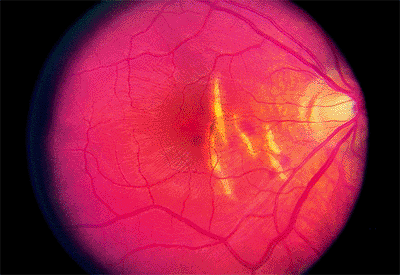 |
Figure 4.30. Multiple choroidal ruptures with subretinal hemorrhages temporal to the disc. |
Clinical Features
Choroidal ruptures are usually temporal to and concentric to the optic disc (Fig. 4.30). They are usually accompanied by a subretinal hemorrhage, which may initially completely or partially mask the rupture. Because the overlying retina is usually intact, no nerve fiber bundle visual field defects are seen. A late but uncommon complication is CNV.
Management
There is no treatment for acute choroidal rupture. Nonsubfoveal CNV responds well to laser photocoagulation.
Avulsed Optic Nerve
Rarely, direct trauma to the eye or a blow to the back of the head tears the lamina cribrosa, resulting in avulsion of the optic nerve (Fig. 4.31).
Clinical Features
A gap is seen where the neural tissue of the optic nerve retracts back into the optic nerve sheath. Retinal and vitreous hemorrhages are detected.
Management
There is no treatment for avulsed optic nerve.
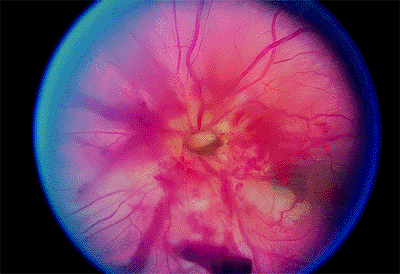 |
Figure 4.31. Avulsed optic nerve. |
Purtscher's Retinopathy
Severe compression injury to the head or chest can result in complement-activated coagulation of leukocytes and other forms of microemboli which can occlude the retinal capillaries.
Clinical Features
Purtscher's retinopathy usually is bilateral. Fundus examination reveals multiple cotton-wool spots in the distribution of the radial peripapillary capillaries (Fig. 4.32). Multiple, superficial hemorrhages may also be present. The visual prognosis is guarded. Optic atrophy often develops to varying degrees. Poor vision results from macular infarction and/or optic nerve dysfunction.
Similar findings can be seen in patients with collagen-vascular disease, pancreatitis in chronic alcoholics, patients on renal dialysis, and any systemic disease state that results in widespread microemboli.
Management
There is no treatment for Purtscher's retinopathy.
Solar Retinopathy
Solar retinopathy is retinal damage caused by excessive exposure to light. Unlike laser burns which are thermal, solar retinopathy is a phototoxic lesion. In a phototoxic injury, the release of free radicals and lysosomal enzymes damages photoreceptor membranes. Blue and near-ultraviolet light have been found to be the most harmful wavelengths.
Clinical Features
Solar retinopathy is usually bilateral, but it can occur asymmetrically because the patient squints one eye. Initially, the fundus is normal. Later, there is a deep yellowish discoloration of the retina. In mild cases, the results of fluorescein angiography are normal, the photoreceptors and pigment epithelium regenerate, and the visual acuity and fundus return to normal. In moderate cases, there is a permanent depigmentation of the central pigment epithelium, and the visual acuity may be mildly reduced (Fig. 4.33). In severe cases, there is marked pigment clumping and only partial recovery of visual acuity.
Mild cases of solar retinopathy resemble adult foveomacular
P.186
dystrophy, and the patient's history is important in differentiating them. Solar retinopathy is usually seen in younger patients.
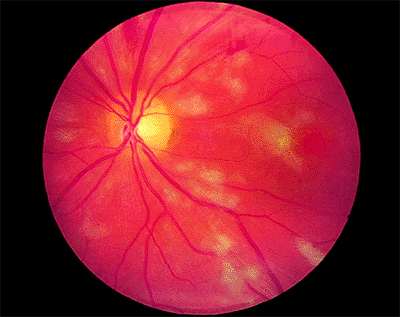 |
Figure 4.32. Multiple superficial nerve fiber layer infarctions (cotton-wool spots) along with some intraretinal hemorrhages in a patient with Purtscher's retinopathy. |
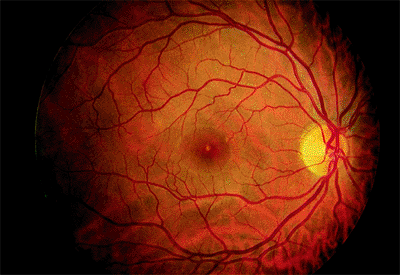 |
Figure 4.33. Small area of pigment epithelial atrophy in a patient who, as part of a religious cult, routinely looked directly at the sun. |
Management
There is no treatment for solar retinopathy.
Valsalva Maculopathy
Valsalva retinopathy is a condition in which an acute rise in intrathoracic or intraabdominal pressure against a closed glottis (i.e., Valsalva's maneuver) causes a marked increase in retinal venous pressure, which ruptures superficial capillaries, resulting in one or more superficial retinal hemorrhages.
Clinical Features
The typical patient complains of a sudden decrease in vision after coughing, sneezing, straining at stool, or heavy lifting. The hemorrhages are typically round and small (Fig. 4.34A) but may be as large as 1 to 3 disc areas. Fluorescein angiography shows no leakage (Fig. 4.34B).
A solitary subinternal membrane hemorrhage may also accompany posterior vitreous detachment. The ophthalmologist must carefully examine the peripheral retina to rule out tears.
Management
No treatment is necessary. The hemorrhages clear spontaneously, with no residual loss of vision.
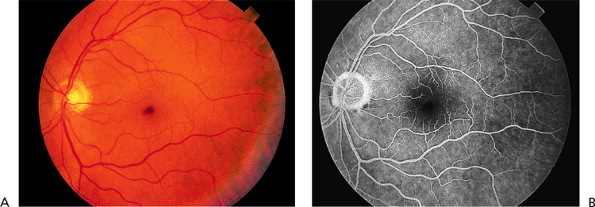 |
Figure 4.34. A: Small, central, superficial hemorrhage in a patient who noticed decreased vision after heavy lifting. B: The fluorescein angiogram shows a blockage of background fluorescence. |
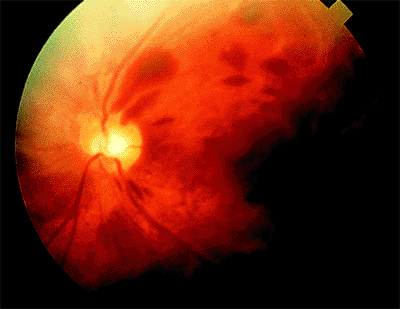 |
Figure 4.35. Terson's syndrome. Multiple superficial retinal hemorrhages are present in the posterior pole. (From Regillo CD. Posterior segment manifestations of systemic trauma. In: Regillo CD, Brown GC, Flynn HW. Vitreoretinal disease: The essentials. New York: Thieme; 1999:537, with permission.) |
Terson's Syndrome
Terson's syndrome is a condition in which intraocular hemorrhage occurs as a result of either spontaneous or trauma-induced acute intracranial bleeding.
Clinical Features
The most common intraocular finding is multiple intraretinal hemorrhages. They are almost always bilateral, superficial, and concentrated in the posterior pole (Fig. 4.35). Significant vitreous hemorrhage can also occur. Visual acuity loss is often proportional to the extent of retinal and/or vitreous hemorrhage and, in many cases, the degree of intraocular hemorrhage is directly related to the severity of the intracranial hemorrhage. Late-appearing ocular sequelae include macular pucker and traction retinal detachments.
Some degree of intraocular hemorrhage is seen in 20% to 40% of patients with acute intracranial hemorrhages. The most common type of intracranial bleed associated with Terson's syndrome is a subarachnoid hemorrhage, the source of which is most often a spontaneous rupture of an anterior communicating aneurysm.
Management
No treatment is necessary. The retinal
P.187
hemorrhages clear spontaneously and vision recovery is often good. Vitrectomy is sometimes employed for nonclearing vitreous hemorrhage or the relatively rare late sequelae.
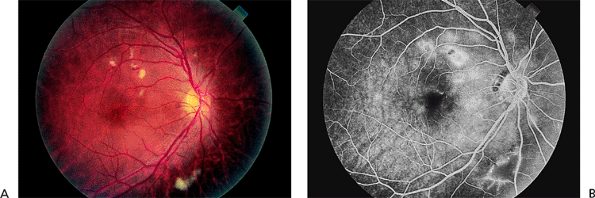 |
Figure 4.36. A: Multiple cotton-wool spots in a patient with radiation retinopathy. B: The late phase of the fluorescein angiogram shows leakage around the cotton-wool spots and from abnormal capillaries. |
Radiation Retinopathy
Radiation retinopathy is an ischemic injury to the retina caused by excessive radiation-induced damage to the retinal capillaries. A typical dose is 3,000 to 3,500 cGy, but as little as 1,500 cGy may suffice.
Clinical Features
The retinal signs are usually first noticed 6 months to 3 years after exposure. Initially, the patients are asymptomatic or have mildly decreased vision. Microaneurysms, hard exudates, soft exudates, superficial re-tinal hemorrhages, macular edema, neovascularization, and optic neuropathy may occur (Fig. 4.36A). Fluorescein angiography may reveal widespread capillary nonperfusion (Fig. 4.36B).
The retinal findings may closely resemble those of diabetic retinopathy. The key to the correct diagnosis is the history of radiation exposure, even if the radiation oncologist insists that the eyes were adequately shielded. In radiation retinopathy, when extensive capillary nonperfusion is found on fluorescein angiography, there are many fewer microaneurysms than would be expected in diabetic retinopathy.
Management
No treatment is known to prevent radiation retinopathy development or progression, although neovascularization may be treated with panretinal laser photocoagulation. The benefit of focal laser treatment is unproven.
Peripheral Retinal Lesions
Paving Stone Degeneration
Paving stones are small, discrete areas of ischemic atrophy of the outer retina. Histopathologic analysis shows attenuation or absence of the choriocapillaris, absence of the RPE, absence of the outer retinal layers, and an adhesion between the remaining inner layers and Bruch's membrane.
Clinical Features
Paving stones are white and are often surrounded by a rim of hyperplastic RPE (Fig. 4.37). They occur singly or in groups and are sometimes confluent. The paving stones usually are anterior to the equator and are most commonly found in the inferior retina.
Management
Paving stones do not require treatment because they are not a predisposing factor to retinal breaks. They limit the spread of retinal detachment.
Lattice Degeneration
Lattice degeneration is focal atrophy of the inner retina with discontinuity of the internal limiting membrane and liquefaction of the overlying vitreous. There are strong vitreous adhesions at the edges of the lesion. Lattice degeneration increases in frequency with increasing axial length of the globe. It is found in 5% to 10% of the general population.
Clinical Features
The lesions are usually anterior to the equator and are elliptical, with the long axis circumferentially oriented (Fig. 4.38). Less commonly, the lesions accompany retinal blood vessels and have a radial (i.e., meridional) orientation. Blood vessels within the lattice often appear white or hyalinized. Sometimes, the underlying pigment epithelium is hyperplastic (i.e., pigmented lattice pattern). Between 20% and 40% of retinal detachments are complications of lattice degeneration. The causative breaks are atrophic holes
P.188
within the lattice lesions or, more commonly, traction tears posterior to or at the ends of the lattice.
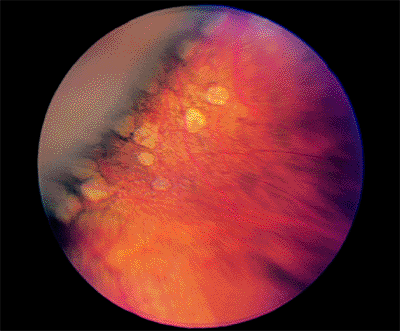 |
Figure 4.37. Peripheral paving stones. |
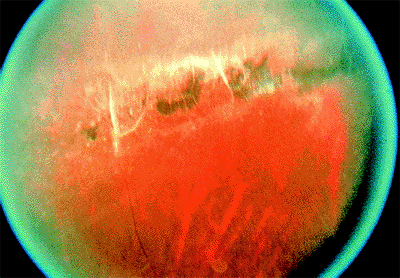 |
Figure 4.38. Patch of lattice degeneration with sclerosed-appearing vessels and hyperpigmentation. |
Management
Long-term follow-up studies have conclusively demonstrated that prophylactic treatment of asymptomatic lattice degeneration with and without atrophic holes is unnecessary. However, if a patient presents with a retinal detachment in one eye, many retinal surgeons treat lattice degeneration in the fellow eye.
Vitreoretinal Tufts
Vitreoretinal tufts are small foci of vitreous or zonular traction.
Clinical Features
Vitreoretinal tufts are slightly elevated (Fig. 4.39) and may have a cystic appearance. They can be surrounded by hyperplastic pigment epithelium. When posterior vitreous detachment occurs, vitreoretinal tufts may be the focus of a retinal tear.
Because they are elevated, vitreoretinal tufts may be difficult to differentiate from flap tears. Examination with indirect ophthalmoscopy and scleral depression or a Goldmann three-mirror lens is helpful.
Management
Asymptomatic vitreoretinal tufts do not require treatment.
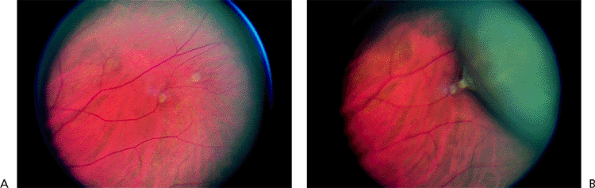 |
Figure 4.39. A: Two vitreoretinal tufts in the periphery. B: On scleral depression, the vitreous traction can be seen tenting the retina. |
Retinal Detachment
Posterior Vitreous Detachment
Posterior vitreous detachment (PVD) is the separation of the vitreous from the posterior portion of the retina (Fig. 4.40).
Clinical Features
The symptoms of posterior vitreous detachment are brief flashes of light or floaters, which are vitreous collagen fibers, hemorrhage, or epipapillary glial tissue torn from the optic disc (Weiss ring) (Fig. 4.41). Posterior vitreous detachment occasionally causes peripapillary hemorrhage. At least one retinal tear is found in 15% of patients with acute posterior vitreous detachment. A tear is found in two thirds of patients with vitreous hemorrhage. In patients without vitreous hemorrhage, a break is found in only 2% to 4%.
When the vitreous is firmly attached to the retina, as at a vitreoretinal tuft, a meridional fold, or lattice degeneration, posterior vitreous detachment may tear the retina (Fig. 4.40). When a piece of retina is torn free from the adjacent retina, the tear is called operculated (Fig. 4.42). When the torn retina remains adherent to the adjacent retina, the tear is called a flap or horseshoe tear (Fig. 4.43). Flap tears are more likely to progress to retinal detachment than are operculated tears, because continuing vitreous traction helps to separate the retina from the underlying pigment epithelium.
Other causes of photopsias, such as migraine, and of vitreous hemorrhage must be ruled out.
Management
The fundus must be carefully inspected to rule out retinal tears. In the setting of a recent PVD, retinal tears are treated with transscleral cryotherapy or laser photocoagulation.
Acute Rhegmatogenous Retinal Detachment
In acute rhegmatogenous retinal detachment, liquefied vitreous gains access to the subretinal space through a retinal break (Greek: rhegma) and separates the sensory retina from the pigment epithelium (Fig. 4.44).
Clinical Features
In 97% of patients with rhegmatogenous
P.189
P.190
retinal detachment, a retinal break is found (Fig. 4.45). In the other 3%, it is presumed to be present. Vitreous hemorrhage is common in cases caused by a flap tear. In cases of recent onset, the detached retina is opaque and corrugated (Fig. 4.46). If the macula is detached, it frequently develops cystoid macular edema that is sometimes difficult to differentiate from a full-thickness macular hole. The subretinal fluid is usually nonshifting, but will undulate with eye movements. The intraocular pressure is often decreased in proportion to the area of detached retina, but may be increased with chronic retinal detachments.
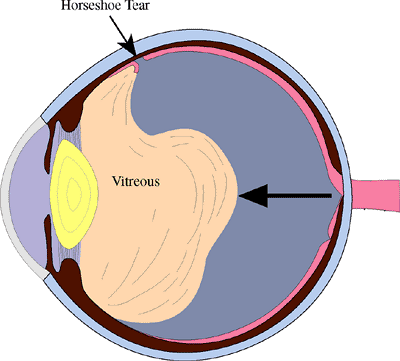 |
Figure 4.40. Posterior vitreous detachment. As the vitreous collapses, traction on areas of vitreoretinal adhesion may pull a strip of retina anteriorly, causing a flap or horseshoe tear. |
 |
Figure 4.41. Weiss ring in a patient with posterior vitreous detachment. |
 |
Figure 4.42. A: Acute operculated hole with a small rim of subretinal fluid. B: Chronic operculated hole with a pigmented demarcation line. |
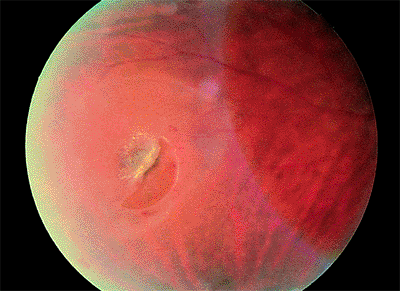 |
Figure 4.43. Flap ( horseshoe ) retinal tear surrounded by subretinal fluid. |
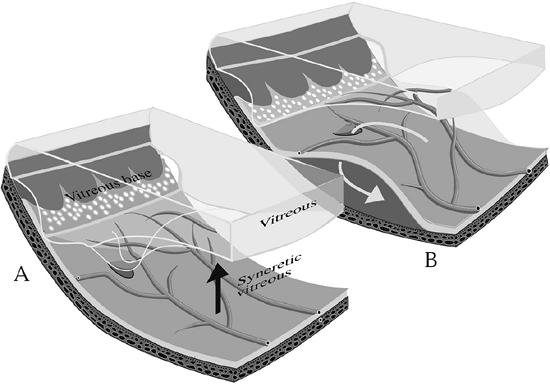 |
Figure 4.44. Primary retinal detachment. A: A posterior vitreous detachment produces traction on the peripheral retina, sometimes causing tears or holes. B: Fluid passes through the tear, dissecting through the subretinal space and creating a retinal detachment. |
Nonrhegmatogenous retinal detachment, retinoschisis, and choroidal detachment must be ruled out.
Management
For uncomplicated, primary rhegmatogenous retinal detachments, treatment approaches include scleral buckling (the gold standard), pneumatic retinopexy, and sometimes vitrectomy. Those complicated by vitreous hemorrhage, giant tears (Fig. 4.47), or significant proliferative vitreoretinopathy (see below) typically require vitrectomy, with or without scleral buckling.
Rhegmatogenous Retinal Detachment With Proliferative Vitreoretinopathy
When the retina detaches, pigment epithelial cells separate from Bruch's membrane, float through the vitreous, and proliferate on the inner retinal surface, the outer surface, or both. They also proliferate on vitreous strands. Glial cells also may proliferate on both retinal surfaces. The RPE and glial cells can form membranes that contract. In proliferative vitreoretinopathy, the proliferation and contraction are sufficient to cause clinically apparent signs.
Clinical Features
Signs of proliferative vitreoretinopathy include increased vitreous cells, posterior rolling of the edge of retinal breaks (Fig. 4.48), fixed folds (Fig. 4.49), equatorial vitreous traction, and subretinal bands.
Management
Rhegmatogenous retinal detachments with very early or limited proliferative vitreoretinopathy often
P.191
can be repaired with a scleral buckling procedure alone. Most cases, however, require vitrectomy techniques. Long-acting gases or silicone oil are commonly needed. Perfluorocarbon liquids are useful intraoperative adjuncts to facilitate retinal reattachment. Relaxing retinotomies are sometimes required to get the retina to reattach.
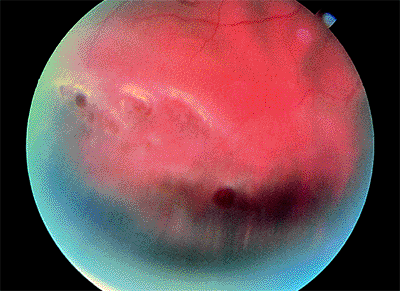 |
Figure 4.45. Retinal detachment caused by atrophic holes in lattice degeneration. |
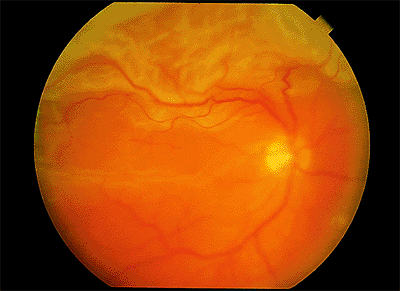 |
Figure 4.46. Corrugated, opaque appearance of the detached retina in a patient with rhegmatogenous retinal detachment. |
 |
Figure 4.47. Equator-plus photograph showing a 6 clock-hour giant retinal tear with a rolled-over retina covering the optic disc. |
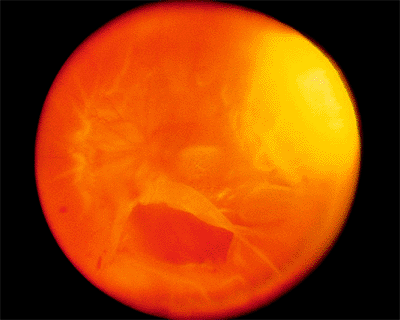 |
Figure 4.48. Equator-plus photograph of a patient with a total retinal detachment with proliferative vitreoretinopathy. There is a large inferior tear with a posteriorly rolled edge. The preequatorial area appears thin, because equatorial vitreous traction is pulling the peripheral retina centrally. |
Longstanding Retinal Detachment Without Proliferative Vitreoretinopathy
In rhegmatogenous retinal detachment caused by small breaks or by a dialysis, the detachment may remain localized indefinitely or progress slowly.
Clinical Features
The retina atrophies and becomes more transparent (Fig. 4.50). Demarcation lines may develop (Fig. 4.51). The underlying RPE becomes depigmented. RPE cells float through the vitreous cavity and proliferate on the outer surface of the retina (i.e., subretinal precipitates) or on vitreous strands (i.e., tobacco dust ) without progressing to proliferative vitreoretinopathy. In a
P.192
few cases, large pockets of fluid, called macrocysts, form in the retina. The intraocular pressure is often elevated.
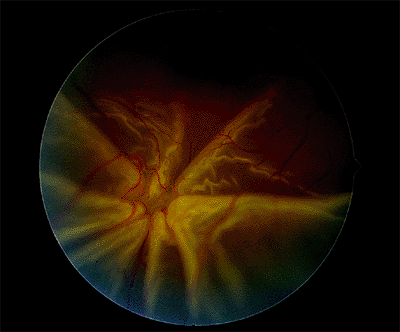 |
Figure 4.49. Fixed fold in a patient with a rhegmatogenous retinal detachment and proliferative vitreoretinopathy. |
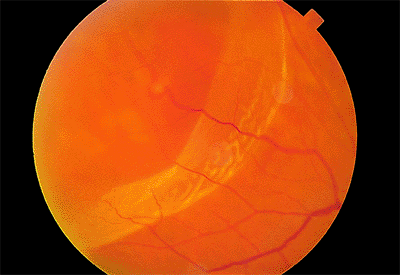 |
Figure 4.50. Rhegmatogenous retinal detachment caused by a small break along a retinal vein. Peripherally, the retina is thin where the detachment was localized for a long time. Posteriorly, the retina was more recently detached and is more opaque and corrugated than the peripheral area. |
Longstanding retinal detachment may be difficult to differentiate from senile retinoschisis, and long-term retinal detachment is part of the differential diagnosis of unilateral glaucoma.
Management
Cases with thick demarcation lines can usually be followed by observation alone. However, detachments with no or only a thin demarcation line usually require treatment. For very limited detachments, laser demarcation can be a useful alternative to surgical reattachment
Exudative Retinal Detachment
Exudative retinal detachments are caused by excessive leakage of fluid from the choroid or the retina. Common causes are neoplasms, inflammatory conditions (e.g., Harada's disease, posterior scleritis), nanophthalmos, uveal effusion syndrome, sympathetic ophthalmia, bullous CSCR, malignant hypertension, and Coats' disease.
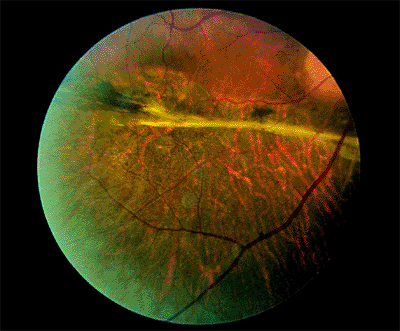 |
Figure 4.51. After repair of a longstanding retinal detachment, atrophic pigment epithelium and a thick demarcation line can be seen inferiorly. |
Clinical Features
In cases caused by tumors, the subretinal fluid initially overlies or surrounds the lesion. Later, the subretinal fluid gravitates (i.e., shifting fluid ) to the most dependent part of the eye, usually forming two bullae inferiorly (Fig. 4.52). No retinal breaks occur.
Rhegmatogenous retinal detachment must be ruled out. If the retina can be seen immediately behind the lens, the detachment usually is exudative. Other findings depend on the cause of the detachment. Unlike rhegmatogenous detachments, one would not expect to find vitreous hemorrhage, vitreous pigment cells ( tobacco dust ), or, of course, a retinal break.
Management
Treatment is directed at the underlying condition (e.g., corticosteroids for inflammatory etiologies, radiation therapy for choroidal tumors, etc.). Visual prognosis is variable and depends on factors related to both the retinal detachment (e.g., extent and chronicity) and the inciting condition (e.g., location and curability).
Traction Retinal Detachment
In traction retinal detachment, the retina is pulled into the vitreous cavity by transvitreal (anteroposterior) traction. The most common causes are proliferative retinopathies, such as diabetic retinopathy and old penetrating injuries.
Clinical Features
Clinically, no breaks are found. The detached retina is smooth, immobile, and concave toward the pupil (Fig. 4.53), and it does not extend to the ora serrata. The examiner must remember that patients with proliferative retinopathy can also have combined traction and rhegmatogenous retinal detachment in which there may be corrugations and in which the retina is convex toward the pupil (Fig. 4.54).
Management
The traction must be released by vitrectomy in which various membrane peeling techniques are employed. Localized, extramacular detachments do not necessarily progress and, therefore, may be observed. Traction detachments that threaten to detach or already extend into the macula require surgery.
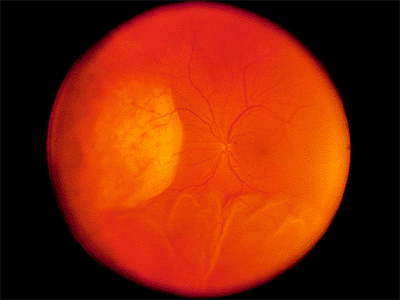 |
Figure 4.52. Large amelanotic malignant melanoma nasally with an inferior exudative retinal detachment. |
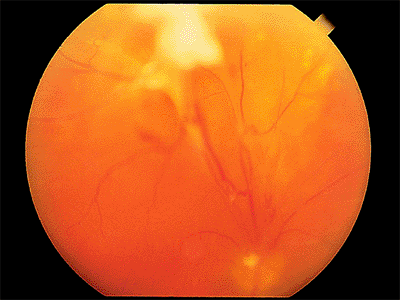 |
Figure 4.53. Superior traction retinal detachment in a patient with proliferative diabetic retinopathy. |
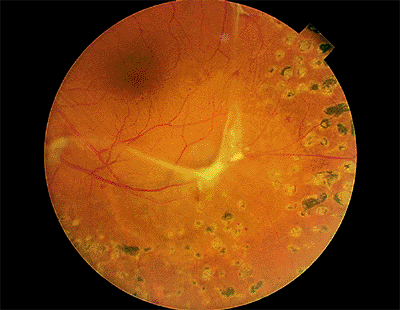 |
Figure 4.54. Combined early traction and rhegmatogenous retinal detachment. A small hole exists just inferior to the fibrovascular proliferation. |
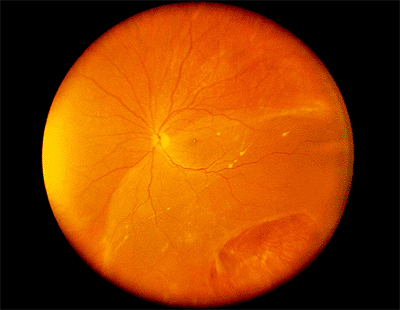 |
Figure 4.55. Equator-plus photograph showing a longstanding inferotemporal retinal detachment with a retinal dialysis from blunt eye trauma. |
 |
Figure 4.56. Avulsed vitreous base after blunt ocular trauma. |
P.193
Traumatic Retinal Detachment
Clinical Features
There are five mechanisms by which blunt trauma can cause retinal breaks. The first type of traumatic break is caused by compression of the eye along its anteroposterior axis, with resultant horizontal expansion. This can result in true dialysis, which is a separation of the retina from the nonpigmented epithelium of the pars plana at the ora serrata (Fig. 4.55), or in linear tears along the anterior border of the vitreous base, the posterior border of the vitreous base, or both. Such tears are most common superonasally and inferotemporally and may be accompanied by an avulsed vitreous base (Fig. 4.56), which is pathognomonic of blunt trauma. The second type, posterior stretch tears (Fig. 4.57), are caused by the same mechanism. The third type of traumatic break is caused by traction on a distant focus of vitreoretinal traction, such as lattice degeneration or a vitreoretinal traction tuft. The fourth type is caused by focal retinal necrosis, in which a foreign object strikes the sclera directly under the retina (Fig. 4.58). These breaks are characterized by ragged edges. The fifth type, a macular hole, may follow severe Berlin's edema or be related to late vitreous contracture. Because traumatic retinal breaks usually occur in young patients with little liquid vitreous, the retinal detachments they cause typically progress slowly,
P.194
with many of the features of longstanding retinal detachment (Fig. 4.55).
 |
Figure 4.57. Traumatic stretch tear. |
 |
Figure 4.58. Necrotic tear caused by a direct blow to the eye. |
Management
Traumatic retinal breaks and detachments are treated like other retinal breaks and detachments.
Choroidal Detachment
Accumulation of fluid or blood in the suprachoroidal space elevates the choroid, pigment epithelium, and retina, creating what is called a choroidal detachment. The most common cause is hypotony during or after intraocular surgery. Serous choroidal detachments often are found in eyes with longstanding retinal detachment. Severe, often hemorrhagic, choroidal detachments that form during or after eye surgery are most common in elderly patients with myopia and glaucoma.
Clinical Features
Choroidal detachments have the same color as adjacent normal pigment epithelium. They may have a slightly corrugated appearance (Fig. 4.59). The pars plana may be seen without scleral depression.
Choroidal detachments can be mistaken for rhegmatogenous retinal detachment and choroidal melanoma. In choroidal detachments, no breaks are found, and choroidal detachments have a denser, more solid appearance than retinal detachments. Serous choroidal detachments transilluminate.
 |
Figure 4.59. Superotemporal choroidal detachments after cataract surgery. The pars plana can be seen easily without scleral depression. |
Management
When hypotony is the cause of the choroidal detachment, the underlying cause, such as a cleft or a wound leak, must be identified and treated. Choroidal detachments, even large ones, often resolve spontaneously. In some cases, there can be a very shallow or flat anterior chamber, secondary intraocular pressure elevation, or severe pain, especially when there is suprachoroidal blood. These associated problems often represent indications for surgical drainage.
Retinoschisis
Senile Retinoschisis
Senile retinoschisis is a peripheral splitting of the retina into two layers. The split is most common in the outer plexiform layer (Fig. 4.60). The cavity contains hyaluronic acid.
Clinical Features
Retinoschisis is usually seen in hyperopic eyes and is most commonly located inferotemporally and superotemporally. Prominent cystoid degeneration occurs at the ora serrata. The inner wall has a smooth surface and is dome shaped (Fig. 4.61). It may have sheathed retinal vessels and white opacities called snowflakes, which are the footplates of M ller's cell columns. Inner wall holes may be found. The outer wall has a pocked appearance and may have round holes (Fig. 4.62), with or without a pigmented demarcation line. The complications of senile retinoschisis are loss of peripheral visual field and rhegmatogenous retinal detachment. When outer wall holes occur alone, the detachments typically advance slowly, and demarcation lines around the holes are common. If inner and outer wall holes are present, the two layers of the schisis may collapse together, and the detachment may look like a typical rhegmatogenous retinal detachment and progress rapidly.
Factors that help to differentiate retinoschisis from a longstanding retinal detachment without proliferative vitreoretinopathy are the absence of a demarcation line, absence of underlying RPE degeneration, absence of tobacco dust, and an absolute scotoma on visual field testing.
Management
Because outer wall breaks rarely cause clinical retinal detachment, the only indication for treatment of retinoschisis is symptomatic, progressive retinal detachment.
Congenital or X-Linked Retinoschisis
Congenital retinoschisis is an X-linked recessively inherited condition in which the basic abnormality is splitting of the nerve fiber layer.
Clinical Features
The hallmark of congenital retinoschisis is the stellate or spoke-wheel macula (Fig. 4.63), which superficially resembles cystoid macular edema but does not
P.195
P.196
show leakage on fluorescein angiography. Half of the patients have peripheral retinoschisis (inferotemporal, 90%; inferonasal, 58%) in which large inner wall holes (Fig. 4.64) with unsupported blood vessels are common. The ERG reflects the widespread inner retinal damage. Characteristically, the a wave is normal, and the b wave is decreased. The ERG is abnormal whether or not there are appreciable peripheral schisis cavities.
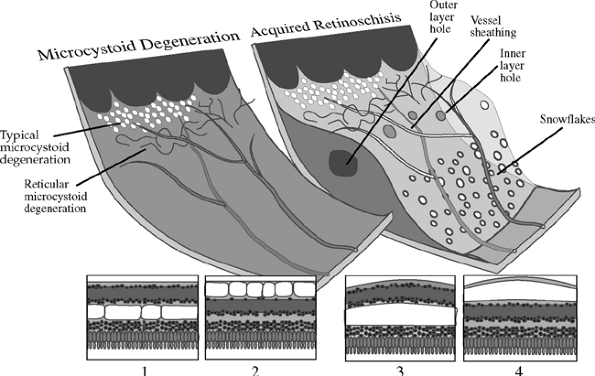 |
Figure 4.60. In typical microcystoid degeneration, pockets of hyaluronic acid form in the outer plexiform layer (1). Reticular microcystoid degeneration is characterized by a cystic degeneration of the nerve fiber layer (2). In typical retinoschisis, a continuous split in the outer plexiform layer is thought to be caused by a consolidation of the hyaluronic pockets of typical microcystoid degeneration (3). Reticular microcystoid degeneration may lead to reticular retinoschisis when a continuous separation in the nerve fiber layer forms (4). Round holes in the inner and outer retinal layers can occasionally lead to a rhegmatogenous retinal detachment. |
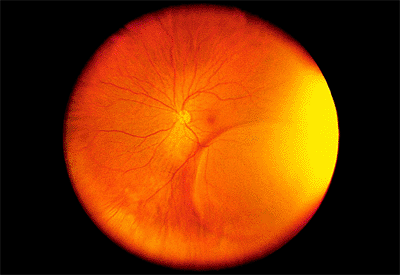 |
Figure 4.61. Equator-plus photograph showing a dome-shaped area of elevation of the inner retinal layer in a patient with senile retinoschisis. |
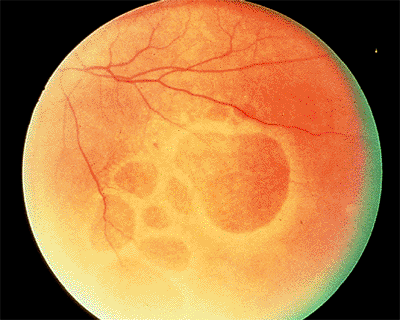 |
Figure 4.62. Large outer wall holes in an eye with senile retinoschisis. |
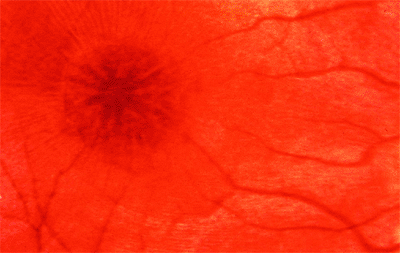 |
Figure 4.63. Spoke-wheel cystoid macular changes in a patient with juvenile retinoschisis. |
The most common reasons for the initial presentation are decreased vision (45%), strabismus (18%), vitreous hemorrhage (15%), and a positive family history (15%). Retinal detachment and vitreous hemorrhage is rare. The visual acuity is only moderately decreased, usually to the 20/70 level. After 20 years of age, the vision usually remains stable.
The only condition in which the macula resembles juvenile retinoschisis is the Goldmann-Favre syndrome.
Management
Because prophylactic photocoagulation to limit the spread of the area of schisis or to prevent retinal detachment is more likely to aggravate the condition than to stabilize it, this approach is not indicated. Surgery is indicated for the rare, secondary complications of retinal detachment or nonclearing vitreous hemorrhage.
 |
Figure 4.64. Large inner wall defect in a patient with juvenile retinoschisis. An unsupported retinal vessel bridges the gap. |
Toxic Retinopathies
Chloroquine Retinopathy
Excessive ingestion of chloroquine causes diffuse retinal deterioration. Traditionally, the damage was thought to be caused by progressive binding of chloroquine by melanin in the RPE cells, but later evidence indicated that the ganglion cells are also directly damaged. The cumulative dose of drug is the most important predictor of retinopathy. Less than 100 g causes no toxicity. When it is greater than 300 g, 70% of patients manifest some signs of toxicity. Plaquenil (hydroxychloroquine) retinopathy is similar to chloroquine retinopathy, but Plaquenil is much less toxic. Plaquenil toxicity is rarely seen with a total dose of less than 700 g or an average daily dose of 400 mg or less.
Clinical Features
The earliest signs of retinopathy are small scotomata on visual field testing, a supranormal EOG, an abnormal photostress test result, and decreased color vision. Because visual acuity is typically affected later, it cannot be relied on to detect early toxicity. Similarly, in an eye with early toxic reactions, the fundus may be normal. The earliest fundus abnormalities are small areas of depigmentation of the RPE (Fig. 4.65). By the time the classic bull's-eye maculopathy is evident (Fig. 4.66), considerable damage has been done.
The end-stage fundus appearance of chloroquine retinopathy resembles a bull's-eye pattern. The most common causes of a bull's-eye are Stargardt's disease, cone dystrophy, and AMD.
Management
The only treatment is to discontinue the drug. Although many patients have some recovery of vision, a few have damage that continues to progress.
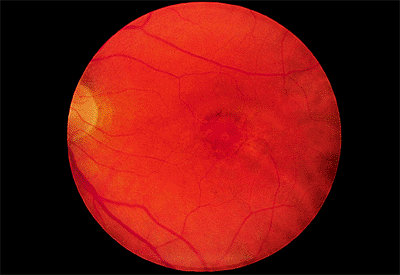 |
Figure 4.65. Early chloroquine retinopathy with a small bull's-eye lesion. |
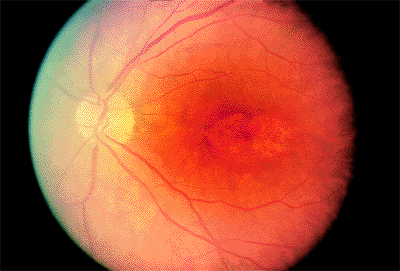 |
Figure 4.66. Fully developed bull's-eye lesion in a patient with chloroquine retinopathy. |
P.197
Thioridazine Toxicity
There are two types of thioridazine (Mellaril) toxicity. In the acute form, large doses (e.g., 2,000 mg/day or more) are administered to psychotic patients, who experience an acute decrease in central vision. In the chronic form, patients who take thioridazine for long periods at lower doses (800 to 1,000 mg/day) have peripheral visual field loss, but they usually retain good central vision.
Clinical Features
The acute type of thioridazine toxicity is characterized by widespread stippling of the RPE, which includes the macula (Fig. 4.67A). In the chronic type, there are circumscribed areas of RPE loss (Fig. 4.67B).
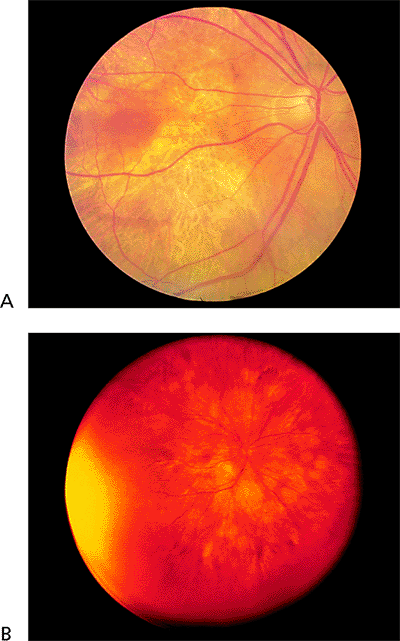 |
Figure 4.67. A: Posterior pole pigment epithelial atrophy in a patient with Mellaril (thioridazine hydrochloride) retinopathy. B: An equator-plus photograph shows multiple large areas of pigment epithelial atrophy. |
In the acute type, the diagnosis is usually easy because of the history of ingestion of massive doses of thioridazine. The chronic type may resemble gyrate atrophy. The main difference is that the areas of atrophy in thioridazine toxicity are not as pronounced as those in gyrate atrophy.
Management
The drug must be discontinued with any signs of retinal toxicity.
Talc Retinopathy
In chronic intravenous drug abusers, talc particles gradually occlude pulmonary capillaries. Pulmonary shunts develop, allowing particles to gain access to the systemic circulation and to the eye, where they occlude retinal capillaries.
Clinical Features
In most cases, the refractile particles can be seen in the macula (Fig. 4.68). Macular capillary occlusions cause hypoxia and edema with decreased vision. Widespread areas of capillary nonperfusion in the periphery can lead to peripheral and optic disc neovascularization and to vitreous hemorrhage.
Proliferative talc retinopathy must be differentiated from other causes of peripheral neovascularization, such as diabetes, sarcoidosis, and Eales disease. Talc maculopathy can resemble radiation and diabetic retinopathies. The patient's history and recognition of the talc particles are important. The differential diagnosis of crystalline retinopathy in general includes canthaxanthine, tamoxifen, nitrofurantoin, primary and secondary oxalosis, cystinosis, and Bietti's disease.
Management
Patients with neovascularization and vitreous hemorrhage should be treated with photocoagulation.
Tamoxifen Retinopathy
Tamoxifen retinopathy is a rare form of crystalline retinopathy occasionally seen in patients taking the drug for extended periods of time.
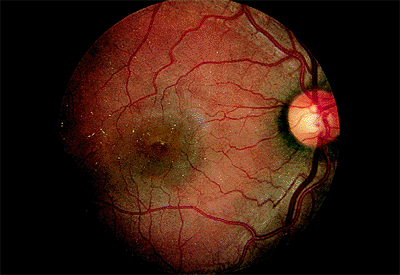 |
Figure 4.68. Multiple talc particles are present in the macula of a patient who was a longstanding intravenous drug abuser. |
P.198
Clinical Features
. Examination reveals subtle, yellow-white dots in the central macula with or without vision loss or other evidence of visual dysfunction. Corneal opacities and optic neuropathy have also been reported.
Management
The drug should be discontinued at the first signs of any visual dysfunction.
Albinism
Albinism can be divided into two somewhat overlapping types: oculocutaneous and ocular. In oculocutaneous albinism, which is inherited autosomal recessively, there is decreased melanin pigment in each melanosome. It can be subdivided into a tyrosinase-positive subtype, in which the skin, hair, and eyes have some pigment, and a tyrosinase-negative subtype, in which there is no pigment. In ocular albinism, the cutaneous findings are minimal. The total number of melanosomes is decreased, although each melanosome may be fully pigmented, and macromelanosomes are present. The disorder may have an X-linked recessive pattern of inheritance.
Clinical Features
The sine qua non of albinism is foveal hypoplasia (Fig. 4.69), which is the absence of the foveal reflex, absence of the yellow macula lutea pigment, absence of the normal hyperpigmentation of the foveal pigment epithelium, and failure of the retinal vasculature to create a foveal avascular zone. Other findings include congenitally subnormal vision, nystagmus, photophobia, iris transillumination, and a hypopigmented fundus. The visual acuity is usually markedly decreased, and the patients have searching nystagmus. Supranormal amplitudes are found on the ERG and EOG.
Women who are carriers of the X-linked variety often show partial iris transillumination. They may also have peripheral RPE mosaicism or a salt and pepper fundus. The visual functions of carriers are normal.
In some patients with lightly pigmented skin, the RPE may also be hypopigmented (i.e., blond fundus), but visual function and the fovea are normal.
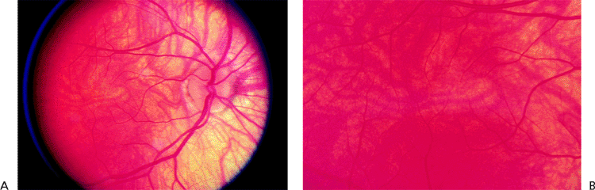 |
Figure 4.69. A: Depigmentation of the fundus occurs in albinism. B: Macular hypoplasia is present in the same patient. |
Management
Periodic examination, genetic counseling, and low vision support constitute management.
Tapetoretinal Diseases
Retinitis Pigmentosa
Retinitis pigmentosa (RP) is characterized by progressive degeneration of the rods and cones. In most cases, there is associated migration of pigment epithelial cells into the retina. The disorder can have autosomal recessive, X-linked recessive, and autosomal dominant patterns of inheritance. The autosomal recessive and X-linked forms have the earliest onset (i.e., early childhood) and the worst prognosis. Patients with the autosomal dominant form may be symptom free until middle age. Genetic analysis has shown that the primary defect in most affected individuals is in the gene that codes for rhodopsin. There are two variants of RP: type I, in which the rods are affected earlier than the cones, and type II, in which the cones are affected earlier than the rods.
Clinical Features
In type I RP, the first symptoms are decreased scotopic vision and decreased peripheral vision. Central visual acuity is initially normal and may remain so, but it can be decreased because of macular atrophy (Fig. 4.70), cystoid macular edema, or the typical posterior subcapsular cataract.
Fundus findings include vitreous cells and opacities, narrowed arteries, diffuse depigmentation of the RPE, and comma-shaped intraretinal proliferation of pigmented cells (i.e., bone spicules ) that may be sparse or densely packed (Figs. 4.71 and 4.72). The waxy pallor of the optic disc is a late sign.
The diagnosis of type I RP is usually made by clinical examination alone. A careful family history can be helpful, as can examination of family members to detect the carrier state. The examiner looks for isolated patches of bone spicules, splotchy pigmentation of the RPE (i.e., salt and pepper pattern), or a bronze sheen in the macula. The diagnosis can be confirmed by an ERG. In the earliest stages of
P.199
the disease, the scotopic ERG is more affected than the photopic. In late stages, the ERG is usually nonrecordable.
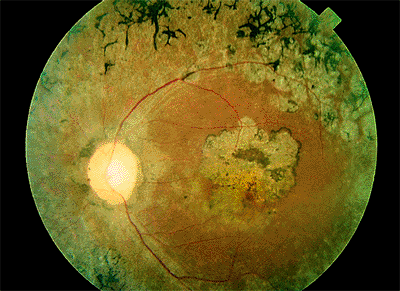 |
Figure 4.70. Narrowed arteries, pale optic disc, bone spicules, and atrophic macula can be seen in a patient with retinitis pigmentosa. |
In type II RP, patients are usually not night blind until the visual field has contracted to less than 10 . Half of the patients have no intraretinal pigment deposition (i.e., retinitis pigmentosa sine pigmento). Nevertheless, they have cells in the vitreous, and the pigment epithelium has a paler appearance than normal. In early cases, the photopic ERG is more severely affected than is the scotopic.
Other causes of night blindness, such as vitamin A deficiency, congenital stationary night blindness, and the RP systemic syndromes, must be ruled out. Certain entities such as congenital syphilis and rubella have a salt and pepper appearance that may be confused with RP, but there is no migration of pigment cells into the sensory retina, and the ERG is usually normal or near normal. The unilateral RP probably does not exist, although there may be pigment migration into the sensory retina. Most cases are caused by blunt trauma, uveitis, and regressed, longstanding retinal detachment.
Management
If a significant cataract is present, cataract extraction with placement of a posterior chamber lens often provides a significant improvement in visual function. Cystoid macular edema may respond to acetazolamide (500 mg/day) with some degree of improved visual acuity. Unfortunately, the beneficial effect is temporary for some patients. A prospective randomized trial showed that daily administration of 15,000 IU of vitamin A slowed the rate of deterioration detected by ERG, but there was no demonstrable benefit for visual acuity or the visual fields.
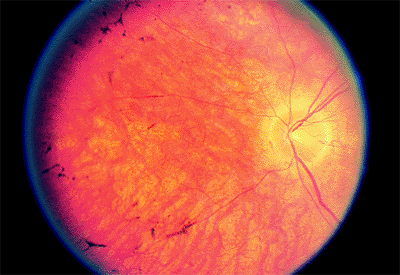 |
Figure 4.71. Narrowed arteries, pale or washed-out retinal pigment epithelium, and a few bone spicules. |
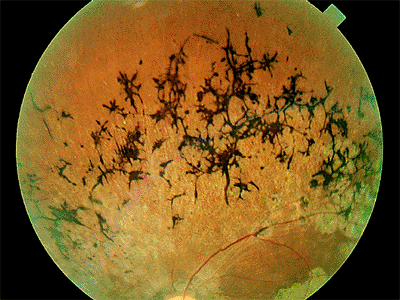 |
Figure 4.72. Densely packed bone spicules. |
Sector Retinitis Pigmentosa
In sector retinitis pigmentosa, one area of the fundus has the prominent bone spicule and vascular changes of classic RP. The inferior retina is most commonly affected, and the changes are bilateral and symmetric. The remaining retina looks normal or almost normal, but careful electrophysiologic testing shows some impairment. Sector RP is inherited autosomal dominantly.
Retinitis Pigmentosa Syndromes
Laurence-Moon-Bardet-Biedl Syndrome
Laurence-Moon-Bardet-Biedl syndrome is an inherited condition that is characterized by an atypical pigmentary retinopathy. Affected individuals may also have mental retardation, hypogenitalism, short stature, polydactyly, obesity, and spastic paraplegia.
Refsum's Syndrome
Refsum's syndrome is caused by an inborn error of metabolism. In rhodopsin metabolism, vitamin A is esterified to palmitic acid, which is metabolized to phytanic acid. Because phytanic acid -hydroxylase is deficient, phytanic acid accumulates in RPE cells, causing the sensory retina to deteriorate and resulting in an atypical type of RP (Fig. 4.73). Systemic findings include cerebellar ataxia, peripheral neuropathy, nerve deafness, and anosmia. There is also increased cerebrospinal fluid protein without pleocytosis. Patients may benefit from a diet low in phytanic acid.
 |
Figure 4.73. Irregular bone spicule-type pigmentation in the periphery of a patient with Refsum's syndrome. |
P.200
Bassen-Kornzweig Syndrome
Bassen-Kornzweig syndrome is caused by failure to produce -lipoprotein, with resultant malabsorption and vitamin A deficiency. The vitamin A deficiency causes night blindness with retinal degeneration. Bone spicules are usually not seen. Systemic findings include steatorrhea, acanthocytosis, ataxic neuropathy, and growth retardation. It must be stressed that the abnormalities caused by this condition can be reversed with vitamin A therapy if the diagnosis is made before significant retinal degeneration has occurred.
Spielmeyer-Vogt-Batten-Mayou Syndrome
Spielmeyer-Vogt-Batten-Mayou syndrome is characterized by seizures, progressive dementia, ataxia, and an RP-like syndrome with a bull's-eye macula. The diagnosis can be confirmed by the finding of vacuolation of peripheral lymphocytes and by metachromasia of skin fibroblasts in cell culture.
Progressive External Ophthalmoplegia
Progressive external ophthalmoplegia is a mitochondrially inherited disease characterized by gradual paresis of the extraocular muscles and eyelids. There is ophthalmoplegia (without diplopia) and ptosis. The severity of retinal involvement varies. The most common variant is benign, with salt and pepper RPE changes and normal visual function. The less common variant progresses to findings similar to those of RP. Systemic findings include facial weakness, dysphagia, small stature, limb girdle myopathy, and cardiac conduction defects. Affected patients may have complete heart block (Kearns-Sayre syndrome.)
Other Retinitis Pigmentosa Related Syndromes
There are many syndromes in which the typical RP is associated with systemic abnormalities. The most common is Usher's syndrome, in which RP is associated with deafness.
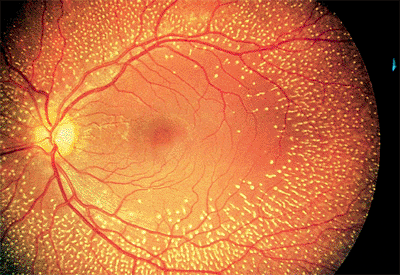 |
Figure 4.74. Multiple dot-like lesions at the level of the pigment epithelium in a patient with fundus albipunctatus. |
Fundus Albipunctatus
Fundus albipunctatus is a descriptive term used to describe eyes that have myriad, small, white subretinal dots (Fig. 4.74). There are two variants. The first, retinitis punctata albescens, progresses to produce findings similar to those of retinitis pigmentosa. The second, albipunctate dystrophy, is a form of congenital stationary night blindness.
In patients with albipunctate dystrophy, the number of white dots may increase or decrease. The amplitude of the scotopic ERG is usually subnormal, but if the patient's eyes are patched and allowed to dark adapt for 3 hours, the amplitude typically recovers to normal. The same is true for dark adaptation, which may take hours to reach the final rod threshold. The visual acuity is usually normal, but there may be late macular atrophy.
Choroideremia
Choroideremia is an X-linked recessive tapetoretinal degeneration that becomes symptomatic in patients between 4
P.201
and 20 years of age. The primary defect is probably in the RPE. The symptoms are similar to those of RP. The fundus, unlike that seen in RP, has no pigment clumping. In its early stages, choroideremia is characterized by patchy atrophy of the peripheral RPE and choriocapillaris. Later, the RPE and choriocapillaris are completely absent, except in the macula (Fig. 4.75). The diagnosis is made by fundus examination, family history, and examination of carrier women who typically have a salt and pepper fundus.
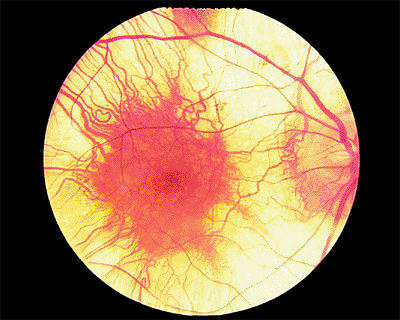 |
Figure 4.75. Central remaining island of pigment epithelium and choriocapillaris in a patient with advanced choroideremia. |
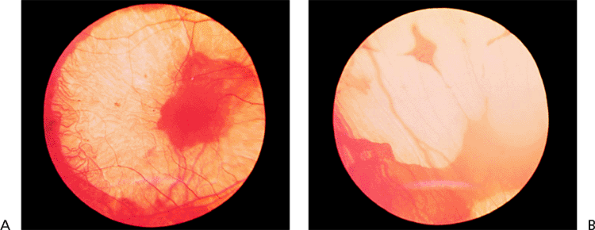 |
Figure 4.76. A: Scalloped areas of pigment epithelium and choriocapillaris dropout in a patient with gyrate atrophy. B: Peripheral scalloped areas of choriocapillaris and pigment epithelial dropout in the same patient. |
Gyrate Atrophy
Gyrate atrophy is an autosomal recessive disease caused by an error in metabolism that results in hyperornithinemia. The age of onset is between 10 and 40 years. Its symptoms are similar to RP. The fundus has well-defined scalloped areas of absent choriocapillaris and RPE with an abrupt transition between normal and atrophic areas (Fig. 4.76). Patients and carriers can be diagnosed by measurement of ornithine ketoacid transaminase in cultures of skin fibroblasts. Lowering of serum ornithine with oral pyridoxine (vitamin B6) therapy has been shown to slow the rate of retinal degeneration.
 |
Figure 4.77. Vitreous bands in a patient with Stickler's syndrome. |
Stickler's Syndrome
Stickler's syndrome is an autosomal dominant condition whose hallmark is an optically empty vitreous, except for a thin layer of cortical vitreous behind the lens and bands of collagen that traverse the vitreous cavity and adhere to the retina (Fig. 4.77). Systematic abnormalities include orofacial abnormalities, such as midfacial flattening and cleft palate, and joint abnormalities, such as hyperextensibility, enlargement, and arthritis. Retinal detachment is common. Repair is frequently difficult, because the retinal breaks are at multiple levels in areas of circumferential and radial lattice degeneration (Fig. 4.78) and because there is a strong tendency for proliferative vitreoretinopathy. Other ocular abnormalities include retinoschisis, pigmentary retinal degeneration with a depressed ERG, cataract, glaucoma, optic atrophy, and high myopia.
Leber's Congenital Amaurosis
Leber's congenital amaurosis is a severe retinal dystrophy manifested at birth or early in infancy. The ERG pattern is nonrecordable. The fundus examination initially may be
P.202
normal, or there may be a central area of atrophic RPE sometimes referred to as a macular coloboma (Fig. 4.79). Later, there is some degree of nonspecific, diffuse RPE mottling and sometimes intraretinal pigment deposits giving a retinitis pigmentosa like picture. Affected patients usually are hyperopic and have pendular nystagmus. Keratoconus is also common. Mental retardation is sometimes present.
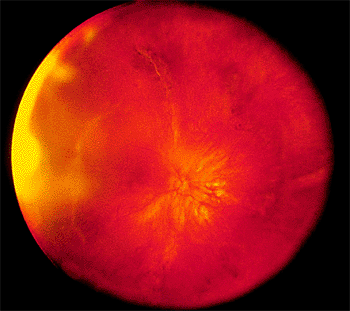 |
Figure 4.78. Equator-plus photograph showing multiple areas of radial and circumferential lattice in a patient with Stickler's syndrome. |
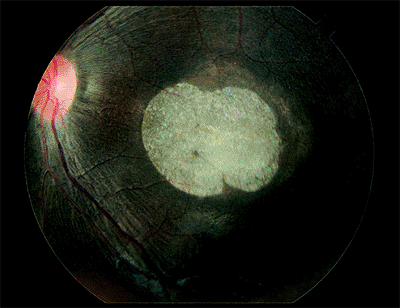 |
Figure 4.79. Macular atrophy and narrowed retinal arteries are seen in a patient with Leber's congenital amaurosis. |
Metabolic Diseases
The sphingolipidoses are a group of diseases caused by a deficiency of the lysosomal enzymes necessary for the degeneration of gangliosides, cerebrosides, and sphingomyelin, which are collectively called sphingolipids. They are important components of cell membranes, especially in nervous tissue.
Patients with Tay-Sachs disease have mental retardation and experience early death. The sphingolipids accumulate in the ganglion cells of the retina, rendering them opaque. Because ganglion cells are more numerous in the perifoveolar retina than elsewhere and are absent in the foveola, the typical fundus finding is a cherry red spot (Fig. 4.80).
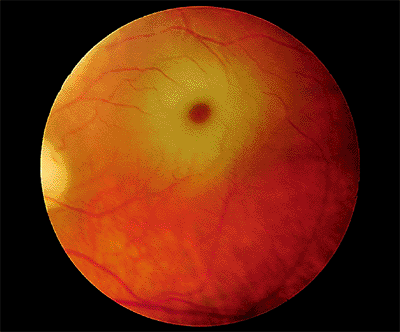 |
Figure 4.80. Cherry red spot in the macula of a patient with Tay-Sachs disease. |
Niemann-Pick disease type B is also called the sea-blue histiocyte syndrome or chronic Niemann-Pick disease. It is the mildest form of the several varieties of Niemann-Pick and is commonly not diagnosed until the late teens or early adult years. Systemic findings include massive hepatosplenomegaly, infiltration of the lungs, and foam cells in the bone marrow. Unlike many of the other sphingolipidoses, there is no mental retardation and no functional involvement of the central nervous system. The typical ocular finding is a macular halo.
Bibliography
American Academy of Ophthalmology. Retina and vitreous: Basic and clinical science course, section 12. San Francisco: Foundation of the AAO; 2000.
Armstrong JD, Meyer D, Xu S, et al. Long-term follow-up of Stargardt's disease and fundus favimaculatus. Ophthalmology 1998;105:448.
Benson WE, Shakin J, Sarin LK. Blunt trauma. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 3. Philadelphia: JB Lippincott; 1992:1.
Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol 1993;111:761.
Bird AC, Bressler NM, Bressler SB, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. Surv Ophthalmol 1995;39:367.
Bird AC. Retinal photoreceptor dystrophies. LI Edward Jackson memorial lecture. Am J Ophthalmol 1995;119:543.
Bird AC. Inherited outer retinal dystrophies. In: Tasman WS. Clinical decisions in medical retinal disease. St. Louis: Mosby; 1994:29.
Bressler NM, Bressler SB, Fine SL. Age-related macular degeneration. Surv Ophthalmol 1988;32:375.
Brown GC. Congenital fundus abnormalities. In: Regillo CD, Brown GC, Flynn HW. Vitreoretinal disease: The essentials. New York: Thieme; 1999:581.
Brown GC. Radiation retinopathy. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 3. Philadelphia: JB Lippincott; 1992:1.
Byer NE. Long-term natural history of lattice degeneration of the retina. Ophthalmology 1989;96:1396.
Byer NE. Lattice degeneration of the retina. Surv Ophthalmol 1979;22:41.
Byer NE. Long-term natural history study of senile retinoschisis with implications for management. Ophthalmology 1986;93:1127.
Byer NE. The natural history of asymptomatic retinal breaks. Ophthalmology 1982;89:1033.
Carr RE. The generalized heredoretinal disorders. In: Regillo CD, Brown GC, Flynn HW. Vitreoretinal disease: The essentials. New York: Thieme; 1999:307.
Chu TG, Cano MR, Green RL, Liggett PE, Lean JS. Massive suprachoroidal hemorrhage with central retinal apposition: a clinical and echographic study. Arch Ophthalmol 1991;109:1575.
Clarkson JF, Altman RD. Angioid streaks. Surv Ophthalmol 1982;26:235.
Condon GP, Brownstein S, Wang NS, et al. Congenital hereditary (juvenile X-linked) retinoschisis. Histopathologic and ultrastructural findings in three eyes. Arch Ophthalmol 1986;104:576.
Cruess AF, Schachat AP, Nicholl J, et al. Chloroquine retinopathy: is fluorescein angiography necessary? Ophthalmology 1985;92:127.
P.203
Dabbs TR, Skjodt K. Prevalence of angioid streaks and other ocular complications of Paget's disease of bone. Br J Ophthalmol 1990;74:579.
Davis MD. Natural history of retinal breaks without detachment. Arch Ophthalmol 1974;92:183.
Deutman AF. Electro-oculography in families with vitelliform dystrophy of the fovea: detection of the carrier state. Arch Ophthalmol 1969;81:305.
Eagle RC, Lucier AC, Bernardino VB, et al. Retinal pigment epithelial abnormalities in fundus flavimaculatus. Ophthalmology 1980;87:1189.
Easterbrook M. Dose relationships in patients with early chloroquine retinopathy. J Rheumatol 1987;14:472.
Ficker L, Vafidis G, While A, Leaver P. Long-term follow-up of a prospective trial of argon laser photocoagulation in the treatment of central serous retinopathy. Br J Ophthalmol 1988;72:829.
Fishman GA, Baca W, Alexander KR, et al. Visual acuity in patients with Best vitelliform macular dystrophy. Ophthalmology 1993;100:1665.
Fishman GA, Farber M, et al. Visual acuity loss in patients with Stargardt's macular dystrophy. Ophthalmology 1987;94:809.
Fishman GA, Glenn AM, Gilbert LD. Rebound of macular edema with continued use of methazolamide in patients with retinitis pigmentosa. Arch Ophthalmol 1993;111:1640.
Flannery JG, Bird AC, Farber DB, Weleber RG, Bok D. A histopathologic study of a choroideremia carrier. Invest Ophthalmol Vis Sci 1990;31:229.
Folk JC, Arrindell EL, Klugman MR. The fellow eye of patients with phakic lattice retinal detachment. Ophthalmology 1989;96:72.
Gass JDM, Joondeph BC. Observations concerning patients with suspected impending macular holes. Am J Ophthalmol 1990;109:638.
Gass JDM. Diseases causing choroidal exudation and hemorrhagic localized detachment of the retina and retinal pigment epithelium. In: Gass JDM. Stereoscopic atlas of macular diseases. Diagnosis and treatment. 4th ed. St. Louis: Mosby; 1997:49.
Gass JDM. Macular dysfunction caused by vitreous and vitreoretinal interface abnormalities. In: Gass JDM. Stereoscopic atlas of macular diseases. Diagnosis and treatment. 4th ed. St. Louis: Mosby; 1997:903.
Gass JDM. Reappraisal of biomicroscopic classification of stages of development of a macular hole. Am J Ophthalmol 1995;119:752.
Gladstone GJ, Tasman W. Solar retinitis after minimal exposure. Arch Ophthalmol 1978;96:1368.
Greaves AH, Sarks JP, Sarks SH. Adult vitelliform macular degeneration: a clinical spectrum. Aust N Z J Ophthalmol 1990;18:171.
Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman lecture. Ophthalmology 1993;100:1519.
Greven CM, Moreno RJ, Tasman W. Unusual manifestations of X-linked retinoschisis. Trans Am Ophthalmol Soc 1990;88:211.
Grossniklaus HE, Green WR. Pathologic findings in pathologic myopia. Retina 1992;12:127.
Guerry RK, Ham WT, Mueller HA. Light toxicity in the posterior segment. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 3. Philadelphia: JB Lippincott; 1992:1.
Ham WT Jr, Mueller HA, Ruffolo JJ Jr, et al. Basic mechanisms underlying the production of photochemical lesions in the mammalian retina. Curr Eye Res 1984;3:165.
Hassan AS, Johnson MW, Schneiderman TE, et al. Management of submacular hemorrhages with intravitreal tPA injection and pneumatic displacement. Ophthalmology 1999;106:1900.
Hampton GR, Kohen D, Bird AC. Visual prognosis of disciform degeneration in myopia. Ophthalmology 1983;90:923.
Heckenlively JR, Arden GB. Principles and practice of clinical electrophysiology of vision. St. Louis: Mosby-Year Book; 1991.
Heckenlively JR. Retinitis pigmentosa. Philadelphia: JB Lippincott; 1988.
Hilton GF. Late serosanguineous detachment of the macula after traumatic choroidal rupture. Am J Ophthalmol 1975;799:997.
Ho AC. Miscellaneous macular degenerations. In: Regillo CD, Brown GC, Flynn HW. Vitreoretinal disease: The essentials. New York: Thieme; 1999:241.
Jalkh AE, Weiter JJ, Trempe CL, et al. Choroidal neovascularization in degenerative myopia: role of laser photocoagulation. Ophthalmic Surg 1987;18:721.
Jampol LM, Setogawa T, Rednam KRV, Tso MOM. Talc retinopathy in primates: a model of ischemic retinopathy. I. Clinical studies. Arch Ophthalmol 1981;99:1273.
Kaiser-Kupfer MI, Caruso RC, Valle D. Gyrate atrophy of the choroid and retina. Long-term reduction of ornithine slows retinal degeneration. Arch Ophthalmol 1991;109:1539.
Klein R, Klein BEK, Jensen SE, et al. The five-year incidence and progression of age-related maculopathy. The Beaver Dam study. Ophthalmology 1997;104:7.
Kozart DM. Anatomic correlates of the retina. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 3. Philadelphia: JB Lippincott; 1994:1.
Krill AE, Deutman AF, Fishman M. The cone degenerations. Doc Ophthalmol 1973;35:1.
Kriss A, Russell-Eggitt I, Harris CM, Lloyd IC, Taylor D. Aspects of albinism. Ophthalmic Paediatr Genet 1992;13:89.
Leonard RE 2nd, Smiddy WE, Flynn HW Jr, et al. Long-term visual outcomes in patients with successful macular hole surgery. Ophthalmology 1997;104:1648.
Levine R, Brucker AJ, Robinson F. Long-term follow-up of idiopathic central serous chorioretinopathy by fluorescein angiography. Ophthalmology 1989;96:854.
Lim JI, Enger C, Fine SL. Foveomacular dystrophy. Am J Ophthalmol 1994;117:1.
Macular Photocoagulation Study Group. Argon laser photocoagulation for neovascular maculopathy. Five-year results from randomized clinical trials. Arch Ophthalmol 1991;109:1109.
Macular Photocoagulation Study Group. Five-year follow-up of fellow eyes of patients with age-related macular degeneration and unilateral extrafoveal choroidal neovascularization. Arch Ophthalmol 1993;111:1189.
Macular Photocoagulation Study Group. Persistent and recurrent neovascularization after krypton laser photocoagulation for neovascular lesions of age-related macular degeneration. Arch Ophthalmol 1990;108:825.
Macular Photocoagulation Study Group. Pretreatment fundus characteristics as predictors of recurrent choroidal neovascularization. Arch Ophthalmol 1991;109:1193.
Macular Photocoagulation Study Group. Risk factors for choroidal neovascularization in the second eye of patients with juxtafoveal or subfoveal choroidal neovascularization secondary to age-related macular degeneration. Arch Ophthalmol 1997;115:741.
Macular Photocoagulation Study Group. Persistent and recurrent neovascularization after krypton laser photocoagulation for neovascular lesions of ocular histoplasmosis. Arch Ophthalmol 1989;107:344.
Mansour AM. Systemic associations of angioid streaks. J Intern Ophthalmol Clin 1991;31:61.
Margherio RR, Cox MS Jr, Trese MT, et al. Removal of epimacular membranes. Ophthalmology 1985;92:1075.
McCulloch C, Arshinoff S. Choroideremia and gyrate atrophy. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 3. Philadelphia: JB Lippincott; 1992:1.
McDonald HR, Schatz H, Johnson RN, Madeira D. Acquired macular disease. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 3. Rev ed. Philadelphia: JB Lippincott; 1994: chap 23.
Mittra RA, Mieler WF. Drug and light ocular toxicity. In: Regillo CD, Brown GC, Flynn HW. Vitreoretinal disease: The essentials. New York: Thieme; 1999:545.
P.204
Murphy SB, Jackson WB, Pare JAP. Talc retinopathy. Can J Ophthalmol 1978;13:152.
O'Connell SR, Bressler NM. Age-related macular degeneration. In: Regillo CD, Brown GC, Flynn HW. Vitreoretinal disease: The essentials. New York: Thieme; 1999:213.
O'Donnell FE Jr, Green WR. The eye in albinism. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 4. Philadelphia: JB Lippincott; 1992:1.
Ogden TE. Topography of the retina. In: Ryan SJ, Ogden TE. Retina. 2nd ed. St. Louis: CV Mosby; 1994:32.
Park SS, Sigelman J, Gragoudas ES. Retina. In: Tasman W, Jaeger EA. Foundations of clinical ophthalmology. Philadelphia: JB Lippincott; 1994:1.
Pece A, Avanza P, Galli L, Brancato R. Laser photocoagulation of choroidal neovascularization in angioid streaks. Retina 1997;17:12.
Petrukhin K, Koisti MJ, Bakall B, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet 1998;19:241.
Regillo CD, Blade KA, Custis PH, O'Connell SR. Evaluating persistent and recurrent choroidal neovascularization: the role of indocyanine green angiography. Ophthalmology 1998;105:1821.
Regillo CD, Benson WE. Retinal detachment: Diagnosis and management. 3rd ed. Philadelphia: Lippincott-Raven; 1998.
Regillo CD, Benson WE, Maguire JI, Annesley WH. Indocyanine green angiography and occult choroidal neovascularization. Ophthalmology 1994;101:280.
Regillo CD. Update on photodynamic therapy. Curr Opin Ophthalmol 2000;11:166.
Regillo CD. The present role of indocyanine green angiography in ophthalmology. Curr Opin Ophthalmol 1999;10:189.
Regillo CD, Custis PH. Surgical management of retinoschisis. Curr Opin Ophthalmol 1997;8:80.
Regillo CD, Tasman WS, Brown GC. Surgical management of complications associated with X-linked retinoschisis. Arch Ophthalmol 1993;111:1080.
Regillo CD. Posterior segment manifestations of systemic trauma. In: Regillo CD, Brown GC, Flynn HW. Vitreoretinal disease: The essentials. New York: Thieme; 1999:537.
Robertson DM, Ilstrup D. Direct, indirect, and sham laser photocoagulation in the management of central serous chorioretinopathy. Am J Ophthalmol 1983;95:457.
Ryan EH, Gilbert HD. Results of surgical treatment of recent-onset full-thickness macular holes. Arch Ophthalmol 1994;1112:1545.
Sanborn EG, Gonder R, Goldberg RE, Benson WE, Kessler S. Evulsion of the optic nerve: a clinicopathological study. Can J Ophthalmol 1984;19:10.
Sarks SH. Drusen and their relationship to senile macular degeneration. Aust J Ophthalmol 1980;8:117.
Schatz H, Yannuzzi LA, Gitter ER. Subretinal neovascularization following argon laser photocoagulation treatment for central serous chorioretinopathy. Complication or misdiagnosis? Trans Am Acad Ophthalmol Otolaryngol 1977;88:893.
Schubert HD. Ocular embryology. In: Regillo CD, Brown GC, Flynn HW. Vitreoretinal disease: The essentials. New York: Thieme; 1999:3.
Schultz PN, Sobol WM. Angioid streaks and pseudoxanthoma elasticum. JAMA 1991;265:45.
Silicone Study Group. Vitrectomy with silicone oil or perfluoropropane gas in eyes with severe proliferative vitreoretinopathy: results of a randomized clinical trial: Silicone Study report 2. Arch Ophthalmol 1992;110:780.
Sipperley JO, Quigley HA, Gass JDM. Traumatic retinopathy in primates: the explanation of commotio retinae. Arch Ophthalmol 1978;96:2267.
Smiddy WE, Flynn HW Jr, Nicholson DH, et al. Results and complications in treated retinal breaks. Am J Ophthalmol 1991;112:623.
Smiddy WE, Michels RG, Green WR. Morphology, pathology, and surgery of idiopathic vitreoretinal macular disorders. A review. Retina 1990;10:288.
Smith RE, Kelley JS, Harbin TS. Late macular complications of choroidal ruptures. Am J Ophthalmol 1974;77:650.
Song MK, Small KW. Macular dystrophies. In: Regillo CD, Brown GC, Flynn HW. Vitreoretinal disease: The essentials. New York: Thieme; 1999:293.
Steinmetz RL, Garner A, Maguire JI, Bird AC. Histopathology of incipient fundus flavimaculatus. Ophthalmology 1991;98:953.
Stickler GB, Belau PG, Farrell FJ, et al. Hereditary progressive arthro-ophthalmopathy. Mayo Clin Proc 1965;40:433.
Straatsma BR, Foos RY, Feman SS. Degenerative diseases of the peripheral retina. In: Tasman W, Jaeger EA. Duane's Clinical Ophthalmology, vol 3. Philadelphia: JB Lippincott; 1992:1.
Tasman W. Hyaloideoretinopathies. In: Regillo CD, Brown GC, Flynn HW. Vitreoretinal disease: The essentials. New York: Thieme; 1999:365.
Thomas MS, Grand MG, Williams DF, et al. Surgical management of subfoveal choroidal neovascularization. Ophthalmology 1992;99:952.
Thompson JT, Sjaarda RN, Lansing MB. The results of vitreous surgery for chronic macular hole. Retina 1997;17:493.
Tornambe PE, Hilton GF, and the Retinal Detachment Study Group. Pneumatic retinopexy: a multicenter randomized controlled clinical trial comparing pneumatic retinopexy with scleral buckling. Ophthalmology 1989;96:772.
Treatment of Age-related Macular Degeneration With Photodynamic Therapy (TAP) Study Group. Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin. One-year results of 2 randomized clinical trials TAP report 1. Arch Ophthalmol 1999;117:1329.
Tse DT, Ober RR. Talc retinopathy. Am J Ophthalmol 1980;90:624.
Vander JF, Duker JS, Jaeger EA. Miscellaneous diseases of the fundus. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 3. Philadelphia: JB Lippincott; 1992:1.
Vander JF. Choroidal neovascularization associated with myopic macular degeneration. In: Tasman WS. Clinical decisions in medical retinal disease. St. Louis: Mosby; 1994.
Vine AK, Shatz H. Adult-onset foveomacular pigment epithelial dystrophy. Am J Ophthalmol 1980;89:680.
Welch JC, Spaeth G, Benson WE. Massive suprachoroidal hemorrhage. Follow-up and outcome of 30 cases. Ophthalmology 1988;95:1202.
Wendel RT, Patel AC, Kelly NE, Salzano TC, Wells JW, Novack GD. Vitreous surgery for macular holes. Ophthalmology 1993;100:1671.
Wray S, Boustany R-MN. Ophthalmic manifestations of defects in metabolism. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 3. Philadelphia: JB Lippincott; 1992:1.
Yannuzzi LA, Slakter JS, Sorenson JA, Guyer DR, Orlock DA. Digital indocyanine green videoangiography and choroidal neovascularization. Retina 1992;12:191.
EAN: 2147483647
Pages: 17