17 - Gynecology
Editors: McPhee, Stephen J.; Papadakis, Maxine A.; Tierney, Lawrence M.
Title: Current Medical Diagnosis & Treatment, 46th Edition
Copyright 2007 McGraw-Hill
> Table of Contents > 20 - Arthritis & Musculoskeletal Disorders
function show_scrollbar() {}
20
Arthritis & Musculoskeletal Disorders
David B. Hellmann MD, MACP
John H. Stone MD, MPH
Diagnosis & Evaluation
Examination of the Patient
In the patient with arthritis, the two clinical clues most helpful for diagnosis are the joint pattern and the presence or absence of extra-articular manifestations. The joint pattern is defined by the answers to three questions: (1) Is inflammation present? (2) How many joints are involved? and (3) What joints are affected? Joint inflammation is manifested by redness, warmth, swelling, and morning stiffness of at least 30 minutes' duration. Both the number of affected joints and the specific sites of involvement affect the differential diagnosis (Table 20-1). Some diseases gout, for example are characteristically monarticular, whereas other diseases, such as rheumatoid arthritis, are chiefly polyarticular. The location of joint involvement can also be distinctive. Only two diseases frequently cause prominent involvement of the distal interphalangeal (DIP) joint: osteoarthritis and psoriatic arthritis. Extra-articular manifestations such as fever, rash, nodules, or neuropathy narrow the differential diagnosis further (see Table 20-1).
Arthrocentesis & Examination of Joint Fluid
Synovial fluid examination (Table 20-2) may provide specific diagnostic information in joint disease. Contraindications to arthrocentesis include infection of the overlying skin, bleeding disorder, or inability of the patient to cooperate. For patients who are receiving long-term anticoagulation therapy with warfarin, joints can be aspirated if the international normalized ratio (INR) is less than 3.0. In such patients, use of a small-gauge needle (eg, 22F or 25F) and application of firm pressure to the aspiration site are prudent measures. Most large joints are easily aspirated (Figure 20-1).
A. Types of Studies
1. Gross examination
If fluid is opaque, a Gram stain is indicated. If bloody, a bleeding disorder, trauma, or traumatic tap is most likely.
2. Microscopic examination
Compensated polarized light microscopy identifies and distinguishes monosodium urate (gout, negatively birefringent) and calcium pyrophosphate (pseudogout, positive birefringent) crystals.
3. Culture
Bacterial cultures as well as special studies for gonococci, tubercle bacilli, or fungi are ordered as appropriate.
B. Interpretation
Although synovial fluid analysis is diagnostic in infectious or microcrystalline arthritis, there is considerable overlap in the cytologic and biochemical values obtained in these and other diseases (Table 20-3). These studies do make possible, however, a differentiation according to severity of inflammation. Inflammatory joint fluids have more than 3000 white blood cells per microliter, of which 50% or more are polymorphonuclear neutrophils (Table 20-2). Noninflammatory fluids usually have less than 3000/mcL white cells and less than 25% polymorphonuclear neutrophils. Synovial fluid glucose and protein levels add little information.
Table 20-1. Diagnostic value of the joint pattern. | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Degenerative & Crystal- Induced Arthritis
Degenerative Joint Disease (Osteoarthritis)
![]() Essentials of Diagnosis
Essentials of Diagnosis
A degenerative disorder without systemic manifestations.
Commonly secondary to other articular disease.
Pain relieved by rest; morning stiffness brief; articular inflammation minimal.
X-ray findings: narrowed joint space, osteophytes, increased density of subchondral bone, bony cysts.
P.827
General Considerations
Osteoarthritis, the most common form of joint disease, is chiefly a disease of aging. Ninety percent of all people have radiographic features of osteoarthritis in weight-bearing joints by age 40. Symptomatic disease also increases with age.
This arthropathy is characterized by degeneration of cartilage and by hypertrophy of bone at the articular margins. Inflammation is usually minimal. Hereditary and mechanical factors may be involved in the pathogenesis.
Degenerative joint disease is divided into two types: (1) primary, which most commonly affects some or all of the following: the terminal interphalangeal joints (Heberden's nodes) and less commonly the proximal interphalangeal (PIP) joints (Bouchard's nodes), the metacarpophalangeal (MCP) and carpometacarpal joints of the thumb, the hip, the knee, the metatarsophalangeal (MTP) joint of the big toe, and the cervical and lumbar spine; and (2) secondary, which may occur in any joint as a sequela to articular injury resulting from either intra-articular (including rheumatoid arthritis) or extra-articular causes. The injury may be acute, as in a fracture; or chronic, as that due to occupational overuse of a joint, metabolic disease (eg, hyperparathyroidism, hemochromatosis, ochronosis), or neurologic disorders (tabes dorsalis; see below). Obesity is a risk factor for knee osteoarthritis and probably for the hip. Recreational running does not increase the incidence of osteoarthritis, but participation in competitive contact sports does. Jobs requiring frequent bending and carrying increase the risk of knee osteoarthritis.
Clinical Findings
A. Symptoms and Signs
The onset is insidious. Initially, there is articular stiffness, seldom lasting more than 15 minutes; this develops later into pain on motion of the affected joint and is made worse by activity or weight bearing and relieved by rest. Deformity may be absent or minimal; however, bony enlargement of the interphalangeal joints is occasionally prominent, and flexion contracture or varus deformity of the knee is not unusual. There is no ankylosis, but limitation of motion of the affected joint or joints is common. Crepitus may often
P.828
be felt in the joint. Joint effusion and other articular signs of inflammation are mild. There are no systemic manifestations.
Table 20-2. Examination of joint fluid. | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
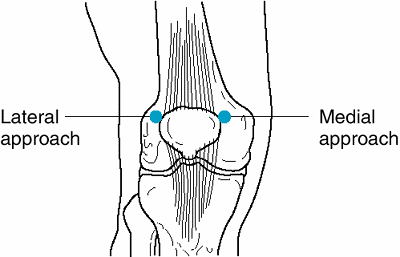 |
Figure 20-1. Aspiration of the knee joint. The knee joint the most commonly aspirated joint can be entered either medially or laterally. The patient should be supine, with the leg fully extended. Apply pressure on the side of the joint opposite to the puncture site to assist in directing the needle toward the bulging synovium. From the lateral approach, the needle (held parallel to the examining table) is directed medially, just beneath the patella, into the suprapatellar space. From the medial approach, the needle (held parallel to the examining table) is introduced between the patella and the medial condyle and advanced upward and laterally, beneath the patella and into the joint space. (Reproduced, with permission, from Nicoll D et al: Pocket Guide to Diagnostic Tests. McGraw-Hill, 1997. ) |
B. Laboratory Findings
Osteoarthritis does not cause elevation of the erythrocyte sedimentation rate (ESR) or other laboratory signs of inflammation.
C. Imaging
Radiographs may reveal narrowing of the joint space; sharpened articular margins; osteophyte formation and lipping of marginal bone; and thickened, dense subchondral bone. Bone cysts may also be present.
Differential Diagnosis
Because articular inflammation is minimal and systemic manifestations are absent, degenerative joint disease should seldom be confused with other arthritides. The distribution of joint involvement in the hands also helps distinguish osteoarthritis from rheumatoid arthritis. Osteoarthritis chiefly affects the DIP and PIP joints and spares the wrist and MCP joints (except at the thumb); rheumatoid arthritis involves the wrists and MCP joints and spares the DIP joints. Furthermore, the joint enlargement is bony-hard and cool in osteoarthritis but spongy and warm in rheumatoid arthritis. Skeletal symptoms due to degenerative changes in joints especially in the spine may cause coexistent metastatic neoplasia, osteoporosis, multiple myeloma, or other bone disease to be overlooked.
Prevention
Weight reduction reduces the risk of developing symptomatic knee osteoarthritis. Maintaining normal
P.829
vitamin D levels may reduce the occurrence and progression of osteoarthritis, in addition to being important for bone health.
Table 20-3. Differential diagnosis by joint fluid groups. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
Treatment
A. General Measures
For patients with mild to moderate osteoarthritis of weight-bearing joints, a supervised walking program may result in clinical improvement of functional status without aggravating the joint pain. Weight loss can also improve the symptoms.
B. Analgesic and Anti-inflammatory Drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) (see Table 5-5) are more effective (and more toxic) than acetaminophen for osteoarthritis of the knee or hip. Their superiority is most convincing in those with severe disease. Patients with mild disease should start with acetaminophen (2.6 4 g/d). Glucosamine and chondroitin sulfate are also effective and safe for knee osteoarthritis; glucosamine may even reduce progression of knee osteoarthritis. NSAIDs should be considered for patients who do not respond to acetaminophen, chondroitin sulfate, and glucosamine. (See discussion of NSAID toxicity in the section on treatment of rheumatoid arthritis.) High doses of NSAIDs, as used in more inflammatory arthritides, are unnecessary.
For many patients, it is possible eventually to reduce the dosage or limit use of drugs to periods of exacerbation. For patients with knee osteoarthritis and effusion, intra-articular injection of triamcinolone (20 40 mg) may obviate the need for analgesics or NSAIDs. Corticosteroid injections up to four times a year appear to be safe. Intra-articular injections of sodium hyaluronate reduce symptoms moderately in some patients. Capsaicin cream 0.025% applied twice daily can also reduce knee pain without NSAIDs. Doxycycline, though not FDA approved for treating osteoarthritis, has shown promise in reducing the progression of knee osteoarthritis.
C. Surgical Measures
Total hip replacement provides excellent symptomatic and functional improvement when that joint is seriously afflicted, as indicated by severely restricted walking and pain at rest, particularly at night. Knee replacement is also effective. Arthroscopic surgery for knee osteoarthritis is ineffective. Experimental techniques to repair focal cartilage loss in the knee by autologous chondrocyte transplantation are promising. However, the indications for and limitations of this procedure require further definition.
Prognosis
Marked disability is less common in patients with osteoarthritis than in those with rheumatoid arthritis, but symptoms may be quite severe and limit activity considerably (especially with involvement of the hips, knees, and cervical spine).
Bjordal JM et al: Non-steroidal anti-inflammatory drugs, including cyclo-oxygenase-2 inhibitors in osteoarthritis knee pain: meta-analysis of randomised placebo controlled trials. BMJ 2004;329:1317.
Brandt KD et al: Effects of doxycycline on progression of osteoarthritis: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum 2005;52:2015.
Felson DT: Clinical practice. Osteoarthritis of the knee. N Engl J Med 2006;354:841.
Fransen M: Dietary weight loss and exercise for obese adults with knee osteoarthritis: modest weight loss targets, mild exercise, modest effects. Arthritis Rheum 2004;50:1366.
Schumacher HR et al: Injectable corticosteroids in treatment of arthritis of the knee. Am J Med 2005;118:1208.
Witt C et al: Acupuncture in patients with osteoarthritis of the knee: a randomised trial. Lancet 2005;366:136.
Crystal Deposition Arthritis
1. Gouty Arthritis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Acute onset, typically nocturnal and usually monarticular, often involving the first MTP joint.
Polyarticular involvement more common in patients with longstanding disease.
Identification of urate crystals in joint fluid or tophi is diagnostic.
Dramatic therapeutic response to NSAIDs.
With chronicity, urate deposits in subcutaneous tissue, bone, cartilage, joints, and other tissues.
General Considerations
Gout is a metabolic disease of heterogeneous nature, often familial, associated with abnormal amounts of urates in the body and characterized early by a recurring acute arthritis, usually monarticular, and later by chronic deforming arthritis. The associated hyperuricemia is due to overproduction or underexcretion of uric acid sometimes both. The disease is especially common in Pacific islanders, eg, Filipinos and Samoans. It is rarely caused by a specifically determined genetic aberration (eg, Lesch-Nyhan syndrome). Secondary gout, which may have a heritable component, is related to acquired causes of hyperuricemia, eg, medication use (especially diuretics, cyclosporine, low-dose aspirin, and niacin), myeloproliferative disorders, multiple myeloma, hemoglobinopathies, chronic renal disease, hypothyroidism, psoriasis, sarcoidosis, and lead poisoning (Table 20-4). Alcohol ingestion promotes hyperuricemia by increasing
P.830
urate production and decreasing the renal excretion of uric acid. Finally, hospitalized patients frequently suffer attacks of gout because of changes in diet, fluid intake, or medications that lead either to rapid reductions or increases in the serum urate level.
Table 20-4. Origin of hyperuricemia. | ||
|---|---|---|
|
About 90% of patients with primary gout are men, usually over 30 years of age. In women, the onset is typically postmenopausal. The characteristic lesion is the tophus, a nodular deposit of monosodium urate monohydrate crystals, with an associated foreign body reaction. These are found in cartilage, subcutaneous and periarticular tissues, tendon, bone, the kidneys, and elsewhere. Urates have been demonstrated in the synovial tissues (and fluid) during acute arthritis; indeed, the acute inflammation of gout is believed to be activated by the phagocytosis by polymorphonuclear cells of urate crystals with the ensuing release from the neutrophils of chemotactic and other substances capable of mediating inflammation. The precise relationship of hyperuricemia to gouty arthritis is still obscure, since chronic hyperuricemia is found in people who never develop gout or uric acid stones. Rapid fluctuations in serum urate levels, either increasing or decreasing, are important factors in precipitating acute gout. The mechanism of the late, chronic stage of gouty arthritis is better understood. This is characterized pathologically by tophaceous invasion of the articular and periarticular tissues, with structural derangement and secondary degeneration (osteoarthritis).
Uric acid kidney stones are present in 5 10% of patients with gouty arthritis. Hyperuricemia correlates highly with the likelihood of developing stones, with the risk of stone formation reaching 50% in patients with a serum urate level above 13 mg/dL. Chronic urate nephropathy is caused by the deposition of monosodium urate crystals in the renal medulla and pyramids. Although progressive renal failure occurs in a substantial percentage of patients with chronic gout, the role of hyperuricemia in causing this outcome is controversial, because many patients with gout have numerous confounding risk factors for renal failure (eg, hypertension, alcohol use, lead exposure, and other risk factors for vascular disease).
Unless there is a rapid breakdown of cellular nucleic acid following aggressive treatment of leukemia or lymphoma, uric acid-lowering drugs need not be instituted until arthritis, renal calculi, or tophi become apparent. Asymptomatic hyperuricemia should not be treated.
Clinical Findings
A. Symptoms and Signs
The acute arthritis is characterized by its sudden onset, frequently nocturnal, either without apparent precipitating cause or following rapid fluctuations in serum urate levels. Either increases or decreases in the serum urate level can precipitate a gout attack. Common precipitants are alcohol excess (particularly beer), changes in medications that affect urate metabolism, and in the hospitalized patient fasting before medical procedures. The MTP joint of the great toe is the most susceptible joint ( podagra ), although others, especially those of the feet, ankles, and knees, are commonly affected. Gouty attacks may develop in periarticular soft tissues such as the arch of the foot. Hips and shoulders are rarely affected. More than one joint may occasionally be affected during the same attack; in such cases, the distribution of the arthritis is usually asymmetric. As the attack progresses, the pain becomes intense. The involved joints are swollen and exquisitely tender and the overlying skin tense, warm, and dusky red. Fever is common and may reach 39 C. Local desquamation and pruritus during recovery from the acute arthritis are characteristic of gout but are not always present. Tophi may be found in the external ears, hands, feet, olecranon, and prepatellar bursas. They usually develop years after the initial attack of gout.
Asymptomatic periods of months or years commonly follow the initial acute attack. After years of recurrent severe monarthritis attacks of the lower extremities and untreated hyperuricemia, gout can evolve into a chronic, deforming polyarthritis of
P.831
upper and lower extremities that mimics rheumatoid arthritis.
B. Laboratory Findings
The serum uric acid is elevated (> 7.5 mg/dL) in 95% of patients who have serial measurements during the course of an attack. However, a single uric acid determination is normal in up to 25% of cases, so it does not exclude gout, especially in patients taking uricopenic drugs. During an acute attack, the ESR and white cell count are frequently elevated. Material aspirated from a tophus shows the typical crystals of sodium urate and confirms the diagnosis. Further confirmation is obtained by identification of sodium urate crystals by compensated polariscopic examination of wet smears prepared from joint fluid aspirates. Such crystals are negatively birefringent and needle-like and may be found free or in neutrophils.
C. Imaging
Early in the disease, radiographs show no changes. Later, punched-out erosions with an overhanging rim of cortical bone ( rat bite ) develop. When these are adjacent to a soft tissue tophus, they are diagnostic of gout.
Differential Diagnosis
Acute gout is often confused with cellulitis. Bacteriologic studies usually exclude acute pyogenic arthritis. Pseudogout is distinguished by the identification of calcium pyrophosphate crystals (strong positive birefringence) in the joint fluid, usually normal serum uric acid, the x-ray appearance of chondrocalcinosis, and the relative therapeutic ineffectiveness of colchicine.
Chronic tophaceous arthritis may resemble chronic rheumatoid arthritis; gout is suggested by an earlier history of monarthritis and is established by the demonstration of urate crystals in a suspected tophus. Likewise, hips and shoulders are generally spared in tophaceous gout. Biopsy may be necessary to distinguish tophi from rheumatoid nodules. An x-ray appearance similar to that of gout may be found in rheumatoid arthritis, sarcoidosis, multiple myeloma, hyperparathyroidism, or Hand-Sch ller-Christian disease. Chronic lead intoxication may result in attacks of gouty arthritis (saturnine gout); abdominal pain, peripheral neuropathy, renal insufficiency, and basophilic stippling of red cells are clues to the diagnosis.
Treatment
A. Acute Attack
Arthritis is treated first and hyperuricemia weeks or months later, if at all. Sudden reduction of serum uric acid often precipitates further episodes of gouty arthritis.
1. NSAIDs
These drugs (see Table 5-5) are the treatment of choice for acute gout. Traditionally, indomethacin has been the most frequently used agent, but all of the other newer NSAIDs are probably equally effective. Indomethacin is initiated at a dosage of 25 50 mg orally every 8 hours and continued until the symptoms have resolved (usually 5 10 days). Active peptic ulcer disease, impaired renal function, and a history of allergic reaction to NSAIDs are contraindications. For patients at high risk for upper gastrointestinal bleeding, a cyclooxygenase type 2 (COX-2) inhibitor may be an appropriate first choice for management of an acute gout attack. Long-term use of COX-2 inhibitors is not advised because of the association with increased risk of cardiovascular events, which has led to the removal of some drugs from the US market (eg, rofecoxib and valdecoxib).
2. Colchicine
Colchicine is no longer recommended for the treatment of acute gout flares. Its use during the intercritical period to prevent gout attacks is discussed below.
3. Corticosteroids
Corticosteroids often give dramatic symptomatic relief in acute episodes of gout and will control most attacks. They are most useful in patients with contraindications to the use of NSAIDs. If the patient's gout is monarticular, intra-articular administration (eg, triamcinolone, 10 40 mg depending on the size of the joint) is most effective. For polyarticular gout, corticosteroids may be given intravenously (eg, methylprednisolone, 40 mg/d tapered over 7 days) or orally (eg, prednisone, 40 60 mg/d tapered over 7 days). Gouty and septic arthritis can coexist, albeit rarely. Therefore, joint aspiration and Gram stain with culture of synovial fluid should be performed before corticosteroids are given.
B. Management Between Attacks
Treatment during symptom-free periods is intended to minimize urate deposition in tissues, which causes chronic tophaceous arthritis, and to reduce the frequency and severity of recurrences.
1. Diet
Potentially reversible causes of hyperuricemia are a high-purine diet, obesity, alcohol consumption, and use of certain medications (see below). Beer consumption appears to confer a higher risk of gout than does whiskey or wine. Higher levels of meat and seafood consumption are associated with increased risks of gout, whereas a higher level of dairy products consumption is associated with a decreased risk. Although dietary purines usually contribute only 1 mg/dL to the serum uric acid level, moderation in eating foods with high purine content is advisable (Table 20-5). A high liquid intake and, more importantly, a daily urinary output of 2 L or more will aid urate excretion and minimize urate precipitation in the urinary tract.
Table 20-5. The purine content of foods.1 | ||
|---|---|---|
|
2. Avoidance of hyperuricemic medications
Thiazide and loop diuretics inhibit renal excretion of uric acid and should be avoided in patients with gout. Similarly, low doses of aspirin (< 3 g daily) aggravate hyperuricemia, as does niacin.
3. Colchicine
Patients with a single episode of gout who are willing to lose weight and stop drinking alcohol are at low risk for another attack and unlikely to benefit from chronic medical therapy. In contrast, older individuals
P.832
with mild chronic renal failure who require diuretic use and have a history of multiple attacks of gout are more likely to benefit from pharmacologic treatment. In general, the higher the uric acid level and the more frequent the attacks, the more likely that chronic medical therapy will be beneficial.
There are two indications for daily colchicine administration. First, colchicine can be used to prevent future attacks. For the person who has mild hyperuricemia and occasional attacks of gouty arthritis, chronic colchicine prophylaxis may be all that is needed. The usual dose is 0.6 mg either once or twice a day. Patients who have coexisting moderate renal insufficiency or heart failure should take colchicine only once a day in order to avoid the peripheral neuromyopathy that can complicate the use of higher doses. Second, colchicine can also be used when uricosuric drugs or allopurinol (see below) are started, to suppress attacks precipitated by abrupt changes in the serum uric acid level.
4. Reduction of serum uric acid
Indications for a urate lowering intervention include frequent acute arthritis not controlled by colchicine prophylaxis, tophaceous deposits, or renal damage. Hyperuricemia with infrequent attacks of arthritis may not require treatment; asymptomatic hyperuricemia should not be treated. If instituted, the goal of medical treatment is to maintain the serum uric acid at or below 5 mg/dL, which should prevent crystallization of urate.
Two classes of agents may be used to lower the serum uric acid the uricosuric drugs and allopurinol (neither is of value in the treatment of acute gout). The choice of one or the other depends on the result of a 24-hour urine uric acid determination. A value under 800 mg/d indicates undersecretion of uric acid, which is amenable to uricosuric agents if renal function is preserved. Patients with more than 800 mg of uric acid in a 24-hour urine collection are overproducers and require allopurinol.
a. Uricosuric drugs
These drugs, which block the tubular reabsorption of filtered urate thereby reducing the metabolic urate pool, prevent the formation of new tophi and reduce the size of those already present. When administered concomitantly with colchicine, they may lessen the frequency of recurrences of acute gout. The indication for uricosuric treatment is the increasing frequency or severity of acute attacks. Uricosuric agents are ineffective in patients with renal insufficiency, with a serum creatinine of more than 2 mg/dL.
The following uricosuric drugs may be used: (1) Probenecid, 0.5 g orally daily initially, with gradual increase to 1 2 g daily; or (2) sulfinpyrazone, 50 100 mg orally twice daily initially, with gradual increase to 200 400 mg twice daily. Hypersensitivity to either with fever and rash occurs in 5% of cases; gastrointestinal complaints are observed in 10%. Probenecid also inhibits the excretion of penicillin, indomethacin, dapsone, and acetazolamide.
Precautions with uricosuric drugs. It is important to maintain a daily urinary output of 2000 mL or more in order to minimize the precipitation of uric acid in the urinary tract. This can be further prevented by giving alkalinizing agents (eg, potassium citrate, 30 80 mEq/d orally) to maintain a urine pH of above 6.0. Uricosuric drugs are avoided in patients with a history of uric acid nephrolithiasis. Aspirin in moderate doses antagonizes the action of uricosuric agents, but low doses (325 mg or less per day) do not; doses greater than 3 g daily are themselves uricosuric.
b. Allopurinol
The xanthine oxidase inhibitor allopurinol promptly lowers plasma urate and urinary uric acid concentrations and facilitates tophus mobilization. The drug is of special value in uric acid overproducers; in tophaceous gout; in patients unresponsive to the uricosuric regimen; and in gouty patients with uric acid renal stones. It should be used in low doses in patients with renal insufficiency and is not indicated in asymptomatic hyperuricemia. The most frequent adverse effect is the precipitation of an acute gouty attack. However, the most common sign of hypersensitivity to allopurinol (occurring in 2% of cases) is a pruritic rash that may progress to toxic epidermal necrolysis. Vasculitis and hepatitis are other rare complications.
The initial daily dose of allopurinol is 300 mg/d for patients who have normal renal function and who are taking prophylactic colchicine. In the absence of prophylactic
P.833
colchicine, the initial dose should be 100 mg/d orally. The dose of allopurinol can be increased in a week if needed to achieve the desired serum uric acid level of 5.0 mg/dL. Successful treatment usually requires a dose of 300 400 mg of allopurinol daily. The maximum daily dose is 800 mg. Allopurinol can be used in renal disease, but the dose must be reduced to decrease the chance of side effects.
Allopurinol interacts with other drugs. The combined use of allopurinol and ampicillin causes a drug rash in 20% of patients. Allopurinol can increase the half-life of probenecid, while probenecid increases the excretion of allopurinol. Thus, a patient taking both drugs may need to use slightly higher than usual doses of allopurinol and lower doses of probenecid. Allopurinol potentiates the effect of azathioprine. If allopurinol cannot be avoided, the dose of azathioprine should be reduced by 75% before allopurinol is started.
Febuxostat, a new xanthine oxidase inhibitor, is being evaluated in phase three trials. Patients who have had hypersensitivity reactions to allopurinol, the only xanthine oxidase inhibitor now on the market, appear to tolerate febuxostat.
C. Chronic Tophaceous Arthritis
With rigorous medical compliance, allopurinol shrinks tophi and in time can lead to their disappearance. Resorption of extensive tophi requires maintaining a serum uric acid below 5 mg/dL, which may be achievable only with concomitant use of allopurinol and a uricosuric agent. Surgical excision of large tophi offers mechanical improvement in selected deformities.
D. Gout in the Transplant Patient
Hyperuricemia and gout commonly develop in many transplant patients because they have decreased renal function and require drugs that inhibit uric acid excretion (especially cyclosporine and diuretics). Treating these patients is challenging: NSAIDs are usually contraindicated because of renal impairment; intravenous colchicine is generally avoided for acute gout because of its narrow therapeutic index (particularly in renal dysfunction); and corticosteroids are already being used. Often the best approach for monarticular gout after excluding infection is injecting corticosteroids into the joint (see above). For polyarticular gout, increasing the dose of systemic corticosteroid may be the only alternative. Since transplant patients often have multiple attacks of gout, long-term relief requires lowering the serum uric acid with allopurinol. (Renal impairment seen in many transplant patients makes uricosuric agents ineffective.) Allopurinol doses should be lowered in patients with renal dysfunction and adjusted according to their effect on the serum uric acid level.
Prognosis
Without treatment, the acute attack may last from a few days to several weeks. The intervals between acute attacks vary up to years, but the asymptomatic periods often become shorter if the disease progresses. Chronic gouty arthritis occurs after repeated attacks of acute gout, but only after inadequate treatment. The younger the patient at the onset of disease, the greater the tendency to a progressive course. Destructive arthropathy is rarely seen in patients whose first attack is after age 50.
Patients with gout are anecdotally thought to have an increased incidence of hypertension, renal disease (eg, nephrosclerosis, interstitial nephritis, pyelonephritis), diabetes mellitus, hypertriglyceridemia, and atherosclerosis.
Becker MA et al: Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med 2005; 353:2450.
Choi HK et al: Alcohol intake and risk of incident gout in men: a prospective study. Lancet 2004;363:1277.
Choi HK et al: Pathogenesis of gout. Ann Intern Med 2005;143: 499.
Terkeltaub RA: Clinical practice. Gout. N Engl J Med 2003;349: 1647.
2. Chondrocalcinosis & Pseudogout
Chondrocalcinosis is the presence of calcium-containing salts in articular cartilage. Diagnosed radiologically, it may be familial and is commonly associated with a wide variety of metabolic disorders, eg, hemochromatosis, hyperparathyroidism, ochronosis, diabetes mellitus, hypothyroidism, Wilson's disease, and gout. Pseudogout (also called calcium pyrophosphate dihydrate [CPPD] deposition disease) is most often seen in persons age 60 or older, is characterized by acute, recurrent and rarely chronic arthritis involving large joints (most commonly the knees and the wrists) and is almost always accompanied by chondrocalcinosis of the affected joints. Other joints frequently affected are the MCPs, hips, shoulders, elbows, and ankles. Involvement of the DIP and PIP joints is no more common in CPPD deposition disease than in other age-matched controls. Pseudogout, like gout, frequently develops 24 48 hours after major surgery. Identification of calcium pyrophosphate crystals in joint aspirates is diagnostic of pseudogout. With light microscopy, the rhomboid-shaped crystals differ from the needle-shaped gout crystals. A red compensator is used for positive identification, since pseudogout crystals are blue when parallel and yellow when perpendicular to the axis of the compensator. Urate crystals give the opposite pattern. X-ray examination shows not only calcification (usually symmetric) of cartilaginous structures but also signs of degenerative joint disease (osteoarthritis). Unlike gout, pseudogout is usually associated with normal serum urate levels and is not dramatically improved by colchicine.
Treatment of chondrocalcinosis is directed at the primary disease, if present. Some of the NSAIDs (salicylates, indomethacin, naproxen, and other drugs) are helpful in the treatment of acute episodes. Patients at
P.834
increased risk for upper gastrointestinal bleeding may use a COX-2 inhibitor to treat acute attacks of pseudogout. Long-term use of COX-2 inhibitors is not advised because of the association with increased risk of cardiovascular events, which has led to the removal of some drugs from the US market (eg, rofecoxib and valdecoxib). Colchicine, 0.6 mg orally twice daily, is more effective for prophylaxis than for acute attacks. Aspiration of the inflamed joint and intra-articular injection of triamcinolone, 10 40 mg, depending on the size of the joint, are also of value in resistant cases.
Wise CM: Crystal-associated arthritis in the elderly. Clin Geriatr Med 2005;21:491.
Pain Syndromes
Neck Pain
![]() Essentials of Diagnosis
Essentials of Diagnosis
Most chronic neck pain is caused by degenerative joint disease and responds to conservative approaches.
Whiplash, the most common type of traumatic injury to the neck, responds to early mobilization.
Serious erosive disease of joints in the neck that may lead to neurologic complications sometimes occurs in rheumatoid arthritis and occasionally in ankylosing spondylitis; the usual joint involved in these disorders is the atlantoaxial joint (C1 2).
General Considerations
At any point in time, about 15% of adults are experiencing neck pain. The prevalence of neck pain peaks at age 50 and develops more commonly in women than in men. A large group of articular and extra-articular disorders are characterized by pain that may involve simultaneously the neck, shoulder girdle, and upper extremity. Diagnostic differentiation may be difficult. Some represent primary disorders of the cervicobrachial region; others are local manifestations of systemic disease. It is frequently not possible to make a specific diagnosis.
Clinical Findings
A. Symptoms and Signs
Neck pain may be limited to the posterior region or, depending on the level of the symptomatic joint, may radiate segmentally to the occiput, anterior chest, shoulder girdle, arm, forearm, and hand. It may be intensified by active or passive neck motions. The general distribution of pain and paresthesias corresponds roughly to the involved dermatome in the upper extremity. Radiating pain in the upper extremity is often intensified by hyperextension of the neck and deviation of the head to the involved side. Limitation of cervical movements is the most common objective finding. Neurologic signs depend on the extent of compression of nerve roots or the spinal cord. Compression of the spinal cord may cause paraparesis or paraplegia.
B. Imaging
The radiographic findings depend on the cause of the pain; many plain radiographs are completely normal in patients who have suffered an acute cervical strain. Loss of cervical lordosis is often seen but is nonspecific. In osteoarthritis, comparative reduction in height of the involved disk space is a frequent finding. The most common late radiographic finding is osteophyte formation anteriorly, adjacent to the disk; other chronic abnormalities occur around the apophysial joint clefts, chiefly in the lower cervical spine.
Use of advanced imaging techniques is indicated in the patient who has severe pain of unknown cause that fails to respond to conservative therapy or in the patient who has evidence of myelopathy. MRI is more sensitive than CT in detecting disk disease, extradural compression, and intramedullary cord disease. CT is preferable for demonstration of fractures.
Differential Diagnosis & Treatment
The causes of neck pain include acute and chronic cervical strain or sprains, herniated nucleus pulposus, osteoarthritis, ankylosing spondylitis, rheumatoid arthritis, fibromyalgia, osteomyelitis, neoplasms, polymyalgia rheumatica, compression fractures, and functional disorders.
A. Nonspecific Neck Pain
In the absence of trauma or evidence of infection, malignancy, neurologic findings, or systemic inflammation, the patient can be treated conservatively. Conservative therapy can include rest, analgesics, or physical therapy.
B. Acute Cervical Musculotendinous Strain
Cervical strain is generally caused by mechanical postural disorders, overexertion, or injury (eg, whiplash). Acute episodes are associated with pain, decreased cervical spine motion, and paraspinal muscle spasm, resulting in stiffness of the neck and loss of motion. Muscle trigger points can often be localized. After whiplash injury, patients often experience not only neck pain but also shoulder girdle discomfort and headache. Management includes administration of analgesics. Soft cervical collars are commonly recommended, but evidence suggests they may delay recovery. Acupuncture, manipulation,
P.835
or physical therapy can help some patients, but the precise role of these treatments is not well established. Corticosteroid injection into cervical facet joints is ineffective. Gradual return to full activity is encouraged.
C. Herniated Nucleus Pulposus
Rupture or prolapse of the nucleus pulposus of the cervical disks into the spinal canal causes pain radiating at a C6 7 level. When intra-abdominal pressure is increased by coughing, sneezing, or other movements, symptoms are aggravated, and cervical muscle spasm may occur. Neurologic abnormalities include decreased reflexes of the deep tendons of the biceps and triceps and decreased sensation and muscle atrophy or weakness in the forearm or hand. Cervical traction, bed rest, and other conservative measures are usually successful. Radicular symptoms usually respond to conservative therapy, including NSAIDs, activity modification, intermittent cervical traction, and neck immobilization. Cervical epidural corticosteroid injections may help those who fail conservative therapy. Surgery is indicated for unremitting pain and progressive weakness despite a full trial of conservative therapy and if a surgically correctable abnormality is identified by MRI or CT myelography. Surgical decompression achieves excellent results in 70 80% of such patients.
D. Arthritic Disorders
Cervical spondylosis (degenerative arthritis) is a collective term describing degenerative changes that occur in the apophysial joints and intervertebral disk joints, with or without neurologic signs. Osteoarthritis of the articular facets is characterized by progressive thinning of the cartilage, subchondral osteoporosis, and osteophytic proliferation around the joint margins. Degeneration of cervical disks and joints may occur in adolescents but is more common after age 40. Degeneration is progressive and is marked by gradual narrowing of the disk space, as demonstrated by x-ray. Osteocartilaginous proliferation occurs around the margin of the vertebral body and gives rise to osteophytic ridges that may encroach upon the intervertebral foramina and spinal canal, causing compression of the neurovascular contents.
Osteoarthritis of the cervical spine is often asymptomatic but may cause diffuse neck pain. A minority of patients with neck pain also suffer from radicular pain or myelopathy. Myelopathy develops insidiously and is manifested by sensory dysfunction and clumsy hands. Some patients also complain of unsteady walking, urinary frequency and urgency, or electrical shock sensations with neck flexion or extension (Lhermitte's sign). Weakness, sensory loss, and spasticity with exaggerated reflexes develop below the level of spinal cord compression. Amyotrophic lateral sclerosis, multiple sclerosis, syringomyelia, spinal cord tumors, and tropical spastic paresis from HTLV-1 infection can mimic myelopathy from cervical arthritis. The mainstay of conservative therapy is immobilizing the cervical spine with a collar. With moderate to severe neurologic symptoms, surgical treatment is indicated.
Atlantoaxial subluxation may occur in patients with either rheumatoid arthritis or ankylosing spondylitis. Inflammation of the synovial structures resulting from erosion and laxity of the transverse ligament can lead to neurologic signs of spinal cord compression. Treatment may vary from use of a cervical collar or more rigid bracing to operative treatment, depending on the degree of subluxation and neurologic progression. Surgical treatment for stabilization of the cervical spine is a last resort.
E. Other Disorders
Osteomyelitis and neoplasms are discussed below. Osteoporosis is discussed in Chapter 26.
Hendriks EJ et al: Prognostic factors for poor recovery in acute whiplash patients. Pain 2005;114:408.
Hoving JL et al: Manual therapy, physical therapy, or continued care by a general practitioner for patients with neck pain. A randomized, controlled trial. Ann Intern Med 2002;136: 713.
White P et al: Acupuncture versus placebo for the treatment of chronic mechanical neck pain: a randomized, controlled trial. Ann Intern Med 2004;141:911.
Thoracic Outlet Syndromes
Thoracic outlet syndromes result from compression of the neurovascular structures supplying the upper extremity. Symptoms and signs arise from intermittent or continuous pressure on elements of the brachial plexus and the subclavian or axillary vessels (veins or arteries) by a variety of anatomic structures of the shoulder girdle region. The neurovascular bundle can be compressed between the anterior or middle scalene muscles and a normal first thoracic rib or a cervical rib. Most commonly thoracic outlet obstruction is caused by sagging of the shoulder girdle resulting from aging, obesity, or pendulous breasts. Faulty posture, occupation, or thoracic muscle hypertrophy from physical activity (eg, weight-lifting, baseball pitching) may be other predisposing factors.
Thoracic outlet obstruction presents in most patients with some combination of four symptoms involving the upper extremity, namely pain, numbness, weakness, and swelling. The predominant symptoms depend on whether the obstruction chiefly affects neural or vascular structures. The onset of symptoms is usually gradual but can be sudden. Some patients spontaneously notice aggravation of symptoms with specific positioning of the arm. Pain radiates from the point of compression to the base of the neck, the axilla, the shoulder girdle region, arm, forearm, and hand. Paresthesias are common and distributed to the volar aspect of the fourth and fifth digits. Sensory symptoms may be aggravated at night or by prolonged use of the extremities. Weakness and muscle atrophy are the principal motor abnormalities. Vascular symptoms consist of arterial ischemia
P.836
characterized by pallor of the fingers on elevation of the extremity, sensitivity to cold, and, rarely, gangrene of the digits or venous obstruction marked by edema, cyanosis, and engorgement.
Reflexes are usually not altered. When the site of compression is between the upper rib and clavicle, partial obliteration of subclavian artery pulsation may be demonstrated by abduction of the arm to a right angle with the elbow simultaneously flexed and rotated externally at the shoulder so that the entire extremity lies in the coronal plane. Neck or arm position has no effect on the diminished pulse, which remains constant in the subclavian steal syndrome.
Chest radiography will identify patients with cervical rib (although most patients with cervical ribs are asymptomatic). MRI with the arms held in different positions is useful in identifying sites of impaired blood flow. Intra-arterial or venous obstruction is confirmed by angiography. Determination of conduction velocities of the ulnar and other peripheral nerves of the upper extremity may help localize the site of their compression.
Thoracic outlet syndrome must be differentiated from osteoarthritis of the cervical spine, tumors of the superior pulmonary sulcus, cervical spinal cord, or nerve roots, and periarthritis of the shoulder.
Treatment is directed toward relief of compression of the neurovascular bundle. Greater than 95% of patients can be treated successfully with conservative therapy consisting of physical therapy and avoiding postures or activities that compress the neurovascular bundle. Some women will benefit from a support bra. Operative treatment, required by less than 5% of patients, is more likely to relieve the neurologic rather than the vascular component that causes symptoms.
Brantigan CO et al: Diagnosing thoracic outlet syndrome. Hand Clin 2004;20:27.
Divi V et al: Thoracic outlet decompression for subclavian vein thrombosis: experience in 71 patients. Arch Surg 2005;140: 54.
Low Back Pain
Low back pain is experienced at some time by up to 80% of the population. The differential diagnosis is broad and includes muscular strain, primary spine disease (eg, disk herniation, degenerative arthritis), systemic diseases (eg, metastatic cancer), and regional diseases (eg, aortic aneurysm). A precise diagnosis cannot be made in the majority of cases. Even when anatomic defects such as vertebral osteophytes or a narrowed disk space are present, clinical disease cannot be assumed since such defects are common in asymptomatic patients. The majority of patients will improve in 1 4 weeks and need no evaluation beyond the initial history and physical examination. The diagnostic challenge is to identify those patients who require more extensive or urgent evaluation.
In practice, this means identifying those patients with pain caused by (1) infection, (2) cancer, (3) inflammatory back disease such as ankylosing spondylitis, (4) or nonrheumatologic conditions, especially expanding aortic aneurysm. Significant or progressive neurologic deficits also require identification. If there is no evidence of these problems, conservative therapy is called for.
1. Clinical Approach to Diagnosis
General History & Physical Examination
Low back pain is a final common pathway of many processes; the pain of vertebral osteomyelitis, for example, is not very different in quality and intensity from that due to back strain of the weekend gardener. Historical factors of importance include smoking, weight loss, age over 50, and cancer, all of which are risk factors for vertebral body metastasis. Osteomyelitis most frequently occurs in adults with a history of recurrent urinary tract infections and is especially common in diabetics.
Previous peptic ulcer disease suggests that a patient's back pain is due to a penetrating ulcer. A history of a cardiac murmur should raise concern about endocarditis, since back pain is a not uncommon manifestation. A background of renal stones might indicate another cause of referred back pain.
History of the Back Pain
Certain qualities of a patient's pain can indicate a specific diagnosis. Low back pain radiating down the buttock and below the knee suggests a herniated disk causing nerve root irritation. Other conditions including sacroiliitis, facet joint degenerative arthritis, spinal stenosis, or irritation of the sciatic nerve from a wallet also cause this pattern.
The diagnosis of disk herniation is further suggested by physical examination (see below) and confirmed by imaging techniques. Disk herniation can be asymptomatic, so its presence does not invariably link it to the symptom.
Low back pain at night, unrelieved by rest or the supine position, suggests the possibility of malignancy, either vertebral body metastasis (chiefly from prostate, breast, lung, multiple myeloma, or lymphoma) or a cauda equina tumor. Similar pain can also be caused by compression fractures (from osteoporosis or myeloma).
Symptoms of large or rapidly evolving neurologic deficits identify patients who need urgent evaluation for possible cauda equina tumor, epidural abscess or, rarely, massive disk herniation. Even with a herniated disk and nerve root impingement, pain is the most prominent symptom; numbness and weakness are less commonly reported and when present are of the magnitude consistent with compression of a single nerve root. Thus, symptoms of bilateral leg weakness (from multiple lumbar nerve root compressions) or of saddle area anesthesia, bowel or bladder incontinence, or impotence (indicating multiple sacral nerve root compressions) indicate a cauda equina process.
P.837
Low back pain that worsens with rest and improves with activity is characteristic of ankylosing spondylitis or other seronegative spondyloarthropathies, especially when the onset is insidious and begins before age 40. Most degenerative back diseases produce precisely the opposite pattern, with rest alleviating and activity aggravating the pain. Low back pain causing the patient to writhe occurs in renal colic but can also indicate a leaking aneurysm. The pain associated with pseudoclaudication from lumbar spinal stenosis is discussed below.
Physical Examination of the Back
Several physical findings should be sought because they help identify those few patients who need more than conservative management.
Neurologic examination of the lower extremities will detect the small deficits produced by disk disease and the large deficits complicating such problems as cauda equina tumors. A positive straight leg raising test indicates nerve root irritation. The examiner performs the test on the supine patient by passively raising the patient's ipsilateral leg. The test is positive if radicular pain is produced with the leg raised 60 degrees or less. It has a specificity of 40% but is 95% sensitive in patients with herniation at the L4 5 or L5-S1 level (the sites of 95% of disk herniations). It can be falsely negative, especially in patients with herniation above the L4 5 level. The crossed straight leg sign is positive when raising the contralateral leg reproduces the sciatica. It has a sensitivity of 25% but is 90% specific for disk herniation.
Detailed examination of the sacral and lumbar nerve roots, especially L5 and S1, is essential for detecting neurologic deficits associated with back pain. Disk herniation produces deficits predictable for the site involved (Table 20-6). Deficits of multiple nerve roots suggest a cauda equina tumor or an epidural abscess, both requiring urgent evaluation and treatment.
Measurement of spinal motion in the patient with acute pain is rarely of diagnostic utility and usually simply confirms that pain limits motion. An exception is the decreased range of motion in multiple regions of the spine (cervical, thoracic, and lumbar) in a diffuse spinal disease such as ankylosing spondylitis. By the time the patient has such limits, however, the diagnosis is usually straightforward.
Table 20-6. Neurologic testing of lumbosacral nerve disorders. | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
If back pain is not severe and does not itself limit motion, Schober's test of lumbar motion is helpful in early diagnosis of ankylosing spondylitis. To perform this test, two marks are made, one 10 cm above S1 and another 5 cm below. The patient then bends forward as far as possible, and the distance between the points is measured. Normally, the distance increases at least 5 cm. Anything less indicates reduced lumbar motion, which in the absence of severe pain is most commonly due to ankylosing spondylitis or other seronegative spondyloarthropathies.
Palpation of the spine usually does not yield diagnostic information. Point tenderness over a vertebral body is reported to suggest osteomyelitis, but this association is uncommon. A step-off noted between the spinous process of adjacent vertebral bodies may indicate spondylolisthesis, but the sensitivity of this finding is extremely low. Tenderness of the soft tissues overlying the greater trochanter of the hip is a manifestation of trochanteric bursitis.
Inspection of the spine is not often of value in identifying serious causes of low back pain. The classic posture of ankylosing spondylitis is a late finding. Scoliosis of mild degree is not associated with an increased risk of clinical back disease. Cutaneous neurofibromas can identify the rare patient with nerve root encasement.
Examination of the hips should be part of the complete examination. While hip arthritis usually produces groin pain, some patients have buttock or low back symptoms.
Further Examination
If the history and physical examination do not suggest the presence of infection, cancer, inflammatory back disease, major neurologic deficits, or pain referred from abdominal or pelvic disease, further evaluation can be eliminated or deferred while conservative therapy is tried. The great majority of patients will improve with conservative care over 1 4 weeks.
Regular radiographs of the lumbosacral spine give 20 times the radiation dose of a chest radiograph and provide limited, albeit important, information. Oblique films double the radiation dose and are not routinely needed. Radiographs can provide evidence of vertebral body osteomyelitis, cancer, fractures, or ankylosing spondylitis. Degenerative changes in the lumbar spine are ubiquitous in patients over 40 and do not prove clinical disease. Plain radiographs have very low sensitivity or specificity for disk disease. Thus, plain radiographs are warranted promptly for patients suspected of having infection, cancer, or fractures; selected other patients who do not improve after 2 4 weeks of conservative therapy are also candidates. The Agency for Health Care Policy and Research guidelines for obtaining lumbar radiographs are summarized in Table 20-7.
Table 20-7. AHRQ criteria for lumbar radiographs in patients with acute low back pain. | ||
|---|---|---|
|
P.838
MRI provides exquisite anatomic detail but is reserved for patients who are considering surgery or have evidence of a systemic disease. For example, MRI is needed urgently for any patient in whom an epidural mass or cauda equina tumor is suspected but not for a patient believed to have a routine disk herniation, since most will improve over 4 6 weeks of conservative therapy. Noncontrast CT does not image cauda equina tumors or other intradural lesions, and if used instead of MRI it must include intrathecal contrast.
Radionuclide bone scanning has limited usefulness. It is most useful for early detection of vertebral body osteomyelitis or osteoblastic metastases. The bone scan is normal in multiple myeloma because lytic lesions do not take up isotope.
2. Management
While any management plan must be individualized, key elements of most conservative treatments for back pain include analgesia and education. Analgesia can usually be provided with NSAIDs (see Table 5-5), but severe pain may require opioids (see Table 5-6). Rarely does the need for opioids extend beyond 1 2 weeks.
Diazepam, cyclobenzaprine, carisoprodol, and methocarbamol have been prescribed as muscle relaxants, though their sedative effects may limit their use. They should be reserved for patients who do not respond to NSAIDs and should also be limited to courses of 1 2 weeks. Their use should be avoided in older patients, who are at risk for falling. All patients should be taught how to protect the back in daily activities ie, not to lift heavy objects, to use the legs rather than the back when lifting, to use a chair with arm rests, and to rise from bed by first rolling to one side and then using the arms to push to an upright position. Back manipulation for benign, mechanical low back pain appears safe and as effective as therapies provided by physicians.
Rest and back exercises, once thought to be cornerstones of conservative therapy, are now known to be ineffective for acute back pain. Advice to rest in bed is less effective than advice to remain active. No bed rest with continuation of ordinary activities as tolerated is superior to 2 days of bed rest, 7 days of bedrest, and back mobilizing exercises. Similarly, for acute back pain, exercise therapy is not effective. The value of corsets or traction is dubious. Epidural corticosteroid injections can provide short-term relief of sciatica but do not improve functional status or reduce the need for surgery. In double-blind studies, repeated injections have been no more effective than a single injection. For chronic low back pain, yoga is as effective as a back exercise program and more effective than a self-care book. Corticosteroid injections into facet joints are ineffective for chronic low back pain.
Surgical consultation is needed urgently for any patient with a major or evolving neurologic deficit. Surgery for disk disease is indicated when there is documentation of herniation by imaging, persistent pain, and a consistent neurologic deficit that has failed to respond to 4 6 weeks of conservative therapy. Percutaneous lumbar discectomy, performed under local anesthesia, is a safe and effective (up to 75%) alternative to laminectomy. The percutaneous procedure is contraindicated in the presence of tumor, infection, spondylolisthesis, foraminal stenosis, loose disk fragments, or severe facet joint arthritis.
Complaints without objective findings suggest a psychological role in symptom formation. Treatment includes reassurance and nonopioid analgesics.
Arden NK et al: A multicentre randomized controlled trial of epidural corticosteroid injections for sciatica: the WEST study. Rheumatology 2005;44:1399.
Cherkin DC et al: A review of the evidence for the effectiveness, safety, and cost of acupuncture, massage therapy, and spinal manipulation for back pain. Ann Intern Med 2003;138:898.
Hagen K et al: Bed rest for acute low-back pain and sciatica. Cochrane Database Syst Rev 2004:(4);CD001254.
Manheimer E et al: Meta-analysis: acupuncture for low back pain. Ann Intern Med 2005;142:651.
Sherman KJ et al: Comparing yoga, exercise, and a self-care book for chronic low back pain: a randomized, controlled trial. Ann Intern Med 2005;143:849.
Speed C: Low back pain. BMJ 2004;328:1119.
van Poppel MN et al: An update of a systematic review of controlled clinical trials on the primary prevention of back pain at the workplace. Occup Med (Lond) 2004;54:345.
Lumbar Spinal Stenosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Most patients are older than 60 years.
Presenting symptom is often back pain radiating to the buttocks and thighs.
Pain often interferes with walking and worsened by lumbar extension.
Back and leg pain often associated with numbness and paresthesia.
Preservation of pedal pulses helps exclude vascular claudication.
Diagnosis best confirmed by MRI.
P.839
General Considerations
Lumbar spinal stenosis, defined as narrowing of the spinal canal with compression of the nerve roots, may be congenital or (more commonly) acquired. It most frequently results from enlarging osteophytes at the facet joints, hypertrophy of the ligamentum flavum, and protrusion or bulging of intervertebral disks. Lumbar spinal stenosis may produce symptoms by directly compressing nerve roots or by compressing nutrient arterioles that supply the nerve roots.
Clinical Findings
A. Symptoms and Signs
Since age is the greatest risk factor for spinal degenerative changes, most patients with lumbar spinal stenosis are over 60 years old. Patients typically complain of either leg pain or trouble walking. The pain may originate in the low back but will extend below the buttock into the thigh in nearly 90% of patients. In approximately 50% of patients, the pain will extend below the knee. The pain is often a combination of aching and numbness, which characteristically worsens with walking. The pain can also be brought on by prolonged standing. Not infrequently the symptoms are bilateral. Many patients are more troubled by poor balance, unsteadiness of gait, or leg weakness that develops as they walk. Some describe these neuroclaudication symptoms as developing spaghetti legs or walking like a drunk sailor. Because the lumbar spinal canal volume increases with back flexion and decreases with extension, some patients observe that they have fewer symptoms walking uphill than down. The back and lower extremity examination in patients with lumbar spinal stenosis is often unimpressive. Fewer than 10% have a positive straight leg raise sign, 25% have diminished deep tendon reflexes, and 60% have slight proximal weakness. Walking with the patient may reveal unsteadiness, although usually the patient's perception of gait disturbance is greater than that of an observer.
B. Imaging
The diagnosis of spinal stenosis in a patient with symptoms is best confirmed by MRI.
Differential Diagnosis
The onset of symptoms with standing, the location of the maximal discomfort to the thighs, and the preservation of pedal pulses help distinguish the pseudoclaudication of spinal stenosis from true claudication caused by vascular insufficiency. Distinguishing spinal stenosis from disk herniation can be challenging since both conditions can produce pain radiating down the back of the leg. Features that favor spinal stenosis are the gradual onset of symptoms, the marked exacerbation with walking, and the amelioration of symptoms with sitting or lumbar flexion. Complaints of bilateral aching in the buttocks associated with stiffness may make some practitioners consider the diagnosis of polymyalgia rheumatica. However, patients with lumbar spinal stenosis do not have the shoulder or neck symptoms characteristic of polymyalgia rheumatica.
Treatment
Weight loss and exercises aimed at reducing lumbar lordosis, which aggravates symptoms of spinal stenosis, can help. Lumbar epidural corticosteroid injections provide some immediate relief for about 50% of patients and more sustained relief for approximately 25%. When disabling symptoms persist, decompressive laminectomy provides at least short-term relief in approximately 80%.
Chang Y et al: The effect of surgical and nonsurgical treatment on longitudinal outcomes of lumbar spinal stenosis over 10 years. J Am Geriatr Soc 2005;53:785.
Sengupta DK et al: Lumbar spinal stenosis. Treatment strategies and indications for surgery. Orthop Clin North Am 2003; 34:281.
Fibromyalgia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Most frequent in women aged 20 50.
Chronic widespread musculoskeletal pain syndrome with multiple tender points.
Fatigue, headaches, numbness common.
Objective signs of inflammation absent; laboratory studies normal.
Partially responsive to exercise, tricyclic antidepressants.
General Considerations
Fibromyalgia is one of the most common rheumatic syndromes in ambulatory general medicine affecting 3 10% of the general population. It shares many features with the chronic fatigue syndrome, namely, an
P.840
increased frequency among women aged 20 50, absence of objective findings, and absence of diagnostic laboratory test results. While many of the clinical features of the two conditions overlap, musculoskeletal pain predominates in fibromyalgia whereas lassitude dominates the chronic fatigue syndrome.
The cause is unknown, but sleep disorders, depression, viral infections, and aberrant perception of painful stimuli have all been proposed. Fibromyalgia can be a rare complication of hypothyroidism, rheumatoid arthritis or, in men, sleep apnea.
Clinical Findings
The patient complains of chronic aching pain and stiffness, frequently involving the entire body but with prominence of pain around the neck, shoulders, low back, and hips. Fatigue, sleep disorders, subjective numbness, chronic headaches, and irritable bowel symptoms are common. Even minor exertion aggravates pain and increases fatigue. Physical examination is normal except for trigger points of pain produced by palpation of various areas such as the trapezius, the medial fat pad of the knee, and the lateral epicondyle of the elbow.
Differential Diagnosis
Fibromyalgia is a diagnosis of exclusion. A detailed history and repeated physical examination can obviate the need for extensive laboratory testing. Rheumatoid arthritis and systemic lupus erythematosus (SLE) present with objective physical findings or abnormalities on routine testing, including the ESR and C-reactive protein. Thyroid function tests are useful, since hypothyroidism can produce a secondary fibromyalgia syndrome. Polymyositis produces weakness rather than pain. The diagnosis of fibromyalgia probably should be made hesitantly in a patient over age 50 and should never be invoked to explain fever, weight loss, or any other objective signs. Polymyalgia rheumatica produces shoulder and pelvic girdle pain, is associated with anemia and an elevated ESR, and occurs after age 50. Hypophosphatemic states, such as oncogenic osteomalacia, should also be included in the differential diagnosis of musculoskeletal pain unassociated with physical findings. In contrast to fibromyalgia, oncogenic osteomalacia usually produces pain in only a few areas and is associated with a low serum phosphate level.
Treatment
Patient education is essential. Patients can be comforted that they have a diagnosable syndrome treatable by specific though imperfect therapies and that the course is not progressive. There is modest efficacy of amitriptyline, fluoxetine, chlorpromazine, or cyclobenzaprine. Amitriptyline is initiated at a dosage of 10 mg orally at bedtime and gradually increased to 40 50 mg depending on efficacy and toxicity. Fewer than 50% of the patients experience a sustained improvement. Exercise programs are also beneficial. NSAIDs are generally ineffective. Tramadol and acetaminophen combinations have ameliorated symptoms modestly in short-term trials. Opioids and corticosteroids are ineffective and should not be used to treat fibromyalgia. Acupuncture is also ineffective.
Prognosis
All patients have chronic symptoms. With treatment, however, many do eventually resume increased activities. Progressive or objective findings do not develop.
Assefi NP et al: A randomized clinical trial of acupuncture compared with sham acupuncture in fibromyalgia. Ann Intern Med 2005;143:10.
Bennett RM et al: Tramadol and acetaminophen combination tablets in the treatment of fibromyalgia pain: a double-blind, randomized, placebo-controlled study. Am J Med 2003;114:537.
Goldenberg DL et al: Management of fibromyalgia syndrome. JAMA 2004;292:2388.
Carpal Tunnel Syndrome
An entrapment neuropathy, carpal tunnel syndrome is a painful disorder caused by compression of the median nerve between the carpal ligament and other structures within the carpal tunnel. The contents of the tunnel can be increased by organic lesions such as synovitis of the tendon sheaths or carpal joints, recent or malhealed fractures, tumors, and occasionally congenital anomalies. Even though no anatomic lesion is apparent, flattening or even circumferential constriction of the median nerve may be observed during operative section of the ligament. The disorder may occur in pregnancy, is seen in individuals with a history of repetitive use of the hands, and may follow injuries of the wrists. There is a familial type of carpal tunnel syndrome in which no etiologic factor can be identified.
Carpal tunnel syndrome can also be a feature of many systemic diseases: rheumatoid arthritis and other rheumatic disorders (inflammatory tenosynovitis); myxedema, localized amyloidosis in chronic renal failure, sarcoidosis, and leukemia (tissue infiltration); acromegaly; and hyperparathyroidism.
Clinical Findings
Pain, burning, and tingling in the distribution of the median nerve (the palmar surface of the thumb and first two and a half fingers) is the initial symptom. Aching pain may radiate proximally into the forearm and occasionally proximally to the shoulder and over the neck and chest. Pain is exacerbated by manual activity, particularly by extremes of volar flexion or dorsiflexion of the wrist. It is most bothersome at night. Impairment of sensation in the median nerve distribution may or may not be demonstrable. Subtle disparity
P.841
between the affected and opposite sides can be shown by testing for two-point discrimination or by requiring the patient to identify different textures of cloth by rubbing them between the tips of the thumb and the index finger. Tinel's or Phalen's sign may be positive. (Tinel's sign is tingling or shock-like pain on volar wrist percussion; Phalen's sign is pain or paresthesia in the distribution of the median nerve when the patient flexes both wrists to 90 degrees with the dorsal aspects of the hands held in apposition for 60 seconds.) The carpal compression test, in which numbness and tingling are induced by the direct application of pressure over the carpal tunnel, may be more sensitive and specific than the Tinel and Phalen tests. Muscle weakness or atrophy, especially of the thenar eminence, appears later than sensory disturbances. Specific examinations include electromyography and determinations of segmental sensory and motor conduction delay. Sensory conduction delay is evident before motor delay.
Differential Diagnosis
This syndrome should be differentiated from other cervicobrachial pain syndromes, from compression syndromes of the median nerve in the forearm or arm, and from mononeuritis multiplex. When left-sided, it may be confused with angina pectoris.
Treatment
Treatment is directed toward relief of pressure on the median nerve. When a causative lesion is discovered, it is treated appropriately. Otherwise, patients in whom causative carpal tunnel syndrome is suspected should modify their hand activities and have the affected wrist splinted for 2 6 weeks. NSAIDs can also be added. Patients should be referred to a specialist for injection of corticosteroid into the carpal tunnel or for operation when they do not improve or when thenar muscle atrophy or weakness develops. Muscle strength returns gradually, but complete recovery cannot be expected when atrophy is pronounced.
Katz JN et al: Clinical practice. Carpal tunnel syndrome. N Engl J Med 2002;346:1807.
Ly-Pen D et al: Surgical decompression versus local steroid injection in carpal tunnel syndrome: a one-year, prospective, randomized, open, controlled clinical trial. Arthritis Rheum 2005;52:612.
MacDermid JC et al: Clinical diagnosis of carpal tunnel syndrome: a systematic review. J Hand Ther 2004;17:309.
Dupuytren'S Contracture
This relatively common disorder is characterized by hyperplasia of the palmar fascia and related structures, with nodule formation and contracture of the palmar fascia. The cause is unknown, but the condition has a genetic predisposition and occurs primarily in white men over 50 years of age. The incidence is higher among alcoholic patients and those with chronic systemic disorders (especially cirrhosis). It is also associated with systemic fibrosing syndrome, which includes Peyronie's disease, mediastinal and retroperitoneal fibrosis, and Riedel's struma. The onset may be acute, but slowly progressive chronic disease is more common.
Dupuytren's contracture manifests itself by nodular or cord-like thickening of one or both hands, with the fourth and fifth fingers most commonly affected. The patient may complain of tightness of the involved digits, with inability to satisfactorily extend the fingers, and on occasion there is tenderness. The resulting cosmetic problems may be unappealing, but in general the contracture is well tolerated since it exaggerates the normal position of function of the hand. Fasciitis involving other areas of the body may lead to plantar fibromatosis (10% of patients) or Peyronie's disease (1 2%).
If the palmar nodule is growing rapidly, injections of triamcinolone into the nodule may be of benefit. Surgical intervention is indicated in patients with significant flexion contractures, depending on the location, but recurrence is not uncommon.
Hart MG et al: Clinical associations of Dupuytren's disease. Postgrad Med J 2005;81:425.
Complex Regional Pain Syndrome (Reflex Sympathetic Dystrophy)
Complex regional pain syndrome is a rare disorder of the extremities characterized by autonomic and vasomotor instability. The cardinal symptoms and signs are diffuse pain (characteristically localized to an arm or hand, leg or foot), swelling of the involved extremity, disturbances of color and temperature in the affected limb, dystrophic changes in the overlying skin and nails, and limited range of motion. Use of the former name of this entity, reflex sympathetic dystrophy, is now discouraged because the precise role of the sympathetic nervous system is unclear and dystrophy is not an inevitable sequela of the syndrome. Most cases are preceded by direct physical trauma, often of relatively minor nature, to the soft tissues, bone, or nerve. Any extremity can be involved, but the syndrome most commonly occurs in the hand and is associated with ipsilateral restriction of shoulder motion (shoulder-hand syndrome). The syndrome proceeds through phases: pain, swelling, and skin color and temperature changes develop early and, if untreated, lead to atrophy and dystrophy. The swelling in complex regional pain syndrome is diffuse ( catcher's mitt hand ) and not restricted to joints. Pain is often burning in quality, intense, and often greatly worsened by minimal stimuli such as light touch. The shoulder-hand variant of this disorder sometimes complicates myocardial infarction or injuries to the neck or shoulder. Complex regional pain syndrome may occur after a knee injury or after arthroscopic knee surgery. There are no systemic symptoms. In the early phases of
P.842
the syndrome, bone scans are sensitive, showing diffuse increased uptake in the affected extremity. X-rays eventually reveal severe generalized osteopenia. In the posttraumatic variant, this is known as Sudeck's atrophy. Symptoms and findings are bilateral in some. This syndrome should be differentiated from other cervicobrachial pain syndromes, rheumatoid arthritis, thoracic outlet obstruction, and scleroderma, among others.
Early mobilization after injury, surgery, or myocardial infarction reduces the likelihood of developing the syndrome. In addition to addressing the underlying disorder, treatment is directed toward restoration of function. Physical therapy is the cornerstone of treatment. Many patients will also benefit from drug therapies, especially antidepressant agents (eg, nortriptyline initiated at a dosage of 10 mg orally at bedtime and gradually increased to 40 75 mg at bedtime). In resistant cases, prednisone, 30 40 mg/d orally for 2 weeks and then tapered over 2 weeks, may be effective. Regional nerve blocks and dorsal-column stimulation have also been demonstrated to be helpful. Patients who have restricted shoulder motion may benefit from the treatment described for scapulohumeral periarthritis. The prognosis partly depends on the stage in which the lesions are encountered and the extent and severity of associated organic disease. Early treatment offers the best prognosis for recovery.
Birklein F: Complex regional pain syndrome. J Neurol 2005; 252:131.
Mailis A et al: Sympathectomy for neuropathic pain. Cochrane Database Syst Rev 2003;(2):CD002918.
Mailis-Gagnon A et al: Spinal cord stimulation for chronic pain. Cochrane Database Syst Rev 2004;(3):CD003783.
Bursitis
Inflammation of the synovium-like cellular membrane overlying bony prominences may be secondary to trauma, infection, or arthritic conditions such as gout, rheumatoid arthritis, or osteoarthritis. The most common locations are the subdeltoid, olecranon, ischial, trochanteric, semimembranous-gastrocnemius (Baker's cyst), and prepatellar bursae.
There are several ways to distinguish bursitis from arthritis. Bursitis is more likely than arthritis to begin abruptly and cause focal tenderness and swelling. Olecranon bursitis, for example, causes an oval (or, if chronic, bulbous) swelling at the tip of the elbow, whereas elbow joint inflammation produces more diffuse swelling. Similarly, a patient with prepatellar bursitis has a small focus of swelling over the kneecap but no distention of the knee joint itself. Active and passive ranges of motion are usually much more limited in arthritis than in bursitis. A patient with trochanteric bursitis will have normal internal rotation of the hip, whereas a patient with hip arthritis will not. Bursitis caused by trauma responds to local heat, rest, NSAIDs, and local corticosteroid injections.
Bursitis can result from infection. The two most common sites are the olecranon and prepatellar bursae. Acute swelling and redness at either of these two sites calls for aspiration to rule out infection. The absence of fever does not exclude infection; and one-third of those with septic olecranon bursitis are afebrile. A bursal fluid white blood cell count of greater than 1000/mcL indicates inflammation from infection, rheumatoid arthritis, or gout. In septic bursitis, the white cell count averages over 50,000/mcL. Most cases are caused by Staphylococcus aureus; the Gram stain is positive in two-thirds. Treatment involves antibiotics and repeated aspiration for tense effusions.
Chronic, stable olecranon bursa swelling unaccompanied by erythema or other signs of inflammation does not suggest infection and does not require aspiration. Aspiration of the olecranon bursa in rheumatoid arthritis and in gout runs the risk of creating a chronic drainage site, which can be reduced by using a small needle (25-gauge if possible) and pulling the skin over the bursa before introducing it. Applying a pressure bandage may also help prevent chronic drainage. Surgical removal of the bursa is indicated only for cases in which repeated infections occur. Repetitive minor trauma to the olecranon bursa should be eliminated by avoiding resting the elbow on a hard surface or by wearing an elbow pad.
A bursa can also become symptomatic when it ruptures. This is particularly true for Baker's cyst, whose rupture can cause calf pain and swelling that mimic thrombophlebitis. Ruptured Baker's cysts are imaged easily by sonography or MRI. In most cases, imaging is unnecessary because the cyst and an associated knee effusion are detectable on physical examination. It may be important to exclude a deep venous thrombosis, which can be mimicked by a ruptured Baker's cyst. Treatment of a ruptured cyst includes rest, leg elevation, and injection of triamcinolone, 20 40 mg into the knee (which communicates with the cyst). Rarely, Baker's cyst can compress vascular structures and cause leg edema and true thrombophlebitis.
Cohen SP et al: Corticosteroid injections for trochanteric bursitis: is fluoroscopy necessary? A pilot study. Br J Anaesth 2005; 94:100.
Sports Medicine Injuries
Musculoskeletal problems commonly occur as a result of both serious athletic pursuits and activities of daily living. For most such disorders, the diagnosis is made easily. Physical therapy is an increasingly important adjunct to the management of these disorders.
1. Rotator Cuff Disorders
A substantial majority of shoulder problems stem from disorders of the rotator cuff. The tendons of the rotator cuff form a musculotendinous unit near their insertions into the proximal humerus. As a result of
P.843
years of cumulative irritation, attenuation of these tendons occurs. Among the relevant muscles, the supraspinatus is most often affected. Distinguishing between the various soft tissue disorders that cause shoulder pain is difficult. Rotator cuff tendinitis, subacromial bursitis, partial and complete rotator cuff tears, and calcific tendinitis frequently cause similar symptoms. In addition, these disorders often occur together, although precise distinction is frequently unimportant for purposes of therapy.
Clinical Findings
Patients usually present with nonspecific pain localized to the shoulder, often noticed more at night when lying on the affected side. Locking sensations occur with motion of the shoulder, particularly through abduction. Because of the shared innervation, symptoms are frequently referred down the proximal lateral arm. With rotator cuff tears, the patient may be unable to abduct or flex the shoulder, depending on the site of the tear.
The rotator cuff may be palpated just lateral to the head of the acromion. Maximum tenderness is usually noted over the supraspinatus insertion. The acromioclavicular joint may also be tender if there is accompanying degenerative arthritis in that joint. There may be prominent crepitus. Pain with range of motion is most pronounced between 60 and 120 degrees of abduction, the site of greatest impingement of the rotator cuff tissues between the humerus and coracoacromial arch.
For patients with partial rotator cuff tendon ruptures, the findings are identical to those of chronic tendinitis and bursitis. Patients with partial ruptures often demonstrate mild abduction weakness. With complete ruptures, weakness of abduction (and, to a lesser extent, flexion) is substantial, even though full range of motion may be maintained by the shoulder's accessory rotator muscles. Patients with complete tears usually have positive drop arm signs: inability to sustain passive abduction of the arm to 90 degrees.
Treatment
For most rotator cuff disorders, the central tenets of therapy are rest and abstention from inciting activities. Temporary use of a sling is helpful in enforcing rest. NSAIDs and moist heat afford some relief. For patients with symptoms persisting after 2 weeks of conservative management, injections of a corticosteroid preparation (eg, 1 mL of triamcinolone, 40 mg/mL) mixed with 2 3 mL of lidocaine hydrochloride (1 2%) may be useful. Tears or partial tears of the rotator cuff tendons that are believed to be chronic do not preclude a corticosteroid injection. Most patients obtain significant relief with one injection, but the procedure along with continued rest may be repeated after 2 3 weeks. Physical therapy is valuable for refractory cases if the patient does not maintain the shoulder's normal range of motion, stiffness and impaired function from a frozen shoulder may ensue.
Aside from patients with complete rotator cuff tears, only those who do not improve after months of conservative therapy are candidates for operation. Persistent symptoms may be a sign of complete tear, which may be diagnosed by MRI. Patients with symptoms that continue in the absence of a complete tear, however, sometimes require surgery to excise the inferolateral portion of the acromion, release obstructed soft tissue, and repair partial tendon tears. Depending on the patient's symptoms and functional status, complete tears may not always require surgical repair. Patients over 75 years of age rarely have symptoms or limitations that necessitate surgery. Moreover, surgical repairs are frequently less successful because of the attrition of the rotator cuff that occurs with aging.
Gerdesmeyer L et al: Extracorporeal shock wave therapy for the treatment of chronic calcifying tendonitis of the rotator cuff: a randomized controlled trial. JAMA 2003;290:2573.
Grant HJ et al: Evaluation of interventions for rotator cuff pathology: a systematic review. J Hand Ther 2004;17:274.
Haahr JP et al: Exercises versus arthroscopic decompression in patients with subacromial impingement: a randomised, controlled study in 90 cases with a one year follow up. Ann Rheum Dis 2005;64:760.
Luime JJ et al: Does this patient have an instability of the shoulder or a labrum lesion? JAMA 2004;292:1989.
2. Lateral & Medial Epicondylitis
These disorders are better known by their sports associations: tennis elbow and golf elbow, respectively. Lateral epicondylitis is the more common of the two. The conditions are caused by overuse, and the pain results from minor tears in the tendons of the forearm's extensor and flexor muscles.
Clinical Findings
The patient presents with pain at the site of tendon insertion. Tasks that require grasping and squeezing, such as shaking hands or opening jars, cause pain.
The diagnoses are easily confirmed on physical examination by elicitation of point tenderness over the involved site. The characteristic pain may also be reproduced by extension or flexion of the wrist against pressure. Clenching the fist and extending the wrist against the pressure of the examiner's palm is a useful maneuver for localizing lateral epicondylitis. Medial epicondylitis may be demonstrated by performing the same maneuver in flexion.
Treatment
The use of counterforce straps (bands worn distal to the elbow, over the bulk of the forearm musculature),
P.844
intended to decrease the forces transmitted to the elbow during activity, are inadequate substitutes for rest. NSAIDs are effective in mild cases. Symptoms that persist after 2 weeks of conservative therapy usually respond either to infiltration of triamcinolone, 10 20 mg mixed with 1 2 mL of 1% lidocaine, around the involved epicondyle or to physiotherapy. Results are quicker with corticosteroid injections but longer-lasting with physiotherapy. Botulinum toxin injection can also ameliorate lateral epicondylitis symptoms but runs the risk of causing digital paresis. After the pain and tenderness have subsided, patients may begin a physical therapy program involving daily stretching of the flexor and extensor tendons.
Smidt N et al: Corticosteroid injections, physiotherapy, or a wait-and-see policy for lateral epicondylitis: a randomised controlled trial. Lancet 2002;359:657.
Trudel D et al: Rehabilitation for patients with lateral epicondylitis: a systematic review. J Hand Ther 2004;17:243.
Wong SM et al: Treatment of lateral epicondylitis with botulinum toxin: a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2005;143:793.
3. Patellofemoral Syndrome
Patellofemoral syndrome is among the most common causes of knee complaints in primary care medicine, particularly among adolescent and young adult patients. The syndrome frequently gives rise to the chief complaint of anterior knee pain. A variety of injuries or anatomic abnormalities predispose patients to irregular patellar movements, leading to the patellofemoral syndrome. Such predisposing conditions include imbalance of quadriceps strength, patella alta, recurrent patellar subluxation, direct trauma to the patella, and meniscal injuries. For most of these causes, the therapeutic approach is similar.
Clinical Findings
Patients frequently have difficulty localizing the source of their complaint but generally confirm that the pain is in the front of the knee, around or underneath the patella. Questions regarding specific precipitants of the pain may be useful in establishing the diagnosis. For example, because of the flexion load, patients have difficulty going up or down staircases (down is usually associated with greater symptoms). Another characteristic symptom is the positive theater sign: after remaining seated for a prolonged period, patients describe extreme discomfort with their first few steps after rising. The symptoms improve with further walking. Finally, patients with patellofemoral syndrome often complain of crepitus, joint locking, or sensations of joint instability, all of which lack readily confirmed anatomic explanations.
The physical examination is less useful than the history in establishing the diagnosis, but a significant number of patients have characteristic physical findings. In particular, when the knee is held in slight flexion, gentle pressure against the patella as the patient contracts the quadriceps muscles may reproduce the symptoms. In some cases, with the knee extended and the quadriceps relaxed, the typical pain may be reproduced by digital pressure under the medial or lateral border of the patella, with side-to-side movement of the bone. Inflammatory findings on examination are incompatible with the diagnosis of patellofemoral syndrome and suggest other disorders.
Treatment
Therapy includes avoidance of flexion loads and strengthening of the quadriceps. Referral to a physical therapist is helpful in educating the patient about home exercises. Many patients learn that the most effective therapy is bicycling, with the seat high enough to permit nearly full knee extension with each cycle. Although most cases respond to these interventions and even resolve altogether, some persist for years. Even in the latter, conservative therapy remains the rule; operation is rarely indicated. There is only limited evidence for the effectiveness of NSAIDs in short-term pain reduction associated with the patellofemoral pain syndrome. The evidence supporting the use of glycosaminoglycan polysulfate is conflicting. Bracing is no more effective than a home exercise program.
Heintjes E et al: Pharmacotherapy for patellofemoral pain syndrome. Cochrane Database Syst Rev 2004;(3):CD003470.
Lun VM et al: Effectiveness of patellar bracing for treatment of patellofemoral pain syndrome. Clin J Sport Med 2005;15:235.
4. Overuse Syndromes of the Knee
Runners particularly those who overtrain, fail to stretch prior to running, or do not attain the proper level of conditioning before starting a running program may develop a variety of painful overuse syndromes of the knee. Most of these conditions are forms of tendinitis or bursitis that can be diagnosed on examination. The most common conditions include anserine bursitis, the iliotibial band syndrome, and popliteal and patellar tendinitis.
Clinical Findings
Symptoms resulting from all of these conditions worsen as the patient continues to run and often require cessation of the activity. Anserine bursitis results in pain medial and inferior to the knee joint over the medial tibia. The iliotibial band syndrome and popliteal tenosynovitis may be difficult to differentiate, because the popliteus tendon inserts into the lateral femoral condyle underneath the iliotibial band. Both conditions result in pain on the lateral side of the knee. Patellar tendinitis, a cause of anterior knee discomfort, typically occurs at the tendon's insertion into the patella rather than at its more inferior insertion.
P.845
All of these diagnoses are confirmed by palpation at the relevant sites around the knee. None is associated with joint effusions or other signs of synovitis.
Treatment
Rest and abstention from the associated physical activities for a period of days to weeks are essential. Once pain has subsided, a program of gentle stretching (particularly prior to resuming exercise) may prevent recurrence. Corticosteroid injections with lidocaine may be useful when intense discomfort is present, but caution must be used when injecting corticosteroids into the region of a tendon to avoid rupture.
5. Medial Meniscus Injuries
Tears of the medial meniscus are the most common knee injuries encountered in primary care. Because the medial meniscus is tethered firmly to the underlying tibia, injuries to the medial meniscus occur ten times more commonly than injuries to the lateral meniscus. Both result from a twisting action exerted on the knee joint while the foot is in a weight-bearing position.
Clinical Findings
The injury is heralded by a tearing or popping sensation followed by severe pain. Occasionally, meniscal tears result from seemingly minor trauma. In contrast to ligamentous injuries, in which hemorrhage causes immediate swelling, effusions associated with meniscal injuries accumulate over hours and are typically worse the day after the injury. Several days after resolution (full or partial), the patient may experience joint locking or instability, recurrent swelling with activity, and pain. The sensation of locking, which may result from mechanical blockage by a fragment of torn meniscus, more commonly results from pseudolocking caused by muscle spasm and swelling.
Joint effusion is usually present, frequently accompanied by a ballottable patella. Tenderness may be localized to the medial joint line, and range of motion in the knee may be restricted. In patients without an acutely painful, swollen knee, McMurray's test may suggest the diagnosis. This test is performed with the patient supine and the hip and knee in full flexion. The examiner has one hand on the involved knee and the other on the ipsilateral foot. As the foot is externally rotated, the examiner extends the patient's knee. The presence of a snap (palpable or audible) suggests a medial meniscus lesion. MRI is the optimal test for confirming the diagnosis if plain films exclude other conditions.
Treatment
Initial management is conservative, with elevation of the joint and application of a compression dressing and ice. Weight bearing should be minimized for the first few days after the injury but may be resumed slowly thereafter. The patient should perform quadriceps-strengthening exercises under the instruction of a physical therapist. Surgery is reserved for patients with symptoms that recur upon resumption of normal activities or for patients with irreducible locking caused by mechanical problems.
Jackson JL et al: Evaluation of acute knee pain in primary care. Ann Intern Med 2003;139:575.
6. Ankle Sprains
Ankle sprains are among the most common of all sports injuries. Most sprains involve the lateral ligament complex, particularly the anterior talofibular ligament. In more severe injuries, the calcaneofibular ligament may also be involved. If both of these ligaments are ruptured, the injury results in significant joint instability and is classified as a grade III (severe) sprain. (Grades I and II correspond to mild and moderate injuries, respectively.) This section reviews only the type of ankle sprain resulting from inversion (varus) injuries, which account for 85% of all sprains.
Clinical Findings
Varus sprains include a spectrum of severity, ranging from slight loss of function to injuries in which the swelling is prompt, the pain prominent, and weight bearing impossible. A history of hearing a pop at the time of injury is frequently associated with the latter.
Hemorrhage resulting from torn ligaments and damaged peroneal muscle tendons may cause substantial ecchymosis. Tenderness is typically present at the site of injury, and the associated swelling may be considerable. Stability of the anterior talofibular and calcaneofibular ligaments should be assessed with the anterior drawer sign: With the foot held in slight plantar flexion, the examiner cups the patient's heel with one hand and the patient's shin with the other. The examiner then applies gentle anterior force in the plane of the patient's foot. Excessive anterior motion of the foot constitutes a positive test (grade III sprain). Plain radiographs exclude associated bony injury.
Treatment
Most ankle sprains even grade III are treated identically. The acronym RICE (rest, ice, compression, elevation) applies more accurately to ankle sprains than to any other injury. Early application of a compression dressing is essential to control swelling and provide stability to the traumatized joint. An Aircast ankle brace may be more effective than an elastic support bandage. Weight bearing should be minimal, with liberal use of crutches. Elevation of the ankle for several days hastens functional recovery by diminishing pain and swelling, and ice is also helpful (alternating 30 minutes on, 30 minutes off). The ice should be applied on top of the compression dressing and not against the skin both because close apposition of the dressing to the skin is critical to control swelling and because
P.846
direct application of ice is uncomfortable and even deleterious to the skin. Referral to a physical therapist may expedite recovery. Patients should be informed that symptoms from lateral ankle sprains may take weeks or months to resolve, and that this period will be prolonged by premature attempts to bear weight on the injured ankle. Surgical repairs of ruptured lateral ligaments provide excellent outcomes but are usually necessary only in cases of chronically unstable joints.
Boyce SH et al: Management of ankle sprains: a randomised controlled trial of the treatment of inversion injuries using an elastic support bandage or an Aircast ankle brace. Br J Sports Med 2005;39:91.
7. Plantar Fasciitis
The most common cause of foot pain in outpatient medicine is plantar fasciitis, which results from constant strain on the plantar fascia at its insertion into the medial tubercle of the calcaneus. Although certain inflammatory disorders such as the seronegative spondyloarthropathies predispose patients to enthesopathies, the majority of cases occur in patients with no associated disease. Most occur as the result of excessive standing and improper footwear.
Clinical Findings
Patients with plantar fasciitis report severe pain on the bottoms of their feet in the morning the first steps out of bed in particular but the pain subsides after a few minutes of ambulation.
The diagnosis may be confirmed by palpation over the plantar fascia's insertion on the medial heel. Radiographs have no role in the diagnosis of this condition heel spurs frequently exist in patients without plantar fasciitis, and most symptomatic patients do not have heel spurs.
Treatment
Treatment consists of imposing an interval of days without prolonged standing and the use of arch supports. Arch supports give relief by requiring the arches to bear more of the patient's weight, thus unloading the plantar enthesis. NSAIDs may provide some relief. In severe cases, a corticosteroid with lidocaine injection (small volume no more than a total of 1.5 mL) directly into the most tender area on the sole of the foot is helpful. Rare patients require release of the plantar fascia from its attachment site at the os calcis.
Autoimmune Diseases
The autoimmune disorders are a protean group of acquired diseases in which genetic factors also play a role. They have in common widespread immunologic alterations and often share features of generalized inflammation.
Because of overlapping clinical features, differentiation among autoimmune diseases may be challenging, particularly in their early stages. These illnesses share certain clinical features, and differentiation among them is often difficult because of this. Common findings include synovitis, pleuritis, myocarditis, endocarditis, pericarditis, peritonitis, vasculitis, myositis, skin rash, and nephritis. Laboratory tests may reveal Coombs-positive hemolytic anemia, thrombocytopenia, leukopenia, immunoglobulin excesses or deficiencies, antinuclear antibodies (which include antibodies to many nuclear constituents, including DNA and extractable nuclear antigen), rheumatoid factors, cryoglobulins, false-positive serologic tests for syphilis, other antiphospholipid antibodies, elevated muscle enzymes, and hypocomplementemia.
Some of the laboratory abnormalities found in autoimmune diseases (eg, false-positive serologic tests for syphilis, rheumatoid factor) occur in asymptomatic individuals. These changes may also be demonstrated in certain asymptomatic relatives of patients with connective tissue diseases, in older persons, in patients using certain medications, and in patients with chronic infectious diseases.
Rheumatoid Arthritis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Prodromal systemic symptoms of malaise, fever, weight loss, and morning stiffness.
Onset usually insidious and in small joints; progression is centripetal and symmetric; deformities common.
Radiographic findings: juxta-articular osteoporosis, joint erosions, and narrowing of the joint spaces.
Rheumatoid factor usually present. The anticyclic citrullinated peptide (CCP) test also has a high sensitivity and specificity for rheumatoid arthritis.
Extra-articular manifestations: subcutaneous nodules, pleural effusion, pericarditis, lymphadenopathy, splenomegaly with leukopenia, and vasculitis.
General Considerations
Rheumatoid arthritis is a chronic systemic inflammatory disease of unknown cause, chiefly affecting synovial membranes of multiple joints. The disease has a wide clinical spectrum with considerable variability in
P.847
joint and extra-articular manifestations. The prevalence in the general population is 1 2%; female patients outnumber males almost 3:1. The usual age at onset is 20 40 years, although rheumatoid arthritis may begin at any age. Susceptibility to rheumatoid arthritis is genetically determined. Many patients have a class II HLA epitope with an identical five-amino-acid sequence; this is known as the shared epitope. If not treated appropriately, rheumatoid arthritis has a strong tendency to shorten life and cause severe disability. Consequently, early, aggressive treatment is now the standard of care.
The pathologic findings in the joint include chronic synovitis with pannus formation. The pannus erodes cartilage, bone, ligaments, and tendons. In the acute phase, effusion and other manifestations of inflammation are common. In the late stage, organization may result in fibrous ankylosis; true bony ankylosis is rare. In both acute and chronic phases, inflammation of soft tissues around the joints may be prominent and is a significant factor in joint damage.
The microscopic findings most characteristic of rheumatoid arthritis are those of the subcutaneous nodule. This is a granuloma with a central zone of fibrinoid necrosis, a surrounding palisade of radially arranged elongated connective tissue cells, and a periphery of chronic granulation tissue. Pathologic alterations indistinguishable from those of the subcutaneous nodule are occasionally seen in the myocardium, pericardium, endocardium, heart valves, visceral pleura, lungs, sclera, dura mater, spleen, larynx, and other tissues. In the era of more effective treatment, secondary amyloidosis is now very rare.
Clinical Findings
A. Symptoms and Signs
The clinical manifestations of rheumatoid disease are highly variable. Although acute presentations may occur, the onset of articular signs of inflammation is usually insidious, with prodromal symptoms of malaise, weight loss, and vague periarticular pain or stiffness. Symmetric joint swelling with stiffness, warmth, tenderness, and pain are characteristic. Stiffness persisting for more than 30 minutes (and usually many hours) is prominent in the morning, subsiding later in the day. The duration of morning stiffness is a useful indicator of disease activity. Stiffness may recur after daytime inactivity and be much more severe after strenuous activity. Although any joint may be affected, the PIP and MCP joints of the fingers as well as the wrists, knees, ankles, and toes are most often involved. Monarticular disease is occasionally seen early. Synovial cysts and rupture of tendons may occur. Entrapment syndromes are not unusual particularly of the median nerve at the carpal tunnel of the wrist. Palmar erythema is noted occasionally, as are tiny hemorrhagic infarcts in the nail folds or finger pulps. Twenty percent of patients have subcutaneous nodules, most commonly situated over bony prominences but also observed in the bursas and tendon sheaths. Nodules correlate with the presence of rheumatoid factor in serum ( seropositivity ), as do most other extra-articular manifestations. Dryness of the eyes, mouth, and other mucous membranes is found especially in advanced disease (see Sj gren's Syndrome). Other ocular manifestations include episcleritis and scleromalacia, the latter due to scleral nodules and capable of causing retinal detachment. Pericarditis and pleural disease, when present, are frequently silent clinically. Nonspecific pericarditis and pleuritis are found in 25 40% of patients at autopsy. Additional nonspecific lesions associated with rheumatoid arthritis include inflammation of small arteries, pulmonary fibrosis, mononuclear cell infiltration of skeletal muscle and perineurium, and hyperplasia of lymph nodes. Aortitis is a rare late complication that can result in aortic regurgitation or rupture and is usually associated with evidence of rheumatoid vasculitis elsewhere in the body.
After months or years, deformities may occur; the most common are ulnar deviation of the fingers, boutonni re deformity (hyperextension of the DIP joint with flexion of the PIP joint), swan-neck deformity (flexion of the DIP joint with extension of the PIP joint), and valgus deformity of the knee. Atrophy of skin or muscle is common, caused by the combined effects of disease and treatment (particularly prednisone). A small number of patients have splenomegaly and lymph node enlargement. A small subset of patients with rheumatoid arthritis have Felty's syndrome, the occurrence of splenomegaly and neutropenia, usually in the setting of severe, destructive arthritis. Felty's syndrome must be distinguished from the large granular lymphocyte syndrome, with which it shares many features.
B. Laboratory Findings
Serum protein abnormalities are often present. The most specific blood test for rheumatoid arthritis is antibody to cyclic citrullinated peptide (specificity ~95%). However, in early disease the CCP antibody has a sensitivity of only 50%. Rheumatoid factor, an IgM antibody directed against the Fc fragment of IgG, is present in the sera of more than 75% of patients and has a specificity of approximately 90%. High titers of rheumatoid factor are commonly associated with severe rheumatoid disease. Titers may also be significantly elevated in a number of diverse conditions, including syphilis, sarcoidosis, infective endocarditis, tuberculosis, leprosy, and parasitic infections. The prevalence of seropositivity also rises with age in healthy individuals. Asymptomatic relatives of patients with autoimmune diseases are more likely to be rheumatoid factor positive as well. Antinuclear antibodies are demonstrable in 20% of patients, although their titers are lower than in SLE.
During both the acute and chronic phases, the ESR and the immune globulins (most commonly IgM and IgG) are typically elevated. A moderate hypochromic normocytic anemia is common. The white cell count
P.848
is normal or slightly elevated, but leukopenia may occur, often in the presence of splenomegaly (eg, Felty's syndrome). The platelet count is often elevated, roughly in proportion to the severity of overall joint inflammation. Joint fluid examination is valuable, reflecting abnormalities that are associated with varying degrees of inflammation. (See Tables 20-1 and 20-2.)
C. Imaging
Of all the laboratory tests, x-ray changes are the most specific for rheumatoid arthritis. X-rays performed during the first 6 months of symptoms, however, are usually normal. The earliest changes occur in the wrists or feet and consist of soft tissue swelling and juxta-articular demineralization. Later, diagnostic changes of uniform joint space narrowing and erosions develop. The erosions are often first evident at the ulnar styloid and at the juxta-articular margin, where the bony surface is not protected by cartilage. Diagnostic changes also occur in the cervical spine, with C1 2 subluxation, but these changes usually take many years to develop. Both MRI and ultrasonography are more sensitive than radiographs in detecting bony and soft tissue changes in rheumatoid arthritis. However, the value of MRI and ultrasound in early diagnosis relative to that of plain radiographs, particularly given cost considerations, has not been established.
Differential Diagnosis
The differentiation of rheumatoid arthritis from other joint conditions and immune-mediated disorders can be difficult. However, certain clinical features are helpful. Osteoarthritis, for example, spares the wrist and the MCP joints, in contrast to rheumatoid arthritis. Degenerative joint disease (osteoarthritis) is not associated with constitutional manifestations and the joint pain is characteristically relieved by rest, unlike the morning stiffness of rheumatoid arthritis. Signs of articular inflammation, prominent in rheumatoid arthritis, are usually minimal in degenerative joint disease. Although gouty arthritis is almost always intermittent and monarticular in the early years, it may evolve with time into a chronic polyarticular process that mimics rheumatoid arthritis. Gouty tophi resemble rheumatoid nodules both in typical location and appearance. The early history of intermittent monarthritis and the presence of synovial urate crystals are distinctive features of gout. Septic arthritis can be distinguished by chills and fever, demonstration of the causative organism in joint fluid, and the frequent presence of a primary focus elsewhere, eg, gonococcal arthritis. Septic arthritis can complicate rheumatoid arthritis and should be considered whenever a patient with rheumatoid arthritis has one joint inflamed out of proportion to the rest. Lyme disease typically involves only one joint, most commonly the knee, and is associated with positive serologic tests (see Chapter 34). Human parvovirus B19 infection in adults can occasionally mimic rheumatoid arthritis. The mean age at onset is approximately 35 years. Arthralgias are much more prominent than arthritis, and rash on the cheeks, torso, or extremities is common. The patients are rheumatoid factor-negative, do not have erosions, and have IgM antibodies to human parvovirus B19 infection.
Malar rash, photosensitivity, discoid skin lesions, alopecia, high titer antibodies to double-stranded DNA, glomerulonephritis, and central nervous system abnormalities point to the diagnosis of SLE. Polymyalgia rheumatica occasionally causes polyarthralgias in patients over age 50, but these patients remain rheumatoid factor-negative and have chiefly proximal muscle pain and stiffness, centered on the shoulder and hip girdles. Rheumatic fever is characterized by the migratory nature of the arthritis, an elevated antistreptolysin titer, and a more dramatic and prompt response to aspirin; carditis and erythema marginatum may occur in adults, but chorea and subcutaneous nodules virtually never do. Finally, a variety of cancers produce paraneoplastic syndromes, including polyarthritis. One form is hypertrophic pulmonary osteoarthropathy most often produced by lung and gastrointestinal carcinomas, characterized by a rheumatoid-like arthritis associated with clubbing, periosteal new bone formation, and a negative rheumatoid factor. Diffuse swelling of the hands with palmar fasciitis occurs in a variety of cancers, especially ovarian carcinoma.
Treatment
A. Basic Program (Nonpharmacologic Management)
The primary objectives in treating rheumatoid arthritis are reduction of inflammation and pain, preservation of function, and prevention of deformity. Patient satisfaction and the success of therapy depend on how effectively the clinician utilizes the nonpharmacologic measures outlined in the following paragraphs.
1. Education and emotional factors
The clinician should explain the disease, describe its fluctuations, and involve the patient and family in decisions about therapy.
2. Physical and occupational therapies
Physical and occupational therapists understand nonpharmacologic treatments of arthritis (described below) and can effectively teach them. The therapist can develop a program the patient can follow at home, with only periodic monitoring.
3. Systemic rest
The amount of systemic rest required depends on the presence and severity of inflammation. With mild inflammation, 2 hours of rest each day may suffice. In general, rest should be continued until significant improvement is sustained for at least 2 weeks; thereafter, the program may be liberalized. However, the increase of physical activity must proceed gradually and with appropriate support for any involved weight-bearing joints.
P.849
4. Articular rest
Decrease of articular inflammation may be expedited by joint rest. Relaxation and stretching of the hip and knee muscles, to prevent flexion contractures, can be accomplished by having the patient lie in the prone position for 15 minutes several times daily. Sitting in a flexed position for prolonged periods is a poor form of joint rest. Appropriate adjustable supports provide rest for inflamed weight-bearing joints, relieve spasm, and may reduce deformities, soft tissue contracture, or instability of the ligaments. The supports must be removable to permit daily range of motion and exercise of the affected extremities (see below).
5. Exercise
Exercises are designed to preserve joint motion, muscular strength, and endurance. Initially, for inflammatory disease, passive range of motion and isometric exercises (such as straight leg raising) are best tolerated. In hydrotherapy, the buoyancy of water permits maximum isotonic and isometric exercise with no more stress on joints than active range of motion exercises. As tolerance for exercise increases and the activity of the disease subsides, progressive resistance exercises may be introduced. Patients should follow the general rule of eliminating any exercise that produces increased pain 1 hour after the exercise has ended.
6. Heat and cold
These are used primarily for their muscle-relaxing and analgesic effects. Radiant or moist heat is generally most satisfactory. Exercise may be better performed after exposure to heat. Some patients derive more relief of joint pain from local application of cold.
7. Assistive devices
Patients with significant hip or knee arthritis may benefit from having an elevated toilet, a gripping bar, or a cane. Patients hold the cane in the hand opposite to the affected knee or hip, thus leaning away from the affected joint. Crutches or walkers may be needed for patients with more extensive disease.
8. Splints
Splints may provide joint rest, reduce pain, and prevent contracture, but their use should be guided by certain principles.
Night splints of the hands or wrists (or both) should maintain the extremity in the position of optimum function. The elbow and shoulder lose motion so rapidly that other local measures and corticosteroid injections are usually preferable to splints.
The best splint for the hip is prone-lying for several hours a day on a firm bed. For the knee, prone-lying may suffice, but splints in maximum tolerated extension are frequently needed. Ankle splints are of the simple right-angle type.
Splints should be applied for the shortest period needed, should be made of lightweight materials for comfort, and should be easily removable for range-of-motion exercises once or twice daily to prevent loss of motion.
Corrective splints, such as those for overcoming knee flexion contractures, should be used under the guidance of a clinician familiar with their proper use.
Note: Avoidance of prolonged sitting or knee pillows may decrease the need for splints.
9. Weight loss
For overweight patients, achieving ideal body weight will reduce the wear and tear placed on arthritic joints of the lower extremities.
B. Nonsteroidal Anti-inflammatory Drugs
The first drug used to treat rheumatoid arthritis is an NSAID. These agents have analgesic and anti-inflammatory effects but do not prevent erosions or alter disease progression. A number of NSAIDs are available, including aspirin, ibuprofen, naproxen, sulindac, diclofenac, nabumetone, etodolac, ketoprofen, celecoxib, and others.
NSAIDs work in arthritis by the same mechanism that causes side effects: inhibition of COX, the enzyme that converts arachidonic acid to prostaglandins. Although prostaglandins play important roles in promoting inflammation and pain, they also help maintain homeostasis in several organs especially the stomach, where prostaglandin E serves as a local hormone responsible for gastric mucosal cytoprotection. The discovery that COX exists in two isomers COX-1 (which is expressed continuously in many cells and is responsible for the salutary effects of prostaglandins) and COX-2 (which is induced by cytokines and expressed in inflammatory tissues) was initially of little practical consequence since all traditional NSAIDs inhibit both isomers.
However, selective COX-2 inhibitors (eg, celecoxib) are FDA-approved for the treatment of osteoarthritis and rheumatoid arthritis. Compared with traditional NSAIDs, COX-2 inhibitors are as effective for treating rheumatoid arthritis but less likely in some circumstances to cause upper gastrointestinal tract adverse events (eg, obstruction, perforation, hemorrhage, or ulceration). Long-term use of COX-2 inhibitors, particularly in the absence of concomitant aspirin use, has been associated with an increased risk of cardiovascular events, leading to the removal of some COX-2 inhibitors from the market and intense scrutiny of all drugs in that class.
1. Gastrointestinal side effects
In terms of efficacy, all NSAIDs appear equivalent. Anecdotes suggest that indomethacin is more effective than other NSAIDs for ankylosing spondylitis (see below). For rheumatoid arthritis, no NSAID is convincingly more effective than another.
For traditional NSAIDs that inhibit both COX-1 and COX-2, gastrointestinal side effects, such as gastric ulceration, perforation, and gastrointestinal hemorrhage, are the most common serious side effects. The overall rate of bleeding with NSAID use in the general population is low (1:6000 users or less) but is increased by long-term use, higher NSAID dose, concomitant corticosteroids or anticoagulants, the presence of rheumatoid arthritis, history of peptic ulcer disease or alcoholism, and age over 70. Twenty-five percent of all hospitalizations and deaths from peptic ulcer disease result from traditional NSAID therapy. Each year, 1:1000 patients with rheumatoid arthritis
P.850
will require hospitalization for NSAID-related gastrointestinal bleeding or perforation. Although all traditional NSAIDs can cause massive gastrointestinal bleeding, the risk may be higher with indomethacin and piroxicam, probably because these drugs preferentially inhibit COX-1 in the stomach.
There are two approaches to reducing the gastrointestinal toxicity of NSAIDs. One approach is to use a traditional NSAID and add either a proton pump inhibitor (eg, omeprazole 20 mg orally daily), famotidine 40 mg orally twice daily, or misoprostol. The efficacy of misoprostol is limited by its poor tolerability; 20% of patients discontinue the medication because of diarrhea and bloating associated with full doses (200 mcg orally four times daily). Giving smaller amounts of misoprostol (eg, 100 mcg four times daily) or using less frequent dosing (eg, 200 mcg twice daily) improves tolerance while reducing efficacy only modestly. Misoprostol is an abortifacient and is contraindicated in patients who are or might become pregnant. Sucralfate, antacids, and ranitidine either do not work or do not work as well as proton pump inhibitors. The expense of proton pump inhibitors and misoprostol dictates that their use should be reserved for patients with risk factors for NSAID-induced gastrointestinal toxicity (noted above). Patients who have recently recovered from an NSAID-induced bleeding gastric ulcer appear to be at high risk for rebleeding (about 5% in 6 months) when an NSAID is reintroduced, even if prophylactic measures such as proton pump inhibitors are used. NSAIDs can also affect the lower intestinal tract, causing perforation or aggravating inflammatory bowel disease.
Acute liver injury from NSAIDs is rare, occurring in about 1 of every 25,000 patients using these agents. Having rheumatoid arthritis or taking sulindac may increase the risk.
2. Renal side effects
All of the NSAIDs, including aspirin and the COX-2 inhibitors, can produce renal toxicity, including interstitial nephritis, nephrotic syndrome, prerenal azotemia, and aggravation of hypertension. Hyperkalemia due to hyporeninemic hypoaldosteronism may also be seen. The risk of renal toxicity is low but is increased by age over 60, a history of renal disease, congestive heart failure, ascites, and diuretic use. COX-2 inhibitors appear to cause as much nephrotoxicity as traditional NSAIDs.
3. Platelet effects
All NSAIDs except the nonacetylated salicylates and the COX-2 inhibitors interfere with platelet function and prolong bleeding time. For all older NSAIDs except aspirin, the effect on bleeding time resolves as the drug is cleared. Aspirin irreversibly inhibits platelet function, so the bleeding time effect resolves only as new platelets are made. COX-2 inhibitors, which differ from other NSAIDs in not inhibiting platelet function, do not increase the risk of bleeding with surgical procedures as most NSAIDs do. Unfortunately, this failure to inhibit platelets is now known to lead to increased risks of myocardial infarction and stroke, particularly when the medications are used in high doses for prolonged periods of time. Patients requiring low-dose aspirin who are not compliant have a greater risk of developing a thrombotic event while taking a coxib than while taking a traditional NSAID. Whether combination therapy with low-dose aspirin and a coxib maintains the gastrointestinal advantage of selective COX-2 inhibitors is not yet known.
Although groups of patients with rheumatoid arthritis respond similarly to NSAIDs, individuals may respond differently an NSAID that works for one patient may not work for another. Thus, if the first NSAID chosen is not effective after 2 3 weeks of use, another should be tried.
C. Additional Drugs
Disease-modifying antirheumatic drugs (DMARDs) should be started as soon as the diagnosis of rheumatoid disease is certain.
1. Methotrexate
Methotrexate is usually the treatment of choice for patients with rheumatoid arthritis who do not respond to NSAIDs. Methotrexate is generally well tolerated and often produces a beneficial effect in 2 6 weeks compared with the 2- to 6-month onset of action for drugs such as gold, penicillamine, and antimalarials. The usual initial dose is 7.5 mg of methotrexate orally once weekly. If the patient has tolerated methotrexate but has not responded in 1 month, the dose can be increased to 15 mg orally once per week. The maximal dose is approximately 25 mg/wk. The most frequent side effects are gastric irritation and stomatitis. If needed to minimize gastrointestinal toxicity, methotrexate can be administered by subcutaneous or intramuscular injection. A severe, potentially life-threatening interstitial pneumonitis occurs rarely and usually responds to cessation of the drug and institution of corticosteroids. Hepatotoxicity with fibrosis and cirrhosis is another important toxic effect that appears to be very rare, with a risk of approximately 1:1000 after 5 years of methotrexate therapy. Still, methotrexate is contraindicated in a patient with any form of chronic hepatitis. Diabetes, obesity, and renal disease increase the risk of hepatotoxicity. Liver function tests should be monitored every 4 8 weeks, along with the complete blood count, serum creatinine, and serum albumin. Heavy alcohol use increases the hepatotoxicity, so patients should be advised to drink alcohol in extreme moderation, if at all. In a patient with no risk factors for hepatotoxicity, liver biopsy is not needed initially but is performed if aminotransferase levels are elevated, despite dosage reduction, in 6 out of 12 monthly determinations or if the serum albumin falls below normal. Cytopenia due to bone marrow suppression and infection are other important potential problems. The risk of developing cytopenia is much higher in patients with a serum creatinine of 2 mg/dL or higher. Side effects, including hepatotoxicity, may be reduced by prescribing either daily folate (1 mg) or weekly leucovorin calcium (2.5 5 mg taken 24 hours
P.851
after the dose of methotrexate). Methotrexate is associated with an increased risk of B cell lymphomas, some of which resolve following the discontinuation of the medication. The combination of methotrexate and other folate antagonists, such as trimethoprim-sulfamethoxazole, should be used cautiously, since pancytopenia can result. Probenecid should also be avoided since it increases methotrexate drug levels and toxicity.
2. Tumor necrosis factor inhibitors
Inhibitors of tumor necrosis factor (TNF) are fulfilling the aim of targeted therapy for rheumatoid arthritis. These medications are frequently added to the treatment of patients who have not responded adequately to methotrexate, and increasingly are added simultaneously with methotrexate for patients with poor prognostic factors. TNF a cytokine central to the inflammatory cascade in rheumatoid arthritis activates lymphocytes and leukocytes, stimulates the elaboration of other cytokines, and is found in elevated concentrations in rheumatoid synovium. Three inhibitors are in use: etanercept, infliximab, and adalimumab. Etanercept, a soluble recombinant TNF receptor:Fc fusion protein, is administered at a dosage of 25 mg subcutaneously twice weekly or 50 mg once per week. Infliximab, a chimeric monoclonal antibody, is administered at a dosage of 3 10 mg/kg intravenously initially and then repeated after 2, 6, 10, and 14 weeks. Adalimumab, a recombinant monoclonal antibody that binds to TNF receptor sites, and is given at a dosage of 40 mg subcutaneously every other week. Each drug produces substantial improvement in more than 60% of patients. Each is usually very well tolerated. Minor irritation at injection sites is the most common side effect of etanercept and adalimumab. Rarely, nonrecurrent leukopenia develops in patients. The use of TNF blockers reduces the need for prednisone, thereby avoiding or attenuating some of the major side effects of corticosteroids. TNF plays a physiologic role in combating many types of infection; TNF inhibitors have been associated with an increased risk of certain opportunistic infections, such as tuberculosis. Screening for latent tuberculosis (see Chapter 9) is now recommended before the initiation of TNF blockers. It is prudent to suspend TNF blockers when a fever or other manifestations of a clinically important infection develops in a patient. Demyelinating neurologic complications that resemble multiple sclerosis have been reported rarely in patients taking TNF inhibitors, but the true magnitude of this risk likely quite small has not been determined with precision. Contrary to expectation, TNF inhibitors were not effective in the treatment of congestive heart failure. The use of infliximab, in fact, was associated with increased morbidity in a congestive heart failure trial. Consequently, TNF inhibitors should be used with extreme caution in patients with congestive heart failure. Infliximab can rarely cause anaphylaxis and induce anti-DNA antibodies (but rarely clinically evident SLE). A final concern about TNF inhibitors is the expense, which is more than $10,000 per year.
3. Antimalarials
Hydroxychloroquine sulfate is the antimalarial agent most often used against rheumatoid arthritis. Monotherapy with hydroxychloroquine should be reserved for patients with mild disease because only 25 50% will respond and in some of those cases only after 3 6 months of therapy. Hydroxychloroquine is often used in combination with other conventional DMARDs, particularly methotrexate and sulfasalazine. The advantage of hydroxychloroquine is its comparatively low toxicity, especially at a dosage of 200 400 mg/d orally (not to exceed 6.5 mg/kg/d). The most important reaction, pigmentary retinitis causing visual loss, is rare at this dose. Ophthalmologic examinations every 6 12 months are required when this drug is used for long-term therapy. Other reactions include neuropathies and myopathies of both skeletal and cardiac muscle, which usually improve when the drug is withdrawn.
4. Corticosteroids
These drugs usually produce an immediate and dramatic anti-inflammatory effect in rheumatoid arthritis, and they may be able to slow the rate of bony destruction. However, their multitudinous side effects greatly limit their long-term use, particularly in doses > 5.0 7.5 mg/d orally of prednisone.
Corticosteroids may be used on a short-term basis to tide patients over acute disabling episodes, to facilitate other treatment measures (eg, physical therapy), or to manage serious extra-articular manifestations (eg, pericarditis, necrotizing scleritis). They may also be indicated for active and progressive disease that does not respond favorably to conservative management and when there are contraindications to or therapeutic failure of methotrexate, gold salts, or other disease-modifying agents.
No more than 10 mg of prednisone or equivalent per day is appropriate for articular disease. Many patients do reasonably well on 5 7.5 mg daily. (The use of 1 mg tablets, to facilitate doses of < 5 mg/d, is encouraged.) When the corticosteroids are to be discontinued, they should be tapered gradually on a planned schedule appropriate to the duration of treatment. All patients receiving long-term corticosteroid therapy should take measures to prevent osteoporosis.
Intra-articular corticosteroids may be helpful if one or two joints are the chief source of difficulty. Intra-articular triamcinolone, 10 40 mg depending on the size of the joint to be injected, may be given for symptomatic relief, but not more than four times a year.
5. Sulfasalazine
This drug is a second-line agent for rheumatoid arthritis. It is usually introduced at a dosage of 0.5 g orally twice daily and then increased each week by 0.5 g until the patient improves or the daily dose reaches 3 g. Side effects, particularly neutropenia and thrombocytopenia, occur in 10 25% and are serious in 2 5%. Sulfasalazine also causes hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency, so a G6PD level should be checked before initiating sulfasalazine. Patients taking sulfasalazine should have complete blood counts monitored
P.852
every 2 4 weeks for the first 3 months, then every 3 months.
6. Leflunomide
Leflunomide, a pyrimidine synthesis inhibitor, is also FDA-approved for treatment of rheumatoid arthritis. It can be used alone or combined with methotrexate. Administration is started at a dosage of 100 mg/d orally for 3 days followed by a maintenance dosage of 20 mg daily. The most frequent side effects are diarrhea, rash, reversible alopecia, and hepatotoxicity. Some patients experience dramatic unexplained weight loss. The drug is carcinogenic, teratogenic, and has a half-life of 2 weeks. Thus, it is contraindicated in premenopausal women or in men who wish to father children.
7. Minocycline
Minocycline is more effective than placebo for rheumatoid arthritis. It is reserved for early, mild cases, since its efficacy is modest, and it works better during the first year of rheumatoid arthritis. The mechanism of action is not clear, but tetracyclines do have anti-inflammatory properties, including the ability to inhibit destructive enzymes such as collagenase. The dosage of minocycline is 200 mg daily. Adverse effects are uncommon except for dizziness, which occurs in about 10%.
8. Combination therapy
Certain combinations of two or more DMARDs have been shown to be effective in treating rheumatoid arthritis. The most common combination therapy has been methotrexate with one of the anti-TNF agents. Combination therapy has been traditionally reserved for patients who have not responded adequately to individual agents. Recent studies suggest that combination therapy produces superior results to sequential monotherapy. If the benefits of combination therapy and the long-term safety of biologic agents are confirmed, then combination therapy will become the initial treatment of choice for patients with rheumatoid arthritis.
D. Other Therapies
Anakinra, a recombinant form of human IL-1 receptor antagonist, may be given to adult patients who have failed one or more DMARDs. Anakinra can be used alone or in combination with DMARDs other than TNF-blocking agents. This drug must be administered daily by subcutaneous injection. The efficacy of anakinra is generally less than that of TNF inhibitors. Because anakinra has been associated with an increased incidence of serious infection, it should be discontinued whenever suggestive symptoms develop.
Experiments have documented the efficacy of blocking T cell activation with proteins that prevent the interaction of T cells and antigen presenting cells. Abatacept, a recombinant protein made by fusing a fragment of the Fc domain of human IgG with the extracellular domain of a T cell inhibitory molecule (CTLA4), is a new medication based on this approach that has received FDA approval for use in rheumatoid arthritis. Finally, clinical trials of therapies designed to deplete B cells in rheumatoid arthritis are under way.
E. Surgical Measures
See section on Some Orthopedic Procedures for Arthritic Joints at the end of this chapter.
Course & Prognosis
Determining the best initial treatment is difficult because patients in whom rheumatoid arthritis is suspected can follow two widely divergent courses. Of all patients who present with polyarthritis that appears to be (but probably is not) rheumatoid arthritis, 50 75% experience remission within 2 years. These patients are often negative for antibody to cyclic citrullinated peptide or rheumatoid factor, have good functional status even during disease activity, and are commonly seen in community practices but rarely in academic centers. Clearly, conservative therapy makes good sense for this patient population.
For patients whose joint symptoms persist beyond 2 years, the outcome is not so favorable. Patients in this group are known to have shorter longevity, dying on average 10 15 years earlier than people without rheumatoid arthritis. The impact of more effective therapies, such as the combination of methotrexate and biologic agents, on these data is not clear. The most common causes of death are infection, respiratory or renal failure, and gastrointestinal disease, many of which can be attributed in large part to complications of conventional DMARD use. In recent years, there has been a growing appreciation of the impact of cardiovascular mortality in rheumatoid arthritis, a complication likely associated with the disease process itself as well as the adverse effects of treatment. Factors that identify those at particular risk for early death include positive rheumatoid factor, poor functional status, more than 30 inflamed joints, and extra-articular manifestations (eg, rheumatoid lung disease). These patients, then, need aggressive therapy early, since extensive bone damage can occur during the first 2 years.
Since some patients with polyarthritis will remit and others will sustain substantial damage within the first 2 years, a guidepost for early decisions about aggressive treatment is needed. Patients who have polyarthritis lasting more than 12 weeks and who are seropositive for rheumatoid factor and cyclic citrullinated peptide are those at greatest risk for having persistent disease and are appropriate candidates for aggressive therapy. Although genetic risk factors for developing rheumatoid arthritis have been identified, it has not been possible in this way to consistently identify those patients needing aggressive therapy.
Edwards JC et al: Efficacy of B cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med 2004;350:2572.
Genovese MC et al: Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med 2005;353:1114.
Goekoop-Ruiterman YPM et al: Clinical and radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): a randomized, controlled trial. Arthritis Rheum 2005;52:3381.
P.853
O'Dell JR: Therapeutic strategies for rheumatoid arthritis. N Engl J Med 2004;350:2591.
Quinn MA et al: Very early treatment with infliximab in addition to methotrexate in early, poor-prognosis rheumatoid arthritis. Arthritis Rheum 2005;52:27.
Raza K et al: Predictive value of antibodies to cyclic citrullinated peptide in patients with very early inflammatory arthritis. J Rheumatol 2005;32:231.
Wassenberg S et al: Very low-dose prednisolone in early rheumatoid arthritis retards radiographic progression over two years: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum 2005;52:3371.
Adult Still'S Disease
Still's disease is considered a variant of rheumatoid arthritis in which high spiking fevers are much more prominent, especially at the outset, than arthritis. This syndrome also occurs in adults. Most adults are in their 20s or 30s; onset after age 60 is rare. The fever is dramatic, often spiking to 40 C, associated with sweats and chills, and then plunging to several degrees below normal. Many patients initially complain of sore throat. An evanescent salmon-colored nonpruritic rash, chiefly on the chest and abdomen, is a characteristic feature. However, the rash can easily be missed since it often appears only with the fever spike. Many patients also have lymphadenopathy and pericardial effusions. Joint symptoms are mild or absent in the beginning, but a destructive arthritis, especially of the wrists, may develop months later. Anemia and leukocytosis, with white blood counts sometimes exceeding 40,000/mcL, are the rule. Ferritin levels are exceptionally high (> 3000 mg/mL) in more than 70% of cases of adult Still's disease for reasons that are not clear. A low percentage (< 20%) of serum ferritin that is glycosylated may be even more specific for adult Still's disease. Although the diagnosis requires exclusion of other causes of fever, the diagnosis of adult Still's disease is strongly suggested by the fever pattern, sore throat, and the classic rash. About half of the patients respond to high-dose aspirin (eg, 1 g three times orally daily) or other NSAIDs, and half require prednisone, sometimes in doses greater than 60 mg/d orally. TNF inhibitors may be helpful for some patients with refractory adult Still's disease, but most patients treated with these agents achieve only partial remissions. More complete and dramatic responses have been achieved with the IL-1 receptor antagonist anakinra.
Fautrel B et al: Tumor necrosis factor-alpha blocking agents in refractory adult Still's disease: An observational study of 20 cases. Ann Rheum Dis 2005;64:262.
Fitzgerald AA et al: Rapid responses to anakinra in patients with refractory adult-onset Still's disease. Arthritis Rheum 2005; 52:1794.
Systemic Lupus Erythematosus
![]() Essentials of Diagnosis
Essentials of Diagnosis
Occurs mainly in young women.
Rash over areas exposed to sunlight.
Joint symptoms in 90% of patients. Multiple system involvement.
Depression of hemoglobin, white blood cells, platelets.
Glomerulonephritis, central nervous system disease, and complications of antiphospholipid antibodies are major sources of disease morbidity.
Serologic findings: antinuclear antibodies (100%), anti-native DNA antibodies (approximately two-thirds), and low serum complement levels (particularly during disease flares).
General Considerations
SLE is an inflammatory autoimmune disorder that may affect multiple organ systems. Many of its clinical manifestations are secondary to the trapping of antigen-antibody complexes in capillaries of visceral structures or to autoantibody-mediated destruction of host cells (eg, thrombocytopenia). The clinical course is marked by spontaneous remission and relapses. The severity may vary from a mild episodic disorder to a rapidly fulminant, life-threatening illness.
The prevalence of SLE is influenced by many factors, including gender, race, and genetic inheritance. About 85% of patients are women. Sex hormones appear to play some role; most cases develop after menarche and before menopause. Among older individuals, the gender distribution is more equal. Race is also a factor, as SLE occurs in 1:1000 white women but in 1:250 black women. Familial occurrence of SLE has been repeatedly documented, and the disorder is concordant in 25 70% of identical twins. If a mother has SLE, her daughters' risks of developing the disease are 1:40 and her sons' risks are 1:250. Aggregation of serologic abnormalities (positive antinuclear antibody) is seen in asymptomatic family members, and the prevalence of other rheumatic diseases is increased among close relatives of patients. The importance of specific genes in SLE is emphasized by the high frequency of certain HLA haplotypes, especially DR2 and DR3, and null complement alleles. Genes that regulate programmed cell death (apoptosis) also appear to be important in the pathogenesis of SLE.
Before making a diagnosis of SLE, it is imperative to ascertain that the condition has not been induced by a drug. A host of pharmacologic agents have been implicated as causing a lupus-like syndrome, but only a few cause the disorder with appreciable frequency (Table 20-8). Procainamide, hydralazine, and isoniazid are the best-studied drugs. While antinuclear antibody tests and other serologic findings become positive in many persons receiving these agents, clinical manifestations occur in only a few.
Table 20-8. Drugs associated with lupus erythematosus. | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
Four features of drug-induced lupus separate it from SLE: (1) the sex ratio is nearly equal; (2) nephritis and central nervous system features are not ordinarily
P.854
present; (3) hypocomplementemia and antibodies to native DNA are absent; and (4) the clinical features and most laboratory abnormalities often revert toward normal when the offending drug is withdrawn.
The diagnosis of SLE should be suspected in patients having a multisystem disease with serologic positivity (eg, antinuclear antibody, false-positive serologic test for syphilis). Differential diagnosis includes rheumatoid arthritis, systemic vasculitis, scleroderma, inflammatory myopathies, viral hepatitis, sarcoidosis, acute drug reactions, and drug-induced lupus.
The diagnosis of SLE can be made with reasonable probability if 4 of the 11 criteria set forth in Table 20-9 are met. These criteria, developed as guidelines for the inclusion of patients in research studies, do not supplant clinical judgment in the diagnosis of SLE.
Clinical Findings
A. Symptoms and Signs
The systemic features include fever, anorexia, malaise, and weight loss. Most patients have skin lesions at some time; the characteristic butterfly (malar) rash affects fewer than half of patients. Other cutaneous manifestations are discoid lupus, typical fingertip lesions, periungual erythema, nail fold infarcts, and splinter hemorrhages. Alopecia is common. Mucous membrane lesions tend to occur during periods of exacerbation. Raynaud's phenomenon, present in about 20% of patients, often antedates other features of the disease.
Joint symptoms, with or without active synovitis, occur in over 90% of patients and are often the earliest manifestation. The arthritis is seldom deforming; erosive changes are almost never noted on radiographs. Subcutaneous nodules are rare.
Ocular manifestations include conjunctivitis, photophobia, transient or permanent monocular blindness, and blurring of vision. Cotton-wool spots on the retina (cytoid bodies) represent degeneration of nerve fibers due to occlusion of retinal blood vessels.
Pleurisy, pleural effusion, bronchopneumonia, and pneumonitis are frequent. Restrictive lung disease is often demonstrated. Alveolar hemorrhage is rare, but potentially life-threatening.
The pericardium is affected in the majority of patients. Cardiac failure may result from myocarditis and hypertension. Cardiac arrhythmias are common. Atypical verrucous endocarditis of Libman-Sacks is usually clinically silent but occasionally can produce acute or chronic valvular incompetence most commonly mitral regurgitation and can serve as a source of emboli.
Mesenteric vasculitis occasionally occurs in SLE and may closely resemble polyarteritis nodosa, including
P.855
the presence of aneurysms in medium-sized blood vessels. Abdominal pain (particularly postprandial), ileus, peritonitis, and perforation may result.
Table 20-9. Criteria for the classification of SLE. (A patient is classified as having SLE if any 4 or more of 11 criteria are met.) | ||
|---|---|---|
|
Table 20-10. Frequency (%) of autoantibodies in rheumatic diseases. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Neurologic complications of SLE include psychosis, organic brain syndrome, seizures, peripheral and cranial neuropathies, transverse myelitis, and strokes. Severe depression and psychosis are sometimes exacerbated by the administration of large doses of corticosteroids. Several forms of glomerulonephritis may occur, including mesangial, focal proliferative, diffuse proliferative, and membranous (see Chapter 22). Some patients may also have interstitial nephritis. With appropriate therapy, the survival rate even for patients with serious renal disease (proliferative glomerulonephritis) is favorable, albeit a substantial portion of patients with severe lupus nephritis still eventually require renal replacement therapy.
B. Laboratory Findings
(Tables 20-10, 20-11, and 19-2.) SLE is characterized by the production of many different autoantibodies, some of which produce specific laboratory abnormalities (eg, hemolytic anemia). Antinuclear antibody tests are sensitive but not specific for SLE ie, they are positive in virtually all patients with lupus but are positive also in many patients with nonlupus conditions such as rheumatoid arthritis, various forms of hepatitis, and interstitial lung disease. Antibodies to double-stranded DNA and to Sm are specific for SLE but not sensitive, since they are present in only 60% and 30% of patients, respectively. Depressed serum complement a finding suggestive of disease activity often returns toward normal in remission. Anti-double-stranded DNA antibody levels also correlate with disease activity in some patients; anti-Sm levels do not.
Three types of antiphospholipid antibodies occur (Table 20-11). The first causes the biologic false-positive
P.856
tests for syphilis; the second is the lupus anticoagulant, which despite its name is a risk factor for venous and arterial thrombosis and miscarriage. It is most commonly identified by prolongation of the activated partial thromboplastin time, an in vitro phenomenon related to antibody reacting to phospholipid in the test materials. Anticardiolipin antibodies are the third type of antiphospholipid antibodies. In many cases, the anticardiolipin antibody appears to be directed at a serum cofactor ( 2-glycoprotein-I) rather than at phospholipid itself. Abnormality of urinary sediment is almost always found in association with renal lesions. Showers of red blood cells, with or without casts, and proteinuria (varying from mild to nephrotic range) are frequent during exacerbation of the disease; these usually abate with remission.
Table 20-11. Frequency of laboratory abnormalities in systemic lupus erythematosus. | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Treatment
Patient education and emotional support, as described for rheumatoid arthritis, are especially important for patients with lupus. Patients with drug-induced SLE usually respond to withdrawal of the offending agent. Since the various manifestations of idiopathic SLE affect prognosis differently and since SLE activity often waxes and wanes, drug therapy both the choice of agents and the intensity of their use must be tailored to match disease severity. Patients with photosensitivity should be cautioned against sun exposure and should apply a protective lotion to the skin while out of doors. Skin lesions often respond to the local administration of corticosteroids. Minor joint symptoms can usually be alleviated by rest and NSAIDs.
Antimalarials (hydroxychloroquine) may be helpful in treating lupus rashes or joint symptoms that do not respond to NSAIDs. When these are used, the dose should not exceed 400 mg/d ( 6.5 mg/kg/d), and biannual monitoring for retinal changes is recommended. Drug-induced neuropathy and myopathy may be erroneously ascribed to the underlying disease. The androgenic corticosteroid danazol may be effective therapy for thrombocytopenia not responsive to corticosteroids. Dehydroepiandrosterone (DHEA) has a therapeutic role comparable to that of the antimalarial agents in the treatment of SLE, but its side effects (particularly acne) are troubling to some patients.
Corticosteroids are required for the control of certain complications. (Systemic corticosteroids are not usually given for minor arthritis, skin rash, leukopenia, or the anemia associated with chronic disease.) Glomerulonephritis, hemolytic anemia, pericarditis or myocarditis, alveolar hemorrhage, central nervous system involvement, and thrombotic thrombocytopenic purpura all require corticosteroid treatment and often other interventions as well. Forty to 60 mg of oral prednisone is often needed initially; however, the lowest dose of corticosteroid that controls the condition should be used. Central nervous system lupus may require higher doses of corticosteroids than are usually given; however, corticosteroid psychosis may mimic lupus cerebritis, in which case reduced doses are appropriate. Immunosuppressive agents such as cyclophosphamide, mycophenolate mofetil, and azathioprine are used in cases resistant to corticosteroids. Treatment of severe lupus nephritis includes an induction phase and a maintenance phase. Cyclophosphamide, which improves renal survival but not patient survival, was for many years the standard treatment for both phases of lupus nephritis. More recently, mycophenolate mofetil, which appears to be more effective and less toxic than cyclophosphamide, is emerging as the treatment of choice for many patients with lupus nephritis. Very close follow-up is needed to watch for potential side effects when immunosuppressants are given; these agents should be administered by physicians experienced in their use. When cyclophosphamide is required, gonadotropin-releasing hormone analogs can be given to protect a woman against the risk of premature ovarian failure. For patients with the antiphospholipid syndrome the presence of antiphospholipid antibodies and compatible clinical events anticoagulation is the treatment of choice. Moderate intensive anticoagulation with warfarin to achieve an INR of 2.0 3.0 is as effective as more intensive regimens. Pregnant patients with recurrent fetal loss associated with antiphospholipid antibodies should be treated with low-molecular-weight heparin plus aspirin.
Course & Prognosis
The prognosis for patients with systemic lupus appears to be considerably better than older reports implied. From both community settings and university centers, 10-year survival rates exceeding 85% are routine. In most patients, the illness pursues a relapsing and remitting course. Corticosteroids, often needed in doses of 40 mg/d or more during severe flares, can usually be tapered to low doses (5 10 mg/d) when the disease is inactive. However, there are some in whom the disease pursues a virulent course, leading to serious impairment of vital structures such as lung, heart, brain, or kidneys, and the disease may lead to death. With improved control of lupus activity and with increasing use of corticosteroids and immunosuppressive drugs, the mortality and morbidity patterns in lupus have changed. Mortality in SLE shows a bimodal pattern. In the early years after diagnosis, infections especially with opportunistic organisms are the leading cause of death, followed by active SLE, chiefly due to renal or central nervous system disease. In later years, accelerated atherosclerosis, attributed in part to corticosteroid use, becomes a major cause of death. Indeed, the incidence of myocardial infarction is five times higher in persons with SLE than in the general population. Therefore, it is especially important for SLE patients to avoid smoking and to minimize other conventional risk factors for atherosclerosis (eg, hypercholesterolemia, hypertension, obesity, and inactivity). Patients with SLE should receive influenza vaccination every year and pneumococcal vaccination every 5
P.857
years. Since some reports indicate that SLE patients have a higher risk of developing malignancy, preventive cancer screening recommendations should be followed assiduously. With more patients living longer, it has become evident that avascular necrosis of bone, affecting most commonly the hips and knees, is responsible for substantial morbidity. Still, the outlook for most patients with SLE has become increasingly favorable.
Arbuckle MR et al: Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med 2003;349:1499.
Asanuma Y et al: Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003;349:2407.
Ginzler EM: Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005;353:2219.
Petri M et al: Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005;353:2550.
Somers EC et al: Use of a gonadotropin-releasing hormone analog for protection against premature ovarian failure during cyclophosphamide therapy in women with severe lupus. Arthritis Rheum 2005;52:2761.
Antiphospholipid Syndrome
![]() Essentials of Diagnosis
Essentials of Diagnosis
Hypercoagulability, with recurrent thromboses in either the venous or arterial circulation.
Thrombocytopenia is common.
Pregnancy complications, specifically pregnancy losses after the first trimester.
Lifelong anticoagulation with warfarin is recommended currently for patients with serious complications of this syndrome, as recurrent events are common.
General Considerations
A primary antiphospholipid antibody syndrome (APS) is diagnosed in patients who have recurrent venous or arterial occlusions, recurrent fetal loss, or thrombocytopenia in the presence of antiphospholipid antibodies but not other features of SLE. In fewer than 1% of patients with antiphospholipid antibodies, a potentially devastating syndrome known as the catastrophic antiphospholipid syndrome occurs, leading to diffuse thromboses, thrombotic microangiopathy, and multiorgan system failure.
Clinical Findings
A. Symptoms and Signs
Patients are often asymptomatic until suffering a thrombotic complication of this syndrome. Thrombotic events may occur in either the arterial or venous circulations. Thus, deep venous thromboses, pulmonary emboli, cerebrovascular accidents are typical clinical events among patients with the APS. Budd-Chiari syndrome, cerebral sinus vein thrombosis, myocardial or digital infarctions, and other thrombotic events also occur. A variety of other symptoms and signs are often attributed to the APS, including mental status changes, livedo reticularis, skin ulcers, microangiopathic nephropathy, and cardiac valvular dysfunction typically mitral regurgitation that may mimic Libman-Sacks endocarditis. Livedo reticularis is strongly associated with the subset of patients with APS in whom arterial ischemic events develop.
B. Laboratory Findings
As noted in the discussion of SLE, three types of antiphospholipid antibody are believed to contribute to this syndrome: (1) anti-cardiolipin antibodies; (2) a lupus anticoagulant that prolongs certain coagulation tests (see below); and (3) an antibody associated with a biologic false-positive test for syphilis (see Table 10-11). Anti-cardiolipin antibodies are typically measured with enzyme immunoassays for either IgG or IgM. In general, IgG anti-cardiolipin antibodies are believed to be more pathologic than IgM. A clue to the presence of a lupus anticoagulant, which may occur in individuals who do not have SLE, may be detected by a prolongation of the partial thromboplastin time (which, paradoxically, is associated with a thrombotic tendency rather than a bleeding risk). A finding more sensitive for a lupus anticoagulant, however, is prolongation of a specialized coagulation assay known as the Russell viper venom time (RVVT). In the presence of a lupus anticoagulant, the RVVT is prolonged and does not correct with mixing studies. With the last type of antiphospholipid antibody, the patient has a positive rapid plasma reagin (RPR), but negative specific anti-treponemal assays.
Differential Diagnosis
The exclusion of other autoimmune disorders, particularly those in the SLE spectrum, is essential, as such disorders may be associated with additional complications requiring alternative treatments. Other genetic or acquired conditions associated with hypercoagulability such as protein C, protein S, factor V Leiden, and antithrombin III deficiency should be excluded. The thrombocytopenia associated with APS must be distinguished from immune thrombocytopenic purpura. Catastrophic APS has a broad differential, including sepsis, pulmonary-renal syndromes, systemic vasculitis, disseminated intravascular coagulation, and thrombotic thrombocytopenic purpura.
Treatment
Present recommendations for anticoagulation are to treat patients with warfarin to maintain an INR of 2.0 3.0. Patients who have recurrent thrombotic events on
P.858
this level of anticoagulation may require higher INRs (> 3.0), but the bleeding risk increases substantially with this degree of anticoagulation. Guidelines indicate that patients with APS should be treated with anticoagulation for life. Because of the teratogenic effects of warfarin, subcutaneous heparin and baby aspirin is the usual approach to prevent pregnancy complications in women with APS. In patients with catastrophic APS, a three-pronged approach is taken in the acute setting: intravenous heparin, high doses of corticosteroids, and either intravenous immune globulin or plasmapheresis.
Finazzi G et al: A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J Thromb Haemost 2005;3:848.
Frances C et al: Dermatologic manifestations of the antiphospholipid syndrome: two hundred consecutive cases. Arthritis Rheum 2005;52:1785.
Trilolo G et al: Randomized study of subcutaneous low molecular weight heparin plus aspirin versus intravenous immunoglobulin in the treatment of recurrent fetal loss associated with antiphospholipid antibodies. Arthritis Rheum 2003;48:728.
Turiel M et al: Five-year follow-up by transesophageal echocardiographic studies in primary antiphospholipid syndrome. Am J Cardiol 2005;96:574.
Systemic Sclerosis (Scleroderma)
![]() Essentials of Diagnosis
Essentials of Diagnosis
Widespread thickening of skin in diffuse systemic sclerosis, including truncal involvement, with areas of increased pigmentation and depigmentation.
Thickening confined to the face and neck, distal arms, feet and hands in limited systemic sclerosis.
Raynaud's phenomenon and antinuclear antibodies are present in virtually all patients.
Systemic features of dysphagia, hypomotility of gastrointestinal tract, pulmonary fibrosis, and cardiac and renal involvement.
General Considerations
Systemic sclerosis is a chronic disorder characterized by diffuse fibrosis of the skin and internal organs. The causes of systemic sclerosis are not known, but autoimmunity, fibroblast dysregulation, graft-versus-host disease from fetal lymphocytes retained in the maternal circulation, and occupational exposure to silica have been implicated. Symptoms usually appear in the third to fifth decades, and women are affected two to three times as frequently as men.
Two forms of systemic sclerosis are generally recognized: limited (80% of patients) and diffuse (20%). Two bedside clues help distinguish the two subsets. First, in the CREST syndrome (representing calcinosis cutis, Raynaud's phenomenon, esophageal motility disorder, sclerodactyly, and telangiestasia), hardening of the skin (scleroderma) is limited to the face and hands. In contrast, in diffuse scleroderma, the skin changes also involve the trunk and proximal extremities. Second, tendon friction rubs, especially frequent over the wrists, ankles, and knees, occur uniquely (but not universally) in diffuse scleroderma. In general, patients with CREST syndrome have better outcomes than those with diffuse disease, largely because renal failure or interstitial lung disease rarely develops in patients with limited disease. Cardiac disease is also more characteristic of diffuse systemic sclerosis. Curiously, however, patients with limited disease are more susceptible to digital ischemia, leading to finger loss and to life-threatening pulmonary hypertension. Gastrointestinal involvement (gut hypomotility) may occur in both forms of systemic sclerosis, leading to such potentially serious complications as pseudoobstruction.
Clinical Findings
A. Symptoms and Signs
Most frequently, the disease makes its appearance in the skin, although visceral involvement may precede the cutaneous features. Polyarthralgia and Raynaud's phenomenon (present in 90% of patients) are early manifestations. Subcutaneous edema, fever, and malaise are common. With time the skin becomes thickened and hidebound, with loss of normal folds. Telangiectasia, pigmentation, and depigmentation are characteristic. Ulceration about the fingertips and subcutaneous calcification are seen. Dysphagia due to esophageal dysfunction is common and results from abnormalities in motility and later from fibrosis. Fibrosis and atrophy of the gastrointestinal tract cause hypomotility, and malabsorption results from bacterial overgrowth. Large-mouthed diverticuli occur in the jejunum, ileum, and colon. Diffuse pulmonary fibrosis and pulmonary vascular disease are reflected in restrictive lung physiology and low diffusing capacities. Cardiac abnormalities include pericarditis, heart block, myocardial fibrosis, and right heart failure secondary to pulmonary hypertension. Scleroderma renal crisis, resulting from obstruction of smaller renal blood vessels, is a marker for a poor outcome even though many cases can now be treated effectively with angiotensin-converting enzyme inhibitors.
B. Laboratory Findings
Mild anemia is often present. Among patients with severe renal disease, the peripheral blood smear can show findings consistent with a microangiopathic hemolytic anemia (because of mechanical damage to red cells from diseased small vessels). Elevation of the ESR is unusual. Proteinuria and cylindruria appear in association with renal involvement. Antinuclear antibody
P.859
tests are nearly always positive, frequently in high titers (Tables 20-10 and 19-2). The scleroderma antibody (SCL-70) directed against topoisomerase III is found in one-third of patients with diffuse systemic sclerosis and in 20% of those with CREST syndrome; an anticentromere antibody is seen in 50% of those with CREST syndrome and in 1% of individuals with diffuse systemic sclerosis (Tables 20-10 and 19-2). Although present in only a minority of patients with diffuse systemic sclerosis, anti-Scl-70 antibodies may portend a poor prognosis, with a high likelihood of serious internal organ involvement (eg, interstitial lung disease). Anticentromere antibodies are highly specific for limited systemic sclerosis, but they also occur occasionally in overlap syndromes. Other scleroderma autoantibodies are those directed against RNA polymerases (anti-RNAP I, II, III) and B23. Anti-RNAP antibodies are associated with diffuse skin changes, cardiac and renal involvement, and increased mortality. Antibodies to B23 are associated with pulmonary hypertension.
Differential Diagnosis
Several diseases with principally cutaneous manifestations may mimic systemic sclerosis in their skin findings. These disorders include morphea and limited systemic sclerosis, two diseases often categorized as forms of localized scleroderma. These disorders are generally limited to circumscribed areas of the skin and usually have excellent outcomes. Morphea is associated with lavender plaques, typically located over the trunk. Linear scleroderma may be associated with atrophy of underlying muscle and bone and may cause deformities that are both cosmetically and functionally disabling.
Eosinophilic fasciitis is a rare disorder presenting with skin changes that resemble diffuse systemic sclerosis. The inflammatory abnormalities, however, are limited to the fascia rather than the dermis and epidermis. Moreover, patients with eosinophilic fasciitis are distinguished from those with systemic scleroderma by the presence of peripheral blood eosinophilia, the absence of Raynaud's phenomenon, the good response to prednisone, and an association (in some cases) with paraproteinemias.
The eosinophilia-myalgia syndrome was first noted in patients who ingested L-tryptophan, an essential amino acid once sold as an over-the-counter remedy for insomnia and premenstrual symptoms (before removal from the market). Since banning of L-tryptophan sales, the eosinophilia-myalgia syndrome has essentially disappeared.
Treatment
Treatment of systemic sclerosis is symptomatic and supportive. There is no medication that is known to alter the underlying disease process. However, interventions for management of specific organ manifestations of this disease have improved substantially. Severe Raynaud's syndrome may respond to calcium channel blockers, eg, long-acting nifedipine, 30 120 mg/d orally, or to losartan, 50 mg/d orally. Intravenous iloprost, a prostacyclin analog that causes vasodilation and platelet inhibition, is moderately effective in healing digital ulcers. Patients with esophageal disease should take medications in liquid or crushed form. Esophageal reflux can be reduced and the risk of scarring diminished by avoidance of late-night meals and elevation of the head of the bed. In addition, proton pump inhibitors (eg, omeprazole, 20 40 mg/d orally), the only drugs that achieve near-complete inhibition of gastric acid production, are remarkably effective for refractory esophagitis. Patients with delayed gastric emptying maintain their weight better if they eat small, frequent meals and remain upright for at least 2 hours after eating. Octreotide, a somatostatin analog, helps some patients with bacterial overgrowth and pseudoobstruction. Malabsorption due to bacterial overgrowth also responds to antibiotics, eg, tetracycline, 500 mg four times orally daily. The hypertensive crises associated with systemic sclerosis renal crisis must be treated early and aggressively (in the hospital) with angiotensin-converting enzyme inhibitors, eg, captopril, 37.5 75 mg/d orally in three divided doses. Prednisone has little or no role in the treatment of scleroderma. Cyclophosphamide, a drug with many important side effects, may improve severe interstitial lung disease. Bosentan, an endothelin receptor antagonist, improves exercise capacity and cardiopulmonary hemodynamics in patients with pulmonary hypertension and helps prevent digital ulceration.
The 9-year survival rate in scleroderma averages approximately 40%. The prognosis tends to be worse in those with diffuse scleroderma, in blacks, in males, and in older patients. In most cases, death results from renal, cardiac, or pulmonary failure. Those persons in whom severe internal organ involvement does not develop in the first 3 years have a substantially better prognosis, with 72% surviving at least 9 years. Breast and lung cancer may be more common in patients with scleroderma.
Girgis RE et al: Long-term outcome of bosentan treatment in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with the scleroderma spectrum of diseases. J Heart Lung Transplant 2005;24:1626.
Herrick AL: Pathogenesis of Raynaud's phenomenon. Rheumatology (Oxford) 2005;44:587.
Ioannidis JP et al: Mortality in systemic sclerosis: an international meta-analysis of individual patient data. Am J Med 2005;118: 2.
Jimenez SA et al: Microchimerism and systemic sclerosis. Curr Opin Rheumatol 2005;17:86.
Korn JH et al: Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum 2004;50:3985.
Steen VD: Autoantibodies in systemic sclerosis. Semin Arthritis Rheum 2005;35:35.
P.860
Steen V et al: Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum 2003;48:516.
Idiopathic Inflammatory Myopathies (Polymyositis & Dermatomyositis)
![]() Essentials of Diagnosis
Essentials of Diagnosis
Bilateral proximal muscle weakness.
Characteristic cutaneous manifestations in dermatomyositis (Gottron's papules, heliotrope rash).
Diagnostic tests: elevated creatine kinase and other muscle enzymes, muscle biopsy, electromyography.
Increased risk of malignancy, particularly in adult dermatomyositis.
Inclusion body myositis can mimic polymyositis but is less responsive to treatment.
General Considerations
Polymyositis and dermatomyositis are systemic disorders of unknown cause whose principal manifestation is muscle weakness. Although their clinical presentations (aside from the presence of certain skin findings in dermatomyositis, some of which are pathognomonic) and treatments are similar, the two diseases are pathologically quite distinct. They affect persons of any age group, but the peak incidence is in the fifth and sixth decades of life. Women are affected twice as commonly as men, and the diseases (particularly polymyositis) also occur more often among blacks than whites. There is an increased risk of malignancy, especially in adult patients with dermatomyositis. Indeed, up to one patient in four with dermatomyositis has an occult malignancy. Malignancies may be evident at the time of presentation with the muscle disease but may not be detected until months afterward in some cases. Rare patients with dermatomyositis have skin disease without overt muscle involvement, a condition termed dermatomyositis sine myositis.
Clinical Findings
A. Symptoms and Signs
Polymyositis may begin abruptly, but the usual presentation is one of gradual and progressive muscle weakness. The weakness chiefly involves proximal muscle groups of the upper and lower extremities as well as the neck. Leg weakness (eg, difficulty in rising from a chair or climbing stairs) typically precedes arm symptoms. In contrast to myasthenia gravis, polymyositis and dermatomyositis do not cause facial or ocular muscle weakness. Pain and tenderness of affected muscles occur in one-fourth of cases, but these are rarely the chief complaints. About one-fourth of patients have dysphagia. In contrast to scleroderma, which affects the smooth muscle of the lower esophagus and can cause a sticking sensation below the sternum, polymyositis or dermatomyositis involves the striated muscles of the upper pharynx and can make initiation of deglutition difficult. Muscle atrophy and contractures occur as late complications of advanced disease. Clinically significant myocarditis is uncommon even though there is often creatine kinase-MB elevation. Patients who are bed-bound from myositis should be screened for respiratory muscle weakness that can be severe enough to cause CO2 retention and to require mechanical ventilation.
In dermatomyositis, the characteristic rash is dusky red and may appear in a malar distribution mimicking the classic rash of SLE. Facial erythema beyond the malar distribution is also characteristic of dermatomyositis. Erythema also occurs over other areas of the face, neck, shoulders, and upper chest and back ( shawl sign ). Periorbital edema and a purplish (heliotrope) suffusion over the eyelids are typical signs. Periungual erythema, dilations of nailbed capillaries, and scaly patches over the dorsum of PIP and MCP joints (Gottron's sign) are highly suggestive. Scalp involvement by dermatomyositis may mimic psoriasis. Infrequently, the cutaneous findings of this disease precede the muscle inflammation by weeks or months. Diagnosing polymyositis in patients over age 70 years can be difficult because weakness may be overlooked or attributed erroneously to idiopathic frailty. Polymyositis can remain undiagnosed or will be misdiagnosed as hepatitis because of elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels. A subset of patients with polymyositis and dermatomyositis develop the antisynthetase syndrome, a group of findings including inflammatory arthritis, Raynaud's phenomenon, interstitial lung disease, and often severe muscle disease associated with certain autoantibodies (eg, anti-Jo1 antibodies).
B. Laboratory Findings
Measurement of serum levels of muscle enzymes, especially creatine kinase and aldolase, is most useful in diagnosis and in assessment of disease activity. Anemia is uncommon. The ESR is not appreciably elevated in half of the patients. Rheumatoid factor is found in a minority of patients. Antinuclear antibodies are present in many patients, and anti-Jo-1 antibodies are seen in the subset of patients who have associated interstitial lung disease (Tables 20-10 and 19-2). Chest radiographs are usually normal, although interstitial fibrosis is occasionally seen. Electromyographic abnormalities consisting of polyphasic potentials, fibrillations, and high-frequency action potentials are helpful in establishing the diagnosis. None of the studies are specific. The search for an occult malignancy should begin with a history and
P.861
physical examination, supplemented with a complete blood count, comprehensive biochemical panel, serum protein electrophoresis, and urinalysis, and should include age- and risk-appropriate cancer screening tests. If these evaluations are unrevealing, more invasive or extensive laboratory evaluations are probably not cost-effective. No matter how extensive the initial screening, some malignancies will not become evident for months after the initial presentation.
C. Muscle Biopsy
Biopsy of clinically involved muscle is the only specific diagnostic test. The pathology findings in polymyositis and dermatomyositis are distinct. Although both include lymphoid inflammatory infiltrates, the findings in dermatomyositis are localized to perivascular regions and there is evidence of humoral and complement-mediated destruction of microvasculature associated with the muscle. In addition to its vascular orientation, the inflammatory infiltrate in dermatomyositis centers on the interfascicular septa and is located around, rather than in, muscle fascicles. A pathological hallmark of dermatomyositis is perifascicular atrophy. In contrast, the pathology of polymyositis characteristically includes endomysial infiltration of the inflammatory infiltrate. Owing to the sometimes patchy distribution of pathologic abnormalities, however, false-negative biopsies sometimes occur in both disorders.
Differential Diagnosis
Muscle inflammation may occur as a component of SLE, systemic sclerosis, Sj gren's syndrome, and overlap syndromes. In those cases, associated findings usually permit the precise diagnosis of the primary condition.
Inclusion body myositis, because of its tendency to mimic polymyositis, is a common cause of treatment-resistant polymyositis. In contrast to the epidemiologic features of polymyositis, however, the typical inclusion body myositis patient is white, male, and over the age of 50. The onset of inclusion body myositis is even more insidious than that of polymyositis or dermatomyositis (eg, occurring over years rather than months), and asymmetric distal motor weakness is common in inclusion body myositis. Creatine phosphokinase (CPK) levels in inclusion body myositis are often minimally elevated and are normal in 25%. Muscle biopsy shows characteristic intracellular vacuoles by light microscopy either tubular or filamentous inclusions in the nucleus or cytoplasm by electron microscopy. Inclusion body myositis is less likely to respond to therapy and is associated with characteristic pathologic findings evident on frozen section or electron microscopy.
Hyperthyroidism and hypothyroidism may both be associated with proximal muscle weakness. Hypothyroidism is associated also with elevations of CPK. Patients with polymyalgia rheumatica are over the age of 50 and in contrast to patients with polymyositis have pain but no objective weakness. Disorders of the peripheral and central nervous systems (eg, chronic inflammatory polyneuropathy, multiple sclerosis, myasthenia gravis, Eaton-Lambert disease, and amyotrophic lateral sclerosis) can produce weakness but are distinguished by characteristic symptoms and neurologic signs and often by distinctive electromyographic abnormalities. A number of systemic vasculitides (polyarteritis nodosa, microscopic polyangiitis, the Churg-Strauss syndrome, Wegener's granulomatosis, and mixed cryoglobulinemia) can produce profound weakness through vasculitic neuropathy. The muscle weakness associated with these disorders, however, is typically distal and asymmetric, at least in the early stages.
Many drugs, including corticosteroids, alcohol, clofibrate, penicillamine, tryptophan, and hydroxychloroquine, can produce proximal muscle weakness. Chronic use of colchicine at doses as low as 0.6 mg twice a day in patients with moderate renal insufficiency can produce a mixed neuropathy-myopathy that mimics polymyositis. The weakness and muscle enzyme elevation reverse with cessation of the drug. HMG-CoA reductase inhibitors, which are frequently used to treat hypercholesterolemia, can cause myopathy and rhabdomyolysis. Although only about 0.1% of patients taking a statin drug alone develop myopathy, concomitant administration of other drugs (especially gemfibrozil, cyclosporine, niacin, macrolide antibiotics, azole antifungals, and protease inhibitors) increases the risk. Statin-induced muscle inflammation resolves after cessation of these medications. Polymyositis can occur as a complication of HIV or HTLV-I infection and with zidovudine therapy as well.
Treatment
Most patients respond to corticosteroids. Often a daily dose of 40 60 mg or more of oral prednisone is required initially. The dose is then adjusted downward according to the response of sequentially observed serum levels of muscle enzymes. Long-term use of corticosteroids is often needed, and the disease may recur or reemerge when they are withdrawn. Patients with an associated neoplasm have a poor prognosis, although remission may follow treatment of the tumor; corticosteroids may or may not be effective in these patients. In patients resistant or intolerant to corticosteroids, therapy with methotrexate or azathioprine may be helpful. Intravenous immune globulin has also been shown to be effective for dermatomyositis resistant to prednisone. TNFs do not appear to be effective in the inflammatory myopathies. Mycophenolate mofetil (1.0 1.5 g twice daily) may be useful as a steroid-sparing agent. B cell depletion therapies eg, rituximab (either 375 mg/m2 administered intravenously once weekly for 4 weeks, or 1 g/wk intravenously for 2 weeks) are a promising avenue of current investigation.
Danko K et al: Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: a longitudinal study of 162 cases. Medicine 2004;83:35.
P.862
Edge JC et al: Mycophenolate mofetil as an effective corticosteroid-sparing therapy for recalcitrant dermatomyositis. Arch Dermatol 2006;142:65.
Troyanov Y et al: Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore) 2005;84:231.
Overlap (Or Mixed) Connective Tissue Disease
Many patients with symptoms and signs compatible with a connective tissue disease have features consistent with more than one type of rheumatic disease. Special attention has been drawn to patients who have overlapping features of SLE, systemic sclerosis, and inflammatory myopathy. Initially, these patients were thought to have a distinct entity ( mixed connective tissue disease ) defined by a specific autoantibody to ribonuclear protein (RNP). In many patients, the manifestations evolve to one predominant disease in time, such as scleroderma, and many patients with antibodies to RNP develop unequivocal SLE. Therefore, overlap connective tissue disease is the preferred designation for patients having features of different rheumatic diseases. Treatments are guided more by the distribution and severity of patients' organ system involvement than by therapies specific to these diverse syndromes.
Bodolay E et al: Five-year follow-up of 665 Hungarian patients with undifferentiated connective tissue disease (UCTD). Clin Exp Rheumatol 2003;21:313.
Wigley FM et al: The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health care level of community-based rheumatologists (the UNCOVER study). Arthritis Rheum 2005;52:2125.
Sj gren's Syndrome
![]() Essentials of Diagnosis
Essentials of Diagnosis
Ninety percent of patients are women; the average age is 50 years.
Dryness of eyes and dry mouth (sicca components) are the most common features; they occur alone or in association with rheumatoid arthritis or other connective tissue disease.
Rheumatoid factor and other autoantibodies common.
Increased incidence of lymphoma.
General Considerations
Sj gren's syndrome, an autoimmune disorder, is the result of chronic dysfunction of exocrine glands in many areas of the body. It is characterized by dryness of the eyes, mouth, and other areas covered by mucous membranes and is frequently associated with a rheumatic disease, most often rheumatoid arthritis. The disorder is predominantly a disease of women, in a ratio of 9:1, with greatest incidence between age 40 and 60 years.
Disorders with which Sj gren's syndrome is frequently associated include rheumatoid arthritis, SLE, primary biliary cirrhosis, scleroderma, polymyositis, Hashimoto's thyroiditis, polyarteritis, and interstitial pulmonary fibrosis. When Sj gren's syndrome occurs without rheumatoid arthritis, HLA-DR2 and -DR3 antigens are present with increased frequency.
Clinical Findings
A. Symptoms and Signs
Keratoconjunctivitis sicca results from inadequate tear production caused by lymphocyte and plasma cell infiltration of the lacrimal glands. Ocular symptoms are usually mild. Burning, itching, and the sensation of having a foreign body or a grain of sand in the eye occur commonly. For some patients, the initial manifestation is the inability to tolerate wearing contact lenses. Many patients with more severe ocular dryness notice ropy secretions across their eyes, especially in the morning. Photophobia may signal corneal ulceration resulting from severe dryness. For most patients, symptoms of dryness of the mouth (xerostomia) dominate those of dry eyes. Patients frequently complain of a cotton mouth sensation and difficulty swallowing foods, especially dry foods like crackers, unless they are washed down with liquids. The persistent oral dryness causes most patients to carry water bottles or other liquid dispensers from which they sip constantly. A few patients have such severe xerostomia that they have difficulty speaking. Persistent xerostomia results often in rampant dental carries; carries at the gum line strongly suggest Sj gren's syndrome. Some patients are most troubled by loss of taste and smell. Parotid enlargement, which may be chronic or relapsing, develops in one-third of patients. Desiccation may involve the nose, throat, larynx, bronchi, vagina, and skin.
Systemic manifestations include dysphagia, vasculitis, pleuritis, obstructive lung disease (in the absence of smoking), neuropsychiatric dysfunction (most commonly peripheral neuropathies), and pancreatitis; they may be related to the associated diseases noted above. Renal tubular acidosis (type I, distal) occurs in 20% of patients. Chronic interstitial nephritis, which may result in impaired renal function, may be seen. A glomerular lesion is rarely observed but may occur secondary to associated cryoglobulinemia.
B. Laboratory Findings
Laboratory findings include mild anemia, leukopenia, and eosinophilia. Polyclonal hypergammaglobulinemia, rheumatoid factor positivity (70%), and antinuclear antibodies (95%) are all common findings.
P.863
Antibodies against the cytoplasmic antigens SS-A and SS-B (also called Ro and La, respectively) are often present in Sj gren's syndrome and tend to correlate with the presence of extraglandular manifestations (Tables 20-10 and 19-2). Thyroid-associated autoimmunity is a common finding among patients with Sj gren's syndrome.
Useful ocular diagnostic tests include the Schirmer test, which measures the quantity of tears secreted. Lip biopsy, a simple procedure, is the only specific diagnostic technique and has minimal risk; if lymphoid foci are seen in accessory salivary glands, the diagnosis is confirmed. Biopsy of the parotid gland should be reserved for patients with atypical presentations such as unilateral gland enlargement.
Treatment & Prognosis
Treatment is symptomatic and supportive. Artificial tears applied frequently will relieve ocular symptoms and avert further desiccation. The mouth should be kept well lubricated. Sipping water frequently or using sugar-free gums and hard candies usually relieves dry mouth symptoms. Pilocarpine (5 mg orally four times daily) and the acetylcholine derivative cevimeline (30 mg orally three times daily) may improve xerostomia symptoms. Atropinic drugs and decongestants decrease salivary secretions and should be avoided. A program of oral hygiene, including fluoride treatment, is essential in order to preserve dentition. If there is an associated rheumatic disease, its systemic treatment is not altered by the presence of Sj gren's syndrome.
Although Sj gren's syndrome may compromise patients' quality of life significantly, the disease is usually consistent with a normal life span. Poor prognoses are influenced mainly by the presence of systemic features associated with underlying disorders, the development in some patients of lymphocytic vasculitis, the occurrence of a painful peripheral neuropathy, and the complication (in a minority of patients) of lymphoma. The patients (3 10% of the total Sj gren's population) at greatest risk for developing lymphoma are those with severe exocrine dysfunction, marked parotid gland enlargement, splenomegaly, vasculitis, peripheral neuropathy, anemia, and mixed monoclonal cryoglobulinemia.
Brito-Zeron P et al: Circulating monoclonal immunoglobulins in Sj gren syndrome: prevalence and clinical significance in 237 patients. Medicine (Baltimore) 2005;84:90.
Garcia-Carrasco M et al: Primary Sj gren syndrome: clinical and immunologic disease patterns in a cohort of 400 patients. Medicine (Baltimore) 2002;81:270.
Ono M et al: Therapeutic effect of cevimeline on dry eye in patients with Sj gren's syndrome: a randomized, double-blind clinical study. Am J Ophthalmol 2004;138:6.
Ramos-Casals M et al: Cutaneous vasculitis in primary Sjogren syndrome: classification and clinical significance of 52 patients. Medicine (Baltimore) 2004;83:96.
Rhabdomyolysis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Associated with crush injuries to muscle, prolonged immobility, drug toxicities, hypothermia, and other causes.
Massive acute elevations of muscle enzymes that peak quickly and usually resolve within days once the inciting injury has been identified and removed.
General Considerations
Defined strictly, rhabdomyolysis is necrosis of skeletal muscle and may be encountered in a wide variety of clinical settings, alone or in concert with other disorders of muscle. When the term rhabdomyolysis is used without being otherwise defined, healthcare providers ordinarily think of the syndrome of crush injury to muscle, associated with myoglobinuria, renal insufficiency, markedly elevated creatine kinase levels and, frequently, multiorgan failure as a consequence of other complications of the trauma. Renal insufficiency in myoglobinuria is caused by tubular injury resulting from excessive quantities of filtered myoglobin. This complication is nearly always associated with hypovolemia. Experimental models of severe rhabdomyolysis in which blood volume and pressure are maintained ordinarily are not associated with acute tubular necrosis. From a practical point of view, however, many patients who suffer crush injuries are indeed volume-contracted, and oliguric renal failure is encountered routinely.
In addition to crush injuries, prolonged immobility, particularly after drug overdose or intoxication and commonly associated with exposure hypothermia, may be associated with rhabdomyolysis. Often there is little evidence for muscle injury on external examination of these patients and specifically, neither myalgia nor myopathy presents. The clue to muscle necrosis in such individuals may be a urinary dipstick testing positive for blood in the absence of red cells in the sediment. This false-positive finding is due to myoglobinuria, which results in a positive reading for blood. Such an abnormality is investigated by serum creatine kinase determination. Other studies elevated in rhabdomyolysis include ALT and lactate dehydrogenase (LDH) and once again, these studies may be obtained for other reasons, such as suspected liver disease or hemolysis. When disproportionately elevated, it is prudent to establish that they are not of muscle origin by confirming them with creatine kinase determination.
A number of other causes of rhabdomyolysis are encountered. Statins, agents used commonly to treat
P.864
hyperlipidemia, are common offenders (see above). The presence of compromised renal and hepatic function, diabetes, and hypothyroidism as well as concomitant use of other medications all increase the risk of rhabdomyolysis in those patients taking statins. Both acute alcohol intoxication and even intramuscular injections may cause some elevation of creatine kinase.
Treatment
Vigorous fluid resuscitation (4 6 L/d, with careful monitoring for fluid overload), mannitol (100 mg/d), and urine alkalinization are suggested early in the course, but definitive evidence for the efficacy of these measures is lacking. On occasion, oliguric tubular necrosis may be converted to a nonoliguric variety, and though the prognosis for recovery of renal function and mortality is the same many clinicians believe it is easier to care for nonoliguric disease because hyperkalemia and pulmonary edema are less important concerns. Myopathic complications of statins usually resolve within several weeks of discontinuing the drug.
Allison RC et al: The other medical causes of rhabdomyolysis. Am J Med Sci 2003;326:79.
Thompson PD et al: Statin-associated myopathy. JAMA 2003;289:1681.
Vasculitis Syndromes
Vasculitis is a heterogeneous group of disorders characterized by the pathologic features of inflammation within the walls of affected blood vessels. The major forms of primary systemic vasculitis are listed in Table 20-12. The first consideration in classifying cases of vasculitis is the size of the major vessels involved: large, medium, or small. The presence of the clinical signs and symptoms shown in Table 20-13 help distinguish among these three groups. After determining the size of the major vessels involved, other issues that contribute to the classification include the following:
Does the process involve arteries, veins, or both?
What are the patient's demographic characteristics (age, gender, ethnicity, smoking status)?
Which organs are involved?
Is there evidence of immune complex deposition?
Is there granulomatous inflammation on tissue biopsy?
Are antineutrophil cytoplasmic antibodies (ANCA) present?
In addition to the disorders considered to be primary vasculitides, there are also multiple forms of vasculitis that are associated with other known underlying conditions. These secondary forms of vasculitis occur in the setting of infections (eg, hepatitis B or C), connective tissue disorders, inflammatory bowel disease, malignancies, and reactions to medications. Only the major primary forms of vasculitis are discussed here.
Table 20-12. Classification scheme of vasculitides according to size of predominant blood vessels involved. | ||
|---|---|---|
|
Polyarteritis Nodosa
![]() Essentials of Diagnosis
Essentials of Diagnosis
Classic polyarteritis nodosa involves only medium-sized vessels (specifically arteries), but involvement of smaller arterioles is sometimes observed.
Clinical findings depend on the arteries involved.
Common symptoms and signs include fever and other constitutional symptoms, abdominal pain, livedo reticularis, mononeuritis multiplex, anemia, and elevated acute phase reactants (ESR or C-reactive protein or both).
Classic polyarteritis nodosa is often associated with a renin-mediated hypertension caused by kidney involvement but spares the lung.
Ten percent of cases are associated with hepatitis B.
Table 20-13. Typical clinical manifestations of large-, medium-, and small-vessel involvement by vasculitis. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
P.865
General Considerations
Polyarteritis nodosa, described in 1866, is acknowledged widely as the first form of vasculitis reported in the medical literature. For many years, all forms of inflammatory vascular disease were termed polyarteritis nodosa. In recent decades, numerous subtypes of vasculitis have been recognized, greatly narrowing the spectrum of vasculitis called polyarteritis nodosa. Although the term is reserved currently for necrotizing arteritis of medium-sized vessels that has a predilection for involving the skin, peripheral nerves, mesenteric vessels (including renal arteries), heart, and brain, the disease can involve most organs. Polyarteritis nodosa is relatively rare, with a prevalence of about 30 per 1 million people. Approximately 10% of cases of polyarteritis nodosa are caused by hepatitis B. Most cases of hepatitis B-associated disease occur within 6 months of hepatitis B infection.
Clinical Findings
A. Symptoms and Signs
The clinical onset is usually insidious, with fever, malaise, weight loss, and other symptoms developing over weeks to months. Pain in the extremities is often a prominent early feature caused by arthralgia, myalgia (particularly affecting the calves), or neuropathy. The combination of mononeuritis multiplex (with the most common finding being foot-drop) and features of a systemic illness is one of the earliest specific clues to the presence of an underlying vasculitis. Polyarteritis nodosa is among the forms of vasculitis most commonly associated with vasculitic neuropathy.
In polyarteritis nodosa, the typical skin findings livedo reticularis, subcutaneous nodules, and skin ulcers reflect the involvement of deeper, medium-sized blood vessels. Digital gangrene is not an unusual occurrence. The most common cutaneous presentation is lower extremity ulcerations, usually occurring near the malleoli. Involvement of the renal arteries leads to a renin-mediated hypertension (much less characteristic of vasculitides involving smaller blood vessels). For unclear reasons, classic polyarteritis nodosa seldom (if ever) involves the lung, with the occasional exception of the bronchial arteries.
Abdominal pain particularly diffuse periumbilical pain precipitated by eating is common but often difficult to attribute to mesenteric vasculitis in the early stages. Nausea and vomiting are common symptoms. Infarction compromises the function of major viscera and may lead to acalculous cholecystitis or appendicitis. Some patients present dramatically with an acute abdomen caused by mesenteric vasculitis and gut perforation or with hypotension resulting from rupture of a microaneurysm in the liver, kidney, or bowel.
Subclinical cardiac involvement is common in polyarteritis nodosa, and overt cardiac dysfunction occasionally occurs (eg, myocardial infarction secondary to coronary vasculitis, or myocarditis).
B. Laboratory Findings
Most patients with polyarteritis nodosa have a slight anemia, and leukocytosis is common. Acute phase reactants are often (but not always) strikingly elevated. A major challenge in making the diagnosis of polyarteritis nodosa, however, is the absence of a disease-specific serologic test (eg, an autoantibody). Patients with classic polyarteritis nodosa are ANCA-negative and may have low titers of rheumatoid factor or antinuclear antibodies, both of which are nonspecific findings. In patients with polyarteritis nodosa, appropriate serologic tests for active hepatitis B infection must be performed.
C. Biopsy and Angiography
The diagnosis of polyarteritis nodosa requires confirmation with either a tissue biopsy or an angiogram. Biopsies of symptomatic sites such as skin (from the edge of an ulcer or the center of a nodule), nerve, or muscle have reasonably high (albeit imperfect) sensitivities. The least invasive tests should usually be obtained first, but biopsy of an involved organ is essential. If performed by experienced physicians, tissue biopsies normally have high benefit-risk ratios because of the importance of establishing the diagnosis. Patients in whom polyarteritis nodosa is suspected eg, on the basis of mesenteric ischemia or new-onset hypertension occurring in the setting of a systemic illness may be diagnosed by the angiographic finding of aneurysmal dilations in the renal, mesenteric, or hepatic arteries. Angiography must be performed cautiously in patients with baseline renal dysfunction.
P.866
Treatment
For polyarteritis nodosa, corticosteroids in high doses (up to 60 mg of oral prednisone daily) may control fever and constitutional symptoms and heal vascular lesions. Pulse methylprednisolone (eg, 1 g intravenously daily for 3 days) may be necessary for patients who are critically ill at presentation. Immunosuppressive agents, especially cyclophosphamide, lower the risk of disease-related death and morbidity among patients who have severe disease. For patients with polyarteritis nodosa associated with hepatitis B, the preferred treatment regimen is a short course of prednisone accompanied by lamivudine (100 mg/d orally) and plasmapheresis (three times a week for up to 6 weeks).
Prognosis
Without treatment, the 5-year survival rate in these disorders is poor on the order of 20%. With appropriate therapy, remissions are possible in many cases and the 5-year survival rate has improved to 60 90%. Poor prognostic factors are renal insufficiency, proteinuria, gastrointestinal ischemia, central nervous system disease, and cardiac involvement. Substantial morbidity and even death may result from adverse effects of cyclophosphamide and corticosteroids. Consequently, these therapies require careful monitoring and expert management. In contrast to many other forms of systemic vasculitis, disease relapses in polyarteritis following the successful induction of remission are the exception rather than the rule, occurring in only about 10% of cases.
Bourgarit A et al: Deaths occurring during the first year after treatment onset for polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: a retrospective analysis of causes and factors predictive of mortality based on 595 patients. Medicine (Baltimore) 2005;84:323.
Guillevin L et al: Hepatitis B virus-associated polyarteritis nodosa: clinical characteristics, outcome, and impact of treatment in 115 patients. Medicine (Baltimore) 2005;84:313.
Polymyalgia Rheumatica & Giant Cell Arteritis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Giant cell (temporal) arteritis is characterized by headache, jaw claudication, polymyalgia rheumatica, visual abnormalities, and a markedly elevated ESR.
The hallmark of polymyalgia rheumatica is pain and stiffness in shoulders and hips lasting for several weeks without other explanation.
General Considerations
Polymyalgia rheumatica and giant cell arteritis probably represent a spectrum of one disease: Both affect the same population (patients over the age of 50), show preference for the same HLA haplotypes, and show similar patterns of cytokines in blood and arteries. Polymyalgia rheumatica and giant cell arteritis also frequently coexist. The important differences between the two conditions are that polymyalgia rheumatica alone does not cause blindness and responds to low-dose (10 20 mg/d) prednisone therapy, whereas giant cell arteritis can cause blindness and large artery complications and requires high-dose therapy (40 60 mg/d).
Clinical Findings
A. Polymyalgia Rheumatica
Polymyalgia rheumatica is a clinical diagnosis based on pain and stiffness of the shoulder and pelvic girdle areas, frequently in association with fever, malaise, and weight loss. In approximately two-thirds of cases, polymyalgia occurs in the absence of giant cell arteritis. Because of the stiffness and pain in the shoulders, hips, and lower back, patients have trouble combing their hair, putting on a coat, or rising from a chair. In contrast to polymyositis and polyarteritis nodosa, polymyalgia rheumatica does not cause muscular weakness either through primary muscle inflammation or secondary to nerve infarction. A few patients have joint swelling, particularly of the knees, wrists, and sternoclavicular joints. Anemia and elevated acute phase reactants (often markedly elevated ESRs, for example) are present in the most cases, but cases of polymyalgia rheumatica occurring with normal acute phase reactants are well-documented. The differential diagnosis of malaise, anemia, and striking acute phase reactant elevations includes rheumatic diseases such as rheumatoid arthritis, systemic vasculitis, multiple myeloma and other malignant disorders, and chronic infections such as bacterial endocarditis and osteomyelitis.
B. Giant Cell Arteritis
Giant cell arteritis is a systemic panarteritis affecting medium-sized and large vessels in patients over the age of 50. The incidence of this disease increases with each decade of life. The mean age at onset is approximately 72 years. Giant cell arteritis is also called temporal arteritis because that artery is frequently involved, as are other extracranial branches of the carotid artery. About 50% of patients with giant cell arteritis also have polymyalgia rheumatica. The classic symptoms suggesting that a patient has arteritis are headache, scalp tenderness, visual symptoms (particularly amaurosis fugax or diplopia), jaw claudication, or throat pain. The temporal artery is usually normal on physical examination but may be nodular, enlarged, tender, or pulseless. Blindness usually results from the syndrome of anterior ischemic optic neuropathy, caused by occlusive arteritis of the posterior ciliary branch of the ophthalmic artery.
P.867
The ischemic optic neuropathy of giant cell arteritis may produce no funduscopic findings for the first 24 48 hours after the onset of blindness.
Asymmetry of pulses in the arms, a murmur of aortic regurgitation, or bruits heard near the clavicle resulting from subclavian artery stenoses identify patients in whom giant cell arteritis has affected the aorta or its major branches. Large vessel involvement characterized chiefly by aneurysm of the thoracic aortic or stenosis of the subclavian, vertebral, carotid, and basilar arteries occurs in approximately 25% of patients with giant cell arteritis, sometimes years after the diagnosis. Forty percent of patients with giant cell arteritis have nonclassic symptoms at presentation, chiefly respiratory tract problems (most frequently dry cough), mononeuritis multiplex (most frequently with painful paralysis of a shoulder), or fever of unknown origin. Giant cell arteritis accounts for 15% of all cases of fever of unknown origin in patients over the age of 65. The fever can be as high as 40 C and is frequently associated with rigors and sweats. In contrast to patients with infection, patients with giant cell arteritis and fever usually have normal white blood cell counts (before prednisone is started). Thus, in an older patient with fever of unknown origin, marked elevations of acute phase reactants, and a normal white blood count, giant cell arteritis must be considered even in the absence of specific features such as headache or jaw claudication. In some cases, instead of having the well-known symptom of jaw claudication, patients complain of vague pain affecting other locations, including the tongue, nose, or ears. Indeed, unexplained head or neck pain in an older patient may signal the presence of giant cell arteritis.
C. Laboratory Findings
Nearly 90% of patients with giant cell arteritis have ESRs > 50 mm/h. The ESR in this disorder is often > 100 mm/h, but cases in which the ESR is low or even normal do occur. In one series, 5% of patients with biopsy-proven giant cell arteritis had ESRs < 40 mm/h. Patients with biopsy-proven giant cell arteritis with normal C-reactive proteins have also been described. Most patients also have a mild normochromic, normocytic anemia and thrombocytosis. The alkaline phosphatase (liver source) is elevated in 20% of patients with giant cell arteritis.
Treatment
A. Polymyalgia Rheumatica
Patients with isolated polymyalgia rheumatica (ie, those not having above the neck symptoms of headache, jaw claudication, scalp tenderness, or visual symptoms) are treated with prednisone, 10 20 mg/d orally. If the patient does not experience a dramatic improvement within 72 hours, the diagnosis should be revisited. Usually after 2 4 weeks of treatment, slow tapering of the prednisone can be attempted. Most patients require some dose of prednisone for a minimum of approximately 1 year; 6 months is too short in most cases. Disease flares are common (50% or more) as prednisone is tapered. The addition of weekly methotrexate may increase the chance of successfully tapering prednisone in some patients.
B. Giant Cell Arteritis
The urgency of early diagnosis and treatment in giant cell arteritis relates to the prevention of blindness. Once blindness develops, it is usually permanent. Therefore, when a patient has symptoms and findings suggestive of temporal arteritis, therapy with prednisone (60 mg/d orally) should be initiated immediately and a temporal artery biopsy performed promptly. Retrospective studies suggest that low-dose aspirin (~ 81 mg/d orally) may reduce the chance of visual loss or stroke in patients with giant cell arteritis and should be added to prednisone in the initial treatment. Although it is prudent to obtain a temporal artery biopsy as soon as possible after instituting treatment, diagnostic findings of giant cell arteritis may still be present 2 weeks (or even considerably longer) after starting corticosteroids. Typically, a positive biopsy shows inflammatory infiltrate in the media and adventitia with lymphocytes, histiocytes, plasma cells, and giant cells. An adequate biopsy specimen is essential (at least 2 cm in length is ideal), because the disease may be segmental. Unilateral temporal artery biopsies are positive in approximately 80 85% of patients, but bilateral biopsies add incrementally to the yield (10 15% in some studies, less in others). Prednisone should be continued in a dosage of 60 mg/d for about 1 month before tapering. When only the symptoms of polymyalgia rheumatica are present, temporal artery biopsy is not necessary.
In adjusting the dosage of corticosteroid, the ESR is a useful but not absolute guide to disease activity. A common error is treating the ESR rather than the patient. The ESR often rises slightly as the prednisone is tapered, even as the disease remains quiescent. Because elderly individuals often have baseline ESRs that are above the normal range, mild ESR elevations should not be an occasion for renewed treatment with prednisone in patients who are asymptomatic. Thoracic aortic aneurysms occur 17 times more frequently in patients with giant cell arteritis than in normal individuals. The aneurysms can develop at any time but typically occur 7 years after the diagnosis of giant cell arteritis is made.
Bongartz T et al: Large-vessel involvement in giant cell arteritis. Curr Opin Rheumatol 2006;18:10.
Gonzalez-Gay MA et al: Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore) 2005;84:269.
Gonzalez-Gay MA et al: Giant cell arteritis: laboratory tests at the time of diagnosis in a series of 240 patients. Medicine (Baltimore) 2005;84:277.
Nesher G et al: Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum 2004;50:1332.
Seo P et al: Large-vessel vasculitis. Arthritis Care Res 2004;51: 128.
P.868
Wegener'S Granulomatosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Originally defined by the triad of upper respiratory tract disease, lower respiratory disease, and glomerulonephritis.
Suspect if mild respiratory symptoms (eg, nasal congestion, sinusitis) are refractory to usual treatment.
Pathology defined by the triad of small vessel vasculitis, granulomatous inflammation, and necrosis.
Ninety percent of patients with severe, active disease have ANCAs, usually directed against proteinase-3 (less commonly against myeloperoxidase).
Renal disease often rapidly progressive without treatment.
General Considerations
Wegener's granulomatosis, which has an estimated incidence of approximately 12 cases per million individuals per year, is the prototype of diseases associated with antineutrophil cytoplasmic antibodies (ANCA). (Other ANCA-associated vasculitides include microscopic polyangiitis and the Churg-Strauss syndrome.) Wegener's granulomatosis is characterized in its full expression by vasculitis of small arteries, arterioles, and capillaries, necrotizing granulomatous lesions of both upper and lower respiratory tract, glomerulonephritis, and other organ manifestations. Without treatment, generalized disease is invariably fatal, with most patients surviving less than 1 year after diagnosis. It occurs most commonly in the fourth and fifth decades of life and affects men and women with equal frequency.
Clinical Findings
A. Symptoms and Signs
The disorder usually develops over 4 12 months, with 90% of patients presenting with upper or lower respiratory tract symptoms or both. Upper respiratory tract symptoms can include nasal congestion, sinusitis, otitis media, mastoiditis, inflammation of the gums, or stridor due to subglottic stenosis. Since many of these symptoms are common, the underlying disease is not often suspected until the patient develops systemic symptoms or the original problem is refractory to treatment. The lungs are affected initially in 40% and eventually in 80%, with symptoms including cough, dyspnea, and hemoptysis. Other early symptoms can include a migratory oligoarthritis with a predilection for large joints; a variety of symptoms related to ocular disease (unilateral proptosis from orbital pseudotumor; red eye from scleritis, episcleritis, anterior uveitis, or peripheral ulcerative keratitis); purpura or other skin lesions; and dysesthesia due to neuropathy. Renal involvement, which develops in three-fourths of the cases, may be subclinical until renal insufficiency is advanced. Fever, malaise, and weight loss are common.
Physical examination can be remarkable for congestion, crusting, ulceration, bleeding, and even perforation of the nasal septum. Destruction of the nasal cartilage with saddle nose deformity occurs late. Otitis media, proptosis, scleritis, episcleritis, and conjunctivitis are other common findings. Newly acquired hypertension, a frequent feature of polyarteritis nodosa, is rare in Wegener's granulomatosis. Venous thrombotic events (eg, deep venous thrombosis and pulmonary embolism) are now recognized to be a common occurrence in Wegener's granulomatosis, at least in part because of the tendency of the disease to involve veins as well as arteries. Vigilance must be maintained for venous thrombotic events.
Although limited forms of Wegener's granulomatosis have been described in which the kidney is spared initially, renal disease will develop in the majority of untreated patients. In such cases, the urinary sediment invariably contains red cells, with or without white cells, and red cell casts. Renal biopsy discloses a segmental necrotizing glomerulonephritis with multiple crescents; this is characteristic but not diagnostic. Pathologists characterize the renal lesion of Wegener's granulomatosis (and other forms of ANCA-associated vasculitis ) as a pauci-immune glomerulonephritis because of the relative absence (compared with immune complex-mediated disorders) of immunoreactants IgG, IgM, IgA, and complement proteins within glomeruli.
B. Laboratory Findings
Most patients have slight anemia, mild leukocytosis, and elevated acute phase reactants. Chest CT is more sensitive than chest radiography; lesions include infiltrates, nodules, masses, and cavities. Often the radiographs prompt concern about lung cancer. Hilar adenopathy is unusual in Wegener's granulomatosis; if present, sarcoidosis, tumor, or infection is more likely. Other common laboratory or radiographic abnormalities include hematuria, red blood cell casts, extensive sinusitis, and even bony sinus erosions.
Histologic features of Wegener's granulomatosis include vasculitis, granulomatous inflammation, geographic necrosis, and acute and chronic inflammation. The full range of pathologic changes are usually evident only on thoracoscopic lung biopsy. Granulomas, observed only rarely in renal biopsy specimens, are found much more commonly on lung biopsy specimens. Nasal biopsies often do not show vasculitis but may show chronic inflammation and other changes which, interpreted by an experienced pathologist, can serve as convincing evidence of the diagnosis.
P.869
Serum tests for ANCA help in the diagnosis of Wegener's granulomatosis and related forms of vasculitis (Tables 20-10 and 19-2). Several different types of ANCA are recognized, but the two subtypes relevant to systemic vasculitis are those directed against proteinase-3 (PR3) and myeloperoxidase (MPO). Antibodies to these two antigens are termed, respectively, PR3-ANCA and MPO-ANCA. The cytoplasmic pattern of immunofluorescence (C-ANCA) caused by PR3-ANCA has a high specificity (> 90%) for either Wegener's granulomatosis or a closely related disease, microscopic polyangiitis (or, less commonly, the Churg-Strauss syndrome). In the setting of active disease, particularly cases in which the disease is severe and generalized to multiple organ systems, the sensitivity of PR3-ANCA is > 95%. A substantial percentage of patients with limited Wegener's granulomatosis disease that does not pose an immediate threat to life and is often confined to the respiratory tract are ANCA-negative. Although ANCA testing may be very helpful when used properly, it does not eliminate the need in most cases for confirmation of the diagnosis by tissue biopsy. Furthermore, ANCA levels correlate erratically with disease activity, and changes in titer should not dictate changes in therapy in the absence of supporting clinical data. The perinuclear (P-ANCA) pattern, caused by MPO-ANCA, is more likely to occur in microscopic polyangiitis or Churg-Strauss but may also be found in Wegener's granulomatosis. Approximately 10 25% of patients with classic Wegener's granulomatosis have MPO-ANCA. Owing to involvement of the same types of blood vessels, similar patterns of organ involvement, and the possibility of failing to identify granulomatous pathology on tissue biopsies because of sampling error, Wegener's granulomatosis is often difficult to differentiate from microscopic polyangiitis. The crucial distinctions between the two disorders are the tendencies for Wegener's granulomatosis to involve the upper respiratory tract (including the ears) and to cause granulomatous inflammation.
All positive immunofluorescence assays for ANCA should be confirmed by enzyme immunoassays for the specific autoantibodies directed against PR3 or MPO.
Treatment
Early treatment is crucial in preventing the devastating end-organ complications of this disease, and often in preserving life. While Wegener's granulomatosis may involve the sinuses or lung for months, once proteinuria or hematuria develops, progression to renal failure can be rapid (over several weeks). Remissions of at least a temporary nature have been induced in more than 90% of patients treated with conventional approaches to therapy, but disease relapses occur in a substantial proportion of those patients who achieve remission. For patients with severe Wegener's granulomatosis, cyclophosphamide remains the standard of care for remission induction. Cyclophosphamide is best given daily by mouth; intermittent high-dose intravenous cyclophosphamide is less effective. Unfortunately, the traditional therapy of oral cyclophosphamide continued for 12 months after the patient achieves remission has resulted in severe toxicity, including a 2.4 times increased risk of all malignancies, a 33-fold increase in bladder cancer, and a 60% chance of ovarian failure. Consequently, the current approach to remission induction in severe cases is to use cyclophosphamide for 3 6 months, followed by a switch to a regimen more likely to be tolerated well. In patients with remission induced by 3 6 months of cyclophosphamide and corticosteroids, azathioprine (up to 2 mg/kg/d orally) has been shown to be as effective as cyclophosphamide in maintaining disease remissions (at least for up to 12 15 months). Before the institution of azathioprine, patients should be tested (through a commercially available blood test) for deficiencies in the level of thiopurine methyltransferase, an enzyme essential to the metabolism of azathioprine. Another current option for remission maintenance is methotrexate, 20 25 mg/wk (administered either PO or IM). Because of its superior side-effect profile, methotrexate is viewed as an appropriate substitute for cyclophosphamide in patients who do not have significant renal dysfunction (of any cause) or immediately life-threatening disease. Treatment with TNF inhibitors, particularly etanercept, does not appear to be effective.
Goek ON et al: Randomized controlled trials in vasculitis associated with anti-neutrophil cytoplasmic antibodies. Curr Opin Rheumatol 2005;17:257.
Merkel PA et al: High incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener's Clinical Occurrence of Thrombosis (WeCLOT) Study. Ann Intern Med 2005;142:620.
Seo P et al: Damage caused by Wegener's granulomatosis and its treatment: prospective data from the Wegener's Granulomatosis Etanercept Trial (WGET). Arthritis Rheum 2005; 52:2168.
Wegener's Granulomatosis Etanercept Trial (WGET) Research Group: Etanercept plus standard therapy for Wegener's granulomatosis. N Engl J Med 2005;352:351.
Microscopic Polyangiitis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Necrotizing vasculitis of small- and medium-sized vessels in both the arterial and venous circulations.
Frequently involves the lung and kidneys with typical complications of alveolar hemorrhage and glomerulonephritis.
Associated with ANCA in three-fourths of all cases, usually anti-myeloperoxidase antibodies (MPO-ANCA) that cause a P-ANCA pattern on immunofluorescence testing. ANCA directed against proteinase-3
P.870
(PR3-ANCA) can also be observed in microscopic polyangiitis.
General Considerations
Microscopic polyangiitis, as its name implies, is the term given to nongranulomatous vasculitis involving small blood vessels. It is often associated with ANCAs that produce a P-ANCA pattern on immunofluorescence testing and are directed against MPO, a constituent of neutrophil granules. Because microscopic polyangiitis may involve medium-sized as well as small blood vessels and because it tends to affect capillaries within the lungs and kidneys, its spectrum overlaps those of both polyarteritis nodosa and Wegener's granulomatosis.
In a minority of cases, microscopic polyangiitis appears to be induced by reactions to medications, particularly propylthiouracil, hydralazine, allopurinol, penicillamine, and sulfasalazine. In rare instances, these drugs induce a systemic vasculitis associated with MPO-ANCA. Such syndromes usually resolve with discontinuation of the offending drug.
Clinical Findings
A. Symptoms and Signs
A wide variety of findings suggesting vasculitis of small blood vessels may develop in microscopic polyangiitis. These include palpable purpura and numerous other signs of cutaneous vasculitis; hematuria, proteinuria, and red blood cell casts in the urine; and pulmonary hemorrhage. The renal lesion is a segmental, necrotizing glomerulonephritis, often with localized intravascular coagulation and the observation of intraglomerular thrombi upon renal biopsy. The pathologic findings in the lung are typically those of capillaritis. Vasculitic neuropathy (mononeuritis multiplex) is also common in microscopic polyangiitis.
Microscopic polyangiitis is the most common cause of pulmonary-renal syndromes, being several times more common than Goodpasture's (antiglomerular basement membrane) syndrome. In a subset of patients with microscopic polyangiitis, interstitial lung fibrosis that mimics usual interstitial pneumonitis is the presenting condition. Microscopic polyangiitis is defined as a pauci-immune necrotizing vasculitis with few or no immune deposits that (1) affects small blood vessels (capillaries, venules, or arterioles); (2) often includes glomerulonephritis and pulmonary capillaritis; and, (3) is often associated with ANCA (antimyeloperoxidase > antiproteinase-3). Distinguishing this disease from Wegener's granulomatosis may be challenging in some cases. Microscopic polyangiitis is not associated with the chronic destructive upper respiratory tract disease often found in Wegener's granulomatosis. Moreover, as noted, a critical difference between the two diseases is the absence of granulomatous inflammation in microscopic polyangiitis. Because their treatments may differ, microscopic polyangiitis must also be differentiated from polyarteritis nodosa. Microscopic polyangiitis usually requires treatment with both cyclophosphamide and corticosteroids.
B. Laboratory Findings
As noted, three-fourths of patients with microscopic polyangiitis are ANCA-positive. Elevated acute phase reactants are also typical of active disease. Careful scrutiny of the urine sediment is essential to excluding active renal disease.
Treatment
In microscopic polyangiitis, patients are likely to require cyclophosphamide because of the urgency in treating pulmonary hemorrhage and glomerulonephritis. Cyclophosphamide may be administered either in an oral daily regimen or via intermittent (usually monthly) intravenous pulses. The typical oral dose is 2 mg/kg/d, but this may need to be decreased substantially for patients with significant renal dysfunction (for example, the dose for patients receiving dialysis is 0.8 1.0 mg/kg/d). For intermittent administration, cyclophosphamide is usually dosed at 750 mg/m2 (500 mg/m2 for those with renal failure). Whenever cyclophosphamide is used, Pneumocystis jiroveci prophylaxis with either single-strength trimethoprim-sulfamethoxazole or dapsone 100 mg daily is essential. Following the induction of remission, azathioprine is a reasonable choice to replace cyclophosphamide.
Prognosis
The key to effecting good outcomes is early diagnosis. Compared with patients who have Wegener's granulomatosis, those who have microscopic polyangiitis are more likely to have significant fibrosis on renal biopsy because of later diagnosis. The likelihood of disease recurrence following remission in microscopic polyangiitis is about 33%.
Bonaci-Nikolic B et al: Antineutrophil cytoplasmic antibody (ANCA)-associated autoimmune diseases induced by antithyroid drugs: comparison with idiopathic ANCA vasculitides. Arthritis Res Ther 2005;7:R1072.
Hogan SL et al: Predictors of relapse and treatment resistance in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis. Ann Intern Med 2005;143:621.
Seo P et al: The antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med 2004;117:39.
Cryoglobulinemia
Vasculitis secondary to cryoglobulinemia occurs in only a small minority of patients with cryoglobulins in their serum, but this diagnosis should be considered when patients present with palpable purpura, especially when recurrent, and peripheral neuropathy. A
P.871
proliferative glomerulonephritis that strongly resembles lupus nephritis may complicate cryoglobulinemia. Abnormal liver function tests, abdominal pain, and pulmonary disease may also occur. The diagnosis is based on a compatible clinical picture and a positive serum test for cryoglobulins. Type I cryoglobulins (monoclonal proteins that lack rheumatoid factor activity) are more commonly seen in lymphoproliferative disease. (Type I cryoglobulins usually cause hyperviscosity syndromes rather than vasculitis.) The diseases most commonly associated with cryoglobulinemic vasculitis are hepatitis C and connective tissues diseases, especially Sj gren's syndrome. Type II (monoclonal antibody with rheumatoid factor activity) and Type III (polyclonal antibody with rheumatoid factor activity) cryoglobulins cause vasculitis. Because 90% of cryoglobulinemia cases are associated with hepatitis C infections, the optimal approach to treatment is viral suppression with interferon- with or without ribavirin. Pegylated forms of interferon- are now the standard of care for mixed cryoglobulinemia because this form of the drug requires less frequent administration. Because immunosuppressive agents may facilitate viral replication, corticosteroids, cyclophosphamide, and other interventions (including plasmapheresis) should be reserved for organ-threatening complications. B cell depletion using agents such as rituximab appears to be a promising avenue of therapy.
Cacoub P et al: PEGylated interferon alfa-2b and ribavirin treatment in patients with hepatitis C virus-related systemic vasculitis. Arthritis Rheum 2005;52:911.
Ferri C et al: Mixed cryoglobulinemia: demographic, clinical, and serologic features and survival in 231 patients. Semin Arthritis Rheum 2004;33:355.
Quartuccio L: Rituximab treatment for glomerulonephritis in HCV-associated mixed cryoglobulinaemia: efficacy and safety in the absence of steroids. Rheumatology (Oxford) 2006;45:842.
Henoch-Sch Nlein Purpura
Henoch-Sch nlein purpura, the most common systemic vasculitis in children, occurs in adults as well. Typical features are palpable purpura, abdominal pain, arthritis, and hematuria. Pathologic features include leukocytoclastic vasculitis with IgA deposition. The cause is not known.
The purpuric skin lesions are typically located on the lower extremities but may also be seen on the hands, arms, trunk, and buttocks. Joint symptoms are present in the majority of patients, the knees and ankles being most commonly involved. Abdominal pain secondary to vasculitis of the intestinal tract is often associated with gastrointestinal bleeding. Hematuria signals the presence of a renal lesion that is usually reversible, although it occasionally may progress to renal insufficiency. Children tend to have more frequent and more serious gastrointestinal vasculitis, whereas adults more often suffer from renal disease. Biopsy of the kidney reveals segmental glomerulonephritis with crescents and mesangial deposition of IgA.
The disease is usually self-limited, lasting 1 6 weeks, and subsides without sequelae if renal involvement is not severe. Chronic courses with persistent or intermittent skin disease are more likely to occur in adults than in children. Corticosteroids (eg, prednisone 20 40 mg/d orally) are effective in treating some disease manifestations of Henoch-Sch nlein purpura but are not universally effective. Some patients with persistent disease appear to benefit from pulse doses of methylprednisolone (1 g/day IV for 3 days). The incremental efficacy of steroid-sparing drugs such as azathioprine and mycophenolate mofetil often used in the setting of renal disease is not known.
Halling SF et al: Henoch Sch nlein nephritis: clinical findings related to renal function and morphology. Pediatr Nephrol 2005;20:46.
Nadrous HF et al: Pulmonary involvement in Henoch-Sch nlein purpura. Mayo Clin Proc 2004;79:1151.
Relapsing Polychondritis
This disease is characterized by inflammatory destructive lesions of cartilaginous structures, principally the ears, nose, trachea, and larynx. Nearly 40% of cases are associated with another disease, especially either other immunologic disorders (such as SLE, rheumatoid arthritis, or Hashimoto's thyroiditis) or cancers (such as multiple myeloma). The disease, which is usually episodic, affects males and females equally. The cartilage is painful, swollen, and tender during an attack and subsequently becomes atrophic, resulting in permanent deformity. Biopsy of the involved cartilage shows inflammation and chondrolysis. Noncartilaginous manifestations of the disease include fever, episcleritis, uveitis, deafness, aortic insufficiency, and rarely glomerulonephritis. In 85% of patients, a migratory, asymmetric, and seronegative arthropathy occurs, affecting both large and small joints and the costochondral junctions.
Prednisone, 0.5 1 mg/kg/d orally, is often effective. Dapsone (100 200 mg/d orally) may also be effective, sparing the need for chronic high-dose corticosteroid treatment. Involvement of the tracheobronchial tree, leading to tracheomalacia, may lead to difficult management issues.
Kent PD et al: Relapsing polychondritis. Curr Opin Rheumatol 2004;16:56.
Beh Et'S Syndrome
![]() Essentials of Diagnosis
Essentials of Diagnosis
Most commonly occurs among persons of Asian, Turkish, or Middle Eastern background, but may affect persons of any demographic profile.
Recurrent, painful aphthous ulcers of the mouth and genitals.
Additional cutaneous findings include erythema nodosum-like lesions, a follicular rash, and the pathergy phenomenon (formation of a sterile pustule at the site of a needle stick).
Either anterior or posterior uveitis. Posterior uveitis may be asymptomatic until significant damage to the retina has occurred.
Variety of neurologic lesions that can mimic multiple sclerosis, particularly through involvement of the white matter of the brainstem.
P.872
General Considerations
Named after the Turkish dermatologist who first described it, this disease is of unknown cause. Essentially all of its protean manifestations, however, are believed to result from vasculitis that may involve all types of blood vessels: small, medium, and large, on both the arterial and venous side of the circulation.
Clinical Findings
A. Symptoms and Signs
The hallmark of Beh et's disease is painful aphthous ulceration in the mouth. These lesions, which usually occur multiply, may be found on the tongue, gums, and inner surfaces of the oral cavity. Genital lesions, similar in appearance, are also common but do not occur in all patients. Other cutaneous lesions of Beh et's disease include tender, erythematous, papular lesions that resemble erythema nodosum. (On biopsy, however, many of these lesions are shown to be secondary to vasculitis rather than septal panniculitis.) These erythema nodosum-like lesions have a tendency to ulcerate, a major difference between the lesions of Beh et's disease and the erythema nodosum seen in cases of sarcoidosis and inflammatory bowel disease. An erythematous follicular rash that occurs frequently on the upper extremities may be a subtle feature of the disease. The pathergy phenomenon is frequently underappreciated (unless the patient is asked); in this phenomenon, sterile pustules develop at sites where needles have been inserted into the skin (eg, for phlebotomy) in some patients.
A nonerosive arthritis occurs in about two-thirds of patients, most commonly affecting the knees and ankles. Eye involvement may be one of the most devastating complications of Beh et's disease. Posterior uveitis, in essence a retinal venulitis, may lead to the insidious destruction of large areas of the retina before the patient becomes aware of visual problems. Anterior uveitis, associated with photophobia and a red eye, is intensely symptomatic. This complication may lead to a hypopyon, the accumulation of pus in the anterior chamber. If not treated properly with mydriatic agents to dilate the pupil and corticosteroid eyedrops to diminish inflammation, the anterior uveitis of Beh et's may lead to synechial formation between the iris and lens, resulting in permanent pupillary distortion.
Central nervous system involvement is another cause of major potential morbidity in Beh et's disease. The central nervous system lesions that may mimic multiple sclerosis radiologically often result in serious disability or death. Findings include sterile meningitis (recurrent meningeal headaches associated with a lymphocytic pleocytosis), cranial nerve palsies, seizures, encephalitis, mental disturbances, and spinal cord lesions. Finally, patients with Beh et's disease have a hypercoagulable tendency that may lead to complicated venous thrombotic events, particularly multiple deep venous thrombosis, pulmonary emboli, cerebral sinus thrombosis, and other problems associated with clotting.
The clinical course may be chronic but is often characterized by remissions and exacerbations.
B. Laboratory Findings
There are no pathognomonic laboratory features of Beh et's disease. Although patients with Beh et's disease often have elevated acute phase reactants, there is no autoantibody or other assay that is distinctive. No markers of hypercoagulability specific to Beh et's have been identified. Beh et's disease is known to have a genetic risk factor (HLA B51), but this gene is neither necessary nor sufficient to cause the disease and is of little help in making the diagnosis or determining prognosis.
Treatment
Corticosteroids (1 mg/kg/d of oral prednisone) are a mainstay of therapy for severe disease manifestations. Azathioprine (2 mg/kg/d orally) may be an effective steroid-sparing agent. Cyclophosphamide (2 mg/kg/d orally) is indicated for severe ocular and central nervous system complications of Beh et's. Both colchicine (0.6 mg once to three times daily orally) and thalidomide (100 mg/d orally) help ameliorate the mucocutaneous findings of Beh et's. TNF inhibition demonstrates some promise as a therapeutic approach, but large studies are lacking to date.
Calamia KT et al: Major vessel involvement in Beh et disease. Curr Opin Rheumatol 2005;17:1.
Kural-Seyahi E et al: The long-term mortality and morbidity of Beh et syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine (Baltimore) 2003;82:60.
Melikoglu M et al: Short-term trial of etanercept in Beh et's disease: a double blind, placebo controlled study. J Rheumatol 2005;32:98.
Primary Angiitis Of The Central Nervous System
Primary angiitis of the central nervous system is a syndrome with several possible causes that produces small
P.873
and medium-sized vasculitis limited to the brain and spinal cord. Biopsy-proved cases have predominated in men who present with a history of weeks to months of headaches, encephalopathy, and multifocal strokes. Systemic signs and symptoms are absent, and routine laboratory tests are usually normal. MRI of the brain is almost always abnormal, and the spinal fluid often reveals a mild lymphocytosis and a modest increase in protein level. Angiograms classically reveal a string of beads pattern produced by alternating segments of arterial narrowing and dilation. However, neither the MRI nor the angiogram appearance is specific for vasculitis. Many conditions, including vasospasm, can produce the same angiographic pattern as vasculitis. Definitive diagnosis requires a compatible clinical picture; exclusion of infection, neoplasm, or metabolic disorder or drug exposure (eg, cocaine) that can mimic primary angiitis of the central nervous system; and a positive brain biopsy. When a patient meeting the above criteria has a positive angiogram without a confirming biopsy, the diagnosis should be considered possible. Angiographically defined cases of central nervous system vasculopathy differ from biopsy-proved cases chiefly involving women who have had an abrupt onset of headaches and stroke (often in the absence of encephalopathy) with normal spinal fluid findings. Many patients who fit this clinical profile and have disease diagnosed by angiography (but not biopsy) may have vasospasm rather than true vasculitis. Such cases may require shorter and less intensive courses of immunosuppression or none at all as opposed to those with biopsy-proved cases. The latter usually improve with prednisone therapy and may require cyclophosphamide. In recent years, cases of central nervous system vasculitis associated with cerebral amyloid angiopathy have been reported. These cases often respond well to corticosteroids, albeit the long-term natural history remains poorly defined.
Scolding NJ et al: Abeta-related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 2005;128:500.
Seronegative Spondyloarthropathies
The seronegative spondyloarthropathies are ankylosing spondylitis, psoriatic arthritis, Reiter's syndrome (also called reactive arthritis), and the arthritis associated with inflammatory bowel disease. These disorders are noted for male predominance, onset usually before age 40, inflammatory arthritis of the spine or the large peripheral joints (or both), uveitis in a significant minority, the absence of autoantibodies in the serum, and a striking association with HLA-B27. Present in only 8% of normal whites and 4% of normal blacks, HLA-B27 is positive in 90% of patients with ankylosing spondylitis and 75% with Reiter's syndrome. HLA-B27 also occurs in 50% of the psoriatic and inflammatory bowel disease patients who have sacroiliitis. Patients with only peripheral arthritis in these latter two syndromes do not show an increase in HLA-B27.
That HLA-B27 itself (and not some other gene) confers susceptibility to these diseases has been demonstrated by experiments with transgenic rats. When the human HLA-B27 gene is expressed in rats, the animals develop a spinal and peripheral arthritis, psoriasiform nail and skin changes, and bowel inflammation. Thus, HLA-B27 is an important risk factor for the spondyloarthropathies. However, some patients with these disorders are HLA-B27-negative, and the great majority of HLA-B27-positive individuals do not develop spondyloarthropathies. The gene is therefore neither necessary nor sufficient to cause spondyloarthropathies.
Infection also appears to play a key role in some of the spondyloarthropathies, especially Reiter's syndrome, which characteristically develops days to weeks after bacterial dysentery or a nongonococcal sexually transmitted infection (see below). The interplay of susceptibility genes and environmental infections is demonstrated by the fact that the risk of developing Reiter's syndrome is 0.2% in the general population, 2% in the HLA-B27 individuals, and 20% in patients with HLA-B27 who become infected with Salmonella, Shigella, or enteric organisms. Despite these gains in our understanding of the importance of HLA-B27 and infection, the precise mechanism by which genes and infection cause spondyloarthropathy is not yet known.
Ankylosing Spondylitis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Chronic low backache in young adults, generally worst in the morning.
Progressive limitation of back motion and of chest expansion.
Transient (50%) or permanent (25%) peripheral arthritis.
Anterior uveitis in 20 25%.
Diagnostic radiographic changes in sacroiliac joints.
Elevated ESR and negative serologic tests for rheumatoid factor.
HLA-B27 testing is most helpful when there is an indeterminate probability of disease.
General Considerations
Ankylosing spondylitis is a chronic inflammatory disease of the joints of the axial skeleton, manifested clinically
P.874
by pain and progressive stiffening of the spine. The age at onset is usually in the late teens or early 20s. The incidence is greater in males than in females, and symptoms are more prominent in men, with ascending involvement of the spine more likely to occur.
Clinical Findings
A. Symptoms and Signs
The onset is usually gradual, with intermittent bouts of back pain that may radiate down the thighs. As the disease advances, symptoms progress in a cephalad direction and back motion becomes limited, with the normal lumbar curve flattened and the thoracic curvature exaggerated. Chest expansion is often limited as a consequence of costovertebral joint involvement. Radicular symptoms due to cauda equina fibrosis may occur years after onset of the disease. In advanced cases, the entire spine becomes fused, allowing no motion in any direction. Transient acute arthritis of the peripheral joints occurs in about 50% of cases, and permanent changes in the peripheral joints most commonly the hips, shoulders, and knees are seen in about 25%.
Spondylitic heart disease, characterized chiefly by atrioventricular conduction defects and aortic insufficiency, occurs in 3 5% of patients with longstanding severe disease. Anterior uveitis is associated in as many as 25% of cases and may be a presenting feature. Pulmonary fibrosis of the upper lobes, with progression to cavitation and bronchiectasis mimicking tuberculosis, may occur, characteristically long after the onset of skeletal symptoms. Constitutional symptoms similar to those of rheumatoid arthritis are absent in most patients.
B. Laboratory Findings
The ESR is elevated in 85% of cases, but serologic tests for rheumatoid factor are characteristically negative. Anemia may be present but is often mild. HLA-B27 is found in 90% of white patients and 50% of black patients with ankylosing spondylitis. Because this antigen occurs in 8% of the healthy white population (and 4% of healthy blacks), it is not a specific diagnostic test.
C. Imaging
The earliest radiographic changes are usually in the sacroiliac joints. In the first few months of the disease process, the sacroiliac changes may be detectable only by CT scanning. Later, erosion and sclerosis of these joints are evident on plain radiographs. Involvement of the apophysial joints of the spine, ossification of the annulus fibrosus, calcification of the anterior and lateral spinal ligaments, and squaring and generalized demineralization of the vertebral bodies may occur in more advanced stages. The term bamboo spine has been used to describe the late radiographic appearance of the spinal column.
Additional radiographic findings include periosteal new bone formation on the iliac crest, ischial tuberosities and calcanei, and alterations of the pubic symphysis and sternomanubrial joint similar to those of the sacroiliacs. Radiologic changes in peripheral joints, when present, tend to be asymmetric and lack the demineralization and erosions seen in rheumatoid arthritis.
Differential Diagnosis
In contrast to ankylosing spondylitis, rheumatoid arthritis predominantly affects multiple, small, peripheral joints of the hands and feet. Rheumatoid arthritis also spares the sacroiliac joints, and has little effect on the rest of the spine except for C1-C2. Finally, rheumatoid arthritis is often associated with rheumatoid nodules and with rheumatoid factor, not with HLA-B27. The history and physical findings of ankylosing spondylitis serve to distinguish this disorder from other causes of low back pain such as disk disease, osteoporosis, soft tissue trauma, and tumors. The most valuable distinguishing radiologic sign of ankylosing spondylitis is the appearance of the sacroiliac joints, although a similar pattern may be seen in Reiter's syndrome and in the arthritis associated with inflammatory intestinal diseases and psoriasis. In ankylosing hyperostosis (diffuse idiopathic skeletal hyperostosis [DISH], Forestier's disease), there is exuberant osteophyte formation. The osteophytes are thicker and more anterior than the syndesmophytes of ankylosing spondylitis, and the sacroiliac joints are not affected. The x-ray appearance of the sacroiliac joints in spondylitis should be distinguished from that in osteitis condensans ilii.
Treatment
A. Basic Program
The general principles of managing chronic arthritis (see above) apply equally well to ankylosing spondylitis. The importance of postural and breathing exercises should be stressed.
B. Drug Therapy
The NSAIDs are used in the treatment of this disorder. Of these, indomethacin appears to be the most effective, though it can be quite toxic. The dosage of indomethacin is usually 25 50 mg orally three times a day, but the smallest effective dose should be used. Indomethacin may produce a variety of untoward reactions, including headache, giddiness, nausea and vomiting, peptic ulcer, renal insufficiency, depression, and psychosis. Other NSAIDs are valuable alternatives and may be used as primary therapy. Sulfasalazine (1000 mg twice daily) is sometimes useful for the peripheral arthritis in patients with spondyloarthropathies but has little symptomatic effect on spinal and sacroiliac joint disease. Curiously, corticosteroids have minimal impact on the arthritis particularly the spondylitis of ankylosing spondylitis. Studies with TNF inhibitors demonstrate that these agents are highly effective in
P.875
both the spinal and peripheral arthritis of ankylosing spondylitis. Either etanercept (25 mg subcutaneously twice a week) or infliximab (5 mg/kg every other month by IV infusion) is reasonable for patients whose symptoms are refractory to physical therapy and other interventions.
Prognosis
Almost all patients have persistent symptoms over decades; rare individuals experience long-term remissions. The severity of disease varies greatly, with about 10% of patients having work disability after 10 years. Developing hip disease within the first 2 years of disease onset presages a worse prognosis. The availability of TNF inhibitors has improved the outlook dramatically for many patients with ankylosing spondylitis, and the early use of these therapies may attenuate many of the long-term disabilities otherwise characteristic of this disease.
Baraliakos X et al: Magnetic resonance imaging examinations of the spine in patients with ankylosing spondylitis before and after therapy with the tumor necrosis factor alpha receptor fusion protein etanercept. Arthritis Rheum 2005;52:1216.
Braun J et al: Persistent clinical response to the anti-TNF-alpha antibody infliximab in patients with ankylosing spondylitis over 3 years. Rheumatology (Oxford) 2005;44:670.
van der Heijde D et al: Efficacy and safety of infliximab in patients with ankylosing spondylitis: Results of a randomized, placebo-controlled trial (ASSERT). Arthritis Rheum 2005; 52:582.
Wanders A et al: Nonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis: A randomized clinical trial. Arthritis Rheum 2005;52: 1756.
Ward MM: Prospects for disease modification in ankylosing spondylitis: Do nonsteroidal antiinflammatory drugs do more than treat symptoms? Arthritis Rheum 2005;52:1634.
Psoriatic Arthritis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Psoriasis precedes onset of arthritis in 80% of cases.
Arthritis usually asymmetric, with sausage appearance of fingers and toes; resembles rheumatoid arthritis; rheumatoid factor is negative.
Sacroiliac joint involvement common; ankylosis of the sacroiliac joints may occur.
Radiographic findings: osteolysis; pencil-in-cup deformity; relative lack of osteoporosis; bony ankylosis; asymmetric sacroiliitis and atypical syndesmophytes.
General Considerations
In 15 20% of patients with psoriasis, arthritis coexists. The patterns or subsets of arthritis that may accompany psoriasis include the following:
Joint disease that resembles rheumatoid arthritis in which polyarthritis is symmetric. Usually, fewer joints are involved than in rheumatoid arthritis.
An oligoarticular form that may lead to considerable destruction of the affected joints.
A pattern of disease in which the DIP joints are primarily affected. Early, this may be monarticular, and often the joint involvement is asymmetric. Pitting of the nails and onycholysis are frequently associated.
A severe deforming arthritis (arthritis mutilans) in which osteolysis is marked.
A spondylitic form in which sacroiliitis and spinal involvement predominate; 50% of these patients are HLA-B27-positive.
Clinical Findings
A. Symptoms and Signs
Although psoriasis usually precedes the onset of arthritis, arthritis precedes or occurs simultaneously with the skin disease in approximately 20% of cases. Arthritis is at least five times more common in patients with severe skin disease than in those with only mild skin findings. Occasionally, however, patients may have a single patch of psoriasis (typically hidden in the scalp, gluteal cleft, or umbilicus) and are unaware of its connection to the arthritis. Thus, a detailed search for cutaneous lesions is essential in patients with arthritis of new onset. Also, the psoriatic lesions may have cleared when arthritis appears in such cases, the history is most useful in diagnosing previously unexplained cases of mono- or oligoarthritis. Nail pitting, a residue of previous psoriasis, is sometimes the only clue.
B. Laboratory Findings
Laboratory studies show an elevation of the ESR, but rheumatoid factor is not present. Uric acid levels may be high, reflecting the active turnover of skin affected by psoriasis. There is a correlation between the extent of psoriatic involvement and the level of uric acid, but gout is no more common than in patients without psoriasis. Desquamation of the skin may also reduce iron stores.
C. Imaging
Radiographic findings are most helpful in distinguishing the disease from other forms of arthritis. There are marginal erosions of bone and irregular destruction of joint and bone, which, in the phalanx, may give the appearance of a sharpened pencil. Fluffy periosteal new bone may be marked, especially at the insertion of muscles and ligaments into bone. Such changes will
P.876
also be seen along the shafts of metacarpals, metatarsals, and phalanges. Paravertebral ossification occurs, which may be distinguished from ankylosing spondylitis by the absence of ossification in the anterior aspect of the spine.
Treatment
Treatment regimens are symptomatic. NSAIDs are usually sufficient for mild cases. Corticosteroids are less effective in psoriatic arthritis than in other forms of inflammatory arthritis. In addition, they may exacerbate the skin disease during tapers. Antimalarials may also exacerbate psoriasis. In resistant cases, methotrexate may be helpful. For cases with disease that is refractory to methotrexate, etanercept or infliximab is usually effective for both arthritis and psoriatic skin disease. Successful treatment of the skin lesions (eg, by PUVA therapy) commonly though not invariably is accompanied by an improvement in peripheral articular symptoms. Both the cutaneous and the articular findings associated with psoriasis respond dramatically in most cases to TNF inhibitors. Alefacept, a biologic agent that is administered by subcutaneous injection, blocks the activation and proliferation of memory effector T cells by binding to CD2. This drug is a promising new treatment for psoriatic arthritis as well as cutaneous disease.
Antoni CE et al: Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT). Arthritis Rheum 2005;52:1227.
Gottlieb AB: Alefacept for psoriasis and psoriatic arthritis. Ann Rheum Dis 2005;64(Suppl 4):iv58.
Reactive Arthritis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Fifty to 80 percent of patients are HLA-B27-positive.
Oligoarthritis, conjunctivitis, urethritis, and mouth ulcers most common features.
Usually follows dysentery or a sexually transmitted infection.
General Considerations
Reactive arthritis (formerly called Reiter's syndrome) is a clinical tetrad of urethritis, conjunctivitis (or, less commonly, uveitis), mucocutaneous lesions, and aseptic arthritis. It occurs most commonly in young men, is associated with HLA-B27 in 80% of white patients and 50 60% of blacks, and often follows infection (see above).
Clinical Findings
A. Symptoms and Signs
Most cases of reactive arthritis develop within days or weeks after either a dysenteric infection (with Shigella, Salmonella, Yersinia, Campylobacter) or a sexually transmitted infection (with Chlamydia trachomatis or perhaps Ureaplasma urealyticum). Whether the inciting infection is sexually transmitted or dysenteric does not affect the subsequent manifestations but does influence the gender ratio: The ratio is 1:1 after enteric infections but 9:1 with male predominance after sexually transmitted infections.
Although affected joints are culture-negative, fragments of putative organisms have been identified by polymerase chain reaction studies on synovial fluid. The exact role of infection remains unclear.
The arthritis is most commonly asymmetric and frequently involves the large weight-bearing joints (chiefly the knee and ankle); sacroiliitis or ankylosing spondylitis is observed in at least 20% of patients, especially after frequent recurrences. Systemic symptoms including fever and weight loss are common at the onset of disease. The mucocutaneous lesions may include balanitis, stomatitis, and keratoderma blennorrhagicum, indistinguishable from pustular psoriasis. Involvement of the fingernails in Reiter's syndrome may also mimic psoriatic changes. Carditis and aortic regurgitation may occur. While most signs of the disease disappear within days or weeks, the arthritis may persist for several months or even years. Recurrences involving any combination of the clinical manifestations are common and are sometimes followed by permanent sequelae, especially in the joints.
B. Imaging
Radiographic signs of permanent or progressive joint disease may be seen in the sacroiliac as well as the peripheral joints.
Differential Diagnosis
Gonococcal arthritis can initially mimic reactive arthritis, but the marked improvement after 24 48 hours of antibiotic administration and the culture results distinguish the two disorders. Rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis must also be considered. By causing similar oral, ocular, and joint lesions, Beh et's disease may also mimic reactive arthritis. The oral lesions of reactive arthritis, however, are typically painless, in contrast to those of Beh et's.
The association of reactive arthritis and HIV has been debated, but evidence now indicates that it is equally common in sexually active men regardless of HIV status.
Treatment
NSAIDs have been the mainstay of therapy. Antibiotics given at the time of a nongonococcal sexually transmitted infection reduce the chance that the individual will develop this disorder. Unfortunately, once reactive arthritis has developed, antibiotics do not alleviate symptoms. Patients
P.877
who fail NSAIDs and tetracycline may respond to sulfasalazine, 1000 mg orally twice daily, or to methotrexate, 7.5 20 mg orally per week. Anti-TNF agents (etanercept, infliximab, adalimumab) are effective options in most patients with disease that is refractory to more conventional therapies.
Colmegna I et al: Recent advances in reactive arthritis. Curr Rheumatol Rep 2005;7:201.
Arthritis & Inflammatory Intestinal Diseases
One-fifth of patients with inflammatory bowel disease have arthritis, making it second only to anemia as the most common extraintestinal manifestation. Arthritis complicates Crohn's disease somewhat more frequently than it does ulcerative colitis. In both diseases, two distinct forms of arthritis occur. The first is peripheral arthritis usually a nondeforming asymmetric oligoarthritis of large joints in which the activity of the joint disease parallels that of the bowel disease. The arthritis usually begins months to years after the bowel disease, but occasionally the joint symptoms develop earlier and may be prominent enough to cause the patient to overlook intestinal symptoms. The second form of arthritis is a spondylitis that is indistinguishable by symptoms or x-ray from ankylosing spondylitis and follows a course independent of the bowel disease. About 50% of these patients are HLA-B27-positive.
Controlling the intestinal inflammation usually eliminates the peripheral arthritis. The spondylitis often requires NSAIDs, which need to be used cautiously since these agents may activate the bowel disease in a few patients. Range-of-motion exercises as prescribed for ankylosing spondylitis can be helpful.
About two-thirds of patients with Whipple's disease experience arthralgia or arthritis, most often an episodic, large-joint polyarthritis. The arthritis usually precedes the gastrointestinal manifestations by years. In fact, the arthritis resolves as the diarrhea develops. Thus, Whipple's disease should be considered in the differential diagnosis of unexplained episodic arthritis.
Infectious Arthritis*
Nongonococcal Acute Bacterial (Septic) Arthritis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Sudden onset of acute arthritis, usually monarticular, most often in large weight-bearing joints and wrists.
Previous joint damage or injection drug abuse common risk factors.
Infection with causative organisms commonly found elsewhere in body.
Joint effusions are usually large, with white blood counts commonly > 50,000/mcL.
General Considerations
Nongonococcal acute bacterial arthritis is a disease of an abnormal host. The key risk factors are persistent bacteremia (eg, injection drug use, endocarditis) and damaged joints (eg, rheumatoid arthritis). S aureus is the most common cause of nongonococcal septic arthritis, followed by group A and group B streptococci. Gram-negative septic arthritis, once rare, has become more common, especially in injection drug users and in other immunocompromised hosts. Escherichia coli and Pseudomonas aeruginosa are the most common gram-negative isolates in adults.
The widespread use of arthroscopy and prosthetic joint surgery has also increased the frequency of septic arthritis. In the latter conditions, Staphylococcus epidermidis is the usual offending organism. Pathologic changes include varying degrees of acute inflammation, with synovitis, effusion, abscess formation in synovial or subchondral tissues, and, if treatment is not adequate, articular destruction.
Clinical Findings
A. Symptoms and Signs
The onset is usually sudden, with acute pain, swelling, and heat of one joint most frequently the knee. Other commonly affected sites are the hip, wrist, shoulder, and ankle. Unusual sites, such as the sternoclavicular or sacroiliac joint, can be involved in injection drug users. Chills and fever are common but are absent in up to 20% of patients. Infection of the hip usually does not produce apparent swelling but results in groin pain greatly aggravated by walking.
B. Laboratory Findings
Blood cultures are positive in approximately 50% of patients. The leukocyte count of the synovial fluid exceeds 50,000/mcL and often 100,000/mcL, with 90% or more polymorphonuclear cells. Synovial fluid glucose is usually low. Gram stain of the synovial fluid is positive in 75% of staphylococcal infections and in 50% of gram-negative infections.
C. Imaging
Radiographs are usually normal early in the disease, but evidence of demineralization may be present within days of onset. Bony erosions and narrowing of
P.878
the joint space followed by osteomyelitis and periostitis may be seen within 2 weeks.
Differential Diagnosis
The septic course with chills and fever, the acute systemic reaction, the joint fluid findings, evidence of infection elsewhere in the body, and the evidence of response to appropriate antibiotics are diagnostic of bacterial arthritis. Gout and pseudogout are excluded by the failure to find crystals on synovial fluid analysis. Acute rheumatic fever and rheumatoid arthritis commonly involve many joints; Still's disease may mimic septic arthritis, but laboratory evidence of infection is absent. Pyogenic arthritis may be superimposed on other types of joint disease, notably rheumatoid arthritis. Indeed, septic arthritis must be excluded (by joint fluid examination) in any patient with rheumatoid arthritis who has a joint strikingly more inflamed than the other joints.
Treatment
Prompt systemic antibiotic therapy of any septic arthritis should be based on the best clinical judgment of the causative organism and the results of smear and culture of joint fluid, blood, urine, or other specific sites of potential infection. If the organism cannot be determined clinically, treatment should be started with bactericidal antibiotics effective against staphylococci, pneumococci, and gram-negative organisms.
Frequent (even daily) local aspiration is indicated when synovial fluid rapidly reaccumulates and causes symptoms. Immediate surgical drainage is reserved for septic arthritis of the hip, because that site is inaccessible to repeated aspiration. For most other joints, surgical drainage by arthroscopy or open technique is used only if medical therapy fails over 2 4 days to improve the fever and the synovial fluid volume, white blood count, and culture results. Pain can be relieved with local hot compresses and by immobilizing the joint with a splint or traction. Rest, immobilization, and elevation are used at the onset of treatment. Early active motion exercises within the limits of tolerance will hasten recovery.
Prognosis
The outcome of septic arthritis depends largely on the antecedent health of the patient, the causative organism (eg, S aureus bacterial arthritis is associated with a poor functional outcome in about 40% of cases), and the promptness of treatment. Five to 10 percent of patients with an infected joint die of respiratory complications of sepsis. The mortality rate is 30% for patients with polyarticular sepsis. Bony ankylosis and articular destruction commonly also occur if treatment is delayed or inadequate.
Nolla JM et al: Group B streptococcus (Streptococcus agalactiae) pyogenic arthritis in nonpregnant adults. Medicine (Baltimore) 2003;82:119.
Ross JJ et al: Sternoclavicular septic arthritis: review of 180 cases. Medicine (Baltimore) 2004;83:139.
Zimmerli W et al: Prosthetic-joint infections. N Engl J Med 2004;351:1645.
Gonococcal Arthritis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Prodromal migratory polyarthralgias.
Tenosynovitis most common sign.
Purulent monarthritis in 50%.
Characteristic skin rash.
Most common in young women during menses or pregnancy.
Symptoms of urethritis frequently absent.
Dramatic response to antibiotics.
General Considerations
In contrast to nongonococcal bacterial arthritis, gonococcal arthritis usually occurs in otherwise healthy individuals. Host factors, however, influence the expression of the disease: Gonococcal arthritis is two to three times more common in women than in men, is especially common during menses and pregnancy, and is rare after age 40. Gonococcal arthritis is also common in male homosexuals, whose high incidence of asymptomatic gonococcal pharyngitis and proctitis predisposes them to disseminated gonococcal infection. Some of the signs of disseminated gonococcal infection may result from an immunologic reaction to nonviable fragments of the organism's cell wall; this may explain the frequent inability to culture organisms from skin and joint lesions. Recurrent disseminated gonococcal infection should prompt testing of the patient's CH50 level to evaluate for a congenital deficiency of the terminal complement components C7 and C8.
Clinical Findings
A. Symptoms and Signs
One to 4 days of migratory polyarthralgias involving the wrist, knee, ankle, or elbow is the most common initial course. Thereafter, two patterns emerge, one (60% of patients) characterized by tenosynovitis and the other (40%) by purulent monarthritis, most frequently involving the knee. Less than half of patients have fever, and less than one-fourth have any genitourinary symptoms. Most patients will have asymptomatic but highly characteristic skin lesions that usually consist of two to ten small necrotic pustules distributed over the extremities, especially the palms and soles.
P.879
B. Laboratory Findings
The peripheral blood leukocyte count averages about 10,000 cells/mcL and is elevated in less than one-third of patients. The synovial fluid white blood cell count, however, is typically over 50,000 cells/mcL. The synovial fluid Gram stain is positive in one-fourth of cases and culture in less than half. Positive blood cultures are seen in 40% of patients with tenosynovitis and virtually never in patients with suppurative arthritis. Urethral, throat, and rectal cultures should be done in all patients, since they are often positive in the absence of local symptoms. Culturing Neisseria gonorrhoeae is facilitated by rapid transport to the microbiology laboratory, inoculation on appropriate media, and incubation in carbon dioxide.
C. Imaging
Radiographs are usually normal or show only soft tissue swelling.
Differential Diagnosis
Reactive arthritis can produce acute monarthritis in a young person but is distinguished by negative cultures, sacroiliitis, and failure to respond to antibiotics. Lyme disease involving the knee is less acute, does not show positive cultures, and may be preceded by known tick exposure and characteristic rash. The synovial fluid analysis will exclude gout, pseudogout, and nongonococcal bacterial arthritis. Rheumatic fever and sarcoidosis can produce migratory tenosynovitis but have other distinguishing features. Infective endocarditis with septic arthritis can mimic disseminated gonococcal infection.
Treatment
In most cases, patients in whom gonococcal arthritis is suspected should be admitted to the hospital to confirm the diagnosis, to exclude endocarditis, and to start treatment. While outpatient treatment has been recommended in the past, the rapid rise in gonococci resistant to penicillin makes initial inpatient treatment advisable. Approximately 4 5% of all gonococcal isolates produce a -lactamase that confers penicillin resistance. An additional 15 20% of gonococcal species have chromosomal mutations that result in relative resistance to penicillin. Therefore, the recommendations for initial treatment of gonococcal arthritis are to give ceftriaxone, 1 g intravenously daily (or cefotaxime, 1 g intravenously every 8 hours); spectinomycin, 2 g intramuscularly every 12 hours, may be used for patients with -lactam allergies). Once improvement from parenteral antibiotics has been achieved for 24 48 hours, patients can be switched to oral cefixime, 400 mg orally twice daily, or, in regions with low rates of quinolone resistance, levofloxacin 500 mg orally daily or ciprofloxacin 500 mg orally twice daily, to complete a 7- to 10-day course.
Prognosis
Generally, gonococcal arthritis responds dramatically in 24 48 hours after initiation of antibiotics so that daily joint aspirations are rarely needed. Complete recovery is the rule.
Bardin T: Gonococcal arthritis. Best Pract Res Clin Rheumatol 2003;17:201.
Rheumatic Manifestations of Hiv Infection
Infection with HIV has been associated with various rheumatic disorders, most commonly arthralgias or reactive arthritis. More rarely myositis, psoriatic arthritis, Sj gren's syndrome, or vasculitis has been reported to occur (see Chapter 31). It is possible that these disorders stem directly from HIV infection itself or from the many other infections that occur in immunodeficient patients. The rheumatic syndromes may follow the diagnosis of AIDS or may precede it by several months. Thus, coexistent HIV infection must be considered in patients presenting with reactive arthritis. The lower extremity joints, especially the knees and ankles, are most commonly affected. Often, as in classic Reiter's syndrome (reactive arthritis), Achilles tendon inflammation (enthesopathy) or knee periarthritis is a prominent and distinguishing feature. Many patients respond to NSAIDs, though a few are unresponsive and develop progressive deformities. In the era of highly active antiretroviral therapies, immunosuppressive medications can be used if necessary in HIV patients, though with reluctance and great caution. Finally, medications used to treat HIV, particularly the protease inhibitors, may be associated with musculoskeletal side effects, chiefly arthralgias.
Calabrese LH et al: Rheumatic complications of human immunodeficiency virus infection in the era of highly active antiretroviral therapy: emergence of a new syndrome of immune reconstitution and changing patterns of disease. Semin Arthritis Rheum 2005;35:166.
Viral Arthritis
Arthritis may be a manifestation of many viral infections. It is generally mild and of short duration, terminating without lasting ill effects. Mumps arthritis may occur in the absence of parotitis. Rubella arthritis, which occurs more commonly in adults than in children, may appear immediately before, during, or soon after the disappearance of the rash. Its usual polyarticular and symmetric distribution mimics that of rheumatoid arthritis. However, the seronegative tests for rheumatoid factor and the rising rubella titers in convalescent serum help confirm the diagnosis. Post-rubella vaccination arthritis may have its onset as long as 6 weeks following vaccination and occurs in all age groups. In adults, arthritis may follow infection with human parvovirus B19 and sometimes mimics rheumatoid arthritis.
P.880
Transient polyarthritis may be associated with type B hepatitis and typically occurs before the onset of jaundice; it may occur in anicteric hepatitis as well. Urticaria or other types of skin rash may be present. Indeed, the clinical picture may be indistinguishable from that of serum sickness. Serum transaminase levels are elevated, and hepatitis B surface antigen is most often present. Serum complement levels are usually low during active arthritis and become normal after remission of arthritis. False-positive tests for rheumatoid factor, when present, disappear within several weeks. The arthritis is mild; it rarely lasts more than a few weeks and is self-limiting and without deformity. Hepatitis C infection may be associated with chronic polyarthralgia or polyarthritis that mimics rheumatoid arthritis, both in the distribution of involved joints and the fact that many patients infected with hepatitis C are rheumatoid factor positive in at least low titers.
Footnote
* Lyme disease is discussed in Chapter 34.
Mariette X: Hepatitis C virus, arthritides, and arthromyalgia. Joint Bone Spine 2003;70:246.
Masuko-Hongo K et al: Virus-associated arthritis. Best Pract Res Clin Rheumatol 2003;17:309.
Infections of Bones
Direct microbial contamination of bones results from open fracture, surgical procedures, gunshot wounds, diagnostic needle aspirations, and therapeutic or self-administered drug injections.
Indirect or secondary infections are first noticed in other areas of the body and extend to bones by hematogenous routes.
Acute Pyogenic Osteomyelitis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Fever and chills associated with pain and tenderness of involved bone.
Aspiration of involved bone is usually diagnostic.
Culture of blood or lesion tissue is essential for precise diagnosis.
ESR often extremely high (eg, > 100 mm/h).
Radiographs early in the course are typically negative.
General Considerations
Osteomyelitis is a serious infection that is often difficult to diagnose and treat. Infection of bone occurs as a consequence of (1) hematogenous dissemination of bacteria, (2) invasion from a contiguous focus of infection, and (3) skin breakdown in the setting of vascular insufficiency.
Clinical Findings
A. Symptoms and Signs
1. Hematogenous osteomyelitis
Osteomyelitis resulting from bacteremia is a disease associated with sickle cell disease, injection drug users, or the elderly. Patients with this form of osteomyelitis often present with sudden onset of high fever, chills, and pain and tenderness of the involved bone. The site of osteomyelitis and the causative organism depend on the host. Among patients with hemoglobinopathies such as sickle cell anemia, osteomyelitis is caused by salmonellae ten times as often as by other bacteria. Osteomyelitis in injection drug users develops most commonly in the spine. Although in this setting S aureus is most common, gram-negative infections, especially P aeruginosa and Serratia species, are also frequent pathogens. Rapid progression to epidural abscess causing fever, pain, and sensory and motor loss is not uncommon. In older patients with hematogenous osteomyelitis, the most common sites are the thoracic and lumbar vertebral bodies. Risk factors for these patients include diabetes, intravenous catheters, and indwelling urinary catheters. These patients often have more subtle presentations, with low-grade fever and gradually increasing bone pain.
2. Osteomyelitis from a contiguous focus of infection
Prosthetic joint replacement, decubitus ulcer, neurosurgery, and trauma most frequently cause soft tissue infections that can spread to bone. S aureus and S epidermidis are the most common organisms. Localized signs of inflammation are usually evident, but high fever and other signs of toxicity are usually absent.
3. Osteomyelitis associated with vascular insufficiency
Patients with diabetes and vascular insufficiency are susceptible to developing a very challenging form of osteomyelitis. The foot and ankle are the most commonly affected sites. Infection originates from an ulcer or other break in the skin that is usually still present when the patient presents but may appear disarmingly unimpressive. Bone pain is often absent or muted by the associated neuropathy. Fever is also commonly absent. One of the best bedside clues that the patient has osteomyelitis is the ability to easily advance a sterile probe through a skin ulcer to bone.
B. Imaging and Laboratory Findings
The plain film is the most readily available imaging procedure to establish the diagnosis of osteomyelitis, but it can be falsely negative early. Early radiographic findings may include soft tissue swelling, loss of tissue planes, and periarticular demineralization of bone. About 2 weeks after onset of symptoms, erosion of bone and alteration of cancellous bone appear, followed by periostitis.
P.881
MRI, CT, and nuclear medicine bone scanning are more sensitive than conventional radiography. MRI is the most sensitive and is particularly helpful in demonstrating the extent of soft tissue involvement. Radionuclide bone scanning is most valuable when osteomyelitis is suspected but no site is obvious. Nuclear medicine studies may also detect multifocal sites of infection. Ultrasound is useful in diagnosing the presence of effusions within joints and extra-articular soft tissue fluid collections but not in detecting bone infections.
Identifying the offending organism is a crucial step in selection of antibiotic therapy. Bone biopsy for culture is required except in those with hematogenous osteomyelitis, who have positive blood cultures. Cultures from overlying ulcers, wounds, or fistulas are unreliable.
Differential Diagnosis
Acute hematogenous osteomyelitis should be distinguished from suppurative arthritis, rheumatic fever, and cellulitis. More subacute forms must be differentiated from tuberculosis or mycotic infections of bone and Ewing's sarcoma or, in the case of vertebral osteomyelitis, from metastatic tumor. When osteomyelitis involves the vertebrae, it commonly traverses the disk a finding not observed in tumor.
Complications
Inadequate treatment of bone infections results in chronicity of infection, and this possibility is increased by delaying diagnosis and treatment. Extension to adjacent bone or joints may complicate acute osteomyelitis. Recurrence of bone infections often results in anemia, a markedly elevated ESR, weight loss, weakness and, rarely, amyloidosis or nephrotic syndrome. Pseudoepitheliomatous hyperplasia, squamous cell carcinoma, or fibrosarcoma may occasionally arise in persistently infected tissues.
Treatment
Most patients require both debridement of necrotic bone and prolonged administration of antibiotics. Patients with vertebral body osteomyelitis and epidural abscess require urgent neurosurgical decompression. Depending on the site and extent of debridement, surgical procedures to stabilize, fill in, cover, or revascularize may be needed. Traditionally, antibiotics have been administered parenterally for at least 4 6 weeks. Oral therapy with quinolones (eg, ciprofloxacin, 750 mg twice daily) for 6 8 weeks has been shown to be as effective as standard parenteral antibiotic therapy for chronic osteomyelitis with susceptible organisms. When treating osteomyelitis caused by S aureus, quinolones are usually combined with rifampin, 300 mg orally twice daily.
Prognosis
If sterility of the lesion is achieved within 2 4 days, a good result can be expected in most cases if there is no compromise of the patient's immune system. However, progression of the disease to a chronic form may occur. It is especially common in the lower extremities and in patients in whom circulation is impaired (eg, diabetics).
Lew D et al: Osteomyelitis. Lancet 2004:24;364:369.
Mycotic Infections of Bones & Joints
Fungal infections of the skeletal system are usually secondary to a primary infection in another organ, frequently the lungs (see Chapter 36). Although skeletal lesions have a predilection for the cancellous portions of long bones and vertebral bodies, the predominant lesion a granuloma with varying degrees of necrosis and abscess formation does not produce a characteristic clinical picture.
Differentiation from other chronic focal infections depends on culture studies of synovial fluid or tissue obtained from the local lesion. Serologic tests provide presumptive support of the diagnosis.
1. Candidiasis
Candidal osteomyelitis most commonly develops in debilitated, malnourished patients undergoing prolonged hospitalization for cancer, neutropenia, trauma, complicated abdominal surgical procedures, or injection drug use. Infected intravenous catheters frequently serve as a hematogenous source. Prosthetic joints can also be infected by Candida.
For susceptible Candida species, fluconazole, 200 mg orally twice daily, is probably as effective as amphotericin B.
Garbino J et al: An unusual cause of vertebral osteomyelitis: Candida species. Scand J Infect Dis 2003;35:288.
2. Coccidioidomycosis
Coccidioidomycosis of bones and joints is usually secondary to primary pulmonary infection. Arthralgia with periarticular swelling, especially in the knees and ankles, occurring as a nonspecific manifestation of systemic coccidioidomycosis, should be distinguished from actual bone or joint infection. Osseous lesions commonly occur in cancellous bone of the vertebrae or near the ends of long bones at tendinous insertions. These lesions are initially osteolytic and thus may mimic metastatic tumor or myeloma.
The precise diagnosis depends on recovery of Coccidioides immitis from the lesion or histologic examination of tissue obtained by open biopsy. Rising titers of complement-fixing antibodies also provide evidence of the disseminated nature of the disease.
Oral azole antifungal agents (fluconazole 200 400 mg daily, or itraconazole 200 mg twice daily) have become
P.882
the treatment of choice for bone and joint coccidioidomycosis. Chronic infection is rarely cured with antifungal agents and may require operative excision of infected bone and soft tissue; amputation may be the only solution for stubbornly progressive infections. Immobilization of joints by plaster casts and avoidance of weight bearing provide benefit. Synovectomy, joint debridement, and arthrodesis are reserved for more advanced joint infections.
3. Histoplasmosis
Focal skeletal or joint involvement in histoplasmosis is rare and generally represents dissemination from a primary focus in the lungs. Skeletal lesions may be single or multiple and are not characteristic.
Steinbach WJ et al: Review of newer antifungal and immunomodulatory strategies for invasive aspergillosis. Clin Infect Dis 2003;37(Suppl 3):S157.
Stratov I: Management of Aspergillus osteomyelitis: report of failure of liposomal amphotericin B and response to voriconazole in an immunocompetent host and literature review. Eur J Clin Microbiol Infect Dis 2003;22:277.
Wheat LJ et al: Histoplasmosis. Infect Dis Clin North Am 2003; 17:1.
Tuberculosis of Bones & Joints
![]() Essentials of Diagnosis
Essentials of Diagnosis
A disease of children, the elderly, or those with HIV infection.
In most cases, a single site of bone or joint is infected.
Spine especially lower thoracic or knee most common sites.
Chest radiographs abnormal in less than half.
General Considerations
Most tuberculous infections in the United States are caused by the human strain of Mycobacterium tuberculosis (see Chapter 9). Infection of the musculoskeletal system is caused by hematogenous spread from a primary lesion of the respiratory tract; it may occur shortly after primary infection or may be seen years later as a disease reactivation. Tuberculosis of the thoracic or lumbar spine (Pott's disease) usually occurs in the absence of extraspinal infection. It is a disease of children in developing nations and of the elderly in the United States. Tuberculosis of peripheral joints is almost always monarticular, with the knee the most common site. Extra-articular tuberculosis occurs in only 20%.
Clinical Findings
A. Symptoms and Signs
The onset of symptoms is generally insidious and not accompanied by general manifestations of fever, sweating, toxicity, or prostration. Pain may be mild at onset, is usually worse at night, and may be accompanied by stiffness. As the disease process progresses, limitation of joint motion becomes prominent because of muscle contractures and joint destruction. The knee is the most commonly involved peripheral joint. Symptoms of pulmonary tuberculosis may also be present.
Local findings during the early stages may be limited to tenderness, soft tissue swelling, joint effusion, and increase in skin temperature about the involved area. As the disease progresses without treatment, muscle atrophy and deformity become apparent. Abscess formation with spontaneous drainage externally leads to sinus formation. Progressive destruction of bone in the spine may cause a hump spine or gibbus deformity, especially in the thoracolumbar region.
B. Laboratory Findings
The precise diagnosis rests on recovery of the acid-fast organism from joint fluid, pus, or tissue specimens. Biopsy of the bony lesion, synovium, or a regional lymph node may demonstrate the characteristic histopathologic picture of caseating necrosis and giant cells.
C. Imaging
There is a latent period between the onset of symptoms and the initial positive radiographic finding. The earliest changes of tuberculous arthritis are those of soft tissue swelling and distention of the capsule by effusion. Subsequently, bone atrophy causes thinning of the trabecular pattern, narrowing of the cortex, and enlargement of the medullary canal. As joint disease progresses, destruction of cartilage, both in the spine and in peripheral joints, is manifested by narrowing of the joint cleft and focal erosion of the articular surface, especially at the margins. Where the lesion is limited to bone, especially in the cancellous portion of the metaphysis, radiography may demonstrate single or multilocular cysts surrounded by sclerotic bone. With spinal tuberculosis, CT scanning is helpful in demonstrating paraspinal soft tissue extensions of the infection (eg, psoas abscess, epidural extension).
Differential Diagnosis
Tuberculosis of the musculoskeletal system must be differentiated from all subacute and chronic infections, rheumatoid arthritis, gout, and, occasionally, osseous dysplasia. In the spine, metastatic tumor may be suggested.
Complications
Destruction of bones or joints may occur in a few weeks or months if adequate treatment is not provided.
P.883
Deformity due to joint destruction, abscess formation with spread into adjacent soft tissues, and sinus formation are common. Paraplegia is the most serious complication of spinal tuberculosis. As healing of severe joint lesions takes place, spontaneous fibrous or bony ankylosis follows.
Treatment (See also Chapter 9)
A. General Measures
General care is especially important when prolonged recumbency is necessary; skillful nursing care must be provided.
B. Chemotherapy
See Chapter 9. Cure with chemotherapy without need for operation may be achieved in most cases, even with extensive disease.
C. Surgical Measures
In acute infections where synovitis is the predominant feature, treatment can be conservative, at least initially. Immobilization by splint or plaster, aspiration, and chemotherapy may suffice to control the infection. Synovectomy may be valuable for less acute hypertrophic lesions that involve tendon sheaths, bursae, or joints.
Wardle N et al: Orthopaedic manifestations of tuberculosis. Hosp Med 2004;65:228.
Arthritis In Sarcoidosis
The frequency of arthritis among patients with sarcoidosis is variously reported between 10% and 35%. It is usually acute in onset, but articular symptoms may appear insidiously and often antedate other manifestations of the disease. Knees and ankles are most commonly involved, but any joint may be affected. Distribution of joint involvement is usually polyarticular and symmetric. The arthritis is commonly self-limited, resolving after several weeks or months and rarely resulting in chronic arthritis, joint destruction, or significant deformity. Sarcoid arthropathy is often associated with erythema nodosum, but the diagnosis is contingent on the demonstration of other extra-articular manifestations of sarcoidosis and, notably, biopsy evidence of noncaseating granulomas. In chronic arthritis, radiographs show typical changes in the bones of the extremities with intact cortex and cystic changes.
Treatment of arthritis in sarcoidosis is usually symptomatic and supportive. Colchicine may be of value. A short course of corticosteroids may be effective in patients with severe and progressive joint disease.
Abril A et al: Rheumatologic manifestations of sarcoidosis. Curr Opin Rheumatol 2004;16:51.
Torralba KD et al: Sarcoid arthritis: a review of clinical features, pathology, and therapy. Sarcoidosis Vasc Diffuse Lung Dis 2003;20:95.
Tumors & Tumor-Like Lesions of Bone
![]() Essentials of Diagnosis
Essentials of Diagnosis
Persistent pain, swelling, or tenderness of a skeletal part.
Pathologic ( spontaneous ) fractures.
Suspicious areas of bony enlargement, deformity, radiodensity, or radiolucency on x-ray.
Histologic evidence of bone neoplasm on biopsy specimen.
General Considerations
Primary tumors of bone are relatively uncommon in comparison with secondary or metastatic neoplasms. They are, however, of great clinical significance because some grow rapidly and metastasize widely.
Although tumors of bone have been categorized classically as primary or secondary, there is some disagreement about which tumors are primary to the skeleton. Tumors of mesenchymal origin that reflect skeletal tissues (eg, bone, cartilage, and connective tissue) and tumors developing in bones that are of hematopoietic, nerve, vascular, fat cell, and notochordal origin should be differentiated from secondary malignant tumors that involve bone by direct extension or hematogenous spread. Because of the great variety of bone tumors, it is difficult to establish a satisfactory simple classification of bone neoplasms.
Clinical Findings
Persistent skeletal pain and swelling, with or without limitation of motion of adjacent joints or spontaneous fracture, are indications for prompt clinical, radiographic, laboratory, and possibly biopsy examination. Radiographs may reveal the location and extent of the lesion and certain characteristics that may suggest the specific diagnosis. The so-called classic radiographic findings of certain tumors (eg, punched-out areas of the skull in multiple myeloma, sun ray appearance of osteogenic sarcoma, and onion peel effect of Ewing's sarcoma), although suggestive, are not pathognomonic. Even a bone tumor's histologic characteristics, considered in isolation, provide incomplete information about the nature of the disease. The age of the patient, the duration of complaints, the site of involvement and the number of bones
P.884
involved, and the presence or absence of associated systemic disease as well as the histologic characteristics must all be considered for proper management.
The possibility of benign developmental skeletal abnormalities, metastatic neoplastic disease, infections (eg, osteomyelitis), posttraumatic bone lesions, or metabolic disease of bone must always be kept in mind. If bone tumors occur in or near the joints, they may be confused with the various types of arthritis, especially monarticular arthritis.
Specific Bone Tumors
Tumors arising from osteoblastic connective tissue include osteoid osteoma and osteosarcoma. Osteoid osteomas are benign tumors seen in children and adolescents. Interstitial laser photocoagulation is the treatment of choice. Osteosarcoma, the most common malignancy of bone, typically occurs in an adolescent who presents with pain or swelling in a bone or joint (especially in or around the knee). Since the symptoms often appear to begin following a sports-related injury, accurate diagnosis may be delayed. Osteosarcoma can also develop in patients with Paget's disease of bone, enchondromatosis, fibrous dysplasia, or hereditary multiple exostoses. Osteosarcomas are treated by resection and chemotherapy, with 5-year survival rates improving from 15% in 1965 to 60% at this time. Fibrosarcomas, which are derived from nonosteoblastic connective tissue, have a prognosis similar to that of the osteogenic sarcomas. Tumors derived from cartilage include enchondromas, chondromyxoid fibromas, and chondrosarcomas. Histologic examination is confirmatory in this group, and the prognosis with appropriate curettement or surgery is generally good.
Other bone tumors include giant cell tumors (osteoclastomas), chondroblastomas, and Ewing's sarcoma. Of these, chondroblastomas are almost always benign. About 50% of giant cell tumors are benign, while the rest may be frankly malignant or recur after excision. Ewing's sarcoma, which affects children, adolescents, and young adults, has a 50% mortality rate in spite of chemotherapy, irradiation, and surgery.
Treatment
Although prompt action is essential for optimal treatment of certain bone tumors, accurate diagnosis is required because of the great potential for harm that may result from temporization, radical or ablative operations, or unnecessary irradiation.
Westhovens R et al: Musculoskeletal manifestations of benign and malignant tumors of bone. Curr Opin Rheumatol 2003;15: 70.
Neurogenic Arthropathy (Charcot'S Joint)
Neurogenic arthropathy is joint destruction resulting from loss or diminution of proprioception, pain, and temperature perception. Although traditionally associated with tabes dorsalis, it is more frequently seen in diabetic neuropathy, syringomyelia, spinal cord injury, pernicious anemia, leprosy, and peripheral nerve injury. Prolonged administration of hydrocortisone by the intra-articular route may also cause Charcot's joint. As normal muscle tone and protective reflexes are lost, secondary degenerative joint disease ensues, resulting in an enlarged, boggy, painless joint with extensive cartilage erosion, osteophyte formation, and multiple loose joint bodies. Radiographic changes may be degenerative or hypertrophic in the same patient.
Treatment is directed against the primary disease; mechanical devices are used to assist in weight bearing and prevention of further trauma. In some instances, amputation becomes unavoidable.
Anderson JJ et al: Bisphosphonates for the treatment of Charcot neuroarthropathy. J Foot Ankle Surg 2004;43:285.
Other Rheumatic Disorders
Rheumatic Manifestations of Cancer
Rheumatologic syndromes may be the presenting manifestations for a variety of cancers. Dermatomyositis in adults, for example, is associated with cancer. In middle-aged or older patients with polyarthritis that mimics rheumatoid arthritis but is associated with new onset of clubbing and periosteal new bone formation, hypertrophic pulmonary osteoarthropathy, a disorder commonly associated with both malignant diseases (eg, lung and intrathoracic cancers) and nonmalignant ones (eg, cyanotic heart disease, cirrhosis, and lung abscess), should be suspected. Palmar fasciitis is characterized by bilateral palmar swelling and finger contraction and may be the first indication of cancer, particularly ovarian carcinoma. Palpable purpura due to leukocytoclastic vasculitis may be the presenting complaint in myeloproliferative disorders. Hairy cell leukemia can be associated with medium-sized vessel vasculitis such as polyarteritis nodosa. Acute leukemia can produce joint pains that are disproportionately severe in comparison to the minimal swelling and heat that are present. Leukemic arthritis complicates approximately 5% of cases. Rheumatic manifestations of myelodysplastic syndromes include cutaneous vasculitis, lupus-like syndromes, neuropathy, and episodic intense arthritis. Erythromelalgia, a painful warmth and redness of the extremities that (unlike Raynaud's) improves with cold exposure or with elevation of the extremity, is often associated with myeloproliferative diseases.
Chakravarty E et al: Rheumatic syndromes associated with malignancy. Curr Opin Rheumatol 2003;15:35.
P.885
Palindromic Rheumatism
Palindromic rheumatism is a disease of unknown cause characterized by frequent recurring attacks (at irregular intervals) of acutely inflamed joints. Periarticular pain with swelling and transient subcutaneous nodules may also occur. The attacks cease within several hours to several days. The knee and finger joints are most commonly affected, but any peripheral joint may be involved. Systemic manifestations other than fever do not occur. Although hundreds of attacks may take place over a period of years, there is no permanent articular damage. Laboratory findings are usually normal. Palindromic rheumatism must be distinguished from acute gouty arthritis and an atypical acute onset of rheumatoid arthritis. In some patients, palindromic rheumatism is a prodrome of rheumatoid arthritis.
Symptomatic treatment with NSAIDs is usually all that is required during the attacks. Hydroxychloroquine may be of value in preventing recurrences.
Avascular Necrosis of Bone
Avascular necrosis of bone is a complication of corticosteroid use, trauma, SLE, pancreatitis, alcoholism, gout, sickle cell disease, and infiltrative diseases (eg, Gaucher's disease). The most commonly affected sites are the proximal and distal femoral heads, leading to hip or knee pain. Many patients with hip disease actually first present with pain referred to the knee. Physical examination will reveal that it is internal rotation of the hip not movement of the knee that is painful. Other commonly affected sites include the ankle, shoulder, and elbow. Initially, radiographs are often normal; MRI, CT scan, and bone scan are all more sensitive techniques. Treatment involves avoidance of weight bearing on the affected joint for at least several weeks. The value of surgical core decompression is controversial. For osteonecrosis of the hip, a variety of procedures designed to preserve the femoral head have been developed for early disease, including vascularized and nonvascularized bone grafting procedures. These procedures are most effective in avoiding or forestalling the need for total hip arthroplasty in young patients who do not have advanced disease. Without a successful intervention of this nature, the natural history of avascular necrosis is usually progression of the bony infarction to cortical collapse, resulting in significant joint dysfunction. Total hip replacement is the usual outcome for all patients who are candidates for that procedure.
Mont MA et al: Outcome of non-vascularized bone grafting for osteonecrosis of the femoral head. Clin Orthop 2003;417: 84.
Shannon BD et al: Femoral osteotomies for avascular necrosis of the femoral head. Clin Orthop Relat Res 2004;418:34.
Some Orthopedic Procedures For Arthritic Joints
Synovectomy
This procedure attempts to eliminate inflammation at a joint by surgically removing as much of the synovium as possible. The procedure has been performed most commonly in patients with rheumatoid arthritis who, despite medical therapy, have a persistent pannus of inflamed synovium (usually around a wrist or a knee). Patients with degenerative diseases do not have marked synovial inflammation and are not candidates for this procedure.
Unfortunately, synovium can regrow. Even in patients with rheumatoid arthritis, the long-term benefits of synovectomy remain unproved.
Arthroplasty
Realigning arthritic joints (arthroplasty) generally does not work as well as complete joint replacement, but it can defer the need for joint replacement. The typical candidate is an adult under 50 years of age who has severe osteoarthritis of one compartment of the knee (typically the medial compartment). Excising a wedge of femur will realign the patient's knee so as to shift weight to the compartment with normal cartilage and thereby eliminate or reduce the patient's pain.
Tendon Rupture
This is a fairly common complication in rheumatoid arthritis and requires immediate orthopedic referral. The most common sites are the finger flexors and extensors, the patellar tendon, and the Achilles tendon.
Arthrodesis
Arthrodesis (fusion) is being used less now than formerly, but a chronically infected, painful joint may be an indication for this surgical procedure.
Total Joint Arthroplasty
In the last 3 decades, remarkable progress has been made in the replacement of severely damaged joints with prosthetic materials. Although many different joints can be replaced, the largest experience and greatest success have been with hip and knee replacement. Indication for total joint arthroplasty is severe pain (usually including pain at rest) accompanied by loss of function and severe destruction of the joint on x-ray. Age is also a consideration, as the durability of artificial joints beyond 10 15 years is limited with older surgical techniques and unproved with newer techniques. Thus,
P.886
patients over 65 are less likely than younger ones to face the challenge of revision.
Whatever the patient's age, success of the replacement depends on the amount of physical stress to which the prosthetic components are subjected. Vigorous impact activity, even with the most advanced biomaterials and design, will result in failure of the prosthesis with time. Revision operations are technically more difficult, and the results may not be as good as with the primary procedure. The patient, therefore, must understand the limitations of joint replacement and the consequences of unrestrained joint usage.
A. Total Hip Arthroplasty
Hip replacement was originally designed for use in patients over 65 years of age with severe osteoarthritis. In these patients usually less active physically the prosthesis not only functioned well but outlasted the patients. Severe arthritis that fails to respond to conservative measures remains the principal indication for hip arthroplasty. Hip arthroplasty may also be indicated in younger patients severely disabled by painful hip disease (eg, rheumatoid arthritis). Contraindications to the operation include active infection and neurotrophic joint disease. Obesity is a relative contraindication. Serious complications may occur in about 1% of patients and include thrombophlebitis, pulmonary embolization, infection, and dislocation of the joint. Extensive experience has now been accumulated, and the short-term results are highly successful in properly selected patients. The long-term success has been limited by loosening of the prosthesis, a complication seen in 30 50% of patients 10 years after replacement with first generation techniques. Although loosening and periprosthetic osteolysis were blamed on the cement, second-generation cementing techniques for prosthetic hips have proved more durable than cementless hips in one out of four cases.
B. Total Knee Arthroplasty
The indications and contraindications for total knee arthroplasty are similar to those for hip arthroplasty. Results are slightly better in osteoarthritis patients than in those with rheumatoid arthritis. Complications are similar to those with hip arthroplasty. The failure rate of knee arthroplasty is slightly higher than that of hip arthroplasty.
Huo MH: What's new in hip arthroplasty. J Bone Joint Surg Am 2003;85-A:1852.
Salvati EA et al: Thromboembolism following total hip replacement. J Long Term Eff Med Implants 2003;13:325.
Sutton PM et al: Treatment of anaemia after joint replacement. A double-blind, randomised, controlled trial of ferrous sulphate versus placebo. J Bone Joint Surg Br 2004;86:31.
EAN: 2147483647
Pages: 49