18 - Obstetrics
Editors: McPhee, Stephen J.; Papadakis, Maxine A.; Tierney, Lawrence M.
Title: Current Medical Diagnosis & Treatment, 46th Edition
Copyright 2007 McGraw-Hill
> Table of Contents > 21 - Fluid & Electrolyte Disorders
function show_scrollbar() {}
21
Fluid & Electrolyte Disorders
Masafumi Fukagawa MD, PhD
Kiyoshi Kurokawa MD, MACP
Maxine A. Papadakis MD
Approach to the Patient
History, Physical Examination, & Basic Laboratory Tests
In many instances, electrolyte disorders are asymptomatic, thus the clinician should never overlook abnormal values in routine laboratory studies. Nevertheless, symptoms such as lethargy, weakness, confusion, delirium, and seizures may develop, especially in the presence of an abnormal serum sodium concentration. Often these symptoms are mistaken for primary neurologic or metabolic disorders. Muscle weakness occurs in patients with severe hypokalemia, hyperkalemia, and hypophosphatemia; confusion, seizures, and coma may develop in those with severe hypercalcemia. Measurement of electrolytes (sodium, potassium, chloride, bicarbonate, calcium, magnesium, and phosphorus) is indicated for any patient with even vague neuromuscular symptoms.
In addition to taking a careful history, the diagnosis and treatment of fluid and electrolyte disorders are based on (1) assessment of total body water and its distribution, (2) serum electrolyte concentrations, (3) urine electrolyte concentrations, and (4) serum osmolality.
A. Body Water
Changes of total body water content are best evaluated by documenting changes in body weight. Table 21-1 shows the sex difference in total body water and the decrease in total body water that occurs with aging. Two-thirds of total body water (40% of body weight) is intracellular fluid (ICF), while one-third (20% of body weight) is extracellular fluid (ECF). Water may be lost from either or both of the fluid compartments. Circulatory and neurologic symptoms, physical examination, and laboratory tests (serum and urine sodium, serum urea nitrogen, serum creatinine) can identify the compartment from which fluid is lost.
One-fourth of extracellular fluid (5% of body weight) is retained within the blood vessels as plasma (effective circulating volume). Effective circulating volume may be assessed by physical examination (blood pressure, pulse rate, jugular vein dilation). In addition to the invasive measurement of central venous pressure or pulmonary wedge pressure, noninvasive measurement of the diameter of the inferior vena cava by ultrasonography may be useful for assessment of effective circulating volume.
B. Serum Electrolytes
Table 21-2 shows the normal values for serum electrolytes. Electrolyte disorders may be suspected by considering the history and underlying disease and the medications the patient is taking.
C. Evaluation of Urine
Urinalysis provides information about underlying renal disorders. Samples also should be obtained for analysis of urine electrolyte abnormalities. An electrolyte concentration in urine is a useful indicator of renal handling of water and the electrolyte, ie, whether the kidney loses or preserves the electrolyte.
In addition to the total urine per day, a spot urine may be used. Simple concentration, or concentration per gram creatinine excretion, is usually sufficient for initial analysis. More precisely, fractional excretion is used. Fractional excretion (FE) of an electrolyte X (FEX) is calculated using a random urine sample with simultaneously obtained serum samples for X and creatinine (Cr).
![]()
D. Serum Osmolality
Serum osmolality (normally 285 295 mosm/kg) can be calculated from the following formula:
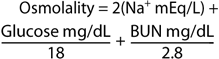
(1 mosm of glucose equals 180 mg/L and 1 mosm of urea nitrogen equals 28 mg/L). Solute concentration is usually expressed in terms of osmolality. The number of particles in solution (ie, osmolytes; either molecules or ions) determines the number of milliosmoles. Each particle has a unit value of 1, so if a substance ionizes, each ion contributes the same amount as a nonionizable molecule. More importantly, permeability of the particle across the cell membrane determines whether it
P.888
acts as a physiologically active osmolyte. Tonicity refers to osmolytes that are impermeable to the cell wall. Since osmolytes do not equilibrate on either side of the cell wall, it is tonicity that leads to osmosis, fluid shifts, stimulation of thirst, and secretion of antidiuretic hormone (ADH). Substances that easily permeate cell membranes (eg, urea, ethanol) are not effective osmolytes and therefore do not cause shifting of fluid in body fluid compartments. For example, glucose in solution is nonionizable. Therefore, 1 mmol of glucose has an osmole concentration of 1 mosm/kg H2O. One millimole of NaCl, however, forms two ions in water (one Na+ and one Cl-) and has an osmole concentration of roughly 2 mosm/kg H2O. Osmoles per kilogram of water is osmolality; osmoles per liter of solution is osmolarity. At the solute concentration of body fluids, the two measurements correspond so closely that they are interchangeable. A discrepancy between actual and calculated osmolality suggests the accumulation of unmeasured osmoles (osmolar gap).
Table 21-1. Total body water (as percentage of body weight) in relation to age and sex. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Riggs JE: Neurologic manifestations of electrolyte disturbances. Neurol Clin 2002;20:157.
Disorders of Sodium Concentration
An abnormal serum sodium concentration does not necessarily imply abnormal sodium balance but rather implies abnormal water balance. Thus, most instances of abnormal sodium concentration are associated with abnormal serum osmolality and shifts of water across the cell membrane. By contrast, abnormal sodium balance results in an edematous state or in volume depletion.
Hyponatremia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Extracellular fluid volume and serum osmolality are important determinants of etiology.
Most cases of hyponatremia result from water imbalance, not sodium imbalance.
Hospitalized patients treated with hypotonic fluid are at increased risk for the development of hyponatremia.
Treatment strategy should be based not only on pathophysiology but on the severity and speed of development.
General Considerations
Hyponatremia (defined as a serum sodium concentration less than 130 mEq/L) is the most common electrolyte abnormality observed in a general hospitalized population, seen in about 2% of patients.
The initial approach to its investigation is the determination of serum osmolality (Figure 21-1).
The Urine Sodium
Although most cases of hyponatremia result from water imbalance, not sodium imbalance, measurement of urine sodium helps distinguish renal from nonrenal causes of hyponatremia. Urine sodium exceeding 20 mEq/L is consistent with renal salt wasting (diuretics, angiotensin-converting enzyme (ACE) inhibitors, mineralocorticoid deficiency, salt-losing nephropathy). Urine sodium less than 10 mEq/L or fractional excretion of sodium less than 1% (unless diuretics have been given) implies avid sodium retention by the kidney to compensate for extrarenal fluid losses from vomiting, diarrhea, sweating, or third-spacing, as with ascites.
Isotonic & Hypertonic Hyponatremia
Isotonic and hypertonic hyponatremia should be initially ruled out by determining serum osmolality, blood lipids, and blood glucose.
Table 21-2. Normal values and mass conversion factors.1 | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
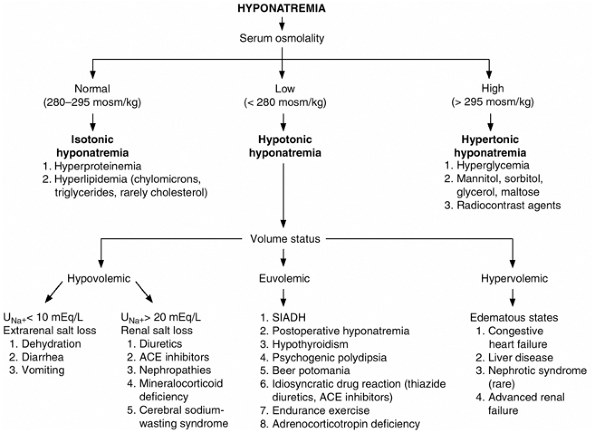 |
Figure 21-1. Evaluation of hyponatremia using serum osmolality and extracellular fluid volume status. ACE = angiotensin-converting enzyme; SIADH = syndrome of inappropriate antidiuretic hormone. (Adapted, with permission, from Narins RG et al: Diagnostic strategies in disorders of fluid, electrolyte and acid-base homeostasis. Am J Med 1982;72:496. ) |
P.889
Isotonic hyponatremia can be seen with hyperlipidemia and hyperproteinemia. Because of marked increases, lipids (chylomicrons; triglycerides, which make the blood visibly lipemic; and very occasionally cholesterol, which may not make the blood visibly lipemic) and proteins (> 10 g/dL, eg, intravenous immunoglobulin therapy) occupy a disproportionately large portion of the plasma volume. Plasma osmolality remains normal because its measurement is unaffected by the lipids or proteins. A decreased volume of water results, so that the sodium concentration in total plasma volume is decreased. Because the sodium concentration in the plasma water is actually normal, hyperlipidemia and hyperproteinemia cause so-called pseudohyponatremia. Most laboratories measure serum electrolytes using ion-specific electrodes and thus avoid misdiagnosis unless dilution of samples is needed before direct measurement.
Hypertonic hyponatremia is most commonly seen with hyperglycemia. When blood glucose becomes acutely elevated, water is drawn from the cells into the extracellular space, diluting the serum sodium. The plasma sodium level falls 2 mEq/L for every 100 mg/dL rise when the glucose concentration is between 200 and 400 mg/dL. If the glucose concentration is above 400 mg/dL, the plasma sodium concentration falls 4 mEq/L for every 100 mg/dL rise in glucose. This dilutional or translocational hyponatremia is not pseudohyponatremia, since the sodium concentration does indeed fall. Infusion of hypertonic solutions containing osmotically active osmoles (eg, mannitol) may also cause hypertonic hyponatremia by drawing water to the extracellular space.
Hypotonic Hyponatremia
Hypotonic hyponatremia is true hyponatremia in a physiologic sense. In this abnormality, water shifts into the cell, usually resulting in increased ICF.
Because the capacity of the kidney to excrete electrolyte-free water is potentially great up to 20 30 L/d in the presence of a normal glomerular filtration rate (GFR) (100 L/d), electrolyte-free water intake must theoretically exceed 30 L/d for hyponatremia to develop. Instead, in hypotonic hyponatremia, retention of electrolyte-free water nearly always occurs because of impaired excretion (renal failure, inappropriate ADH excess, etc).
P.890
Determinations of the urine osmolality and urine sodium are useful diagnostic tools.
Once a diagnosis of hypotonic hyponatremia has been made, an accurate determination of the patient's volume status is essential in directing further evaluation.
A. Hypovolemic Hypotonic Hyponatremia
Hyponatremia with decreased extracellular fluid volume occurs in the setting of renal or extrarenal volume loss (Figure 21-1). Total body sodium is decreased. To maintain intravascular volume, ADH secretion increases, and free water is retained. The drive to replenish intravascular volume overrides the need to sustain normal osmolality; losses of salt and water are replaced by water alone. The combination of low fractional excretion of sodium (< 0.5%) and low fractional urea clearance (< 55%) is the best way to predict improvement with saline therapy. Hyponatremia has been shown to develop in patients with intracranial diseases through renal sodium wasting. Unlike those with syndrome of inappropriate ADH secretion, these patients are hypovolemic, though plasma levels of ADH are inappropriately high for the osmolality. Observations in patients with subarachnoid hemorrhage suggest that the cerebral salt-wasting syndrome is caused by increased secretion of brain natriuretic peptide with suppression of aldosterone secretion.
Treatment consists of replacement of lost volume with isotonic or half-normal (0.45%) saline or lactated Ringer's infusion. The rate of correction must be adjusted to prevent permanent cerebral damage (see below).
B. Euvolemic Hypotonic Hyponatremia
1. Clinical syndromes
a. Syndrome of inappropriate antidiuretic hormone secretion (SIADH)
(Table 21-3.) Hypovolemia physiologically stimulates ADH secretion, so the diagnosis of SIADH is made only if the patient is euvolemic. In SIADH, increased ADH release occurs without osmolality-dependent or volume-dependent physiologic stimulation. Normal regulation of ADH release occurs from both the central nervous system and the chest via baroreceptors and neural input. It follows that the causes of SIADH are disorders affecting the central nervous system structural, metabolic, psychiatric, or pharmacologic or the lungs. Furthermore, some carcinomas, such as small cell lung carcinoma, synthesize ADH. Other states associated with SIADH include administration of drugs that either increase ADH secretion or potentiate its action.
Table 21-3. Causes of syndrome of inappropriate secretion of ADH (SIADH). | ||
|---|---|---|
|
(1) Patterns of abnormal antidiuretic hormone secretion
(a) Random secretion
ADH release is unrelated to osmoregulation. This pattern is seen in carcinomas and central nervous system diseases.
(b) Reset osmostat
This variant is characterized by ADH secretion appropriately suppressed at very low serum osmolalities but with ADH osmoregulation downset to a lower level of normal. Therefore,
P.891
ADH is secreted at a subnormal serum osmolality threshold (< 280 mosm/kg). Appropriate urinary dilution can be attained but at low serum osmolalities. This pattern is seen in the elderly, and in patients with pulmonary processes, tuberculosis, or malnutrition. During pregnancy, the physiologic reset osmostat may suppress osmolality by about 10 mmol/kg of water.
(c) Leak of antidiuretic hormone
In conditions such as basilar skull fractures, low levels of ADH are leaked into the circulation despite hypo-osmolality. If serum osmolality rises to normal, ADH secretion increases appropriately and then continues to respond normally if osmolality further increases.
(2) Clinical features
SIADH is characterized by (1) hyponatremia; (2) decreased osmolality (< 280 mosm/kg) with inappropriately increased urine osmolality (> 150 mosm/kg); (3) absence of cardiac, renal, or liver disease; (4) normal thyroid and adrenal function (see Chapter 26 for thyroid function tests and cosyntropin stimulation test); and (5) urine sodium usually over 20 mEq/L. Natriuresis compensates for the slight increase in volume from ADH secretion. The mechanisms that regulate sodium excretion in response to increases in extracellular volume, such as suppression of the sympathetic nervous and renin-angiotensin systems and increased secretion of atrial natriuretic factor, are preserved and account for the increase in urinary sodium. The expansion of extracellular volume is not large enough to cause clinical hypervolemia, hypertension, or edema. Other changes frequently seen in SIADH include low blood urea nitrogen (BUN) (< 10 mg/dL) and hypouricemia (< 4 mg/dL), which are not only dilutional but result from increased urea and uric acid clearances in response to the volume-expanded state. A high BUN suggests a volume-contracted state, which excludes a diagnosis of SIADH.
b. Hyponatremia after a surgery or procedure
Severe postoperative hyponatremia can develop in 2 days or less after elective surgery in healthy patients, especially premenopausal women. Most have received excessive postoperative hypotonic fluid in the setting of elevated ADH levels related to pain of surgery with continuing excretion of hypertonic urine. Patients awake normally from general anesthesia, but within 2 days develop nausea, headache, seizures, and even respiratory arrest.
Similar mechanisms have been suspected for hyponatremia after colonoscopy. This is not a direct effect of the large volume of liquid-cleansing agents such as polyethylene glycol solution but is due to the diarrhea, nausea, vomiting, and potential volume depletion sometimes produced by these agents. Increased oral water intake or hypotonic fluid administration after colonoscopy (in the presence of elevated ADH) may then result in hyponatremia; occasionally, if thirst is impaired, hypernatremia results.
c. Hypothyroidism
Hyponatremia is not commonly caused by hypothyroidism, but it can occur on occasion with serum sodium levels as low as 105 mEq/L. Water retention is the cause, probably both from inappropriately elevated ADH levels and from alterations in the handling of water by the kidneys.
d. Psychogenic polydipsia and beer potomania
Marked excess free water intake (generally > 10 L/d) may produce hyponatremia. Euvolemia is maintained through the renal excretion of sodium. Urine sodium is therefore generally elevated (> 20 mEq/L), but unlike SIADH, levels of ADH are suppressed. Urine osmolality is appropriately low (< 300 mosm/kg) as the increased free water is excreted. Hyponatremia from bursts of ADH occurs in manic-depressive patients with excess free water intake. Psychogenic polydipsia is observed in patients with psychological problems, and these patients frequently take drugs interfering with water excretion. Similarly, excessive intake of beer, which contains very small amounts of sodium (< 5 mEq/L), can cause severe hyponatremia in cirrhotic patients, who have elevated ADH and often a decreased GFR.
e. Idiosyncratic diuretic reaction
In addition to hyponatremia developing from volume contraction due to diuretic therapy (see above), a less common diuretic-induced hyponatremia can occur in euvolemic patients, typically from thiazides. This syndrome is most often seen in healthy older women (over 70 years of age) often after a few days of therapy. The mechanism for the hyponatremia appears to be a combination of excessive renal sodium loss and water retention.
f. Idiosyncratic ACE inhibitor reactions
ACE inhibitors can cause central polydipsia and increased ADH secretion, both of which result in severe, symptomatic hyponatremia. ACE does not block the conversion of angiotensin I to angiotensin II in the brain. Thus, angiotensin II converted in the brain stimulates thirst and ADH secretion.
g. Endurance exercise hyponatremia
Hyponatremia after endurance exercise (eg, triathlon events) may be caused by a combination of excessive hypotonic fluid intake and continued ADH secretion. Reperfusion of the exercise-induced ischemic splanchnic bed causes delayed absorption of excessive quantities of hypotonic fluid ingested during exercise. Sustained elevation of ADH prevents water excretion in this setting. The retention of hypotonic fluid may be further exacerbated by nonsteroidal anti-inflammatory drugs (NSAIDs) frequently used by athletes. New guidelines encourage runners to drink between 400 mL/h and 800 mL/h as opposed to the previous as much as possible advice. However, the clinical relevance of this new recommendation remains to be determined.
h. Mineralocorticoid-responsive hyponatremia in the elderly
In a subgroup of elderly patients, hyponatremia does not resolve in response to water restriction even though they are euvolemic with high ADH levels. These patients may respond to fludrocortisone treatment.
P.892
i. Adrenal deficiency
This is an important but often overlooked cause of euvolemic hyponatremia. The adrenal insufficiency may be either primary or secondary, due to adrenocorticotropin (ACTH) deficiency. Routine laboratory data may not easily distinguish hyponatremia due to ACTH deficiency from that of SIADH. However, low plasma bicarbonate (or total CO2) levels suggest ACTH deficiency.
j. Methylenedioxymethylamphetamine ( Ecstasy ) abuse
Abuse of 3,4-methylenedioxymethylamphetamine (MDMA), also known as Ecstasy, can lead to severe neurologic symptoms, including seizures, brain edema, and herniation from severe hyponatremia. MDMA and its metabolites have been shown to induce enhanced ADH release from the hypothalamus.
k. Selective serotonin reuptake inhibitors (SSRIs)
SIADH induced by selective serotonin (or epinephrine) reuptake inhibitors, such as fluoxetine, paroxetine, and rofloxacin, is fairly common in geriatric patients. Enhanced secretion or action of ADH may result from increased serotonergic tone.
l. Amiodarone
SIADH during amiodarone-loading has been reported. Hyponatremia usually improves with dose reduction.
m. Hyponatremia in HIV-infected patients
Hyponatremia is seen in up to 50% of patients hospitalized for HIV infection and in 20% of ambulatory AIDS patients, often associated with pneumonia and central nervous system processes. If hyponatremia is present at the time of hospital admission, it is just as likely to be due to hypovolemic gastrointestinal loss as to euvolemic SIADH. However, if hyponatremia develops after hospital admission, most patients have euvolemic SIADH. Infrequently, hypovolemic hyponatremia is due to adrenal insufficiency from infections or ketoconazole toxicity, isolated mineralocorticoid deficiency with hyporeninemic hypoaldosteronism, or an HIV-specific impairment in renal sodium conservation.
2. Treatment
a. Symptomatic hyponatremia
Symptomatic hyponatremia is usually seen in patients with serum sodium levels less than 120 mEq/L. If there are central nervous system symptoms, hyponatremia should be rapidly treated at any level of serum sodium concentration.
(1) Rate and degree of correction
Central pontine myelinolysis may occur from osmotically induced demyelination due to overly rapid correction of serum sodium (an increase of more than 1 mEq/L/h, or 25 mEq/L within the first day of therapy). Hypoxic-anoxic episodes during hyponatremia may contribute to the demyelination. Premenopausal women in whom hyponatremic encephalopathy develops are about 25 times more likely than menopausal women to die or suffer permanent brain damage, suggesting a hormonal role in the pathophysiology of this disorder.
A reasonable approach is to increase the serum sodium concentration by no more than 1 2 mEq/L/h and not more than 25 30 mEq/L in the first 2 days; the rate should be reduced to 0.5 1 mEq/L/h as soon as neurologic symptoms improve. The initial goal is to achieve a serum sodium concentration of 125 130 mEq/L, guarding against overcorrection.
(2) Saline plus furosemide
Hypertonic (eg, 3%) saline with furosemide is indicated for symptomatic hyponatremic patients. If 3% saline without a diuretic is administered to a patient with SIADH, the serum sodium concentration increases temporarily, but euvolemic patients excrete the excess sodium. If furosemide (0.5 1 mg/kg intravenously) is added, however, the kidney cannot concentrate urine even in the presence of high levels of ADH. Infusion of 3% saline is accompanied by excretion of isotonic urine with a net loss of free water. The sodium concentration of 3% saline is 513 mEq/L. To determine how much 3% saline to administer, a spot urinary Na+ is determined after a furosemide diuresis has begun. The excreted Na+ is replaced with 3% saline, empirically begun at 1 2 mL/kg/h and then adjusted based on urinary output and urinary sodium. For example, after administration of furosemide, urine volume may be 400 mL/h and sodium plus potassium excretion 100 mEq/L. The excreted Na+ plus K+ is 40 mEq/h, which is replaced with 78 mL/h of 3% saline (40 mEq/h divided by 513 mEq/L). Free water loss is about 1% of total body water. Therefore, an approximately 1% rise in plasma sodium concentration (1 1.5 mEq/L/h) can be expected. Measurements of plasma sodium should be done approximately every 4 hours and the patient observed closely.
b. Asymptomatic hyponatremia
In asymptomatic hyponatremia, the correction rate of hyponatremia need be no more than 0.5 mEq/L/h. No specific treatment is needed for patients with reset osmostats.
(1) Water restriction
Water intake should be restricted to 0.5 1 L/d. A gradual increase of serum sodium will occur over days.
(2) 0.9% saline
0.9% saline with furosemide may be used in asymptomatic patients whose serum sodium is less than 120 mEq/L. Urinary sodium and potassium losses are replaced as above.
(3) Demeclocycline
Demeclocycline (300 600 mg twice daily) is useful for patients who cannot adhere to water restriction or need additional therapy; it inhibits the effect of ADH on the distal tubule. Onset of action may require 1 week, and concentrating may be permanently impaired. Therapy with demeclocycline in cirrhosis appears to increase the risk of renal failure.
(4) Fludrocortisone
Hyponatremia occurring as part of the cerebral salt-wasting syndrome can be treated with fludrocortisone.
(5) Selective vasopressin V2 antagonist
The renal effect of ADH on water excretion is mediated by the V2 receptor. Oral selective V2 antagonists have been in clinical trials and should become available for treatment of SIADH in the near future.
P.893
C. Hypervolemic Hypotonic Hyponatremia
Hyponatremia with increased extracellular fluid volume is seen when hyponatremia is accompanied by edema-associated disorders such as congestive heart failure, cirrhosis, nephrotic syndrome, and advanced renal disease (Figure 21-1). In congestive heart failure, total body sodium is increased, yet effective circulating volume is sensed as inadequate by baroreceptors. Increased ADH and aldosterone results, with retention of water and sodium.
The urine sodium concentration is generally less than 10 mEq/L unless the patient has been taking diuretics.
Treatment
A. Water Restriction
The treatment of hyponatremia is that of the underlying condition (eg, improving cardiac output in congestive heart failure) and water restriction (to < 1 2 L of water daily).
B. Diuretics and V2 Antagonists
To hasten excretion of water and salt, use of diuretics may be indicated. Because diuretics may worsen hyponatremia, the patient must be cautioned not to increase free water intake. A potential role for V2 antagonists for the treatment of hyponatremia in congestive heart failure is under investigation.
C. Hypertonic (3%) Saline
Hypertonic saline administration is dangerous in volume-overloaded states and is not routinely recommended. In patients with severe hyponatremia (serum sodium < 110 mEq/L) and central nervous system symptoms, judicious administration of small amounts (100 200 mL) of 3% saline with diuretics may be necessary. Emergency dialysis should also be considered.
Adrogue HJ et al: Hyponatremia. N Engl J Med 2000;342:1581.
Castello L et al: Hyponatremia in liver cirrhosis: pathophysiological principles of management. Dig Liver Dis 2005;37:73.
Decaux G et al: Treatment of symptomatic hyponatremia. Am J Med Sci 2003;326:25.
Goh KP: Management of hyponatremia. Am Fam Physician 2004;69:2387.
Goldsmith SR: Current treatments and novel pharmacologic treatments for hyponatremia in congestive heart failure. Am J Cardiol 2005;95(9A):14B.
Hoorn EJ et al: Diagnostic approach to a patient with hyponatremia: traditional versus physiology-based options. QJM 2005; 98:529.
Milionis HJ et al: The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ 2002;166:1056.
Moritz ML et al: The pathophysiology and treatment of hyponatremic encephalopathy: an update. Nephrol Dial Transplant 2003;18:2486.
Hypernatremia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Occurs most commonly when water intake or water supplementation is inadequate, as in patients with altered mental status.
Urine osmolality helps differentiate renal from nonrenal water loss.
General Considerations
An intact thirst mechanism usually prevents hypernatremia (> 145 mEq/L). Thus, whatever the underlying disorder (eg, dehydration, lactulose or mannitol therapy, central and nephrogenic diabetes insipidus), excess water loss can cause hypernatremia only when adequate water intake is not possible, as with unconscious patients.
Rarely, excessive sodium intake may cause hypernatremia. Hypernatremia in primary aldosteronism is mild and usually does not cause symptoms. Hypernatremia in the presence of salt and water overload is uncommon but has been reported in very ill patients in the course of therapy.
Clinical Findings
A. Symptoms and Signs
When dehydration exists, orthostatic hypotension and oliguria are typical findings. Because water shifts from the cells to the intravascular space to protect volume status, these symptoms may be delayed. Hyperthermia, delirium, and coma may be seen with severe hyperosmolality.
B. Laboratory Findings
1. Urine osmolality > 400 mosm/kg
Renal water-conserving ability is functioning.
a. Nonrenal losses
Hypernatremia will develop if water ingestion fails to keep up with hypotonic losses from excessive sweating, exertional losses from the respiratory tract, or through stool water. Lactulose causes an osmotic diarrhea with loss of free water.
b. Renal losses
Whereas diabetic hyperglycemia can cause pseudohyponatremia (see above), progressive volume depletion from the osmotic diuresis of glycosuria can result in true hypernatremia. Osmotic diuresis can occur with the use of mannitol or urea.
2. Urine osmolality < 250 mosm/kg
A dilute urine with osmolality less than 250 mosm/kg with hypernatremia is characteristic of central and nephrogenic diabetes insipidus. Nephrogenic diabetes insipidus, seen with lithium or demeclocycline therapy, after relief of prolonged urinary tract obstruction, or with interstitial
P.894
nephritis, results from renal insensitivity to ADH. Hypercalcemia and hypokalemia may be contributing factors when present.
Treatment
Treatment of hypernatremia is directed toward correcting the cause of the fluid loss and replacing water and, as needed, electrolytes. In response to increases in plasma osmolality, brain cells synthesize solutes or idiogenic osmoles which increase osmotic flow of water back into the brain cells to regulate their volume. This begins 4 6 hours after dehydration and takes several days to reach a steady state. If hypernatremia is too rapidly corrected, the osmotic imbalance may cause water to preferentially enter brain cells, causing cerebral edema and potentially severe neurologic impairment. Fluid therapy should be administered over a 48-hour period, aiming for a decrease in serum sodium of 1 mEq/L/h (1 mmol/L/h). Potassium and phosphate may be added as indicated by serum levels; other electrolytes are also monitored frequently.
A. Choice of Type of Fluid for Replacement
1. Hypernatremia with hypovolemia
Severe hypovolemia should be treated with isotonic (0.9%) saline to restore the volume deficit and to treat the hyperosmolality, since the osmolality of isotonic saline (308 mosm/kg) is often lower than that of the plasma. This should be followed by 0.45% saline to replace any remaining free water deficit. Milder volume deficit may be treated with 0.45% saline and 5% dextrose in water.
2. Hypernatremia with euvolemia
Water drinking or 5% dextrose and water intravenously will result in excretion of excess sodium in the urine. If the GFR is decreased, diuretics will increase urinary sodium excretion but may impair renal concentrating ability, increasing the quantity of water that needs to be replaced.
3. Hypernatremia with hypervolemia
Treatment consists of providing water as 5% dextrose in water to reduce hyperosmolality, but this will expand vascular volume. Thus, loop diuretics such as furosemide (0.5 1 mg/kg) should be administered intravenously to remove the excess sodium. In severe renal insufficiency, hemodialysis may be necessary.
B. Calculation of Water Deficit
When calculating fluid replacement, both the deficit and the maintenance requirements should be added to each 24-hour replacement regimen.
1. Acute hypernatremia
In acute dehydration without much solute loss, free water loss is similar to the weight loss. Initially, 5% dextrose in water may be used. As correction of water deficit progresses, therapy should continue with 0.45% saline with dextrose.
2. Chronic hypernatremia
Water deficit is calculated to restore normal osmolality for total body water. Total body water (TBW) (Table 21-1) correlates with muscle mass and therefore decreases with advancing age, cachexia, and dehydration and is lower in women than in men. Current TBW equals 0.4 0.6 % current body weight.

Adrogue HJ et al: Hypernatremia. N Engl J Med 2000;342:1493.
Fall PJ: Hyponatremia and hypernatremia. A systematic approach to causes and their correction. Postgrad Med 2000;107:75.
Kugler JP et al: Hyponatremia and hypernatremia in the elderly. Am Fam Physician 2000;61:3623.
Lin M et al: Disorders of water imbalance. Emerg Med Clin North Am 2005;23:749.
Hyperosmolar Disorders & Osmolar Gaps
Hyperosmolality With Transient Or No Significant Shift In Water
Urea and alcohol are two substances that readily cross cell membranes and can produce hyperosmolality. Because of its permeant nature, urea has little effect on the shift of water across the cell membrane. Alcohol quickly equilibrates between intracellular and extracellular water, adding 22 mosm/L for every 1000 mg/L of ethanol. This measured hyperosmolality does not produce symptoms by itself because of the equilibrium described, but in any case of stupor or coma in which measured osmolality exceeds that calculated from values of serum Na+ and glucose and BUN, ethanol intoxication should be considered as a possible explanation of the discrepancy (osmolar gap). Toxic alcohol ingestion, particularly methanol or ethylene glycol, also causes an osmolar gap characterized by anion gap metabolic acidosis (see Chapter 39).
The combination of anion gap metabolic acidosis and an osmolar gap exceeding 10 mosm/kg is not specific for toxic alcohol ingestion. Nearly 50% of patients with alcoholic ketoacidosis or lactic acidosis have similar findings, caused in part by elevations of endogenous glycerol, acetone, and acetone metabolites (see Metabolic Acidosis).
Hyperosmolality Associated With Significant Shifts In Water
Increased concentrations of solutes that do not readily enter cells produce a shift of water from the intracellular space to effect a true intracellular dehydration. Sodium and glucose are the solutes commonly involved.
P.895
In these instances, the hyperosmolality does produce symptoms.
Clinical symptoms are mainly referred to the central nervous system. The severity of symptoms depends on the degree of hyperosmolality and rapidity of development. In acute hyperosmolality, symptoms of somnolence and confusion can appear when the osmolality exceeds 320 330 mosm/L, and coma, respiratory arrest, and death can result when it exceeds 340 350 mosm/L.
Chiasson JL et al: Diagnosis and treatment of diabetic ketoacidosis and the hyperglycemic hyperosmolar state. CMAJ 2003; 168:859.
Delaney MF et al: Diabetic ketoacidosis and hyperglycemic hyperosmolar nonketotic syndrome. Endocrinol Metab Clin North Am 2000;29:683.
Stoner GD: Hyperosmolar hyperglycemic state. Am Fam Physician 2005;71:1723.
Disorders of Potassium Concentration
Hypokalemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Severe hypokalemia may induce dangerous arrhythmias and even rhabdomyolysis.
Rule out intracellular potassium shifts.
Assess urinary potassium excretion to rule out renal loss.
When renal loss is suspected, evaluate mineralocorticoid action by urinary sodium and potassium excretion, transtubular [K+] gradient, and plasma aldosterone level.
General Considerations
The total potassium content of the body is 50 mEq/kg, more than 95% of which is intracellular. The plasma potassium concentration is maintained in a narrow range through two main regulating mechanisms: potassium shift between intracellular and extracellular compartments and modulation of renal potassium excretion. A deficit of 4 5 mEq/kg exists for each 1 mEq/L decrement in serum potassium concentration below a level of 4 mEq/L.
Clinical Findings
A. Symptoms and Signs
Muscular weakness, fatigue, and muscle cramps are frequent complaints in mild to moderate hypokalemia. Smooth muscle involvement may result in constipation or ileus. Flaccid paralysis, hyporeflexia, hypercapnia, tetany, and rhabdomyolysis may be seen with severe hypokalemia (< 2.5 mEq/L).
B. Laboratory Findings
The electrocardiogram (ECG) shows decreased amplitude and broadening of T waves, prominent U waves, premature ventricular contractions, and depressed ST segments. Hypokalemia also increases the likelihood of digitalis toxicity. Thus, in patients with heart disease, hypokalemia induced by certain drugs such as 2-adrenergic agonists and diuretics may impose a substantial risk.
Pathophysiology & Diagnosis
Hypokalemia can occur as a result of shifting of potassium intracellularly from the extracellular space, extrarenal potassium loss (or insufficient potassium intake), or renal potassium loss (Table 21-4). Potassium uptake by
P.896
the cell is stimulated by insulin in the presence of glucose. It is also facilitated by -adrenergic stimulation, whereas -adrenergic stimulation blocks it. All of these effects are transient. Self-limited hypokalemia occurs in 50 60% of trauma patients, perhaps related to enhanced release of epinephrine. Profound hypokalemia due to barium or cesium intoxication has been reported that may also be the result of transport of potassium into cells. Hypokalemia in the presence of acidosis suggests profound potassium depletion and requires urgent treatment.
Table 21-4. Causes of hypokalemia. | |
|---|---|
|
Table 21-5. Genetic disorders associated with electrolyte metabolism disturbances. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
The most common cause of hypokalemia, especially in developing countries, is gastrointestinal loss due to infectious diarrhea. The potassium concentration in intestinal secretion is 10 times higher (80 mEq/L) than in gastric juice. Aldosterone, which facilitates urinary potassium excretion through enhanced potassium secretion at the distal renal tubules, is the most important regulator of body potassium content. Urinary potassium concentration is low (< 20 mEq/L) as a result of extrarenal fluid loss (eg, diarrhea, vomiting) and inappropriately high (> 40 mEq/L) with urinary losses (eg, mineralocorticoid excess, Bartter's syndrome, Liddle's syndrome). Various genetic mutations that affect fluid and electrolyte metabolism, including disorders of potassium metabolism, have been reported recently, for which the presence or absence of hypertension may serve as a clue to the diagnosis (Table 21-5). Licorice-induced hypokalemia results from inhibition of 11 -hydroxysteroid dehydrogenase, which inactivates cortisol. Cortisol thus escapes degradation, binds to aldosterone receptors, and exerts aldosterone-like effects. It has been shown that homozygous mutations of the related gene lead to apparent mineralocorticoid excess. For an example, a mutation of a mineralocorticoid receptor increases its affinity to progesterone. In affected patients, hypertension and hypokalemia develop during pregnancy.
The transtubular [K+] gradient (TTKG) is a simple and rapid evaluation of net potassium secretion. TTKG is calculated as follows:
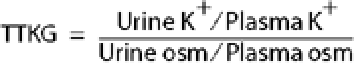
Hypokalemia with a TTKG > 4 suggests renal potassium loss with increased distal K+ secretion. In such cases, plasma renin and aldosterone levels are helpful in differential diagnosis. The presence of nonabsorbed anions, including bicarbonate, also increases TTKG.
Magnesium is an important cofactor for potassium uptake and for maintenance of intracellular potassium levels. Loop diuretics (eg, furosemide) cause substantial renal potassium and magnesium losses. Magnesium depletion should be suspected in refractory hypokalemia despite potassium repletion.
Treatment
The safest way to treat mild to moderate deficiency is with oral potassium, and all potassium formulations are easily absorbed. Dietary potassium is almost entirely coupled
P.897
to phosphate rather than chloride and is therefore not effective in correcting potassium loss associated with chloride depletion, such as from diuretics or vomiting. In the setting of abnormal renal function and mild to moderate diuretic dosage, 20 mEq/d of oral potassium is generally sufficient to prevent hypokalemia, but 40 100 mEq/d over a period of days to weeks is needed to treat hypokalemia and fully replete potassium stores.
Intravenous potassium replacement is indicated for patients with severe hypokalemia and for those who cannot take oral supplementation. For severe deficiency, potassium may be given through a peripheral intravenous line in a concentration that should not exceed 40 mEq/L at rates of up to 40 mEq/L/h. Continuous ECG monitoring is indicated, and the serum potassium level should be checked every 3 6 hours. For the initial administration, avoid glucose-containing fluid to prevent further shifts of potassium into the cells. Magnesium deficiency also needs to be corrected at the same time, particularly in refractory hypokalemia.
Coca SG et al: The cardiovascular implications of hypokalemia. Am J Kidney Dis 2005;45:233.
Cohn JN et al: New guidelines for potassium replacement in clinical practice. Arch Intern Med 2000;160:2429.
Groeneveld JH et al: An approach to the patient with severe hypokalemia: the potassium quiz. QJM 2005;98:305.
Schaefer TJ et al: Disorders of potassium. Emerg Med Clin North Am 2005;23:723.
Welfare W et al: Challenges in managing profound hypokalemia. BMJ 2002;324:269.
Hyperkalemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Hyperkalemia may develop in patients taking ACE inhibitors, angiotensin receptor blockers, potassium-sparing diuretics, or their combination, even with no or only mild renal dysfunction.
The ECG may show peaked T waves, widened QRS and biphasic QRS-T complexes, or may be normal despite life-threatening hyperkalemia.
Measurement of plasma potassium level differentiates potassium leak from blood cells in cases of clotting, leukocytosis, and thrombocytosis from elevated serum potassium.
Rule out extracellular potassium shift from the cells in acidosis and assess renal potassium excretion.
General Considerations
Hyperkalemia usually develops in patients with advanced renal dysfunction but can also develop with no or only mild renal dysfunction. Many cases of hyperkalemia are spurious or associated with acidosis (Table 21-6). The common practice of repeatedly clenching and unclenching a fist during venipuncture may raise the potassium concentration by 1 2 mEq/L by causing acidosis and consequent potassium loss from cells.
Intracellular potassium shifts to the extracellular fluid in hyperkalemia associated with acidosis. Serum potassium concentration rises about 0.7 mEq/L for every decrease of 0.1 pH unit during acidosis. Potassium movement out of cells occurs primarily in metabolic acidosis due to the accumulation of minerals such as NH4Cl or HCl. The inability of the chloride anion to permeate the cell membrane results in the transcellular exchange of H+ for K+. Metabolic acidosis from organic acids (keto acids and lactic acid) does not induce hyperkalemia. Unlike the minerals, these organic acids easily permeate cell membranes and retard Na+-K+-ATPase. The hyperkalemia frequently observed
P.898
in diabetic ketoacidosis is not due to the acidosis but to a combination of the hyperosmolality (the intracellular K+ concentration of the dehydrated cell increases and K+ diffuses extracellularly) and deficiencies of insulin, catecholamines, and aldosterone. Aminocaproic acid, a synthetic amino acid structurally related to lysine and arginine used for the prevention of operative blood loss, may induce shift of potassium. In the absence of acidosis, serum potassium concentration rises about 1 mEq/L when there is a total body potassium excess of 1 4 mEq/kg. However, the higher the serum potassium concentration, the smaller the excess necessary to raise the potassium levels further.
Table 21-6. Causes of hyperkalemia. | ||
|---|---|---|
|
Mineralocorticoid deficiency from Addison's disease (high renin) or chronic kidney disease (low renin) is another cause of hyperkalemia with decreased renal excretion of potassium. Mineralocorticoid resistance due to genetic disorders, interstitial renal disease, or urinary tract obstruction also leads to hypokalemia.
ACE inhibitors or angiotensin receptor blockers, commonly used in patients with congestive heart failure or renal insufficiency, may cause hyperkalemia. Recent trends involving simultaneous use of spironolactone or eplerenone, or -blockers further increases the risk of hyperkalemia. Thiazide or loop diuretics and sodium bicarbonate may be effective in minimizing hyperkalemia. Mild hyperkalemia that recurs often in patients in the absence of ACE inhibitor drug therapy is usually due to type IV renal tubular acidosis (RTA). Heparin inhibits aldosterone production by inhibiting the final enzymatic step in its manufacture in the adrenal glands, and thus can be a cause of hyperkalemia.
Trimethoprim is structurally related to amiloride and triamterene, and all three drugs inhibit renal potassium excretion through suppression of sodium channels in the distal nephron. Serum potassium levels rise progressively over 4 5 days in patients treated with standard or high-dose trimethoprim (combined with sulfamethoxazole or dapsone), especially if there is concurrent renal insufficiency. Over 50% of inpatients taking this drug have potassium levels over 5 mEq/L, and 20% have marked hyperkalemia (> 5.5 mEq/L). The potassium concentration returns to baseline after drug discontinuation.
In addition, it is of note that immunosuppressive drugs such as cyclosporine and tacrolimus can induce hyperkalemia in organ transplant recipients and especially in kidney transplant patients. This is partly due to the suppression of basolateral Na+-K+-ATPase in principal cells. Furthermore, five cases have been recently reported of severe hyperkalemia and cardiovascular disturbances caused by the use of drugs with KATP channel-opening properties (K-channel syndrome), such as nicorandil, cyclosporine, or isoflurane. The hyperkalemia was successfully reversed by the administration of glibenclamide.
Hyperkalemia is commonly seen in HIV-infected patients and has been attributed to impaired renal excretion of potassium due to the use of pentamidine or trimethoprim-sulfamethoxazole or to hyporeninemic hypoaldosteronism.
Clinical Findings
An elevated K+ concentration interferes with normal neuromuscular function to produce muscle weakness and, rarely, flaccid paralysis; abdominal distention and diarrhea may occur. Electrocardiography is not a sensitive method for detecting hyperkalemia, since nearly half of patients with a serum potassium level greater than 6.5 mEq/L will not manifest ECG changes. ECG changes in hyperkalemia include peaked T waves of increased amplitude, widening of the QRS, and biphasic QRS-T complexes. Inhibition of atrial depolarization despite normal conduction through usual pathways may occur. This sinoventricular rhythm resembles a junctional mechanism and occurs because of greater sensitivity of atrial myocytes to hyperkalemia than is the case for ventricular muscle cells. The heart rate may be slow; ventricular fibrillation and cardiac arrest are terminal events.
Treatment
First confirm that the elevated level of serum K+ is genuine. Potassium concentration can be measured in plasma rather than in serum to avoid leakage of potassium out of cells into the serum of the blood sample in the course of clotting, which may be observed in thrombocytosis. Renal dysfunction should be ruled out at the initial assessment.
Treatment consists of withholding potassium and giving cation exchange resins by mouth or enema. Sodium polystyrene sulfonate, 40 80 g/d in divided doses, is usually effective. Emergent treatment of hyperkalemia is indicated if cardiac toxicity or muscular paralysis is present or if the hyperkalemia is severe (serum potassium > 6.5 7 mEq/L) even in the absence of ECG changes. Insulin plus 10 50% glucose (5 10 g of glucose per unit of insulin) may be given to deposit K+ with glycogen in the liver (Table 21-7). Calcium may be given intravenously as an antagonist ion but not when digoxin toxicity is suspected, since calcium may augment the deleterious effects of digoxin on the heart. Transcellular shifts of potassium can also be mediated by 2-adrenergic stimulation. Thus, one or two standard doses of nebulized albuterol can reduce serum K+ 0.5 1 mEq/L within 30 minutes after administration in dialysis patients, and this effect is sustained for at least 2 hours. Sodium bicarbonate can be given intravenously as an emergency measure in severe hyperkalemia; the increase in blood pH results in a shift of K+ into cells. Hemodialysis or peritoneal dialysis may be required to remove K+ in the presence of protracted renal insufficiency. Therapy of the precipitating event proceeds concurrently.
Table 21-7. Treatment of hyperkalemia. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Gross P et al: Hyperkalemia: again. Nephrol Dial Transplant 2004;19:2163.
Halperin ML et al: Potassium. Lancet 1998;352:135.
Kamel KS et al: Controversial issues in the treatment of hyperkalemia. Nephrol Dial Transplant 2003;18:2215.
Palmer BF: Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585.
P.899
Singer M et al: Reversal of life-threatening, drug-related potassium-channel syndrome by glibenclamide. Lancet 2005; 365:1873.
Tamirisa KP et al: Spironolactone-induced renal insufficiency and hyperkalemia in patients with heart failure. Am Heart J 2004;148:971.
Disorders of Calcium Concentration
The normal total plasma (or serum) calcium concentration is 9 10.3 mg/dL. It is ionized calcium (normal: 4.7 5.3 mg/dL) that is physiologically active and is necessary for muscle contraction and nerve function.
Calcium-sensing protein, a receptor-like protein with the special function of detecting extracellular calcium ion concentrations, has been identified in parathyroid cells and in the kidney. Some diseases (eg, familial hypocalcemia and familial hypocalciuric hypercalcemia) associated with disturbed calcium metabolism are due to functional defects of this protein (Table 21-5).
Hypocalcemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Often mistaken as a neurologic disorder.
Check for decreased parathyroid hormone (PTH), vitamin D, or magnesium depletion.
If the ionized calcium level is normal despite a low total serum calcium, calcium metabolism is usually normal.
General Considerations
Development of true hypocalcemia (decreased ionized calcium) implies insufficient action of PTH or active vitamin D. The most common cause of low total serum
P.900
calcium is hypoalbuminemia; correction of serum calcium concentration is needed to accurately reflect the ionized calcium concentration. When albumin is low, serum Ca2+ concentration is depressed in a ratio of 0.8 1 mg of Ca2+ to 1 g of albumin. Thus,
Corrected calcium2+ (mg/dL) = Ca2+ (mg/dL) + 0.8 ~ 1.0 (4 - albumin [g/dL])
Important causes of hypocalcemia are listed in Table 21-8.
The most common cause of hypocalcemia is renal failure, in which decreased production of active vitamin D3 and hyperphosphatemia both play a role (see Chapter 22). Some cases of primary hypoparathyroidism are due to mutation of calcium-sensing protein in which inappropriate suppression of PTH release leads to hypocalcemia (see Chapter 15). Hypocalcemia in pancreatitis is also a marker for severe disease. Elderly hospitalized patients with low ionized serum calcium and hypophosphatemia, with or without an elevated parathyroid level, are likely deficient in vitamin D.
Clinical Findings
A. Symptoms and Signs
Hypocalcemia increases excitation of nerve and muscle cells, primarily affecting the neuromuscular and cardiovascular systems. Extensive spasm of skeletal muscle causes cramps and tetany. Laryngospasm with stridor can obstruct the airway. Convulsions can occur as well as paresthesias of lips and extremities and abdominal pain. Chvostek's sign (contraction of the facial muscle in response to tapping the facial nerve anterior to the ear) and Trousseau's sign (carpal spasm occurring after occlusion of the brachial artery with a blood pressure cuff for 3 minutes) are usually readily elicited. Prolongation of the QT interval (due to lengthened ST segment) predisposes to the development of ventricular arrhythmias. In chronic hypoparathyroidism, cataracts and calcification of basal ganglia of the brain may appear (see Hypoparathyroidism, Chapter 26).
Table 21-8. Causes of hypocalcemia. | ||
|---|---|---|
|
B. Laboratory Findings
Serum calcium concentration is low (< 9 mg/dL). In true hypocalcemia, the ionized serum calcium concentration is also low (< 4.7 mg/dL). Serum phosphate is usually elevated in hypoparathyroidism or end-stage renal failure, whereas it is suppressed in early-stage renal failure or vitamin D deficiency.
Serum magnesium concentration is commonly low, and hypomagnesemia reduces both PTH release and tissue responsiveness to PTH, causing hypocalcemia. In respiratory alkalosis, total serum calcium is normal but ionized calcium is low. The ECG shows a prolonged QT interval.
Treatment1
A. Severe, Symptomatic Hypocalcemia
In the presence of tetany, arrhythmias, or seizures, calcium gluconate 10% (10 20 mL) administered intravenously over 10 15 minutes is indicated. Because of the short duration of action, calcium infusion is usually required. Ten to 15 milligrams of calcium per kilogram body weight, or six to eight 10-mL vials of 10% calcium gluconate (558 744 mg of calcium), is added to 1 L of D5W and infused over 4 6 hours. By monitoring the serum calcium level frequently (every 4 6 hours), the infusion rate is adjusted to maintain the serum calcium level at 7 8.5 mg/dL.
B. Asymptomatic Hypocalcemia
Oral calcium (1 2 g) and vitamin D preparations are used. Calcium carbonate is well tolerated and less expensive than many other calcium tablets. The low serum Ca2+ associated with low serum albumin concentration does not require replacement therapy. If serum Mg2+ is low, therapy must include replacement of magnesium, which by itself will usually correct hypocalcemia.
Ariyan CE et al: Assessment and management of patients with abnormal calcium. Crit Care Med 2004;32(4 Suppl):S146.
Diercks DB et al: Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004;27:153.
P.901
Lyman D: Undiagnosed vitamin D deficiency in the hospitalized patient. Am Fam Physician 2005;71:299.
Hypercalcemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Primary hyperparathyroidism and malignancy-associated hypercalcemia are the most common causes.
Hypercalciuria usually precedes hypercalcemia.
Most often, asymptomatic, mild hypercalcemia ( 11 mg/dL) is due to primary hyperparathyroidism, whereas the symptomatic, severe hypercalcemia ( 14 mg/dL) is due to hypercalcemia of malignancy.
General Considerations
Important causes of hypercalcemia are listed in Table 21-9. Primary hyperparathyroidism and malignancy account for 90% of all cases of hypercalcemia. Primary hyperparathyroidism is the most common cause of hypercalcemia (usually mild) in ambulatory patients. Chronic hypercalcemia (over 6 months) or some manifestation such as nephrolithiasis also suggests a benign cause. Tumor production of PTH-related proteins (PTHrP) is the most common paraneoplastic endocrine syndrome, accounting for most cases of hypercalcemia in inpatients (see Table 40-6). The neoplasm is clinically apparent in nearly all cases when the hypercalcemia is detected, and the prognosis is poor.
Table 21-9. Causes of hypercalcemia. | ||
|---|---|---|
|
Milk-alkali syndrome, which had become rare with the advent of nonabsorbable antacid therapy for ulcer disease, has had a resurgence related to calcium ingestion for prevention of osteoporosis. In the milk-alkali syndrome, massive calcium and vitamin D ingestion can cause hypercalcemic nephropathy. Because of the decreased GFR, retention of the alkali in the calcium antacid occurs and causes metabolic alkalosis, which can be worsened by the vomiting associated with this disorder.
Hypercalcemia also causes nephrogenic diabetes insipidus. Development of polyuria is mediated through activation of calcium-sensing receptors in collecting ducts. Volume depletion further worsens hypercalcemia.
Clinical Findings
A. Symptoms and Signs
Hypercalcemia may affect gastrointestinal, renal, and neurologic function. The focus of the history and physical examination should be on the duration of the process of hypercalcemia and evidence for a neoplasm. Mild hypercalcemia is often asymptomatic. Symptoms usually occur if the serum calcium is above 12 mg/dL and tend to be more severe if hypercalcemia develops acutely. Symptoms irrespective of cause are constipation and polyuria, except in hypocalciuric hypercalcemia, in which polyuria is absent. Other gastrointestinal symptoms may include nausea, vomiting, anorexia, and peptic ulcer disease. Renal colic or hematuria from nephrolithiasis may be present. Polyuria from hypercalciuria-induced nephrogenic diabetes insipidus can result in volume depletion and azotemia. Neurologic manifestations may range from mild drowsiness to weakness, depression, lethargy, stupor, and coma in severe hypercalcemia. Ventricular extrasystoles and idioventricular rhythm occur and can be accentuated by digitalis.
B. Laboratory Findings
A significant elevation of serum calcium is seen; the level must be interpreted in relation to the serum albumin level (see Hypocalcemia, above). A high serum chloride concentration and a low serum phosphate concentration in a ratio > 33 to 1 is suggestive of primary hyperparathyroidism where PTH decreases proximal tubular phosphate reabsorption. A low serum chloride concentration with a high serum bicarbonate concentration, along with elevations of BUN and creatinine, suggests milk-alkali syndrome. The highest serum calcium levels (> 15 mg/dL) generally occur in malignancy. More than 200 mg/d of urinary calcium excretion suggests hypercalciuria; less than 100 mg/d suggests
P.902
hypocalciuria. Hypercalciuric patients such as those with malignancy or those receiving oral active vitamin D therapy may easily develop hypercalcemia in case of volume depletion. Serum phosphate may or may not be low, depending on the cause. Hypocalciuric hypercalcemia occurs in milk-alkali syndrome, thiazide diuretic use, and familial hypocalciuric hypercalcemia.
The chest radiograph may reveal a malignancy or granulomatous disease. The ECG shows a shortened QT interval. Measurements of PTH and PTHrP help distinguish between malignancy-associated hypercalcemia (suppressed PTH, elevated PTHrP) and hyperparathyroidism (elevated PTH).
Treatment
Until the primary disease can be brought under control, renal excretion of calcium with resultant decrease in serum calcium concentration is promoted. Excretion of Na+ is accompanied by excretion of Ca2+.
The tendency in hypercalcemia is toward volume depletion from nephrogenic diabetes insipidus. Therefore, establishing euvolemia and inducing natriuresis by giving saline with furosemide is the emergency treatment of choice. In dehydrated patients with normal cardiac and renal function, 0.45% saline or 0.9% saline can be given rapidly (250 500 mL/h). Intravenous furosemide (20 40 mg every 2 hours) prevents volume overload and enhances Ca2+ excretion. Thiazides can actually worsen hypercalcemia (as can furosemide if inadequate saline is given).
Bisphosphonates are the mainstay of treatment of hypercalcemia of malignancy. They are safe, effective, and normalize calcium in more than 70% of patients, although it may require up to 48 72 hours before their full therapeutic effect is achieved. In emergency cases, dialysis with low or no calcium dialysate may be needed. A calcimimetic agent, cinacalcet hydrochloride, that suppresses PTH secretion and decreases serum calcium concentration holds promise as a future treatment option. See Chapter 40 for a discussion of the treatment of hypercalcemia of malignancy and Chapter 26 for a discussion of the treatment of hypercalcemia of hyperparathyroidism.
Typically, patients with end-stage renal disease who receive long-term dialysis develop hypocalcemia and hyperphosphatemia if they do not receive proper supplementation of calcium and active vitamin D. On the other hand, hypercalcemia can sometimes develop, particularly in the setting of severe secondary hyperparathyroidism, characterized by high levels of PTH and subsequent release of calcium from bone. Therapy may include intravenous vitamin D, which further increases the serum calcium concentration. Another type of hypercalcemia occurs when the PTH levels are low. In this setting, bone turnover is decreased, which results in a low buffering capacity for calcium. When calcium is administered in calcium-containing phosphate binders or in the dialysate, or when vitamin D is administered, hypercalcemia results. Hypercalcemia in dialysis patients usually occurs in the presence of hyperphosphatemia, and severe metastatic calcification, eg, involving blood vessels, may occur. Malignancy should also be considered as a cause of the hypercalcemia.
Footnote
1See also Chapter 26 for discussion of the treatment of hypoparathyroidism.
Bilezikian JP et al: Clinical practice. Asymptomatic primary hyperparathyroidism. N Engl J Med 2004;350:1746.
Caroll MF et al: A practical approach to hypercalcemia. Am Fam Physician 2003;67:1959.
Inzucchi SE: Management of hypercalcemia. Diagnostic workup, therapeutic options for hyperparathyroidism and other common causes. Postgrad Med 2004;115:27.
Sarko J: Bone and mineral metabolism. Emerg Med Clin North Am 2005;23:703.
Schwartz SR et al: Hypercalcemic hypocalciuria: a critical differential diagnosis for hyperparathyroidism. Otolaryngol Clin North Am 2004;37:887.
Disorders of Phosphorus Concentration
In plasma, phosphate is mainly present as inorganic phosphate, and this fraction is very small (< 0.2% of total phosphate). However, body phosphate metabolism is regulated through plasma inorganic phosphate.
Important determinants of plasma inorganic phosphate concentration are its renal excretion, intestinal absorption, and shift between the intracellular and extracellular spaces. In general, the kidney is the most important regulator of the serum phosphate level. PTH decreases the absorption of phosphate in the proximal tubule while 1 25 dihydroxy-vitamin D3 increases tubular phosphate reabsorption. Renal proximal tubular reabsorption of phosphate is decreased by volume expansion, corticosteroid administration, and proximal tubular dysfunction, such as occurs in Fanconi's syndrome due to myeloma or other diseases. Fibroblast growth factor 23 (FGF-23) is an additional phosphaturic hormone. Intestinal absorption of phosphate is facilitated by active vitamin D. PTH, which both stimulates phosphate release from bone and is phosphaturic, can lead to hypophosphatemia and to depletion of bone phosphate store if hypersecretion continues.
Growth hormone, on the other hand, augments proximal tubular reabsorption of phosphate. Cellular phosphate uptake is stimulated by various factors and conditions, including alkalemia, insulin, epinephrine, feeding, hungry bone syndrome, and accelerated cell proliferation.
Phosphorus metabolism and homeostasis are intimately related to calcium metabolism. See sections on metabolic bone disease in Chapter 26.
Hypophosphatemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Severe hypophosphatemia may cause tissue hypooxygenation and even rhabdomyolysis.
Renal loss of phosphate can be diagnosed by measuring urinary phosphate excretion and by calculating maximal tubular phosphate reabsorption rate (TmP/GFR).
PTH is one of the major factors that decrease TmP/GFR, leading to renal loss of phosphate.
P.903
General Considerations
Hypophosphatemia may occur in the presence of normal phosphate stores. Serious depletion of body phosphate stores may exist with low, normal, or high concentrations of phosphorus in serum. Leading causes of hypophosphatemia are listed in Table 21-10.
In the presence of severe hypophosphatemia (1 mg/dL or less), affinity of hemoglobin for oxygen is increased through a decrease in the erythrocyte 2,3-diphosphoglycerate concentration. This impairs tissue oxygenation and thus cell metabolism, which underlies the effects of hypophosphatemia such as muscle weakness or even rhabdomyolysis.
Table 21-10. Causes of hypophosphatemia. | ||
|---|---|---|
|
Severe hypophosphatemia is common and multifactorial in alcoholic patients. In acute alcohol withdrawal, increased plasma insulin and epinephrine along with respiratory alkalosis promote intracellular shift of phosphate. Vomiting, diarrhea, and poor dietary intake contribute to hypophosphatemia. Chronic alcohol use results in a decrease in the renal threshold of phosphate excretion. This renal tubular dysfunction reverses after a month of abstinence. Patients with chronic obstructive pulmonary disease and asthma commonly have hypophosphatemia, attributed to xanthine derivatives causing shifts of phosphate intracellularly and the phosphaturic effects of -adrenergic agonists, loop diuretics, xanthine derivatives, and corticosteroids. The metabolic syndrome, a major contributor to coronary heart disease risk, is associated with low phosphate (and magnesium) levels but the clinical significance of these disturbances is unclear. Refeeding or glucose administration to phosphate-depleted patients may cause fatal hypophosphatemia.
Moderate hypophosphatemia (1.0 2.5 mg/dL) occurs commonly in hospitalized patients and may not reflect decreased phosphate stores. Hypophosphatemia is a potent stimulator of 1 -hydroxylation of vitamin D in the kidney to form active vitamin D. However, in oncogenic osteomalacia, which accompanies various mesenchymal tumors, activation of vitamin D is suppressed in spite of hypophosphatemia. This suppression may be due to overproduction of phosphatonins, such as FGF-23. Serum phosphate levels also decrease transiently after food intake, thus fasting samples are recommended for an accurate analysis.
Clinical Findings
A. Symptoms and Signs
Acute, severe hypophosphatemia (0.1 0.2 mg/dL) can lead to rhabdomyolysis, paresthesias, and encephalopathy (irritability, confusion, dysarthria, seizures, and coma). Respiratory failure or failure to wean from a respirator may occur. Arrhythmias and heart failure are uncommon but serious manifestations. Acute hemolytic anemia has been reported with increased erythrocyte fragility and platelet dysfunction with petechial hemorrhages. There is increased susceptibility to gram-negative sepsis from impaired chemotaxis of leukocytes.
Chronic severe depletion may be manifested by anorexia, pain in muscles and bones, and fractures.
B. Laboratory Findings
Evaluation of urinary phosphate excretion is a useful clue to the diagnosis of hypophosphatemia. A spot urine with > 20 mg/dL of phosphate suggests renal
P.904
phosphate loss. Tubular phosphate reabsorption can be assessed by TmP/GFR.

where serum Pi = serum phosphate concentration
UPi = urine phosphate concentration
UV = urine volume
The normal range of TmP/GFR is 2.5 4.5 mg/dL; lower values indicate urinary phosphate loss. The main factors regulating TmP/GFR are PTH and phosphate intake. Increase of PTH or phosphate intake decreases TmP/GFR, so that more phosphate is excreted into the urine.
Measurement of plasma PTH or PTHrP levels may be helpful. Serum FGF-23 levels can also be measured; however, the clinical usefulness of doing so remains to be established.
Other clinical features may be suggestive of specific causes of hypophosphatemia. Evidence of anemia due to hemolysis may be present (eg, elevated serum lactate dehydrogenase). Rhabdomyolysis results in elevated serum creatine kinase (which contains mostly the MM fraction but also some MB fraction) and, in many cases, myoglobin in the urine. Other values vary according to the cause. Renal glycosuria and hypouricemia together with hypophosphatemia indicate Fanconi's syndrome. In chronic depletion, radiographs and biopsies of bones show changes resembling those of osteomalacia.
Treatment
Treatment is best directed toward prophylaxis by including phosphate in repletion and maintenance fluids. A rapid decline in calcium levels can occur with parenteral administration of phosphate; therefore, when possible, oral replacement of phosphate is preferable. Moderate hypophosphatemia (1.0 2.5 mg/dL) is usually asymptomatic and does not require treatment. The hypophosphatemia in patients with diabetic ketoacidosis will usually correct with normal dietary intake. Chronic hypophosphatemia can be treated with oral phosphate repletion. Phosphate salts are available in skim milk (approximately 1 g [33 mmol]/L). Tablets or capsules of mixtures of sodium and potassium phosphate may be given to provide 0.5 1 g (18 32 mmol) per day. For severe, symptomatic hypophosphatemia (serum phosphorus 1 mg/dL), an infusion should provide 279 310 mg (9 10 mmol)/12 h until the serum phosphorus exceeds 1 mg/dL and the patient can be switched to oral therapy. The infusion rate should be decreased if hypotension occurs. Because the response to phosphate supplementation is not predictable, monitoring of plasma phosphate, calcium, and potassium every 6 hours is necessary. A magnesium deficit often coexists and should be treated simultaneously.
Contraindications to therapy with phosphate salts include hypoparathyroidism, renal insufficiency, tissue damage and necrosis, and hypercalcemia. When hyperglycemia due to any cause is treated, phosphate accompanies glucose into cells, and hypophosphatemia may ensue.
Gaasbeek A et al: Hypophosphatemia: an update on its etiology and treatment. Am J Med 2005;118:1094.
Shiber JR et al: Serum phosphate abnormalities in the emergency department. J Emerg Med 2002;23:395.
Taylor BE et al: Treatment of hypophosphatemia using a protocol based on patient weight and serum phosphorus level in a surgical intensive care unit. J Am Coll Surg 2004;198:198.
Hyperphosphatemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Renal failure is the most common cause.
Hyperphosphatemia in the presence of hypercalcemia imposes a high risk of metastatic calcification.
General Considerations
Chronic renal insufficiency from decreased excretion of phosphorus and decreased renal hydroxylation of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D is the main cause of hyperphosphatemia. Other causes are listed in Table 21-11. Children normally have higher serum phosphate levels than adults.
Clinical Findings
A. Symptoms and Signs
The clinical manifestations are those of the underlying disorder (eg, chronic renal failure) of hypocalcemia. Inadequately treated hyperphosphatemia in chronic renal failure leads to secondary hyperparathyroidism, renal osteodystrophy, and extraosseous calcification of soft tissues.
Table 21-11. Causes of hyperphosphatemia. | |
|---|---|
|
B. Laboratory Findings
In addition to elevated phosphate, blood chemistry abnormalities are those of the underlying disease.
Treatment
In acute and chronic renal failure, dialysis will reduce serum phosphate. Absorption of phosphate can be reduced by administration of calcium carbonate, 0.5 1.5 g three times daily with meals (500 mg tablets). Another phosphate binder is sevelamer hydrochloride, which can be titrated to target phosphorus levels using 800 1600 mg three times daily with meals (400 and
P.905
800 mg tablets and 403 mg capsules). Because this agent does not contain calcium or aluminum, it may be especially useful for patients with hypercalcemia or uremia. Despite its usefulness, it has been suspected of producing mild hyperchloremic metabolic acidosis in patients with chronic kidney disease. The 2004 Calcium Acetate Renagel Evaluation (CARE) study on the treatment of hyperphosphatemia in hemodialysis patients concluded that in the absence of hypercalcemia, calcium therapy is more effective than sevelamer in the control of serum phosphorus and the calcium-phosphate product.
Akizawa T et al: New strategies for the treatment of secondary hyperparathyroidism. Am J Kidney Dis 2003;41(3 Suppl 1): S100.
Friedman EA: Consequences and management of hyperphosphatemia in patients with renal insufficiency. Kidney Int Suppl 2005;(95):S1.
Qunibi WY et al: Treatment of hyperphosphatemia in hemodialysis patients: The Calcium Acetate Renagel Evaluation (CARE Study). Kidney Int 2004;65:1914.
Shiber JR et al: Serum phosphate abnormalities in the emergency department. J Emerg Med 2002;23:39.
Disorders of Magnesium Concentration
The normal plasma concentration is 1.5 2.5 mEq/L, with about one-third bound to protein and two-thirds existing as free cation. Excretion of magnesium ion is via the kidney. Normally, about 3% of magnesium filtered by the glomerulus is excreted in urine. Magnesium exerts physiologic effects on the nervous system resembling those of calcium. Magnesium acts directly upon the myoneural junction.
Altered concentration of Mg2+ in the plasma usually provokes an associated alteration of Ca2+. Hypermagnesemia suppresses secretion of PTH with consequent hypocalcemia. Severe and prolonged magnesium depletion impairs secretion of PTH with consequent hypocalcemia. Hypomagnesemia may impair end-organ response to PTH as well.
Hypomagnesemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Serum concentration of magnesium may not be decreased even in the presence of magnesium depletion. Check urinary magnesium excretion if renal magnesium wasting is suspected.
Causes neurologic symptoms and arrhythmias.
Impairs release of PTH.
General Considerations
Causes of hypomagnesemia are listed in Table 21-12. Normomagnesemia does not exclude magnesium depletion because only 1% of total body magnesium is in the
P.906
extracellular fluid. Nearly 50% of hospitalized patients in whom serum electrolytes are ordered have unrecognized hypomagnesemia. Up to 40% of patients with hypomagnesemia have hypokalemia, and up to 50% have hypocalcemia. Hypomagnesemia and hypokalemia share many etiologies, including diuretics, diarrhea, alcoholism, aminoglycosides, and amphotericin B. Renal potassium wasting also occurs from hypomagnesemia, and is refractory to potassium rleplacement until magnesium is repleted. Hypomagnesemia also suppresses PTH release and causes end-organ resistance to it and to low 1,25-vitamin D levels. This hypocalcemia is also refractory to calcium replacement until the magnesium is repleted. In addition, molecular mechanisms of magnesium wasting have been revealed in some hereditary disorders.
Table 21-12. Causes of hypomagnesemia. | |
|---|---|
|
Clinical Findings
A. Symptoms and Signs
Common symptoms are those of hypokalemia and hypocalcemia, with weakness and muscle cramps. There is marked neuromuscular and central nervous system hyperirritability, with tremors, athetoid movements, jerking, nystagmus, and a positive Babinski response. There may be hypertension, tachycardia, and ventricular arrhythmias. Confusion and disorientation may be prominent features.
B. Laboratory Findings
Urinary excretion of magnesium exceeding 10 30 mg/d or a fractional excretion more than 2% indicates renal magnesium wasting. In calculating fractional excretion of magnesium, since only 30% is protein bound, it follows that 70% of circulating magnesium is filtered by the glomerulus. In addition to hypomagnesemia, hypocalcemia and hypokalemia are often present. The ECG shows a prolonged QT interval, due to lengthening of the ST segment. PTH secretion is often suppressed (see Hypocalcemia, above).
Treatment
Magnesium oxide, 250 500 mg by mouth once or twice daily, is useful for repleting stores in patients with chronic hypomagnesemia. Treatment of symptomatic hypomagnesemia can include an infusion of 1 2 g of magnesium sulfate, followed by an infusion of 6 g magnesium sulfate in at least 1 L of fluids over 24 hours, repeated for up to 7 days to replete magnesium stores. Magnesium sulfate may also be given intramuscularly in a dosage of 200 800 mg/d (8 33 mmol/d) in four divided doses. Serum levels must be monitored daily and dosage adjusted to keep the concentration from rising above 2.5 mmol/L. Tendon reflexes may also be checked, since hypermagnesemia causes hyporeflexia. K+ and Ca2+ replacement may be required as well, but patients with hypokalemia and hypocalcemia of hypomagnesemia do not recover without magnesium supplementation.
Patients with normal renal function can excrete excess magnesium and hypermagnesemia should not develop with replacement dosages. In patients with renal insufficiency, replacement of magnesium should be done cautiously to avoid hypermagnesemia. Reduced doses (50 75% dose reduction) and more frequent monitoring (at least twice daily) are indicated.
Tong GM et al: Magnesium deficiency in critical illness. J Intensive Care Med 2005;20:3.
Topf JM et al: Hypomagnesemia and hypermagnesemia. Rev Endocr Metab Disord 2003;4:195.
Touyz RM: Magnesium in clinical medicine. Front Biosci 2004; 9:1278.
Hypermagnesemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Almost always associated with renal insufficiency and a history of chronic intake of magnesium-containing drugs.
General Considerations
Magnesium excess is almost always the result of renal insufficiency and the inability to excrete what has been taken in from food or drugs, especially the long-term use of antacids and laxatives. Magnesium replacement should be done cautiously in patients with renal insufficiency, and dose reductions up to 75% may be needed to avoid hypermagnesemia.
Clinical Findings
A. Symptoms and Signs
Muscle weakness, decreased deep tendon reflexes, mental obtundation, and confusion are characteristic manifestations. Weakness even flaccid paralysis ileus, urinary retention, and hypotension are noted. There may be respiratory muscle paralysis or cardiac arrest.
B. Laboratory Findings
Serum Mg2+ is elevated. In the common setting of renal insufficiency, concentrations of BUN and of serum creatinine, phosphate, and uric acid are elevated; serum K+ may be elevated. Serum Ca2+ is often low. The ECG shows increased PR interval, broadened QRS complexes, and peaked T waves, probably related to associated hyperkalemia.
Treatment
Treatment is directed toward alleviating renal insufficiency. Calcium acts as an antagonist to Mg2+ and may be given intravenously as calcium chloride, 500 mg or more at a rate of 100 mg (4.5 mmol)/min. Hemodialysis or
P.907
peritoneal dialysis may be necessary to remove the magnesium, particularly when there is severe renal failure.
Long-term use of magnesium-containing drugs, such as magnesium hydroxylate and magnesium sulfate, should be avoided in patients with renal insufficiency.
Topf JM et al: Hypomagnesemia and hypermagnesemia. Rev Endocr Metab Disord 2003;4:195.
Acid-Base Disorders
To assess a patient's acid-base status, measurement of arterial pH, PCO2, and plasma bicarbonate (HCO3-) is needed. Blood gas analyzers directly measure pH and PCO2, and the HCO3- value is calculated from the Henderson-Hasselbalch equation:

The total venous CO2 measurement is a more direct determination of HCO3-. Because of the dissociation characteristics of carbonic acid (H2CO3) at body pH, dissolved CO2 is almost exclusively in the form of HCO3-, and for clinical purposes the total carbon dioxide content is equivalent ( 3 mEq/L) to the HCO3- concentration:
![]()
If precise measurements of oxygenation are not needed or if oxygen saturation obtained from the pulse oximeter is adequate, venous blood gases generally provide useful information for assessment of acid-base balance and can be used interchangeably with arterial blood gases since the arteriovenous differences in pH and PCO2 are small and relatively constant. Venous blood pH is usually 0.03 0.04 units lower than that of arterial blood, and venous blood PCO2 is 7 or 8 mm Hg higher. Calculated HCO3- concentration in venous blood is at most 2 mEq/L higher than that of arterial blood. Serum HCO3- measurement also provides equivalent information to the arterial base deficit in surgical intensive care unit patients. An important exception to the rule of interchangeability between arterial and venous blood gases for determination of acid-base balance is during cardiopulmonary arrest. In this setting, arterial pH may be 7.41 and venous pH 7.15, and arterial blood PCO2 can be 32 mm Hg with a venous blood PCO2 of 74 mm Hg.
Types of Acid-Base Disorders
There are two types of acid-base disorders: respiratory and metabolic. Primary respiratory disorders affect blood acidity by causing changes in PCO2, and primary metabolic disorders are caused by disturbances in the HCO3- concentration. The primary disturbances are usually accompanied by compensatory changes; however, even though these changes attenuate a pH shift from the normal value (7.40), they do not fully compensate for the primary acid-base disorders even if the disorders are chronic. Therefore, if the pH is less than 7.40, the primary process is acidosis (either respiratory or metabolic). If the pH is higher than 7.40, the primary process is either respiratory or metabolic alkalosis. The presence of one disorder with its appropriate compensatory change is a simple disorder.
Mixed Acid-Base Disorders
The presence of more than one simple disorder (not compensatory) is a mixed disorder. Double or triple disorders can coexist but not quadruple ones, because simultaneous respiratory acidosis and alkalosis are not possible.
Clinicians frequently find it difficult to decide if a mixed disorder is present. One useful scheme is to determine if the degree of compensation for the primary disorder is appropriate (Table 21-13). In respiratory disorders, if the magnitude of compensation in HCO3- level differs from what is predicted, the patient has a mixed disorder. Therefore, superimposed metabolic acidosis will decrease HCO3- to lower than the predicted level, and a metabolic alkalosis will increase HCO3- over the predicted value. For example, a patient with chronic respiratory acidosis and PCO2 of 60 mm Hg should have a HCO3- of 31 mEq/L (assuming that normal HCO3- is 24 mEq/L). If the HCO3- is 25 mEq/L, a superimposed metabolic acidosis exists, and if the HCO3- is 45 mEq/L, there is a superimposed metabolic alkalosis. Using data from Table 21-13, similar calculations can be made for primary metabolic disorders.
Furthermore, corrected bicarbonate (cHCO3-), calculated from measured HCO3- plus the increase in anion gap (see box), is useful to assess the superimposed metabolic alkalosis or normal anion gap metabolic acidosis. In increased anion gap acidosis, there must be a mole for mole decrease in HCO3- as anion gap increases. Therefore, an HCO3- value higher or lower than normal (24 mEq/L) indicates the concomitant presence of metabolic alkalosis or normal anion gap acidosis, respectively.
Step-By-Step Analysis of Acid-Base Status
Step 1: Determine the primary (or main) disorder whether it is metabolic or respiratory from blood, pH, HCO3-, and PCO2 values.
Step 2: Determine the presence of mixed acid-base disorders by calculating the range of compensatory responses (Table 21-13).
Step 3: Calculate the anion gap (Table 21-14).
Step 4: Calculate the corrected HCO3- concentration if the anion gap is increased (see above).
Step 5: Examine the patient to determine whether the clinical signs are compatible with the acid-base analysis thus obtained.
Table 21-13. Primary acid-base disorders and expected compensation. | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
P.908
Haber RJ: A practical approach to acid-base disorders. West J Med 1991;155:146.
Herd AM: An approach to complex acid-base problems: keeping it simple. Can Fam Physician 2005;51:226.
Kellum JA: Determinants of plasma acid-base balance. Crit Care Clin 2005;21:329.
Williamson JC: Acid-base disorders: classification and management strategies. Am Fam Physician 1995;52:584.
Metabolic Acidosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Decreased HCO3- with acidemia.
Classified into high anion gap acidosis and normal anion gap acidosis.
The highest anion gap acidoses are seen in lactic acidosis, ketoacidosis, or toxins.
Normal anion gap acidosis is mainly caused by gastrointestinal HCO3- loss or RTA. Urinary anion gap may help distinguish between these causes.
General Considerations
The hallmark of metabolic acidosis is decreased HCO3-, seen also in respiratory alkalosis (see above), but the pH distinguishes between the two disorders. Calculation of the anion gap is useful in determining the cause of the metabolic acidosis (Table 21-14). The anion gap represents the difference between readily measured anions and cations.
In plasma,
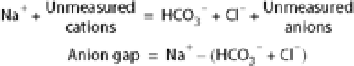
The major unmeasured cations are calcium (1 mEq/L), magnesium (2 mEq/L), -globulins, and potassium (4 mEq/L). The major unmeasured anions are negatively charged albumin (2 mEq/L per g/dL), phosphate (2 mEq/L), sulfate (1 mEq/L), lactate (1 2 mEq/L), and other organic anions (3 4 mEq/L). Traditionally, the normal anion gap has been 12 4 mEq/L.
P.909
With the current generation of autoanalyzers, the reference range may be lower (6 1 mEq/L), primarily from an increase in Cl- values. Despite its usefulness, the serum anion gap can be misleading. Non-acid-base disorders that may contribute to an error in anion gap interpretation include hypoalbuminemia (see below), antibiotic administration (eg, carbenicillin is an unmeasured anion; polymyxin is an unmeasured cation), hypernatremia, or hyponatremia.
Table 21-14. Abnormal anion gap.1 | ||
|---|---|---|
|
Decreased Anion Gap
A decreased anion gap can occur because of a reduction in unmeasured anions or an increase in unmeasured cations.
A. Decreased Unmeasured Anions
If the sodium concentration remains normal but HCO3- and Cl- increase, the anion gap will decrease. This is seen when there are decreased unmeasured anions, especially in hypoalbuminemia, which explains the low anion gap that frequently occurs in patients with hepatic cirrhosis. For every 1 g/dL decline in serum albumin, a 2 mEq/L decrease in anion gap will occur. Thus, without such a correction, the presence of an increased anion gap acidosis may be overlooked in patients who have marked hypoalbuminemia.
B. Increased Unmeasured Cations
If the sodium concentration falls because of addition of unmeasured cations but HCO3- and Cl- remain unchanged, the anion gap will decrease. This is seen in (1) severe hypercalcemia, hypermagnesemia, or hyperkalemia; (2) IgG myeloma, where the immunoglobulin is cationic in 70% of cases; and (3) lithium toxicity.
Jurado RL et al: Low anion gap. South Med J 1998;91:624.
Increased Anion Gap Acidosis (Increased Unmeasured Anions)
The hallmark of this disorder is that metabolic acidosis (thus low HCO3-) is associated with normal serum Cl-, so that the anion gap increases. Normochloremic metabolic acidosis generally results from addition to the blood of nonchloride acids such as lactate, acetoacetate, -hydroxybutyrate, and exogenous toxins. Unmeasured anions such as isocitrate, alpha-ketoglutarate, malate and D-lactate, may further contribute to the anion gap of lactic acidosis, diabetic ketoacidosis, and acidosis of unknown etiology. An exception is uremia, with underexcretion of organic acids and anions.
A. Lactic Acidosis
Lactic acid is formed from pyruvate in anaerobic glycolysis. Therefore, most of the lactate is produced in tissues with high rates of glycolysis, such as gut (responsible for over 50% of lactate production), skeletal muscle, brain, skin, and erythrocytes. Normally, lactate levels remain low (1 mEq/L) because of metabolism of lactate principally by the liver through gluconeogenesis or oxidation via the Krebs cycle. Furthermore, the kidneys metabolize about 30% of lactate.
In lactic acidosis, lactate levels are at least 4 5 mEq/L but commonly 10 30 mEq/L. The mortality rate exceeds 50%. There are two basic types of lactic acidosis, both associated with increased lactate production and decreased lactate utilization. Type A is characterized by hypoxia or decreased tissue perfusion, whereas in type B there is no clinical evidence of hypoxia.
Type A (hypoxic) lactic acidosis is the more common type, resulting from poor tissue perfusion; cardiogenic, septic, or hemorrhagic shock; and carbon monoxide or cyanide poisoning. These conditions not only cause lactic acid production to increase peripherally but, more importantly, hepatic metabolism of lactate to decrease as liver perfusion declines. In addition, severe acidosis impairs the ability of the liver to extract the perfused lactate.
Type B lactic acidosis may be due to metabolic causes, such as diabetes, ketoacidosis, liver disease, renal failure, infection, leukemia, or lymphoma, or it may occur as a result of toxicity from ethanol, methanol, salicylates, isoniazid, or metformin. Propylene glycol, used as a vehicle for intravenous agents such as nitroglycerin, etomidate, and high-dose diazepam, may cause lactic acidosis from liver metabolism. Nutritional problems are important causes of lactic acidosis. Parenteral nutrition without thiamin causes severe refractory lactic acidosis from the deranged metabolism of pyruvate. Patients with short bowel syndrome may develop D-lactic acidosis with encephalopathy due to carbohydrate malabsorption in the intestine and subsequent fermentation by colonic bacteria.
AIDS without AIDS-related lymphoma is associated with type B lactic acidosis. Treatment of HIV patients with nucleoside analog reverse transcriptase inhibitors may cause lactic acidosis due to mitochondrial toxicity.
Idiopathic lactic acidosis, usually in debilitated patients, has an extremely high mortality rate. (For treatment of lactic acidosis, see below and Chapter 27.)
B. Diabetic Ketoacidosis
This metabolic abnormality is characterized by hyperglycemia and metabolic acidosis (pH < 7.25 or plasma bicarbonate < 16 mEq/L). Anion gap metabolic acidosis is the acid-base disturbance generally ascribed to diabetic ketoacidosis:
![]()
where B- is -hydroxybutyrate or acetoacetate. The anion gap should be calculated from the serum electrolytes as measured, since correction of the serum sodium for the dilutional effect of hyperglycemia will incorrectly exaggerate the anion gap. The increased anion gap is due to hyperketonemia (acetoacetate and -hydroxybutyrate) and at times to an increase in
P.910
serum lactate secondary to reduced tissue perfusion and increased anaerobic metabolism. If a rise in anion gap from normal is equal to a fall in HCO3-, a diagnosis of simple metabolic acidosis can be made. However, the presence of concurrent metabolic alkalosis or normal anion gap metabolic acidosis is suggested if the value of the measured HCO3- plus the increase in anion gap (cHCO3-) is higher or lower than the normal value for HCO3-, respectively.
During the recovery phase of diabetic ketoacidosis, anion gap acidosis can be transformed into hyperchloremic non-anion gap acidosis. The mechanism for this is as follows: As GFR increases from NaCl therapy of diabetic ketoacidosis, the retention of Cl- causes a mild decrease in the anion gap from dilution. More importantly, the increased GFR causes the urinary excretion of ketone salts (NaB), which are formed as bicarbonate is consumed:
![]()
The kidney reabsorbs ketone anions poorly but can compensate for the loss of anions (and therefore Na+) by increasing the reabsorption of Cl-. Conversely, even on presentation, patients with diabetic ketoacidosis and normal renal perfusion may have marked ketonuria, severe metabolic acidosis, and only a mildly increased anion gap. Again, the variable relationship between the rise in the anion gap and the fall in the HCO3- can occur with the urinary loss of Na+ or K+ salts of -hydroxybutyrate, which will lower the anion gap without altering the H+ excretion or the severity of the acidosis. Because Ketostix reacts to acetoacetate, less to acetone, and not at all to the predominant keto acid, -hydroxybutyrate, the test may become more positive even as the patient improves owing to the metabolism of hydroxybutyrate. Thus, the patient's clinical status and the reduction of the anion gap are better markers of improvement than monitoring the serum acetone test. Conversely, in the presence of concomitant lactic acidosis, a shift in the redox state can increase -hydroxybutyrate and decrease the readily detectable acetoacetate, thus lowering the nitroprusside reaction.
C. Alcoholic Ketoacidosis
This is a common disorder of chronically malnourished patients who consume large quantities of alcohol daily. Most of these patients have mixed acid-base disorders (10% have a triple acid-base disorder). Although decreased HCO3- is usual, 50% of the patients may have normal or alkalemic pH. Three types of metabolic acidosis are seen in alcoholic ketoacidosis: (1) Ketoacidosis is due to -hydroxybutyrate and acetoacetate excess. (2) Lactic acidosis: Alcohol metabolism increases the NADH:NAD ratio, causing increased production and decreased utilization of lactate. Accompanying thiamin deficiency, which inhibits pyruvate carboxylase, further enhances lactic acid production in many cases. Moderate to severe elevations of lactate (> 6 mmol/L) are seen with concomitant disorders such as sepsis, pancreatitis, or hypoglycemia. (3) Hyperchloremic acidosis from bicarbonate loss in the urine is associated with ketonuria (see above). Metabolic alkalosis occurs from volume contraction and vomiting. Respiratory alkalosis results from alcohol withdrawal, pain, or associated disorders such as sepsis or liver disease. Half of the patients have either hypoglycemia or hyperglycemia. When serum glucose levels are greater than 250 mg/dL, the distinction from diabetic ketoacidosis is difficult. The diagnosis of alcoholic ketoacidosis is supported by the absence of a diabetic history and by no evidence of glucose intolerance after initial therapy.
D. Toxins
(See also Chapter 39.) Multiple toxins and drugs can increase the anion gap by increasing endogenous acid production. Examples include methanol (metabolized to formic acid), ethylene glycol (glycolic and oxalic acid), and salicylates (salicylic acid and lactic acid), which can cause a mixed disorder of metabolic acidosis with respiratory alkalosis. In toluene poisoning, a metabolite hippurate is rapidly excreted by the kidney and may present as a normal anion gap acidosis. Isopropyl alcohol, which is metabolized to acetone, increases the osmolar gap, but not the anion gap.
E. Uremic Acidosis
At GFRs below 20 mL/min, the inability to excrete H+ with retention of acid anions such as PO43- and SO42- results in an increased anion gap acidosis, which rarely is severe. The unmeasured anions replace HCO3- (which is consumed as a buffer). Hyperchloremic normal anion gap acidosis develops in milder cases of renal insufficiency.
Normal Anion Gap Acidosis (Table 21-15)
The hallmark of this disorder is that the low HCO3- of metabolic acidosis is associated with hyperchloremia, so that the anion gap remains normal. The most common causes are gastrointestinal HCO3- loss and defects in renal acidification (renal tubular acidoses). The urinary anion gap can differentiate between these two common causes (see below).
Table 21-15. Hyperchloremic, normal anion gap metabolic acidoses. | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||
A. Gastrointestinal HCO3- Loss
Bicarbonate is secreted in multiple areas in the gastrointestinal tract. Small bowel and pancreatic secretions contain large amounts of HCO3-. Therefore, massive diarrhea or pancreatic drainage can result in HCO3- loss because of increased HCO3- secretion and decreased absorption. Hyperchloremia occurs because the ileum and colon secrete HCO3- in a one-to-one exchange for Cl- by countertransport. The resultant volume contraction causes further increased Cl- retention by the kidney in the setting of decreased anion,
P.911
HCO3-. Patients with ureterosigmoidostomies can develop hyperchloremic metabolic acidosis because the colon secretes HCO3- in the urine in exchange for Cl-.
B. Renal Tubular Acidosis (RTA)
Hyperchloremic acidosis with a normal anion gap and normal (or near normal) GFR, and in the absence of diarrhea, defines RTA. The defect is either inability to excrete H+ (inadequate generation of new HCO3-) or inappropriate reabsorption of HCO3-. Three major types can be differentiated by the clinical setting, urinary pH, urinary anion gap (see below), and serum K+ level. (The term type III renal tubular acidosis is no longer used because of the controversies surrounding its definition.) Recently, the mechanisms of each abnormality have been better elucidated by identifying the responsible molecules and their gene mutations.
1. Classic distal RTA (type I)
This disorder is characterized by hypokalemic hyperchloremic metabolic acidosis and is due to selective deficiency in H+ secretion in intercalated cells in the collecting tubule. Despite acidosis, urinary pH cannot be acidified and is above 5.5, which retards the binding of H+ to phosphate (H+ + HPO42- H2PO4), and thus inhibits titratable acid excretion. Furthermore, urinary excretion of NH4+Cl- is decreased, and the urinary anion gap is positive (see below). Enhanced K+ excretion occurs probably because there is less competition from H+ in the distal nephron transport system. Furthermore, as a response to renal salt wasting, hyperaldosteronism occurs. Nephrocalcinosis and nephrolithiasis frequently accompany this disorder since chronic acidosis decreases tubular calcium reabsorption. The hypercalciuria, alkaline urine, and lowered level of urinary citrate cause calcium phosphate stones and nephrocalcinosis.
Distal RTA develops as a consequence of dysproteinemic syndromes, autoimmune disease, and drugs and toxins such as amphotericin B.
2. Proximal RTA (type II)
Proximal RTA is a hypokalemic hyperchloremic metabolic acidosis due to a selective defect in the proximal tubule's ability to adequately reabsorb filtered HCO3-. Carbonic anhydrase inhibitors (acetazolamide) can cause proximal RTA. About 90% of filtered HCO3- is absorbed by the proximal tubule. The distal nephron has a limited ability to absorb HCO3- but becomes overwhelmed and does not function adequately when there is increased delivery. Eventually, distal delivery of filtered HCO3- declines because the plasma HCO3- level has dropped as a result of progressive urinary HCO3- wastage. When the plasma HCO3- level drops to 15 18 mEq/L, delivery of HCO3- drops to the point where the distal nephron is no longer overwhelmed and can regain function. At that point, bicarbonaturia disappears, and urinary pH can be acidic. Thiazide-induced volume contraction can be used to enhance proximal HCO3- reabsorption, leading to the decrease in distal HCO3- delivery and improvement of bicarbonaturia and renal acidification. The increased delivery of HCO3- to the distal nephron also increases K+ secretion, and hypokalemia results if a patient is loaded with excess HCO3- and K+ is not adequately supplemented. Proximal RTA often exists with other defects of absorption in the proximal tubule, resulting in glucosuria, aminoaciduria, phosphaturia, and uricaciduria. Causes include multiple myeloma with Fanconi's syndrome and nephrotoxic drugs.
3. Hyporeninemic hypoaldosteronemic RTA (type IV)
Type IV is the most common form of RTA in clinical practice. This is the only type characterized by
P.912
hyperkalemic, hyperchloremic acidosis. The defect is aldosterone deficiency or antagonism, which impairs distal nephron Na+ reabsorption and K+ and H+ excretion. Renal salt wasting is frequently present. Relative hypoaldosteronism from hyporeninemia is most commonly found in diabetic nephropathy, tubulointerstitial renal diseases, hypertensive nephrosclerosis, and AIDS. In patients with these disorders, caution must be taken when using drugs that can exacerbate the hyperkalemia, such as ACE inhibitors (which will further reduce aldosterone levels), aldosterone receptor blockers such as spironolactone, and NSAIDs.
C. Dilutional Acidosis
Rapid dilution of plasma volume by 0.9% NaCl may cause a mild hyperchloremic acidosis.
D. Recovery from Diabetic Ketoacidosis
See earlier section, Increased Anion Gap Acidosis (Increased Unmeasured Anion).
E. Posthypocapnia
In prolonged respiratory alkalosis, HCO3- decreases and Cl- increases from decreased renal NH4+Cl- excretion. If the respiratory alkalosis is corrected quickly, PCO2 will increase acutely but HCO3- will remain low until the kidneys can generate new HCO3-, which generally takes several days. In the meantime, the increased PCO2 with low HCO3- causes metabolic acidosis.
F. Hyperalimentation
Hyperalimentation fluids may contain amino acid solutions that acidify when metabolized, such as arginine hydrochloride and lysine hydrochloride.
Urinary Anion Gap to Assess Hyperchloremic Metabolic Acidosis
Increased renal NH4+Cl- excretion to enhance H+ removal is a normal physiologic response to metabolic acidosis. NH3 reacts with H+ to form NH4+, which is accompanied by the anion Cl- for excretion. The normal daily urinary excretion of NH4Cl of about 30 mEq can be increased up to 200 mEq in response to acid load.
Urinary anion gap from a random urine sample ([Na+ + K+] Cl-) reflects the ability of the kidney to excrete NH4Cl as in the following equation:
![]()
where 80 is the average value for the difference in the urinary anions and cations other than Na+, K+, NH4+, and Cl. Therefore, urinary anion gap is equal to (80 NH4+), and thus aids in the distinction between gastrointestinal and renal causes of hyperchloremic acidosis. If the cause of the metabolic acidosis is gastrointestinal HCO3- loss (diarrhea), the renal acidification ability remains normal and NH4Cl excretion increases in response to the acidosis. The urinary anion gap is negative (eg, -30 mEq/L). If the cause is distal RTA, the urinary anion gap is positive (eg, +25 mEq/L), since the basic lesion in the disorder is the inability of the kidney to excrete H+ and thus the inability to increase NH4Cl excretion. In proximal (type II) RTA, the kidney has defective HCO3- reabsorption, leading to increased HCO3- excretion rather than decreased NH4Cl excretion. Thus, the urinary anion gap is often negative in proximal (type II) RTA.
Urinary pH may not as readily differentiate between the two causes. Despite acidosis, if volume depletion from diarrhea causes inadequate Na+ delivery to the distal nephron and therefore decreased exchange with H+, urinary pH may not be lower than 5.3. In the presence of this relatively high urine pH, however, H+ excretion continues due to buffering of NH3 to NH4+, since the pK of this reaction is as high as 9.1. Potassium depletion, which can accompany diarrhea (and surreptitious laxative abuse), may also impair renal acidification. Thus, when volume depletion is present, the urinary anion gap is a better measurement of ability to acidify the urine than urinary pH.
When large amounts of other anions are present in the urine, the urinary anion gap may not be reliable. In such a situation, NH4+ excretion can be estimated using the urinary osmolar gap.
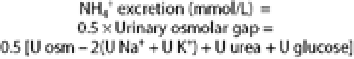
where urinary (U) concentrations and osmolality are in millimoles per liter.
Clinical Findings
A. Symptoms and Signs
Symptoms of metabolic acidosis are mainly those of the underlying disorder. Compensatory hyperventilation is an important clinical sign and may be misinterpreted as a primary respiratory disorder; when severe, Kussmaul respirations (deep, regular, sighing respirations) are seen.
B. Laboratory Findings
Blood pH, serum HCO3-, and PCO2 are decreased. Anion gap may be normal (hyperchloremic) or increased (normochloremic). Hyperkalemia may be seen (see above).
Treatment
A. Increased Anion Gap Acidosis
Treatment is aimed at the underlying disorder, such as insulin and fluid therapy for diabetes and appropriate volume resuscitation to restore tissue perfusion. The metabolism of lactate will produce HCO3- and increase pH. The use of supplemental HCO3- is indicated
P.913
for treatment of hyperkalemia (Table 21-7) and some forms of normal anion gap acidosis but has been controversial for treatment of increased anion gap metabolic acidosis.
Controversy remains about the efficacy and safety of alkali therapy for severe metabolic acidosis. Administration of large amounts of HCO3- may have deleterious effects, including hypernatremia and hyperosmolality. Furthermore, intracellular pH may decrease because administered HCO3- is converted to CO2, which easily diffuses into cells. There, it combines with water to create additional hydrogen ions and worsening of intracellular acidosis. Theoretically, this could impair cellular function, but the clinical significance of this phenomenon is uncertain.
In addition, alkali administration is known to stimulate phosphofructokinase activity, thus exacerbating lactic acidosis via enhanced lactate production. Ketogenesis is also augmented by alkali therapy.
In salicylate intoxication, however, alkali therapy must be started unless blood pH is already alkalinized by respiratory alkalosis, since the increment in pH converts salicylate to more impermeable salicylic acid and thus prevents central nervous system damage. In alcoholic ketoacidosis, thiamin should be given together with glucose to avoid the development of Wernicke's encephalopathy. The amount of HCO3- deficit can be calculated as follows:
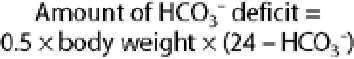
Half of the calculated deficit should be administered within the first 3 4 hours to avoid overcorrection and volume overload. In methanol intoxication, ethanol has been used as a competitive substrate for alcohol dehydrogenase, which metabolizes methanol to formaldehyde. Recently, direct inhibition of alcohol dehydrogenase by fomepizole has been reported. Fomepizole may be used in methanol intoxication in the near future.
B. Normal Anion Gap Acidosis
Treatment of RTA is mainly achieved by administration of alkali (either as bicarbonate or citrate) to correct metabolic abnormalities and prevent nephrocalcinosis and renal failure.
Large amounts of alkali (10 15 mEq/kg/d) may be required to treat proximal RTA because most of the alkali is excreted into the urine, which exacerbates hypokalemia. Thus, a mixture of sodium and potassium salts, such as K-Shohl solution, is preferred. The addition of thiazides may reduce the amount of alkali required, but hypokalemia may develop. Correction of type 1 distal RTA requires a smaller amount of alkali (1 3 mEq/kg/d) and potassium supplementation as needed.
For the treatment of type IV RTA, dietary potassium restriction may be needed and potassium-retaining drugs should be withdrawn. Fludrocortisone may be effective in cases with hypoaldosteronism, but should be used with care, preferably in combination with loop diuretics. In some cases, alkali supplementation (1 3 mEq/kg/d) may be required.
Casaletto JJ: Differential diagnosis of metabolic acidosis. Emerg Med Clin North Am 2005;23:771.
Forni LG et al: Circulating anions usually associated with the Krebs cycle in patients with metabolic acidosis. Crit Care 2005;9:R591.
Hassan H et al: Evaluation of serum anion gap in patients with liver cirrhosis of diverse etiologies. Mt Sinai J Med 2004; 71:281.
Levraut J et al: Treatment of metabolic acidosis. Curr Opin Crit Care 2003;9:260.
Matin MJ et al: Use of serum bicarbonate measurement in place of arterial base deficit in the surgical intensive care unit. Arch Surg 2005;140:745.
Soriano JR: Renal tubular acidosis: the clinical entity. J Am Soc Nephrol 2002;13:2160.
Metabolic Alkalosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
High HCO3- with alkalemia.
Evaluate effective circulating volume by physical examination and check urinary chloride concentration. This will help differentiate saline-responsive metabolic alkalosis from saline-unresponsive alkalosis.
Classification
Metabolic alkalosis is characterized by high HCO3-. The high HCO3- is also seen in chronic respiratory acidosis (see below), but pH differentiates the two disorders. Abnormalities that generate HCO3- within the body are called initiation factors of metabolic alkalosis, whereas abnormalities that promote renal conservation of HCO3- are called maintenance factors. Metabolic alkalosis may remain even after the initiation factors have disappeared.
It is useful to classify the causes of metabolic alkalosis into two groups based on saline responsiveness or urinary Cl-, which are markers for volume status (Table 21-16). Saline-responsive metabolic alkalosis is a sign of extracellular volume contraction, and saline-unresponsive alkalosis implies a volume-expanded state. It is rare for a compensatory increase in PCO2 to exceed 55 mm Hg. A higher value implies a superimposed respiratory acidosis.
Table 21-16. Metabolic alkalosis. | ||||||
|---|---|---|---|---|---|---|
| ||||||
A. Saline-Responsive Metabolic Alkalosis
Saline-responsive metabolic alkalosis is by far the more common disorder. It is characterized by normotensive extracellular volume contraction and hypokalemia. Less frequently, hypotension or orthostatic hypotension
P.914
may be seen. In vomiting or nasogastric suction, for example, loss of acid (HCl) initiates the alkalosis, but volume contraction from loss of Cl- sustains the alkalosis because the decline in GFR causes avid renal Na+ and HCO3- reabsorption. Because there is Cl- depletion from loss of HCl, NaCl, and KCl from the stomach, the available anion is HCO3-, whose reabsorption is increased proximally, and urine pH may remain acidic despite alkalemia (paradoxic aciduria). Renal Cl- reabsorption (as well as Na+ reabsorption) is high, and the urinary Cl- is therefore low (< 10 20 mEq/L). In alkalosis, bicarbonaturia may force Na+ excretion as the accompanying cation even if volume depletion is present. Therefore, urinary Cl- is preferred to urinary Na+ as a measure of extracellular volume. An exception to the usefulness of urinary Cl- is in patients who have recently received diuretics. Their urine may contain high Na+ and Cl- despite extracellular volume contraction. If diuretics are discontinued, the urinary Cl- will decrease.
Metabolic alkalosis is generally associated with hypokalemia. This is due partly to the direct effect of alkalosis per se on renal potassium excretion and partly to secondary hyperaldosteronism from volume depletion. Hypokalemia induced in this fashion further worsens the metabolic alkalosis by increasing bicarbonate reabsorption in the proximal tubule and hydrogen ion secretion in the distal tubule. Administration of KCl will correct the disorder. Repletion of KCl is important to reverse the disorder.
1. Contraction alkalosis
Diuretics can acutely decrease extracellular volume from urinary loss of NaCl and water. There is no associated bicarbonaturia, so that body HCO3- content remains normal. However, plasma HCO3- increases because of extracellular fluid contraction the reverse of what occurs in dilutional acidosis.
2. Posthypercapnia alkalosis
In chronic respiratory acidosis, compensatory increases in HCO3- occur (Table 21-13). Hypercapnia also directly affects the proximal tubule to decrease NaCl reabsorption, which can cause extracellular volume depletion. If PCO2 is corrected rapidly, as with mechanical ventilation, metabolic alkalosis will ensue until adequate bicarbonaturia occurs. Hypovolemia will inhibit bicarbonaturia until Cl- is repleted. Many patients with chronic respiratory acidosis receive diuretics, which further exacerbates the metabolic alkalosis.
B. Saline-Unresponsive Alkalosis
1. Hyperaldosteronism
Primary hyperaldosteronism causes expansion of extracellular volume with hypertension. Metabolic alkalosis with hypokalemia results from the renal mineralocorticoid effect. In an attempt to decrease extracellular volume, high levels of NaCl are excreted, and for that reason the urinary Cl- is high (> 20 mEq/L, often higher). Therapy with NaCl will only increase volume expansion and hypertension and will not treat the underlying problem of mineralocorticoid excess.
2. Alkali administration with decreased GFR
Despite large ingestions of HCO3-, enhanced bicarbonaturia almost always prevents a patient with normal renal function from developing metabolic alkalosis. However, with renal insufficiency, urinary excretion of bicarbonate is inadequate. If large amounts of HCO3- or metabolizable salts of organic acids such as sodium lactate, sodium citrate, or sodium gluconate are consumed, as with intensive antacid therapy, metabolic alkalosis will
P.915
occur. In milk-alkali syndrome, large and sustained ingestion of absorbable antacids and milk causes renal insufficiency from hypercalcemia. Decreased GFR prevents appropriate bicarbonaturia from the ingested alkali, and metabolic alkalosis occurs. Volume contraction from renal hypercalcemic effects further exacerbates the alkalosis.
Clinical Findings
A. Symptoms and Signs
There are no characteristic symptoms or signs. Orthostatic hypotension may be encountered. Weakness and hyporeflexia occur if serum K+ is markedly low. Tetany and neuromuscular irritability occur rarely.
B. Laboratory Findings
The arterial blood pH and bicarbonate are elevated. The arterial PCO2 is increased. Serum potassium and chloride are decreased. There may be an increased anion gap.
Treatment
Mild alkalosis is generally well tolerated. Severe or symptomatic alkalosis (pH > 7.60) requires urgent treatment.
A. Saline-Responsive Metabolic Alkalosis
Therapy for saline-responsive metabolic alkalosis is aimed at correction of extracellular volume deficit. Depending on the degree of hypovolemia, adequate amounts of 0.9% NaCl and KCl should be administered. Discontinuation of diuretics and administration of H2-blockers in patients whose alkalosis is due to nasogastric suction can be useful. If impaired pulmonary or cardiovascular status prohibits adequate volume repletion, acetazolamide, 250 500 mg intravenously every 4 6 hours, can be used. One must be alert to the possible development of hypokalemia, since potassium depletion can be induced by forced kaliuresis via bicarbonaturia. Administration of acid can be used as emergency therapy. HCl, 0.1 mol/L, is infused via a central vein (the solution is sclerosing). Dosage is calculated to decrease the HCO3- level by 50% over 2 4 hours, assuming an HCO3- volume of distribution (L) of 0.5 % body weight (kg). Patients with marked renal insufficiency may require dialysis.
B. Saline-Unresponsive Metabolic Alkalosis
Therapy for saline-unresponsive metabolic alkalosis includes surgical removal of a mineralocorticoid-producing tumor and blockage of aldosterone effect with an ACE inhibitor or with spironolactone. Metabolic alkalosis in primary aldosteronism can be treated only with potassium repletion.
Bartholow C et al: Hypokalemia and metabolic alkalosis: algorithms for combined clinical problem solving. Compr Ther 2000;26:114.
Galla JH: Metabolic alkalosis. J Am Soc Nephrol 2000;11:369.
Respiratory Acidosis (Hypercapnia)
Respiratory acidosis results from decreased alveolar ventilation and subsequent hypercapnia. Pulmonary as well as nonpulmonary disorders can cause hypoventilation. The clinician must be mindful of readily reversible causes of respiratory acidosis, especially opioid-induced central nervous system depression.
Acute respiratory failure is associated with severe acidosis and only a small increase in the plasma bicarbonate. After 6 12 hours, the primary increase in PCO2 evokes a renal compensatory response to generate more HCO3-, which tends to ameliorate the respiratory acidosis. This usually takes several days to complete.
Chronic respiratory acidosis is generally seen in patients with underlying lung disease, such as chronic obstructive pulmonary disease. Urinary excretion of acid in the form of NH4+ and Cl- ions results in the characteristic hypochloremia of chronic respiratory acidosis. When chronic respiratory acidosis is corrected suddenly, especially in patients who receive mechanical ventilation, there is a 2- to 3-day lag in renal bicarbonate excretion, resulting in posthypercapnic metabolic alkalosis.
Clinical Findings
A. Symptoms and Signs
With acute onset, there is somnolence and confusion, and myoclonus with asterixis may be seen. Coma from CO2 narcosis ensues. Severe hypercapnia increases cerebral blood flow and cerebrospinal fluid pressure. Signs of increased intracranial pressure (papilledema, pseudotumor cerebri) may be seen.
B. Laboratory Findings
Arterial pH is low and PCO2 is increased. Serum HCO3- is elevated, but not enough to completely compensate for the hypercapnia. If the disorder is chronic, hypochloremia is seen.
Treatment
Because opioid drug overdose is an important reversible cause of acute respiratory acidosis, naloxone, 0.04 2 mg intravenously (see Chapter 39), is administered to all such patients if no obvious cause for respiratory depression is present. In all forms of respiratory acidosis, treatment is directed at the underlying disorder to improve ventilation.
Epstein SK et al: Respiratory acidosis. Respir Care 2001;46:366.
Madias NE et al: Cross-talk between two organs: how the kidney responds to disruption of acid-base balance by the lung. Nephron Physiol 2003;93:61.
P.916
Respiratory Alkalosis (Hypocapnia)
Respiratory alkalosis, or hypocapnia, occurs when hyperventilation reduces the PCO2, which increases the pH. The most common cause of respiratory alkalosis is hyperventilation syndrome (Table 21-17), but bacterial septicemia and cirrhosis are other common causes. Symptoms in acute respiratory alkalosis are related to decreased cerebral blood flow induced by the disorder. Pregnancy is another cause of chronic respiratory alkalosis, probably from progesterone stimulation of the respiratory center, producing an average PCO2 of 30 mm Hg.
Determination of appropriate compensatory changes in the HCO3- is useful to sort out the presence of an associated metabolic disorder (see above under Mixed Acid-Base Disorders).
As in respiratory acidosis, the changes in HCO3- values are greater if the respiratory alkalosis is chronic (Table 21-13). Although serum HCO3- is frequently below 15 mEq/L in metabolic acidosis, it is unusual to see such a low level in respiratory alkalosis, and its presence would imply a superimposed (noncompensatory) metabolic acidosis.
Table 21-17. Causes of respiratory alkalosis. | ||
|---|---|---|
|
Clinical Findings
A. Symptoms and Signs
In acute cases (hyperventilation), there is light-headedness, anxiety, paresthesias, numbness about the mouth, and a tingling sensation in the hands and feet. Tetany occurs in more severe alkalosis from a fall in ionized calcium. In chronic cases, findings are those of the responsible condition.
B. Laboratory Findings
Arterial blood pH is elevated, and PCO2 is low. Serum bicarbonate is decreased in chronic respiratory alkalosis.
Treatment
Treatment is directed toward the underlying cause. In acute hyperventilation syndrome from anxiety, rebreathing into a paper bag will increase the PCO2. The processes are usually self-limited since muscle weakness caused by hyperventilation-induced alkalemia will suppress ventilation. Sedation may be necessary if the process persists. Rapid correction of chronic respiratory alkalosis may result in metabolic acidosis as PCO2 is increased in the setting of a previous compensatory decrease in HCO3-.
Foster GT et al: Respiratory alkalosis. Respir Care 2001;46:384.
Laffey JG et al: Hypocapnia. N Engl J Med 2002;347:43.
Fluid Management
An average adult whose entire intake is parenteral would require for maintenance 2500 3000 mL of 5% dextrose in 0.2% saline solution (34 mEq Na+ plus 34 mEq Cl-/L). To each liter, 30 mEq of KCl could be added. In 3 L, the total chloride intake would be 192 mEq, which is easily tolerated. Guidelines for gastrointestinal fluid losses are shown in Table 21-18.
Weight loss or gain is the best indication of water balance. Insensible water loss should be further considered in febrile patients. Water loss increases by 100 150 mL/d for each degree of body temperature over 37 C.
In situations requiring maintenance or maintenance plus replacement of fluid and electrolyte by parenteral infusion, the total daily ration should be administered continuously over the 24-hour period to ensure the best utilization by the patient.
If parenteral fluids are the only source of water, electrolytes, and calories for longer than a week, more complex fluids containing amino acids, lipids, trace
P.917
metals, and vitamins may be indicated. (See Total Parenteral Nutrition, Chapter 29.)
Table 21-18. Replacement guidelines for sweat and gastrointestinal fluid losses. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For parenteral alimentation, 620 mg (20 mmol) of phosphorus is required for every 1000 nonprotein kcal to maintain phosphate balance and to ensure anabolic function. For prolonged parenteral fluid maintenance, a daily ration is 620 1240 mg (20 40 mmol) of phosphorus.
EAN: 2147483647
Pages: 49