19 - Allergic Immunologic Disorders
Editors: McPhee, Stephen J.; Papadakis, Maxine A.; Tierney, Lawrence M.
Title: Current Medical Diagnosis & Treatment, 46th Edition
Copyright 2007 McGraw-Hill
> Table of Contents > 22 - Kidney
function show_scrollbar() {}
22
Kidney
Suzanne Watnick MD
Gail Morrison MD
Approach To Renal Disease
Renal disease presents in one of two ways: discovered incidentally during a routine medical evaluation or with evidence of renal dysfunction such as hypertension, edema, nausea, and hematuria. The initial approach in both situations should be to assess the cause and severity of renal abnormalities. In all cases this evaluation includes (1) an estimation of disease duration, (2) a careful urinalysis, and (3) an assessment of the glomerular filtration rate (GFR). The history and physical examination, though equally important, are variable among renal syndromes thus, specific symptoms and signs are discussed under each disease entity. Further diagnostic categorization is according to anatomic distribution: prerenal disease, postrenal disease, and intrinsic renal disease. Intrinsic renal disease can further be divided into glomerular, tubular, interstitial, and vascular abnormalities.
Disease Duration
Renal disease may be acute or chronic. Acute renal failure is worsening of renal function over hours to days, resulting in the retention of nitrogenous wastes (such as urea nitrogen) and creatinine in the blood. Retention of these substances is called azotemia. Chronic renal failure (chronic kidney disease) results from an abnormal loss of renal function over months to years. Differentiating between the two is important for diagnosis, treatment, and outcome. Oliguria is unusual in chronic renal insufficiency. Anemia (from low renal erythropoietin production) is rare in the initial period of acute renal failure. Small kidneys are most consistent with chronic kidney disease, whereas normal to large-size kidneys can be seen with both chronic and acute disease.
Urinalysis
A urinalysis has been likened to a poor man's renal biopsy. The urine is collected in midstream or, if that is not feasible, by bladder catheterization. The urine should be examined within 1 hour after collection to avoid destruction of formed elements. Urinalysis includes a dipstick examination followed by microscopic assessment if the dipstick has positive findings. The dipstick examination measures urinary specific gravity, pH, protein, hemoglobin, glucose, ketones, bilirubin, nitrites, and leukocyte esterase. Microscopy searches for all formed elements crystals, cells, casts, and infecting organisms.
Various findings on the urinalysis are indicative of certain patterns of renal disease. A bland urinary sediment is common, especially in chronic kidney disease and prerenal and postrenal disorders. The presence of hematuria with dysmorphic red blood cells, red blood cell casts, and proteinuria is indicative of glomerulonephritis. Red blood cells are misshapen during passage from the capillary through the glomerular basement membrane into the urinary space of Bowman's capsule.
Casts are composed of Tamm-Horsfall urinary mucoprotein in the shape of the nephron segment where they were formed. Heavy proteinuria and lipiduria are consistent with the nephrotic syndrome. Pigmented granular casts and renal tubular epithelial cells alone or in casts suggest acute tubular necrosis. White blood cells, including neutrophils and eosinophils, white blood cell casts, red blood cells, and small amounts of protein can be found in interstitial nephritis and pyelonephritis (Table 22-1); Wright's stain can detect eosinophiluria. Pyuria alone can indicate a urinary tract infection. Hematuria and proteinuria are discussed more thoroughly below.
Table 22-1. Significance of specific urinary casts. | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Proteinuria
Proteinuria is defined as excessive protein excretion in the urine, generally greater than 150 160 mg/24 h in adults. Significant proteinuria is a sign of an underlying renal abnormality, usually glomerular in origin when greater than 1 g/d. It is typically accompanied by other clinical abnormalities elevated blood urea nitrogen (BUN) and serum creatinine levels, abnormal urinary sediment, or evidence of systemic illness (eg, fever, rash, vasculitis).
There are four primary reasons for development of proteinuria: (1) Functional proteinuria is a benign process stemming from stressors such as acute illness, exercise, and orthostatic proteinuria. The latter condition, generally found in people under age 30 years, results in the excretion of abnormal amounts of urinary protein, typically less than 1 g/d. The orthostatic nature of the proteinuria is confirmed by measuring
P.919
an 8-hour overnight supine urinary protein excretion, which should be less than 50 mg. (2) Overload proteinuria can result from overproduction of circulating, filterable plasma proteins (monoclonal gammopathies), such as Bence Jones proteins associated with multiple myeloma. Urinary protein electrophoresis will exhibit a discrete protein peak. Other examples of overload proteinuria include myoglobinuria in rhabdomyolysis and hemoglobinuria in hemolysis. (3) Glomerular proteinuria results from effacement of epithelial cell foot processes and altered glomerular permeability with an increased filtration fraction of normal plasma proteins. Glomerular diseases exhibit some degree of proteinuria. The urinary electrophoresis will have a pattern exhibiting a large albumin spike indicative of increased permeability of albumin across a damaged glomerular basement membrane (GBM). (4) Tubular proteinuria occurs as a result of faulty reabsorption of normally filtered proteins in the proximal tubule, such as 2-microglobulin and immunoglobulin light chains. Causes include acute tubular necrosis, toxic injury (lead, aminoglycosides), drug-induced interstitial nephritis, and hereditary metabolic disorders (Wilson's disease and Fanconi's syndrome).
Evaluation of proteinuria by urinary dipstick primarily detects albumin and intact globulins, while overlooking positively charged light chains of immunoglobulins. These proteins can be detected by the addition of sulfosalicylic acid to the urine specimen. Precipitation indicates the presence of paraproteins.
The next step and the most reliable way to quantify proteinuria is a 24-hour urine collection. A finding of greater than 150 mg/24 h is abnormal, and greater than 3.5 g/24 h is consistent with nephrotic-range proteinuria. A simpler but less accurate method is to collect a random urine sample. The ratio of urinary protein concentration to urinary creatinine concentration (Uprotein/Ucreatinine) correlates with 24-hour urine protein collection (< 0.2 is normal and corresponds to excretion of less than 200 mg/24 h). If a patient has proteinuria with loss of renal function, renal biopsy may be indicated, particularly if the renal insufficiency is acute in onset. The clinical consequences of proteinuria are discussed in the section on the nephrotic syndrome. The benefit of a urine protein-to-creatinine ratio is the ease of collection and the lack of error from overcollection or undercollection.
In both diabetics and nondiabetics, therapy aimed at reducing proteinuria may also reduce progression of renal disease. Angiotensin-converting enzyme (ACE) inhibitors are effective by lowering efferent arteriolar resistance out of proportion to afferent arteriolar resistance, thereby reducing glomerular capillary pressure and lowering urinary protein excretion. Other effects include alterations of glomerular mesangial proliferation. ACE inhibitors can be used in patients despite compromised GFR as long as significant hyperkalemia does not occur and serum creatinine rises less than 30% and stabilizes over 2 months. Large randomized controlled trials (ie, the RENAAL and IDNT studies) have also proved the benefit of angiotensin II receptor blockers in reducing proteinuria and preventing progression of renal disease in diabetic nephropathy. Recently, head-to-head comparisons of an ACE-I and an angiotensin receptor blocker (ARB) have shown the ARB to be no better than the ACE-I in preventing progression of renal disease in diabetic persons with proteinuria. The consequences of dietary restrictions in patients with proteinuria are discussed in the section on chronic kidney disease.
Hematuria
Hematuria is significant if there are more than three red cells per high-power field. It is usually detected incidentally by the urine dipstick examination or clinically following an episode of macroscopic hematuria. The diagnosis must be confirmed via microscopic examination, as false-positive dipstick tests can be caused by vitamin C, beets and rhubarb, bacteria, and myoglobin. Transient hematuria is common, but in patients under 40 years it is less often of clinical significance.
Hematuria may be due to renal or extrarenal causes. Extrarenal causes are addressed in Chapter 23; most worrisome are urologic malignancies. Renal causes account for approximately 10% of cases and are best considered anatomically as glomerular or nonglomerular. The most common extraglomerular sources include cysts, calculi, interstitial nephritis, and renal neoplasia. Glomerular causes include immunoglobulin A (IgA) nephropathy, thin GBM disease, postinfectious glomerulonephritis, membranoproliferative glomerulonephritis, and systemic nephritic syndromes.
Currently, the United States Health Preventive Services Task Force does not recommend screening for hematuria. See Chapter 23 for evaluation of hematuria.
P.920
Estimation of GFR
The GFR provides a useful index of overall renal function; however, patients with renal disease can actually have a normal or increased GFR. The GFR measures the amount of plasma ultrafiltered across the glomerular capillaries and correlates with the ability of the kidneys to filter fluids and various substances. Daily GFR in normal individuals is variable, with a range of 150 250 L/24 h or 100 120 mL/min/1.73 m2 of body surface area. GFR can be measured indirectly by determining the renal clearance of plasma substances that are not bound to plasma proteins, are freely filterable across the glomerulus, and are neither secreted nor reabsorbed along the renal tubules.
The formula used to determine the renal clearance of a substance is
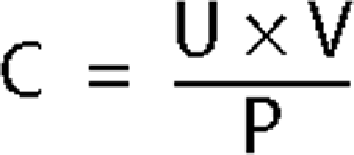
where C is the clearance, U and P are the urine and plasma concentrations of the substance (mg/dL), and V is the urine flow rate (mL/min). Inulin and creatinine clearance are used as markers of GFR. Inulin clearance following a continuous infusion is one of the most accurate methods for measurement of GFR. The cost and the complexity of the administration and analysis of inulin preclude its routine use. In clinical practice, the clearance rate of endogenous creatinine, the creatinine clearance, is the usual means of estimating GFR. Creatinine is a product of muscle metabolism produced at a relatively constant rate and cleared by renal excretion. It is freely filterable by the glomerulus and not reabsorbed by the renal tubules. With stable renal function, creatinine production and excretion are equal; thus, plasma creatinine concentrations remain constant. However, it is not a perfect indicator of GFR for the following reasons: (1) A small amount is normally eliminated by tubular secretion, and the fraction secreted progressively increases as GFR declines (overestimating GFR); (2) with severe renal failure, gut microorganisms degrade creatinine; (3) an individual's meat intake and muscle mass affect baseline plasma creatinine levels; (4) commonly used drugs such as aspirin, cimetidine, probenecid, and trimethoprim reduce tubular secretion of creatinine, increasing the plasma creatinine concentration and falsely indicating renal dysfunction; and (5) the accuracy of the measurement necessitates a stable plasma creatinine concentration over a 24-hour period, so that during the development of and recovery from acute renal failure, the creatinine clearance is of questionable value (Table 22-2).
Table 22-2. Conditions affecting serum creatinine independently of glomerular filtration rate. | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
To measure creatinine clearance, collect a 24-hour urine sample and determine the plasma creatinine level on the same day. An incomplete or prolonged urine collection is a common source of error. One way of estimating the completeness of the collection is to calculate a 24-hour creatinine excretion; the amount should be constant:
Ucr V = 15 20 mg/kg for healthy young women
Ucr V = 20 25 mg/kg for healthy young men
The creatinine clearance (Ccr) is approximately 100 mL/min/1.73 m2 in healthy young women and 120 mL/min/1.73 m2 in healthy young men. The Ccr declines by an average of 0.8 mL/min/yr after age 40 years as part of the aging process, but 35% of subjects in one study had no decline in renal function over 10 years. Because urine collection may be difficult, Ccr can be estimated from the formula of Cockcroft and Gault, which incorporates age, sex, and weight to estimate Ccr from plasma creatinine levels without any urinary measurements:
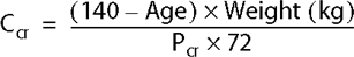
For women, the estimated GFR is multiplied by 0.85 because muscle mass is less. This formula overestimates GFR in patients who are obese or edematous, and is most accurate when normalized for body surface area of 1.73 m2. A more complicated but more accurate assessment can be obtained via the 4-variable Modification of Diet in Renal Disease (MDRD) formula. This requires SCr, gender, race, and age; the formula can be found at http://www.nephron.com under the heading MDRD GFR.
Urea is another index helpful in assessing renal function. It is synthesized mainly in the liver and is the end product of protein catabolism. Urea is freely filtered by the glomerulus, and about 30 70% is reabsorbed in the nephron. Unlike creatinine clearance, which overestimates GFR, urea clearance underestimates GFR. Urea reabsorption may be decreased in well-hydrated patients, whereas dehydration causes increased reabsorption, increasing BUN. A normal BUN:creatinine ratio
P.921
is 10:1. With dehydration, the ratio can increase to 20:1 or higher. Other causes of increased BUN include increased catabolism (gastrointestinal bleeding, cell lysis, and corticosteroid usage), increased dietary protein, and decreased renal perfusion (congestive heart failure, renal artery stenosis) (Table 22-3). Reduced BUN is seen in liver disease and in the syndrome of inappropriate antidiuretic hormone (SIADH) secretion.
Table 22-3. Conditions affecting BUN independently of GFR. | ||
|---|---|---|
|
As patients approach end-stage renal disease (ESRD), a more accurate measure of GFR than creatinine clearance is the average of the creatinine and urea clearances. The creatinine clearance overestimates GFR, as mentioned above, while the urea clearance underestimates GFR. Therefore, an average of the two more accurately approximates the true GFR.
Imaging Studies
Radionuclide Studies
Radionuclide studies can measure renal function. [125I]Iothalamate gives a surprisingly accurate measurement of GFR. It is injected intravenously, excreted renally, and sampled from the venous circulation over time. Technetium-labeled diethylenetriamine pentaacetic acid (99mTc-DTPA) is freely filtered by the glomerulus and not reabsorbed and is used to estimate GFR. Technetium-labeled dimercaptosuccinic acid (99mTc-DMSA) is bound to the tubules and provides an assessment of functional renal mass. [131I]Orthoiodohippurate is secreted into the renal tubules and assesses renal plasma flow (RPF). The indications for nuclear renography are to measure function and flow, to determine the contribution of each kidney to overall renal function, to demonstrate the presence or absence of functioning renal tissue in mass lesions, to detect obstruction, and to evaluate renovascular disease.
Poor flow along with poor function is consistent with acute tubular necrosis or ESRD. Decreased flow to one kidney suggests arterial occlusion of that kidney. To further investigate renal artery stenosis, the test is done both with and without captopril (see Chapter 11).
Ultrasonography
Ultrasonography can identify the thickness and echogenicity of the renal cortex, medulla, and pyramids, and a distended urinary collecting system. Kidney size can be determined; a kidney less than 9 cm in length in an adult indicates significant irreversible renal disease. A difference in size of more than 1.5 cm between the two kidneys is observed in unilateral renal disease. Renal ultrasound is also performed to search for hydronephrosis and obstruction, to characterize renal mass lesions, to screen for autosomal dominant polycystic kidney disease, to evaluate the perirenal space, to localize the kidney for a percutaneous invasive procedure, and to assess postvoiding residual urine volume of the bladder.
Intravenous Urography
The intravenous pyelogram (IVP) had been the standard imaging procedure for evaluating the urinary tract since it provides an assessment of the kidneys, ureters, and bladder. An IVP necessitates the injection of contrast and is relatively contraindicated in patients at increased risk for acute renal failure (eg, diabetes mellitus with serum creatinine greater than 2 mg/dL, severe volume contraction, or prerenal azotemia), chronic kidney disease with serum creatinine greater than 5 mg/dL, and multiple myeloma. IVP is performed to obtain a detailed view of the pelvicaliceal system, assess renal size and shape, detect and localize renal stones, and assess renal function. It is particularly useful in diagnosing certain disorders such as medullary sponge kidney and papillary necrosis. Ultrasonography is replacing IVP to avoid dye administration, and helical CT scanning is replacing IVP for stone evaluation.
CT Scanning
CT scanning is required for further investigation of abnormalities detected by ultrasound or IVP. Although routine CT requires radiographic contrast administration, no contrast is necessary if the reason for the study is to demonstrate hemorrhage or calcifications in the kidneys such as suspected stone disease. Noncontrast helical CT scanning to detect renal stones is 95% sensitive and 98% specific in patients with acute flank pain. Because contrast is filtered by the glomeruli and concentrated in the tubules, there is enhancement of parenchymal tissue, making abnormalities such as cysts or neoplasms easily identified and allowing good visualization of renal vessels and ureters. CT scanning is especially useful for evaluation of solid or cystic lesions in the kidney or the retroperitoneal space, particularly if the ultrasound results are suboptimal.
Mri Scanning
MRI can easily distinguish renal cortex from medulla. Loss of corticomedullary function in a variety of disorders (eg, glomerulonephritis, hydronephrosis, renal
P.922
vascular occlusion, and renal failure) will be evident on MRI. Renal cysts can also be identified by MRI. For some solid lesions, MRI may be superior to CT scans. MRI is indicated as an addition or alternative to CT scanning for staging renal cell cancer and as a substitute for CT scanning in the evaluation of a renal mass, especially for patients in whom contrast is contraindicated; in addition, the adrenals are well imaged. MRI is nearly 100% sensitive and 96 98% specific for the diagnosis of renal artery stenosis.
Arteriography & Venography
Renal arteriography is useful in the evaluation of atherosclerotic or fibrodysplastic stenotic lesions, aneurysms, vasculitis, and renal mass lesions. Venography is the best test for diagnosis of renal vein thrombosis, though CT scanning and MRI are less invasive for this purpose.
Renal Biopsy
Indications for percutaneous needle biopsy include (1) unexplained acute renal failure or chronic kidney disease; (2) acute nephritic syndromes; (3) unexplained proteinuria and hematuria; (4) previously identified and treated lesions to plan future therapy; (5) systemic diseases associated with kidney dysfunction, such as systemic lupus erythematosus, Goodpasture's syndrome, and Wegener's granulomatosis, to confirm the extent of renal involvement and to guide management; (6) suspected transplant rejection, to differentiate it from other causes of acute renal failure; and (7) to guide treatment. If a patient is unwilling to accept therapy based on biopsy findings, the risk of biopsy may outweigh its benefit. Relative contraindications include a solitary or ectopic kidney (exception: transplant allografts), horseshoe kidney, uncorrected bleeding disorder, severe uncontrolled hypertension, renal infection, renal neoplasm, hydronephrosis, ESRD, congenital anomalies, multiple cysts, or an uncooperative patient.
Prior to biopsy, patients should have well-controlled blood pressure; blood work should include a hematocrit, platelet count, prothrombin time, and partial thromboplastin time. After biopsy, hematuria occurs in nearly all patients. Fewer than 10% will have macroscopic hematuria. Patients should remain supine for 4 6 hours postbiopsy. A patient with a 6-hour postbiopsy hematocrit more than 3% lower than baseline should be closely monitored.
Percutaneous kidney biopsies are generally safe. One percent of patients will experience significant bleeding and 0.1 0.3% will require blood transfusions. More than half of patients will have at least a small hematoma. Risk of major bleeding persists up to 72 hours after the biopsy. Any type of anticoagulation therapy should be held for 5 7 days postbiopsy if possible. The risks of nephrectomy and mortality are about 0.06 0.08%. When a percutaneous needle biopsy is technically not feasible and renal tissue is deemed clinically essential, a closed renal biopsy via interventional radiologic techniques or open renal biopsy under general anesthesia can be done.
Barnett AH et al: Diabetics Exposed to Telmisartan and Enalapril Study Group: Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med 2004;351:1952.
Carman TL et al: Noninvasive imaging of the renal arteries. Urol Clin North Am 2001;28:815.
Cohen RA et al: Clinical practice. Microscopic hematuria. N Engl J Med 2003;348:2330.
Johnson CA et al: Clinical practice guidelines for chronic kidney disease in adults: Part II. Glomerular filtration rate, proteinuria, and other markers. Am Fam Physician 2004;70:1091.
Manjunath G et al: Estimating the glomerular filtration rate. Postgrad Med 2001;110:55.
Whittier WL et al: Timing of complications in percutaneous renal biopsy. J Am Soc Nephrol 2004;15:142.
Acute Renal Failure
![]() Essentials of Diagnosis
Essentials of Diagnosis
Sudden increase in BUN or serum creatinine.
Oliguria often associated.
Symptoms and signs depend on cause.
General Considerations
Five percent of hospital admissions and 30% of intensive care unit (ICU) admissions carry a diagnosis of acute renal failure, and it will develop in 25% of hospitalized patients. Acute renal failure is defined as a sudden decrease in renal function, resulting in an inability to maintain fluid and electrolyte balance and to excrete nitrogenous wastes. Serum creatinine is a convenient marker. In the absence of functioning kidneys, serum creatinine concentration will typically increase by 1 1.5 mg/dL daily although with certain conditions, such as rhabdomyolysis, serum creatinine can increase more rapidly. Acute renal failure is now also being termed acute kidney injury since it may be a more appropriately descriptive term.
Clinical Findings
A. Symptoms and Signs
The uremic milieu of acute renal failure can cause nonspecific symptoms. When present, they are often due to azotemia or its underlying cause. Azotemia can cause nausea, vomiting, malaise, and altered sensorium. Hypertension is rare, but fluid homeostasis is often altered. Hypovolemia can cause prerenal disease, whereas hypervolemia
P.923
can result from intrinsic or postrenal disease. Pericardial effusions can occur with azotemia, and a pericardial friction rub can be present. Effusions may result in cardiac tamponade. Arrhythmias occur especially with hyperkalemia. The lung examination may show rales in the presence of hypervolemia. Acute renal failure can cause nonspecific diffuse abdominal pain and ileus as well as platelet dysfunction; thus, bleeding is more common in these patients. The neurologic examination reveals encephalopathic changes with asterixis and confusion; seizures may ensue.
Table 22-4. Classification and differential diagnosis of acute renal failure. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B. Laboratory Findings
Elevated BUN and creatinine are present, though these elevations do not in themselves distinguish acute from chronic renal failure. Hyperkalemia often occurs from impaired renal potassium excretion. The ECG can reveal peaked T waves, PR prolongation, and QRS widening. A long QT segment can occur with hypocalcemia. Anion gap metabolic acidosis (due to decreased organic acid clearance) is often noted. Hyperphosphatemia occurs when phosphorus cannot be secreted by damaged tubules either with or without increased cell catabolism. Hypocalcemia with metastatic calcium phosphate deposition may be observed when the product of calcium and phosphorus exceeds 70 mg/dL. Anemia can occur as a result of decreased erythropoietin production over weeks, and associated platelet dysfunction is typical.
Classification & Etiology
Acute renal failure can be divided into three categories: prerenal azotemia, intrinsic renal disease, and postrenal azotemia. Identifying the cause is the first step toward treating the patient (Table 22-4).
A. Prerenal Azotemia
Prerenal azotemia is the most common cause of acute renal failure, accounting for 40 80% of cases, depending on the population studied. It is due to renal hypoperfusion. This is an appropriate physiologic change. If it can be immediately reversed with restoration of renal blood flow, renal parenchymal damage does not occur. If hypoperfusion persists, ischemia can result, causing intrinsic renal failure.
Decreased renal perfusion can occur in one of three ways: a decrease in intravascular volume, a change in vascular resistance, or low cardiac output. Causes of volume depletion include hemorrhage, gastrointestinal losses, dehydration, excessive diuresis, extravascular space sequestration, pancreatitis, burns, trauma, and peritonitis.
Changes in vascular resistance can occur systemically with sepsis, anaphylaxis, anesthesia, and afterload-reducing drugs. ACE inhibitors prevent efferent renal arteriolar constriction out of proportion to the afferent arteriole; thus, GFR will decrease. Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent afferent arteriolar vasodilation by inhibiting prostaglandin-mediated signals. Thus, in cirrhosis and congestive heart failure, when prostaglandins are recruited to increase renal blood flow, NSAIDs will have particularly deleterious effects. Epinephrine, norepinephrine, high-dose dopamine, anesthetic agents, and cyclosporine also can cause renal vasoconstriction. Renal artery stenosis causes increased resistance and decreased perfusion.
Low cardiac output is a state of low effective renal arterial blood flow. This occurs in states of cardiogenic
P.924
shock, congestive heart failure, pulmonary embolism, and pericardial tamponade. Arrhythmias and valvular disorders can also reduce cardiac output. In the ICU setting, positive pressure ventilation will decrease venous return, also decreasing cardiac output.
When GFR falls acutely, it is important to determine whether acute renal failure is due to prerenal or intrinsic renal causes. The history and physical examination are important, and urinalysis can be helpful. The BUN:creatinine ratio will typically exceed 20:1 due to increased urea reabsorption. In an oliguric patient, another useful index is the fractional excretion of sodium (FENa). With decreased GFR, the kidney will reabsorb salt and water avidly if there is no intrinsic tubular dysfunction. Thus, patients with prerenal failure should have a low fractional excretion percent of sodium (< 1%). The FENa is calculated as follows: FENa = clearance of Na+/GFR = clearance of Na+/creatinine clearance:
![]()
Oliguric states are more accurately assessed with this formula than nonoliguric states because the kidneys do not avidly reabsorb water and sodium in nonoliguric states. (Oliguria is defined as urinary output < 400 500 mL/d.) Diuretics can cause increased sodium excretion. Thus, if the FENa is high within 12 24 hours after diuretic administration, the cause of acute renal failure may not be accurately predicted. Acute renal failure due to glomerulonephritis can have a low FENa because sodium reabsorption and tubular function may not be compromised.
Treatment of prerenal azotemia depends entirely on its cause, but maintenance of euvolemia, attention to serum potassium, and avoidance of nephrotoxic drugs are the benchmarks of therapy. This involves careful assessment of volume status, drug usage, and cardiac function.
B. Postrenal Azotemia
Postrenal azotemia is the least common cause of acute renal failure, accounting for approximately 5 10% of cases, but is important to detect because of its reversibility. It occurs when urinary flow from both kidneys, or a single functioning kidney, is obstructed. Each nephron has an elevated intraluminal pressure, causing a decrease in GFR.
Causes include urethral obstruction, bladder dysfunction or obstruction, and obstruction of both ureters or renal pelvises. In men, benign prostatic hyperplasia is the most common cause. Patients taking anticholinergic drugs are particularly at risk. Bladder, prostate, and cervical cancers as well as retroperitoneal processes and neurogenic bladder can also cause obstruction. Less common causes are blood clots, bilateral ureteral stones, urethral stones or stricture, and bilateral papillary necrosis. In patients with a single functioning kidney, obstruction of a solitary ureter can cause postrenal azotemia.
Patients may be anuric or polyuric and may complain of lower abdominal pain. Obstruction can be constant or intermittent and partial or complete. On examination, the patient may have an enlarged prostate, distended bladder, or mass detected on pelvic examination.
Laboratory examination may initially reveal high urine osmolality, low urine sodium, high BUN:creatinine ratio, and low FENa. These indices are similar to a prerenal picture because extensive intrinsic renal damage has not occurred. After several days, the urine sodium increases as the kidneys fail and are unable to concentrate the urine thus, isosthenuria is present. The urine sediment is generally benign.
Patients with acute renal failure and suspected postrenal azotemia should undergo bladder ultrasonography and bladder catheterization if hydroureter and hydronephrosis are present along with an enlarged bladder. These patients often undergo a postobstructive diuresis, and care should be taken to avoid dehydration. Rarely, obstruction is not diagnosed by ultrasonography. For example, patients with retroperitoneal fibrosis from tumor or radiation may not show dilation of the urinary tract. If suspicion does exist, a CT scan or MRI can establish the diagnosis. Prompt treatment of obstruction within days by catheters, stents, or other surgical procedures can result in complete reversal of the acute process.
C. Intrinsic Renal Failure
Intrinsic renal disorders account for up to 50% of all cases of acute renal failure. Intrinsic (or parenchymal) dysfunction is considered after prerenal and postrenal causes have been excluded. The sites of injury are the tubules, interstitium, vasculature, and glomeruli.
Acute Tubular Necrosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Acute renal insufficiency.
Clinical scenario consistent with diagnosis (ischemic or toxic insult).
Urine sediment with pigmented granular casts and renal tubular epithelial cells.
General Considerations
Acute renal failure due to tubular damage is termed acute tubular necrosis and accounts for 85% of intrinsic acute renal failure. The two major causes of acute tubular necrosis are ischemia and toxin exposure. Ischemia causes tubular damage from states of low perfusion and is often preceded by a state of prerenal azotemia. Ischemic acute renal failure is characterized
P.925
not only by inadequate GFR but also by renal blood flow inadequate to maintain parenchymal cellular formation. This occurs in the setting of prolonged hypotension or hypoxemia, such as dehydration, shock, and sepsis. Major surgical procedures can involve prolonged periods of hypoperfusion, which are exacerbated by vasodilating anesthetic agents.
The other major cause of acute tubular necrosis is nephrotoxin exposure. Exogenous nephrotoxins more commonly cause damage than endogenous nephrotoxins.
A. Exogenous Nephrotoxins
Up to 25% of hospitalized patients receiving therapeutic levels of aminoglycosides sustain some degree of acute tubular necrosis. Nonoliguric renal failure typically occurs after 5 10 days of exposure. Predisposing factors include underlying renal damage, dehydration, and advanced age. Aminoglycosides can remain in renal tissues for up to a month, so renal function may not recover for some time after stopping the medication. Monitoring of peak and trough levels is important, but trough levels are more helpful in predicting renal toxicity. Gentamicin is as nephrotoxic as tobramycin; streptomycin is the least nephrotoxic of the aminoglycosides, likely due to the number of cationic amino side chains present on each molecule. Amphotericin B is typically nephrotoxic after a dose of 2 3 g. This causes severe vasoconstriction with distal tubular damage and can lead to distal renal tubular acidosis with hypokalemia and nephrogenic diabetes insipidus. Vancomycin, acyclovir, and several cephalosporins have been known to cause acute tubular necrosis.
Radiographic contrast media can be directly nephrotoxic. Contrast nephropathy is the third leading cause of new acute renal failure in hospitalized patients. It probably results from the synergistic combination of direct renal tubular epithelial cell toxicity and renal medullary ischemia. Predisposing factors include advanced age, preexisting renal disease (serum creatinine > 2 mg/dL), volume depletion, diabetic nephropathy, congestive heart failure, multiple myeloma, repeated doses of contrast, and recent exposure to other nephrotoxic agents, including NSAIDs and ACE inhibitors. The combination of diabetes mellitus and renal dysfunction poses the greatest risk (15 50%) for contrast nephropathy. Lower volumes of contrast with lower osmolality are recommended in high-risk patients. Toxicity usually occurs 24 48 hours after the radiocontrast study. Nonionic contrast media may be less toxic, but this has never been proved. Prevention should be the goal when using these agents. Patients should be hydrated with 1 L of 0.45% saline over 12 hours both before and after the contrast administration cautiously in patients with preexisting cardiac dysfunction. Neither mannitol nor furosemide offers benefit over saline hydration. In fact, furosemide may lead to increased rates of renal dysfunction in this setting. In some but not all studies, N-acetylcysteine given before and after contrast decreased the incidence of dye-induced nephrotoxicity. Its benefit seems more pronounced in subjects with a lower GFR. Acetylcysteine is a thiol-containing antioxidant with little toxicity whose mechanism of action is unclear. With little harm and possible benefit, administering acetylcysteine 600 mg orally every 12 hours twice, before and after a dye load, for patients at risk for acute renal failure, is a reasonable strategy. Investigators have shown a benefit using sodium bicarbonate rather than normal saline as the isotonic volume expander. Other nephrotoxic agents should be avoided during the day before and after dye administration.
Cyclosporine toxicity is usually dose dependent. It causes distal tubular dysfunction from severe vasoconstriction. Regular blood level monitoring is important to prevent nephrotoxicity. With patients who are taking cyclosporine for renal transplant rejection, kidney biopsy is often necessary to distinguish transplant rejection from cyclosporine toxicity. Renal function usually improves after reducing the dose or stopping the drug.
Other exogenous nephrotoxins include antineoplastics, such as cisplatin and organic solvents, and heavy metals such as mercury, cadmium, and arsenic.
B. Endogenous Nephrotoxins
Endogenous nephrotoxins include heme-containing products, uric acid, and paraproteins. Myoglobinuria as a consequence of rhabdomyolysis leads to acute tubular necrosis. Necrotic muscle releases large amounts of myoglobin, which is freely filtered across the glomerulus. The myoglobin is reabsorbed by the renal tubules, and direct damage can occur. Distal tubular obstruction from pigmented casts and intrarenal vasoconstriction can also cause damage. This type of renal failure occurs in the setting of crush injury, or muscle necrosis from prolonged unconsciousness, seizures, cocaine, and alcohol abuse. Dehydration and acidosis predispose to the development of myoglobinuric acute renal failure. Patients may complain of muscular pain and often have signs of muscle injury. Rhabdomyolysis of clinical importance commonly occurs with a serum creatine kinase (CK) greater than 20,000 50,000 IU/L. One study showed that 58% of patients with acute renal failure from rhabdomyolysis had CK levels greater than 16,000 IU/L. Only 11% of patients without renal failure had CK values greater than 16,000 IU/L. The globin moiety of myoglobin will cause the urine dipstick to read falsely positive for hemoglobin: the urine appears dark brown, but no red cells are present. With lysis of muscle cells, patients also become hyperkalemic, hyperphosphatemic, and hyperuricemic. The mainstay of treatment is hydration. Other adjunctive treatments include mannitol for free radical clearance and diuresis as well as alkalinization of the urine. These modalities have not been proved to change outcomes in human trials.
Hemoglobin can cause a similar form of acute renal tubular necrosis. Massive intravascular hemolysis is seen in transfusion reactions and in certain hemolytic anemias. Reversal of the underlying disorder and hydration are the mainstays of treatment.
P.926
Hyperuricemia can occur in the setting of rapid cell turnover and lysis. Chemotherapy for germ cell neoplasms and leukemia and lymphoma are the primary causes. Acute renal failure occurs with intratubular deposition of uric acid crystals; serum uric acid levels are often greater than 20 mg/dL and urine uric acid levels greater than 600 mg/24 h. A urine uric acid to urine creatinine ratio greater than 1.0 indicates risk of acute renal failure.
Bence Jones protein seen in conjunction with multiple myeloma can cause direct tubular toxicity and tubular obstruction. Other renal complications from multiple myeloma include hypercalcemia and proximal renal tubular acidosis.
Clinical Findings
A. Symptoms and Signs
See Acute Renal Failure.
B. Laboratory Findings
Urinalysis may show evidence of acute tubular damage. The urine may be brown. On microscopic examination, an active sediment may show pigmented granular casts or muddy brown casts. Renal tubular epithelial cells and epithelial cell casts are often present as well (see Table 22-1). Hyperkalemia and hyperphosphatemia are commonly encountered.
Treatment
Treatment is aimed at hastening recovery and avoiding complications. Preventive measures should be taken to avoid volume overload and hyperkalemia. Loop-blocking diuretics have been used in large doses (eg, furosemide in doses ranging from 20 mg to 160 mg orally or intravenously twice daily) to effect adequate diuresis and may help convert oliguric to nonoliguric renal failure. Such a conversion has never been shown to affect outcomes such as mortality, though. One recent retrospective study has shown potentially worse outcomes in patients who receive doses of furosemide, including nonrecovery of renal function and an increased risk of death. A more recent prospective randomized controlled trial has shown no difference between the administration of large doses of diuretics versus placebo on either recovery from acute renal failure or death. Widespread use of diuretics in critically ill patients with acute renal failure should be discouraged. Side effects of supranormal dosing include deafness. This is mainly due to peak furosemide levels and can be avoided by the use of a furosemide drip. A starting dose of 0.2 0.6 mg/kg/h is appropriate, increasing to a maximum of 1 mg/kg/h. A bolus of the hourly dose should be administered at the beginning of treatment. Intravenous thiazide diuretics can be used to augment urinary output; chlorothiazide, 500 mg intravenously every 8 12 hours, is a reasonable choice. Short-term effects also include activation of the renin-angiotensin system. Nutritional support should maintain adequate intake while preventing excessive catabolism. Dietary protein restriction of 0.6 g/kg/d helps prevent metabolic acidosis. Hypocalcemia and hyperphosphatemia can be improved with diet and phosphate-binding agents, such as aluminum hydroxide (500 mg orally with meals) over the short term, calcium carbonate (500 1500 mg orally three times daily), calcium acetate (667 mg, two or three tablets, orally before meals), or sevelamer (800 1600 mg orally three times daily). Hypocalcemia should not be treated in patients with rhabdomyolysis unless they are symptomatic. Hypermagnesemia can occur because of reduced magnesium excretion by the renal tubules, so magnesium-containing antacids and laxatives should be avoided in these patients. Dosages must be adjusted according to the estimated degree of renal impairment for drugs eliminated by the kidney.
Indications for dialysis in acute renal failure from acute tubular necrosis or other intrinsic disorders are as follows: life-threatening electrolyte disturbances (such as hyperkalemia), volume overload unresponsive to diuresis, worsening acidosis, and uremic complications (eg, encephalopathy, pericarditis, and seizures). In gravely ill patients, less severe but worsening abnormalities may also be indications for dialytic support.
Course & Prognosis
The clinical course of acute tubular necrosis is often divided into three phases: initial injury, maintenance, and recovery. The maintenance phase is expressed as either oliguric (urinary output < 500 mL/d) or nonoliguric. Nonoliguric acute tubular necrosis has a better outcome. Conversion from oliguric to nonoliguric states may be attempted but has not been shown to change the prognosis. Drugs such as dopamine and diuretics are sometimes used for this purpose but have not been shown to improve outcomes. Renal dose dopamine (1 3 mcg/kg/min) can increase renal blood flow but can also potentiate arrhythmias and myocardial ischemia. In numerous studies, dopamine use in this setting has been shown to have no benefit. Average duration of the maintenance phase is 1 3 weeks but may be several months. Cellular repair and removal of tubular debris occur during this period. The recovery phase is heralded by diuresis. GFR begins to rise; BUN and serum creatinine fall. Other treatments for acute tubular necrosis, such as atrial natriuretic peptide use, have not proven beneficial. Ongoing randomized controlled studies are looking at the usefulness of intensive versus conventional renal replacement therapy for a survival benefit.
The mortality rate from acute renal failure is 20 50% in medical illness and up to 70% in a surgical setting. Increased mortality is associated with advanced age, severe underlying disease, and multisystem organ failure. Leading causes of death are infections, fluid and electrolyte disturbances, and worsening of
P.927
underlying disease. Mortality rates have not changed significantly over 20 years, making prevention of acute renal failure a high priority.
Interstitial Nephritis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Fever.
Transient maculopapular rash.
Acute renal insufficiency.
Pyuria (including eosinophiluria), white blood cell casts, and hematuria.
General Considerations
Acute interstitial nephritis accounts for 10 15% of cases of intrinsic renal failure. An interstitial inflammatory response with edema and possible tubular cell damage is the typical pathologic finding. Cell-mediated immune reactions prevail over humoral responses. T lymphocytes can cause direct cytotoxicity or release lymphokines that recruit monocytes and inflammatory cells.
Although drugs account for over 70% of cases, acute interstitial nephritis also occurs in infectious diseases, immunologic disorders, or as an idiopathic condition. The most common drugs are penicillins and cephalosporins, sulfonamides and sulfonamide-containing diuretics, NSAIDs, rifampin, phenytoin, and allopurinol. Infectious causes include streptococcal infections, leptospirosis, cytomegalovirus, histoplasmosis, and Rocky Mountain spotted fever. Immunologic entities are more commonly associated with glomerulonephritis, but systemic lupus erythematosus, Sj gren's syndrome, sarcoidosis, and cryoglobulinemia can cause interstitial nephritis.
Clinical Findings
Clinical features can include fever (> 80%), rash (25 50%), arthralgias, and peripheral blood eosinophilia (80%). The urine often contains red cells (95%), white cells, and white cell casts. Proteinuria can be a feature, particularly in NSAID-induced interstitial nephritis, but is usually modest. Eosinophiluria can be detected by Wright's or Hansel's stain.
Treatment & Prognosis
Acute interstitial nephritis often carries a good prognosis. Recovery occurs over weeks to months, but acute dialytic therapy may be necessary in up to one-third of all patients before resolution. Patients rarely progress to ESRD. Those with prolonged courses of oliguric failure and advanced age have a worse prognosis. Treatment consists of supportive measures and removal of the inciting agent. If renal failure persists after these steps, a short course of corticosteroids can be given. Short-term, high-dose methylprednisolone (0.5 1 g/d for 1 4 days) or prednisone (60 mg/d for 1 2 weeks) followed by a prednisone taper can be used in these more severe cases of drug-induced interstitial nephritis.
Glomerulonephritis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Hematuria, dysmorphic red cells, red cell casts, and mild proteinuria.
Dependent edema and hypertension.
Acute renal insufficiency.
General Considerations
Acute glomerulonephritis is a relatively uncommon cause of acute renal failure, accounting for about 5% of cases of intrinsic renal failure. Pathologically, inflammatory glomerular lesions are seen. These include mesangioproliferative, focal and diffuse proliferative, and crescentic lesions. The larger the percentage of glomeruli involved and the more severe the lesion, the more likely it is that the patient will have a poor clinical outcome.
Categorization of acute glomerulonephritis can be done by serologic analysis. Markers include antineutrophil cytoplasmic antibodies (ANCA), anti-GBM antibodies, and other immune markers of disease.
Immune complex deposition usually occurs when moderate antigen excess over antibody production occurs. Complexes formed with marked antigen excess tend to remain in the circulation. Antibody excess with large antigen-antibody aggregates usually results in phagocytosis and clearance of the precipitates by the mononuclear phagocytic system in the liver and spleen. Causes include IgA nephropathy (Berger's disease), peri-infectious or postinfectious glomerulonephritis, endocarditis, lupus nephritis, cryoglobulinemic glomerulonephritis (often associated with hepatitis C virus), and membranoproliferative glomerulonephritis.
Anti-GBM-associated acute glomerulonephritis is either confined to the kidney or associated with pulmonary hemorrhage. The latter is termed Goodpasture's syndrome. Injury is related to autoantibodies aimed against type IV collagen in the GBM rather than to immune complex deposition.
Pauci-immune acute glomerulonephritis is a form of small-vessel vasculitis associated with ANCA, causing primary and secondary renal diseases that do not have direct immune complex deposition or antibody binding. Tissue injury is believed to be due to cell-mediated immune processes. An example is Wegener's
P.928
granulomatosis, a systemic necrotizing vasculitis of small arteries and veins associated with intravascular and extravascular granuloma formation. In addition to glomerulonephritis, these patients can have upper airway, pulmonary, and skin manifestations of disease. Cytoplasmic ANCA (C-ANCA) is both specific (88%) and sensitive (95%) for this entity. Microscopic polyangiitis is another pauci-immune vasculitis causing acute glomerulonephritis. Perinuclear staining (P-ANCA) is the common pattern. ANCA-associated and anti-GBM-associated acute glomerulonephritis can evolve to crescentic glomerulonephritis and often have poor outcomes unless treatment is started early. Both are described more fully below.
Other vascular causes of acute glomerulonephritis include malignant hypertension and the thrombotic microangiopathies such as hemolytic-uremic syndrome (see Chapter 11) and thrombotic thrombocytopenic purpura (see Chapter 13).
Clinical Findings
A. Symptoms and Signs
Patients with acute glomerulonephritis are often hypertensive and edematous, and have an abnormal urinary sediment. The edema is found first in body parts with low tissue tension, such as the periorbital and scrotal regions.
B. Laboratory Findings
Dipstick and microscopic evaluation will reveal evidence of hematuria, moderate proteinuria (usually < 2 g/d), and cellular elements such as red cells, red cell casts, and white cells. Red cell casts are specific for glomerulonephritis, and a detailed search is warranted. Twenty-four hour urine for protein excretion and creatinine clearance quantifies the amount of proteinuria and documents the degree of renal dysfunction. However, in cases of rapidly changing serum creatinine values, the urinary creatinine clearance is an unreliable marker of GFR. FENa is usually low unless renal dysfunction is marked.
Further tests include complement levels (C3, C4, CH50), ASO titer, anti-GBM antibody levels, ANCAs, antinuclear antibody titers, cryoglobulins, hepatitis serologies, blood cultures, renal ultrasound, and occasionally renal biopsy.
Treatment
Depending on the nature and severity of disease, treatment can consist of high-dose corticosteroids and cytotoxic agents such as cyclophosphamide. Plasma exchange can be used in Goodpasture's disease as a temporizing measure until chemotherapy can take effect. Treatment and prognosis for specific diseases are discussed more fully below.
Albright RC Jr: Acute renal failure: a practical update. Mayo Clin Proc 2001;76:67.
Briguori C et al: Contrast agent-associated nephrotoxicity. Prog Cardiovasc Dis 2003;45:493.
Cantarovich F et al: High-dose furosemide for established ARF: a prospective, randomized, double-blind, placebo-controlled, multicenter trial. Am J Kidn Dis 2004;44:402.
Edwards BF: Postoperative renal insufficiency. Med Clin North Am 2001;85:1241.
Esson ML et al: Diagnosis and treatment of acute tubular necrosis. Ann Intern Med 2002;137:744.
Kodner CM et al: Diagnosis and management of acute interstitial nephritis. Am Fam Physician 2003;67:2527.
Malinoski DJ et al: Crush injury and rhabdomyolysis. Crit Care Clin 2004;20:171.
Mehta R et al: Diuretics, mortality, and nonrecovery of renal function in acute renal failure. JAMA 2002;288:2547.
Merten GJ et al: Prevention of contrast-induced nephropathy with sodium bicarbonate: a randomized controlled trial. JAMA 2004;291:2328.
Parmet S et al: JAMA patient page. Acute renal failure. JAMA 2002;288:2634.
Perazella MA: Drug-induced renal failure: update on new medications and unique mechanisms of nephrotoxicity. Am J Med Sci 2003;325:349.
Sauret JM et al: Rhabdomyolysis. Am Fam Physician 2002;65: 907.
Singri N et al: Acute renal failure. JAMA 2003;289:747.
Vinen CS et al: Acute glomerulonephritis. Postgrad Med J 2003;79:206.
Warnock DG: Towards a definition and classification of acute kidney injury. J Am Soc Nephrol 2005;16:3149.
Chronic Kidney Disease
![]() Essentials of Diagnosis
Essentials of Diagnosis
Progressive azotemia over months to years.
Symptoms and signs of uremia when nearing end-stage disease.
Hypertension in the majority.
Isosthenuria and broad casts in urinary sediment are common.
Bilateral small kidneys on ultrasound are diagnostic.
General Considerations
Chronic kidney disease affects up to 20 million Americans, or one in nine adults. Most are unaware of the condition because they remain asymptomatic until the disease has significantly progressed. The National Kidney
P.929
Foundation has developed a new staging system that helps clinicians formulate practice plans (Table 22-5). Over 70% of cases of late-stage chronic kidney disease are due to diabetes mellitus or hypertension. Glomerulonephritis, cystic diseases, and other urologic diseases account for another 12%, and 15% of patients have other or unknown causes. The major causes of chronic renal failure are listed in Table 22-6.
Table 22-5. Stages of chronic kidney disease: a clinical action plan.1,2 | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Chronic kidney disease is rarely reversible and leads to a progressive decline in renal function. This occurs even after an inciting event has been removed. Reduction in renal mass leads to hypertrophy of the remaining nephrons with hyperfiltration, and the GFR in these nephrons is transiently at supranormal levels. These adaptations place a burden on the remaining nephrons and lead to progressive glomerular sclerosis and interstitial fibrosis, suggesting that hyperfiltration may worsen renal function. However, decreased renal mass in kidney donors is not associated with chronic renal failure.
Clinical Findings
A. Symptoms and Signs
The symptoms of chronic kidney disease often develop slowly and are nonspecific (Table 22-7). Individuals can remain asymptomatic until renal failure is far advanced (GFR < 10 15 mL/min). Manifestations include fatigue, weakness, and malaise. Gastrointestinal complaints, such as anorexia, nausea, vomiting, a metallic taste in the mouth, and hiccups, are common. Neurologic problems include irritability, difficulty in concentrating, insomnia, subtle memory defects, restless legs, and twitching. Pruritus is common and difficult to treat. As uremia progresses, decreased libido, menstrual irregularities, chest pain from pericarditis, and paresthesias can develop. Symptoms of drug toxicity especially for drugs eliminated by the kidney increase as renal clearance worsens.
On physical examination, the patient appears chronically ill. Hypertension is common. The skin may be yellow, with signs of easy bruisability. Rarely seen in the dialysis era is uremic frost, a cutaneous reflection
P.930
of ESRD. Uremic fetor is the characteristic fishy odor of the breath. Cardiopulmonary signs may include rales, cardiomegaly, edema, and a pericardial friction rub. Mental status can vary from decreased concentration to confusion, stupor, and coma. Myoclonus and asterixis are additional signs of uremic effects on the central nervous system.
Table 22-6. Major causes of chronic renal failure. | |
|---|---|
|
Table 22-7. Symptoms and signs of uremia. | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The term uremia is used for this clinical syndrome, but the exact cause remains unknown. BUN and serum creatinine are considered markers for unknown toxins, with parathyroid hormone (PTH) believed to be one such toxin.
In any patient with renal failure, it is important to identify and correct all possibly reversible causes. Urinary tract infections, obstruction, extracellular fluid volume depletion, nephrotoxins, hypertension, and congestive heart failure should be excluded (Table 22-8). Any of the above can worsen underlying chronic renal failure.
B. Laboratory Findings
The diagnosis of renal failure is made by documenting elevations of the BUN and serum creatinine concentrations. Further evaluation is needed to differentiate between acute and chronic renal failure. Evidence of previously elevated BUN and creatinine, abnormal prior urinalyses, and stable but abnormal serum creatinine on successive days is most consistent with a chronic process. It is helpful to plot the inverse of serum creatinine (1/SCr) versus time if three or more prior measurements are available; this estimates time to ESRD (Figure 22-1). If the slope of the line acutely declines, new causes of renal failure should be excluded as outlined above. Anemia, metabolic acidosis, hyperphosphatemia, hypocalcemia, and hyperkalemia can occur with both acute and chronic renal failure. The urinalysis shows isosthenuria if tubular concentrating and diluting ability are impaired. The urinary sediment can show broad waxy casts as a result of dilated, hypertrophic nephrons.
C. Imaging
The finding of small echogenic kidneys bilaterally (< 10 cm) by ultrasonography supports a diagnosis of chronic renal failure, though normal or even large kidneys can be seen with chronic renal failure caused by adult polycystic kidney disease, diabetic nephropathy, HIV-associated nephropathy, multiple myeloma, amyloidosis, and obstructive uropathy. Radiologic evidence of renal osteodystrophy is another helpful finding, since x-ray changes of secondary hyperparathyroidism do not appear unless parathyroid levels have been elevated for at least 1 year. Evidence of subperiosteal reabsorption along the radial sides of the digital bones of the hand confirms hyperparathyroidism.
Complications
A. Hyperkalemia
Potassium balance generally remains intact in chronic renal failure until the GFR is less than 10 20 mL/min.
P.931
However, certain states pose an increased risk of hyperkalemia at higher GFRs. Endogenous causes include any type of cellular destruction such as hemolysis and trauma, hyporeninemic hypoaldosteronism (type IV renal tubular acidosis, seen particularly in diabetes mellitus), and acidemic states (0.6 mEq/L elevation in K+ for each 0.1 unit decrease in pH). Exogenous causes include diet (eg, citrus fruits and salt substitutes containing potassium) and drugs that decrease K+ secretion (amiloride, triamterene, spironolactone, NSAIDs, ACE inhibitors) or block cellular uptake ( -blockers).
Table 22-8. Reversible causes of renal failure. | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
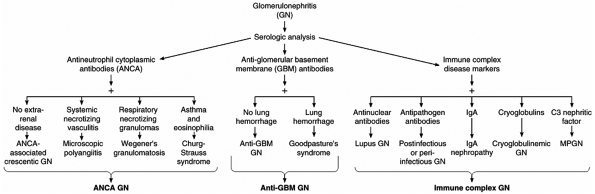 |
Figure 22-1. Decline in renal function plotted against time to end-stage renal disease (ESRD). The solid line indicates the linear decline in renal function over time. The dotted line indicates the approximate time to ESRD. |
Treatment of acute hyperkalemia involves cardiac monitoring, intravenous calcium chloride or gluconate, insulin administration with glucose, bicarbonate, and an orally or rectally administered ion exchange resin (sodium polystyrene sulfonate). The resin exchanges sodium for potassium and can administer a significant sodium load to a patient. -Agonists, such as albuterol, may also be used in acute cases. Chronic hyperkalemia is best treated with dietary potassium restriction (2 g/d) and sodium polystyrene sulfonate when necessary. The usual dose is 15 30 g once a day in juice or sorbitol.
B. Acid-Base Disorders
Damaged kidneys are unable to excrete the 1 mEq/kg/d of acid generated by metabolism of dietary proteins. The resultant metabolic acidosis is primarily due to loss of renal mass. This limits production of ammonia (NH3) and limits buffering of H+ in the urine. (Other causes include decreased filtration of titratable acids such as sulfates and phosphates, decreased proximal tubular bicarbonate resorption, and decreased renal tubular hydrogen ion secretion.) Although patients with chronic renal failure are in positive hydrogen ion balance, the arterial blood pH is maintained at 7.33 7.37 and serum bicarbonate concentration rarely falls below 15 mEq/L. The excess hydrogen ions are buffered by the large calcium carbonate and calcium phosphate stores in bone. This contributes to the renal osteodystrophy of chronic renal failure described below.
The serum bicarbonate level should be maintained at greater than 21 mEq/L according to recently published national guidelines. Alkali supplements include sodium bicarbonate, calcium bicarbonate, and sodium citrate. Citrate salts increase the absorption of dietary aluminum and should be avoided in patients exposed to aluminum. Administration should begin with 20 30 mmol/d of alkali divided into two doses per day and titrated as needed.
C. Cardiovascular Complications
Long-term complications of chronic kidney disease include a high risk of morbidity and mortality of cardiovascular disease in comparison to the general population. Mortality due to a cardiovascular cause accounts for 45% of all deaths of patients receiving dialysis. The precise biologic mechanisms for this are unclear but may have to do with the uremic milieu, underlying coexistent comorbidities, and a hesitancy to perform investigative procedures in patients with chronic kidney disease.
1. Hypertension
As renal failure progresses, hypertension due to salt and water retention usually develops. Hyperreninemic states and exogenous erythropoietin administration can also exacerbate hypertension. Hypertension is the most common complication of ESRD and must be meticulously controlled. Failure to do so can accelerate the progression of renal damage.
Control of hypertension can be achieved with salt and water restriction, weight loss if indicated, and pharmacologic therapy. The ability of the kidney to adjust to variations in sodium and water intake becomes limited as renal failure progresses. An elevated sodium chloride intake leads to congestive heart failure, edema, and hypertension, whereas low salt intake leads to volume contraction and hypotension. A mildly decreased salt diet (4 g/d) can be started, and salt intake should be reduced to 2 g/d if hypertension persists. Initial drug therapy can include ACE inhibitors or angiotensin II receptor blockers (if serum potassium and GFR permit), calcium channel-blocking agents, diuretics, and -blocking agents. The adjunctive drugs that are often needed (eg, clonidine, hydralazine, minoxidil) reflect the difficulty of achieving and maintaining hypertensive control in these patients. Goal blood pressure for patients with chronic kidney disease is less than 130/80 mm Hg.
P.932
2. Pericarditis
With uremia, pericarditis may develop. The cause is believed to be retention of metabolic toxins. Symptoms include chest pain and fever. Pulsus paradoxus can be present. A friction rub may be auscultated, but the lack of a rub does not rule out a significant pericardial effusion. Chest radiography will show an enlarged cardiac silhouette, and an ECG will show characteristic findings as explained in Chapter 10. Cardiac tamponade can occur; these patients have signs of poor cardiac output, with jugular venous distention and lungs clear to auscultation. Pericarditis is an absolute indication for initiation of hemodialysis.
3. Congestive heart failure
Patients with ESRD tend toward a high cardiac output. They often have extracellular fluid overload, shunting of blood through an arteriovenous fistula for dialysis, and anemia. In addition to hypertension, these abnormalities cause increased myocardial work and oxygen demand. Patients with chronic kidney disease may also have accelerated rates of atherosclerosis. All of these factors contribute to left ventricular hypertrophy and dilation, present in 75% of patients starting dialysis. PTH may also play a role in the pathogenesis of the cardiomyopathy of renal failure.
Water and salt intake should be controlled in patients who are oliguric or anuric. Diuretics are of value, though certain thiazides are ineffective when the GFR is less than 10 15 mL/min. Loop diuretics are commonly used, and higher doses are required as renal function declines. Digoxin should be used with caution since it is excreted by the kidney. The proved efficacy of ACE inhibitors in congestive heart failure holds true for patients with chronic renal failure. Despite the risks of hyperkalemia and worsening renal function, ACE inhibitors can be used for patients with a serum creatinine greater than 3 mg/dL with close supervision. Along with angiotensin II receptor blockers, ACE inhibitors have been shown to slow the progression to ESRD, even for patients with advanced chronic kidney disease. (See above section regarding treatment of proteinuria.) Once a patient is receiving dialysis, these risks become less relevant. When an ACE inhibitor or ARB drug is initiated, patients should have serum creatinine and potassium checked within 5 14 days.
D. Hematologic Complications
1. Anemia
The anemia of chronic renal failure is characteristically normochromic and normocytic. It is due primarily to decreased erythropoietin production, which becomes clinically significant when GFR falls below 20 25 mL/min. Many patients are iron deficient as well. Low-grade hemolysis and blood loss from platelet dysfunction or hemodialysis play an additional role.
Recombinant erythropoietin (epoetin alfa) is used in patients whose hematocrits are less than 33%. The effective dose can vary; the starting dose is 50 units/kg (3000 4000 units/dose) once or twice a week. It can be given intravenously (eg, in the hemodialysis patient) or subcutaneously (eg, in any predialysis or dialysis patient). Subcutaneous administration is preferable to intravenous administration because it requires a 33% lower dose for the same effect. Recombinant darbepoietin is also now available. Its administration is less frequent, and intravenous and subcutaneous doses have equivalent effects. Iron stores must be adequate to ensure response. Hemodialysis patients typically require 50 200 mg intravenous iron each month due to expected blood loss at dialysis. Other patients with serum ferritin less than 100 ng/mL or iron saturation less than 20% should also receive iron supplementation. Depending on the clinical situation, iron therapy should be withheld if the serum ferritin is greater than 800 ng/mL. Oral therapy with ferrous sulfate, 325 mg once daily to three times daily, is adequate but not always well tolerated, and gut absorption of iron is impaired in uremic patients. Ferrous fumarate is the best-accepted formulation, and intravenous iron may be used in dialysis patients. Hypertension is a complication of epoetin alfa therapy in about 20% of patients. It develops more abruptly in the patients with the lowest hematocrit values at initiation of therapy, and in those with the greatest rate of rise in hemoglobin. The dosage may require adjustment, or antihypertensive drugs may need to given. Hemoglobin levels should rise no more than 1 g/dL every 3 4 weeks.
2. Coagulopathy
The coagulopathy of chronic kidney disease is mainly caused by platelet dysfunction. Platelet counts are only mildly decreased, but the bleeding time is prolonged. Platelets show abnormal adhesiveness and aggregation. Clinically, patients can have petechiae, purpura, and an increased tendency for bleeding during surgery.
Treatment is required only in patients who are symptomatic. Raising the hematocrit to 30% can reduce bleeding time in many patients. Desmopressin (25 mcg intravenously every 8 12 hours for two doses) is effective and often used in preparation for surgery. It causes release of factor VIII bound to von Willebrand's factor from endothelial cells. Conjugated estrogens, 0.6 mg/kg diluted in 50 mL of 0.9% sodium chloride infused over 30 40 minutes daily, or 2.5 5 mg orally for 5 7 days, have an effect for several weeks. Dialysis improves the bleeding time but does not normalize it. Peritoneal dialysis is preferable to hemodialysis because the latter requires heparin to prevent clotting in the dialyzer. Cryoprecipitate (10 15 bags) is rarely used and lasts less than 24 hours.
E. Neurologic Complications
Uremic encephalopathy does not occur until GFR falls below 10 15 mL/min. Encephalopathy may be due to tertiary hyperparathyroidism, where an elevated PTH level or, rarely, hypercalcemia, can be the culprit. PTH may be one of the uremic toxins. Symptoms begin with difficulty in concentrating and can progress to lethargy, confusion, and coma. Physical findings include nystagmus, weakness, asterixis, and hyperreflexia. These symptoms and signs may improve after initiation of dialysis.
P.933
Neuropathy is found in 65% of patients who receive dialysis or who will need it soon but not until GFR is 10% of normal. Peripheral neuropathies manifest themselves as sensorimotor polyneuropathies (stocking and glove distribution) and isolated or multiple isolated mononeuropathies. Patients can have restless legs, loss of deep tendon reflexes, and distal pain. The earlier initiation of dialysis may prevent peripheral neuropathies, and the response to dialysis is variable. Other neuropathies result in impotence and autonomic dysfunction.
F. Disorders of Mineral Metabolism
The disorders of calcium, phosphorus, and bone are referred to as renal osteodystrophy. The most common disorder is osteitis fibrosa cystica the bony changes of secondary hyperparathyroidism. This affects ~ 50% of patients nearing ESRD. As GFR decreases below 25% of normal, phosphorus excretion is impaired. Hyperphosphatemia leads to hypocalcemia, stimulating secretion of PTH, which has a phosphaturic effect and normalizes serum phosphorus. This continuous process leads to markedly elevated PTH levels and high bone turnover with osteoclastic bone resorption and subperiosteal lesions. Metastatic calcifications, such as tumoral calcinosis, can occur. Radiographically, lesions are most prominent in the phalanges and lateral ends of the clavicles.
Osteomalacia is a form of renal osteodystrophy with low bone turnover (affecting ~ 10% of patients nearing ESRD). With worsening renal function, there is decreased renal conversion of 25-hydroxycholecalciferol to the 1,25-dihydroxy form. Gut absorption of calcium is diminished, leading to hypocalcemia and abnormal bone mineralization. Deposition of aluminum in bone can also lead to osteomalacia. Elevated aluminum levels are seen in patients after years of chronic aluminum hydroxide administration for phosphorus binding. This entity is seen with decreasing frequency because aluminum-based binders are used less in the chronic setting and water used for hemodialysis is now cleared of aluminum.
Adynamic bone disease is a disorder of low bone turnover. More than 25% of patients nearing ESRD show evidence of minimal osteoid and decreased or absent bone remodeling. Its frequency is increasing because of increased use of active vitamin D analogs, which suppress PTH production.
All of the above entities can cause bony pain and proximal muscle weakness. Spontaneous bone fractures can occur that are slow to heal. When the calcium-phosphorus product (serum calcium [mg/dL] serum phosphate [mg/dL]) is above 60 70, metastatic calcifications are commonly seen in blood vessels, soft tissues, lungs, and myocardium. Treatment should begin with dietary phosphorus restriction to 1000 mg/d. Oral phosphorus-binding agents, such as calcium carbonate or calcium acetate, act in the gut and are given in divided doses three or four times daily with meals. These should be titrated to a serum calcium of less than 10 mg/dL (preventing hypercalcemia) and serum phosphorus of 2.7 4.6 mg/dL in patients with a GFR of 15 59 mL/min/1.73 m2 and serum phosphorus of 3.5 5.5 mg/dL in patients with a GFR of less than 15 mL/min/1.73 m2. Sevelemer and lanthanum carbonate are other phosphorus-binding agents that do not contain calcium; they are particularly useful in patients with hypercalcemia, although long-term effects are unknown. Aluminum hydroxide is an effective phosphorus binder but can cause osteomalacia and neurologic complications. It can be used in the acute setting for serum phosphorus greater than 7 mg/dL, but long-term use should be avoided. If aluminum levels are high, chelation with deferoxamine can be effective. Vitamin D or vitamin D analogs should be given with secondary hyperparathyroidism (iPTH more than two to three times normal) if phosphorus levels are less than 5.5 mg/dL and calcium less than 10 mg/dL. Vitamin D suppresses PTH and increases serum calcium and phosphorus levels; both need to be monitored closely to prevent hypercalcemia and hyperphosphatemia. If calcitriol is used, the dosage should be 0.25 0.5 mcg daily or every other day initially. Cinacalcet can be used if elevated serum phosphorus or calcium levels prohibit the use of vitamin D analogs. Cinacalcet is a calcimimetic agent that targets the calcium-sensing receptor on the chief cells of the parathyroid gland.
G. Endocrine Disorders
Circulating insulin levels are higher because of decreased renal insulin clearance. Glucose intolerance can occur in chronic renal failure when GFR is less than 10 20 mL/min. Primarily, this is due to peripheral insulin resistance. Fasting glucose levels are usually normal or only slightly elevated. Therefore, patients can be either hyperglycemic or hypoglycemic depending on the predominant disturbance. Most commonly, diabetic patients require decreased doses of hypoglycemic agents.
Decreased libido and impotence are common in chronic renal failure. Men have decreased testosterone levels; women are often anovulatory. Despite a high degree of infertility, pregnancy can occur particularly in women who are well dialyzed and well nourished. Therefore, contraception is advisable for women who do not wish to become pregnant.
Treatment
A. Dietary Management
Every patient with chronic renal failure should be evaluated by a renal nutritionist. Specific recommendations should be made concerning protein, salt, water, potassium, and phosphorus intake.
1. Protein restriction
Experimental models have shown that protein restriction slows the progression to ESRD; however, this has not been consistently proved in clinical trials. The MDRD Study was meant to clarify the issue, but the results were inconclusive. A subsequent
P.934
meta-analysis of five clinical trials did show a significant benefit but did not control for certain effects such as ACE inhibitor therapy. The benefits of protein restriction in slowing the rate of decline of GFR must be weighed against the risk of cachexia upon the institution of dialysis. Low serum albumin at the start of dialysis is one of the strongest predictors of mortality in this population. In general, protein intake should not exceed 1 g/kg/d, and if protein restriction proves to be beneficial, the level of restriction may be increased to 0.6 0.8 g/kg/d.
2. Salt and water restriction
In advanced renal failure, the kidney is unable to adapt to large changes in sodium intake. Intake greater than 3 4 g/d can lead to edema, hypertension, and congestive heart failure, whereas intake of less than 1 g/d can lead to volume depletion and hypotension. For the nondialysis patient approaching ESRD, 2 g/d of sodium is an initial recommendation. A daily intake of 1 2 L of fluid maintains water balance.
3. Potassium restriction
Restriction is needed once the GFR has fallen below 10 20 mL/min. Patients should receive detailed lists concerning potassium content of foods and should limit their intake to less than 60 70 mEq/d. Normal intake is about 100 mEq/d.
4. Phosphorus restriction
The phosphorus level should be kept below 4.6 mg/dL, with a dietary restriction of 800 1000 mg/d. Foods rich in phosphorus such as cola beverages, eggs, dairy products, and meat should be limited. Below a GFR of 20 30 mL/min, phosphorus binders are usually required. The treatment of hyperphosphatemia is discussed in the section on disorders of mineral metabolism.
5. Magnesium restriction
Magnesium is excreted primarily by the kidneys. Dangerous hypermagnesemia is rare unless the patient ingests medications high in magnesium or receives it parenterally. All magnesium-containing laxatives and antacids are relatively contraindicated in renal failure.
B. Dialysis
When conservative management of ESRD is inadequate, hemodialysis, peritoneal dialysis, and kidney transplantation are alternatives (see below). According to the Kidney Disease Outcomes Quality Initiative (KDOQI) guidelines, dialysis should be started when a patient has a GFR of 10 mL/min or serum creatinine of 8 mg/dL. Diabetic patient should start when the GFR reaches 15 mL/min or serum creatinine is 6 mg/dL. Other indications for dialysis include (1) uremic symptoms, such as pericarditis, encephalopathy, or coagulopathy; (2) fluid overload unresponsive to diuresis; (3) refractory hyperkalemia; (4) severe metabolic acidosis (pH < 7.20); and (5) neurologic symptoms, such as seizures or neuropathy. Preparation for dialysis requires a team approach. Dietitians, social workers, psychiatrists, and transplant surgeons should be involved as well as primary care physicians and nephrologists. The patient and family need early counseling regarding the risks and benefits of therapy. The options of not starting or withdrawing dialysis should be discussed openly.
1. Hemodialysis
Hemodialysis requires a constant flow of blood along one side of a semipermeable membrane with a cleansing solution, or dialysate, along the other. Diffusion and convection allow the dialysate to remove unwanted substances from the blood while adding back needed components. Vascular access for hemodialysis can be accomplished by an arteriovenous fistula (the preferred method) or prosthetic graft. Indwelling catheters should be considered temporary measures. Native fistulas typically last longer than prosthetic shunts but require longer time (6 8 weeks or more after surgical construction) before they can be used. Infection, thrombosis, and aneurysm formation are complications seen more often in grafts than fistulas. Staphylococcus aureus is the most common infecting agent.
Patients typically require hemodialysis three times a week. Sessions last 3 5 hours depending on patient size, type of dialyzer used, and other factors. Periodic measurement of dialysis adequacy should determine the duration of treatment. Home hemodialysis is an option that is becoming less popular because of the need for a trained helper, large equipment, and costs. Nocturnal and daily hemodialysis are other forms of hemodialysis that offer improved outcomes in certain populations. Unfamiliarity with home hemodialysis and the financial impact with most current reimbursement schemes are currently limiting the availability of these modalities.
2. Peritoneal dialysis
With peritoneal dialysis, the peritoneal membrane is the dialyzer. Fluids and solutes move across the capillary bed that lies between the visceral and parietal layers of the membrane into the dialysate. Dialysate enters the peritoneal cavity through a catheter. The most common kind of peritoneal dialysis is continuous ambulatory peritoneal dialysis (CAPD). Patients exchange the dialysate four to six times a day. Continuous cyclic peritoneal dialysis (CCPD) utilizes a cycler machine to automatically perform exchanges at night. The dialysate remains in the peritoneal cavity between exchanges. As with hemodialysis, actual peritoneal dialysis prescriptions are guided by adequacy measurements.
The percentage of dialysis patients using peritoneal dialysis has been decreasing over the past several years. Peritoneal dialysis permits greater patient autonomy; its continuous nature minimizes the symptomatic swings observed in hemodialysis patients; and poorly dialyzable compounds such as phosphates are better cleared, which permits less dietary restriction. The dialysate removes large amounts of albumin, and nutritional status must be closely watched.
The most common complication of peritoneal dialysis is peritonitis. Rates are as high as 0.8 episodes per patient-year. The patient can experience nausea and vomiting, abdominal pain, diarrhea or constipation, and fever. The dialysate can be cloudy and contain
P.935
greater than 100 white cells/mcL of which over 50% are polymorphonuclear neutrophils. S aureus is the most common infecting organism.
The total costs of peritoneal dialysis and hemodialysis are approximately the same. Equipment expenses are less for peritoneal dialysis, but the costs of peritonitis are high. Patients treated with both modalities more often prefer peritoneal dialysis to hemodialysis.
Survival rates on dialysis depend on the underlying disease process. Five-year Kaplan-Meier survival rates vary from 21% for patients with diabetes to 47% for patients with glomerulonephritis. Overall 5-year survival is currently estimated at 36%. Patients undergoing dialysis have an average life expectancy of 3 4 years, but survival for as long as 25 years is seen depending on the disease entity. Studies are conflicting regarding the survival advantage associated with either peritoneal dialysis or hemodialysis.
C. Kidney Transplantation
Up to 50% of all patients with ESRD are suitable for transplantation. Age is becoming less of a barrier. Two-thirds of kidney transplants come from deceased donors, with the remainder from living related or unrelated donors. Immunosuppressive drugs include corticosteroids, azathioprine, mycophenolate mofetil, tacrolimus, cyclosporine, and rapamycin. A patient with a deceased-donor renal transplant typically requires stronger immunosuppression than patients with living related kidney donor transplants. However, this depends to a great extent on the degree of HLA-type matching. The 1- and 5-year kidney graft survival rates are approximately 94% and 76%, respectively, for living related and living unrelated donor transplants and 88% and 65%, respectively, for deceased donor transplants. The average wait for a cadaveric transplant is 2 4 years; this is becoming progressively longer as more people are going onto waiting lists while the deceased donor pool is not expanding. Aside from medication use, the life of a transplanted patient can return to nearly normal.
Prognosis
Mortality is higher for patients undergoing dialysis than for age-matched controls. Yearly mortality is 21.2 deaths per 100 patient-years. The expected remaining lifetime for the age group 55 64 is 22 years, whereas that of the ESRD population is 5 years. The most common cause of death is cardiac dysfunction (45%). Other causes include infection (14%), cerebrovascular disease (6%), and malignancy (4%). Diabetes, age, a low serum albumin, lower socioeconomic status, and inadequate dialysis are all significant predictors of mortality.
For those who require dialysis to sustain life but elect not to undergo dialysis, death ensues within days to weeks. In general, uremia develops and patients lose consciousness prior to death. Arrhythmias can occur as a result of electrolyte imbalance. Volume overload and dyspnea can be managed by volume restriction and opioids as described in Chapter 5. Meticulous efforts at palliative care are essential.
Barry JM: Current status of renal transplantation. Patient evaluations and outcomes. Urol Clin North Am 2001;28: 677.
Block GA et al: Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med 2004;350: 1516.
Bolton WK et al: Preparing the patient for renal replacement therapy. Teamwork optimizes outcomes. Postgrad Med 2002; 111:97.
Collins AJ et al: Cardiovascular disease in end-stage renal disease patients. Am J Kidney Dis 2001;38(4 Suppl 1):S26.
Fan SL et al: Bisphosphonates in renal osteodystrophy. Curr Opin Nephrol Hypertens 2001;10:581.
Go AS et al: Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296.
JAMA patient page. Kidney failure. JAMA 2001;286:2898.
Jardine AG et al: Cardiovascular complications of renal disease. Heart 2001;86:459.
Levey AS et al: National Kidney Foundation: National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med 2003;139:137. Erratum in Ann Intern Med 2003;139:605.
Mathur RV et al: Calciphylaxis. Postgrad Med J 2001;77:557.
Ramanathan V et al: Renal transplantation. Semin Nephrol 2001; 21:213.
Ruggenenti P et al: Progression, remission, regression of chronic renal diseases. Lancet 2001;357:1601.
Smogorzewski MJ: Central nervous dysfunction in uremia. Am J Kidney Dis 2001;38(4 Suppl 1):S122.
Renal Artery Stenosis
The two most common forms of renal artery stenosis are atherosclerotic ischemic renal disease and fibromuscular dysplasia. The prevalence of this condition has been estimated only by autopsy and angiographic studies. Approximately 5% of Americans with hypertension suffer from renal artery stenosis.
Atherosclerotic ischemic renal disease accounts for 67 95% of all cases of renal artery stenosis. It occurs most commonly in those over 45 years of age with a history of atherosclerotic disease. Other risk factors include renal insufficiency, diabetes mellitus, tobacco use, and hypertension.
Clues to diagnosis include refractory hypertension, new-onset hypertension in an older patient, pulmonary edema with poorly controlled blood pressure, and acute renal failure upon starting an ACE inhibitor. In addition to hypertension, physical examination may reveal an audible abdominal bruit on the affected side. Laboratory values can show elevated BUN and serum creatinine levels in the setting of significant renal ischemia, and abdominal ultrasound discloses asymmetric
P.936
kidney size when one renal artery is affected out of proportion to the other.
Three prevailing methods used for screening are Doppler ultrasonography, captopril renography, and magnetic resonance angiography (MRA). Doppler ultrasonography is highly sensitive and specific (> 90% with an experienced ultrasonographer) and relatively inexpensive. However, this method is extremely operator and patient dependent. Measurements of blood flow must be made at the aorta and along each third of the renal artery in order to assess the disease. This test is a poor choice for patients who are obese, unable to lie supine, or have interfering bowel gas patterns.
Captopril renography capitalizes on the difference in renal perfusion with and without ACE inhibitors. A kidney distal to a significant stenosis requires high angiotensin II levels to maintain adequate perfusion. With an ACE inhibitor, perfusion is markedly diminished. The affected kidney enhances less, whereas the unaffected one enhances more in the setting of a captopril challenge. Sensitivity ranges from 75% to 100% and specificity from 60% to 90%. This procedure is not as accurate in moderate to severe renal insufficiency.
MRA is an excellent but expensive way to screen for renal artery stenosis. Sensitivity is 99 100%. Specificity ranges from 71% to 96%. Turbulent blood flow can cause false-positive results.
Renal angiography is the gold standard for diagnosis. CO2 subtraction angiography can be used in place of dye when the risk of dye nephropathy exists eg, in diabetic patients with renal insufficiency. Lesions are most commonly found in the proximal third or ostial region of the renal artery. The risk of atheroembolic phenomena after angiography is not trivial in this population, ranging from 5% to 10%.
Treatment is controversial. Options include medical management, angioplasty with or without stenting, and surgical bypass. Angioplasty might reduce the number of antihypertensive medications but does not significantly change outcome in comparison to patients medically managed. Stenting produces significantly better angioplastic results. However, blood pressure is equally improved, and serum creatinines are similar at 6 months of observation. Angioplasty is equally as effective as, and safer than, surgical revision.
Fibromuscular dysplasia primarily affects young women. Unexplained hypertension in a young woman is reason to screen for this disorder. The noninvasive tests mentioned above should be used for detection. This disorder has a characteristic beads-on-a-string appearance on angiography. Treatment with percutaneous transluminal angioplasty is often curative.
Bloch MJ et al: Clinical insights into the diagnosis and management of renovascular disease. An evidence-based review. Minerva Med 2004;95:357.
Kalra PA et al: Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005;68:293.
Korsakas S et al: Delay of dialysis in end-stage renal failure: prospective study on percutaneous renal artery interventions. Kidney Int 2004;65:251.
Nordmann AJ et al: Balloon angioplasty versus medical therapy for hypertensive patients with renal artery obstruction. Cochrane Database Syst Rev 2003;(3):CD002944.
Safian RD et al: Renal artery stenosis. N Engl J Med 2001;8:344.
Glomerulonephropathies
Abnormalities of glomerular function can be caused by damage to the major components of the glomerulus: the epithelium (podocytes), basement membrane, capillary endothelium, or mesangium. The damage is often manifested as an inflammatory process. A specific histologic pattern of glomerular injury can be seen on renal biopsy, one of the most helpful techniques available for defining the cause of glomerular disease. Clinically, hematuria, proteinuria, hypertension, and a reduced GFR are typical findings of glomerular diseases presenting as nephritic syndromes; heavy proteinuria (> 3.5 g/24 h), hypoalbuminemia, hyperlipidemia, and edema are typical findings of glomerular diseases presenting as nephrotic syndromes.
Classification
Glomerular diseases generally can be classified into one of three major syndromes: nephritic syndrome, nephrotic syndrome, and asymptomatic renal disease. Specific glomerular diseases usually exhibit characteristics of one of the above syndromes, though some can have varying components of all three.
Glomerular diseases can also be classified according to whether they cause only renal abnormalities (primary renal disease) or whether the renal abnormalities result from a systemic disease (secondary renal disease).
Nephritic Syndrome
![]() Essentials of Diagnosis
Essentials of Diagnosis
Edema.
Hypertension.
Hematuria (with or without dysmorphic red cells, red blood cell casts).
General Considerations
Acute glomerulonephritis usually signifies an inflammatory process causing renal dysfunction over days to weeks that may or may not resolve. If the inflammatory
P.937
process is severe, the glomerulonephritis may lead to a greater than 50% loss of nephron function over the course of just weeks to months. Such a process, called rapidly progressive acute glomerulonephritis, can cause permanent damage to glomeruli if not identified and treated rapidly. Prolonged inflammatory changes can result in chronic glomerulonephritis with persistent renal abnormalities that progress to ESRD.
Clinical Findings
A. Symptoms and Signs
Edema is first seen in regions of low tissue pressure such as the periorbital and scrotal areas. Hypertension, if present, is due to volume overload rather than vasoactive substances such as angiotensin II, whose levels are low.
B. Laboratory Findings
1. Serum chemistries
There are no serum chemistries characteristic of nephritic syndrome, but certain special tests are often performed depending on the history and the results of the preliminary evaluation. These include complement levels, antinuclear antibodies (ANA), cryoglobulins, hepatitis serologies, ANCA, anti-GBM antibodies, antistreptolysin O (ASO) titers, and C3 nephritic factor (Figure 22-2).
2. Urinalysis
The urinalysis shows red blood cells. These may be misshapen from traversing a damaged capillary membrane so-called dysmorphic red blood cells. Red blood cell casts and moderate degrees of proteinuria are also characteristic of the urinary sediment. Placing the patient in a lordotic position for an hour increases sensitivity for finding red cell casts in the next urine specimen.
3. Biopsy
Renal biopsy should be considered if there are no other contraindications to biopsy (eg, bleeding disorders, thrombocytopenia, uncontrolled hypertension). Rapidly progressive glomerulonephritis is likely when over 50% of glomeruli contain crescents. The type of disease can be categorized according to the immunofluorescent pattern and appearance on electron microscopy (Table 22-9).
Treatment
Treatment includes aggressive reduction of hypertension and fluid overload and specific therapeutic maneuvers aimed at the underlying cause. Salt and water restriction, diuretic therapy, and possibly dialysis are needed. The inflammatory glomerular injury may require corticosteroids and cytotoxic agents. (See specific diseases discussed below.)
Postinfectious Glomerulonephritis
Postinfectious glomerulonephritis is most often due to infection with nephritogenic group A -hemolytic streptococci, especially type 12. It can occur sporadically or in clusters and during epidemics can account for up to 10% of known streptococcal infections. It commonly appears after pharyngitis or impetigo. Onset occurs within 1 3 weeks after infection (average, 7 10 days).
Other causes of postinfectious glomerulonephritis include bacteremic states such as systemic S aureus infection. Infective endocarditis and shunt infections cause similar lesions.
These are referred to as peri-infectious glomerulonephritides. Viral, fungal, and parasitic causes include hepatitis B or C, cytomegalovirus infection, infectious mononucleosis, coccidioidomycosis, malaria, and toxoplasmosis.
Clinical Findings
A. Symptoms and Signs
The patient is oliguric, edematous, and variably hypertensive.
B. Laboratory Findings
Serum complement levels are low; in postinfectious glomerulonephritis due to group A streptococcal infection, ASO titers can be high unless the immune response has been blunted with previous antibiotic treatment. Classically, the urine is described as cola-colored. Urinary red blood cells, red cell casts, and proteinuria under 3.5 g/d are present. On microscopy, this entity appears as a diffuse proliferative glomerulonephritis. Immunofluorescence shows IgG and C3 in a granular pattern in the mesangium and along the capillary basement membrane. Electron microscopy shows large, dense subepithelial deposits or humps.
Treatment
Treatment for this entity is supportive. Appropriate antibiotics should be used. Antihypertensives, salt restriction, and diuretics should be used if needed. Corticosteroids have not been shown to improve outcome. Prognosis in children is very favorable, but adults are more prone to crescent formation and chronic renal insufficiency. A rapidly progressive glomerulonephritis will develop in less than 5% of adults, and a smaller percentage of adults will progress to ESRD.
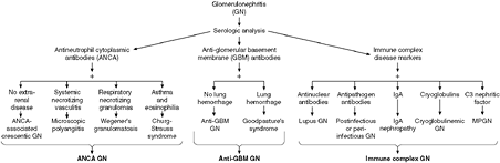 |
Figure 22-2. Serologic analysis of patients with glomerulonephritis. MPGN = membranoproliferative glomerulonephritis. (Reproduced, with permission, from Jennette JC et al: Primer on Kidney Diseases. Academic Press, 1994. ) |
IgA NEPHROPATHY & HENOCH-SCH NLEIN PURPURA
IgA Nephropathy
IgA nephropathy (Berger's disease) is a primary renal disease of IgA deposition in the glomerular mesangium. The inciting cause is unknown, but the same lesion is seen in Henoch-Sch nlein purpura. IgA nephropathy is also associated with hepatic cirrhosis, celiac disease, and infections such as with HIV and cytomegalovirus.
IgA nephropathy is the most common form of acute glomerulonephritis in the United States and is even more prevalent worldwide, particularly in Asia. It
P.938
P.939
is most commonly seen in children and young adults, with males affected two to three times more commonly than females.
Table 22-9. Classification and findings in glomerulonephritis: nephritic syndromes. | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
An episode of gross hematuria is the most common presenting complaint. Frequently, this is associated with an upper respiratory infection (50%), gastrointestinal symptoms (10%), or a flu-like illness (15%). The urine becomes red or cola-colored 1 2 days after onset. In contrast to postinfectious glomerulonephritis, this feature has been called synpharyngitic hematuria since there is no significant latent period. Other findings include asymptomatic microscopic hematuria as an incidental finding and the nephrotic syndrome (see below). Approximately one-third of patients will experience a clinical remission. Forty to 50 percent of patients will have progressive renal insufficiency. The remainder will show chronic microscopic hematuria and a stable serum creatinine. The most unfavorable prognostic indicator is proteinuria greater than 1 g/d; others include hypertension, persistent microscopic hematuria and proteinuria, glomerulosclerosis, and abnormal renal function.
The serum IgA level is increased in up to 50% of patients, and for that reason a normal serum IgA does not rule out the disease. Serum complement levels are usually normal, and renal biopsy is the standard for diagnosis. Glomeruli show a focal glomerulonephritis with diffuse mesangial IgA deposits and proliferation of mesangial cells. IgG and C3 can also be seen in the mesangium of all glomeruli. Skin biopsy often reveals granular deposits of IgA in dermal capillaries of affected patients.
In patients with significant proteinuria (> 1 g/d), ACE inhibitors or ARB drugs should be used to reduce proteinuria and hypertension. The target blood pressure is less than 130/80 mm Hg. In patients with proteinuria of 1.0 3.5 g/d, corticosteroid therapy has proven beneficial. A recent regimen showed a 2% doubling of creatinine after 6 years in the treatment group versus a 21% doubling of creatinine in the control group. The regimen consisted of giving methylprednisolone, 1 g/d intravenously, for 3 days during months 1, 3, and 5, plus prednisone in a dosage of 0.5 mg/kg every other day for 6 months. This was aimed at patients with creatinine clearances greater than 70 mL/min. Other treatments have included fish oil, with variable results in clinical trials. Recent studies that have shown a benefit also show that low doses (2 5 g/d) are just as efficacious as high doses (9 12 g/d). A recent meta-analysis has showed no benefit from fish oil, but there was a trend toward benefit in patients with more proteinuria. There are very few side effects of long-term fish oil administration aside from fishy breath and eructations. Renal transplantation is an excellent option for patients with ESRD, but recurrent disease has been documented in 30% of patients 5 10 years posttransplant. Fortunately, recurrent disease rarely leads to failure of the allograft.
Henoch-Sch nlein Purpura (Anaphylactoid Purpura)
This disease is a leukocytoclastic vasculitis of unknown cause. It is most common in children and has a male
P.940
predominance. It classically presents with palpable purpura, arthralgias, and abdominal symptoms such as nausea, colic, and melena. Purpuric skin lesions are most often found on the lower extremities. Renal insufficiency is common with a nephritic presentation. The renal lesions are identical to those found in IgA nephropathy. Most patients will recover fully over several weeks.
Further details about Henoch-Sch nlein purpura are provided in Chapter 20.
David JC et al: What is the difference between IgA nephropathy and Henoch-Sch nlein purpura nephritis? Kidney Int 2001; 59:823.
Donadio JV et al: IgA nephropathy. N Engl J Med 2002; 347:738.
Gedalia A: Henoch-Sch nlein purpura. Curr Rheumatol Rep 2004;6:195.
Julian BA et al: IgA nephropathy: an update. Curr Opin Nephrol Hypertens 2004;13:171.
Lang MM et al: Identifying poststreptococcal glomerulonephritis. Nurse Pract 2001;26:34.
Pauci-Immune Glomerulonephritis (Anca-Associated)
Pauci-immune glomerular lesions are seen with Wegener's granulomatosis, Churg-Strauss disease, and microscopic polyangiitis. All are small-vessel vasculitides. Wegener's granulomatosis also involves granulomatous inflammation of the respiratory tract with a necrotizing vasculitis of small and medium-sized vessels. Microscopic polyangiitis (polyarteritis) is similar to Wegener's granulomatosis without granulomatous inflammation, but both commonly exhibit a necrotizing glomerulonephritis. ANCA-associated glomerulonephritis can also present as a primary renal lesion. The pathogenesis of these entities is unknown, but more than 80% of pauci-immune glomerulonephritis is associated with antineutrophil cytoplasmic antibodies.
Clinical Findings
A. Symptoms and Signs
Symptoms of a systemic inflammatory disease, including fever, malaise, and weight loss, may be present. In addition to hematuria and proteinuria from glomerular inflammation, some patients exhibit purpura from dermal capillary involvement and mononeuritis multiplex from nerve arteriolar involvement. Ninety percent of patients with Wegener's granulomatosis will have upper or lower respiratory tract symptoms with nodular lesions that can cavitate and bleed.
B. Laboratory Findings
Serologically, ANCA subtype analysis can be done. A cytoplasmic pattern (C-ANCA) is specific for antiproteinase-3 antibodies, while a perinuclear pattern (P-ANCA) is specific for antimyeloperoxidase antibodies. Over 90% of patients with Wegener's syndrome will have C-ANCA; the remainder can have a P-ANCA pattern. Microscopic angiitis will have either a P-ANCA or C-ANCA pattern about 80% of the time. Pathologically, the small vessels and glomeruli will lack immune deposits (pauci-immune); however, a cell-mediated immune response is often seen. Necrotizing lesions and crescents signify a rapidly progressive glomerulonephritis.
Treatment
Treatment should be instituted early if aggressive disease is suspected. High doses of corticosteroids (methylprednisolone, 1 2 g/d for 3 days, followed by prednisone, 1 mg/kg for 1 month, with a slow taper over the next 6 months) and cytotoxic agents (cyclophosphamide, 0.5 1.0 g/m2 intravenously or 1.5 2 mg/kg orally for 3 6 months tapered over 1 year) are recommended for controlling end-organ damage. Intravenous cyclophosphamide is likely associated with fewer side effects and is just as efficacious as corticosteroids. Without treatment, prognosis is extremely poor, but with the above regimen, complete remission can be achieved in about 75% of patients. The addition of plasmapheresis does not seem to improve outcomes. Prognosis depends mainly on the extent of glomerular involvement before treatment is started. ANCA levels can be monitored to help determine the efficacy of treatment.
Anti-Glomerular Basement Membrane Glomerulonephritis & Goodpasture'S Syndrome
Goodpasture's syndrome is defined by the clinical constellation of glomerulonephritis and pulmonary hemorrhage; injury to both is mediated by anti-GBM antibodies. Up to one-third of patients with anti-GBM glomerulonephritis have no evidence of lung injury. Anti-GBM-associated glomerulonephritis accounts for about 10% of patients with rapidly progressive acute glomerulonephritis. The incidence in males is approximately six times that in females, and the disease occurs most commonly in the second and third decades but has a wide range. It has been associated with influenza A infection, hydrocarbon solvent exposure, and HLA-DR2 and -B7 antigens.
Clinical Findings
A. Symptoms and Signs
The onset of disease is preceded by an upper respiratory tract infection in 20 60% of cases. Patients experience hemoptysis, dyspnea, and possible respiratory failure. Hypertension and edema are seen as components of the nephritic syndrome.
B. Laboratory Findings
Laboratory evaluation can show iron deficiency anemia, and complement levels are normal. Sputum contains
P.941
hemosiderin-laden macrophages. Chest radiographs can show shifting pulmonary infiltrates due to pulmonary hemorrhage. The diffusion capacity of carbon monoxide is markedly increased. Diagnosis is confirmed by finding circulating anti-GBM antibodies, which are positive in over 90% of patients.
Treatment
The treatment of choice is a combination of plasma exchange therapy to remove circulating antibodies and administration of immunosuppressive drugs to prevent formation of new antibodies and control the inflammatory response. Corticosteroids are given initially in pulse doses of prednisone or methylprednisolone, 1 2 g/d for 3 days, then 1 mg/kg/d. Cyclophosphamide is administered intravenously at a dose of 0.5 1.0 g/m2 or orally at a dosage of 2 3 mg/kg/d. Daily plasmapheresis is performed for up to 2 weeks. A poorer prognosis exists in patients with oliguria and a serum creatinine greater than 6 7 mg/dL. Anti-GBM antibody levels should decrease as the clinical course improves.
Cryoglobulin-Associated Glomerulonephritis
Essential (mixed) cryoglobulinemia is a disorder associated with cold-precipitable immunoglobulins (cryoglobulins). Glomerular disease results from the precipitation of cryoglobulins in glomerular capillaries. The cause is typically an underlying infection such as hepatitis B and C or other occult viral, bacterial, and fungal infections.
Patients exhibit necrotizing skin lesions in dependent areas, arthralgias, fever, and hepatosplenomegaly. Serum complement levels are depressed. Rheumatoid factor is often elevated when cryoglobulins are present. Rapidly progressive glomerulonephritis is seen on pathologic examination with the presence of crescents.
Treatment consists of aggressively treating the underlying infection. Pulse corticosteroids, plasma exchange, and cytotoxic agents can be used. Interferon- (IFN- ) has been shown to benefit patients with hepatitis C-related cryoglobulinemia.
Booth AD et al: Pan-Thames Renal Research Group: Outcome of ANCA-associated renal vasculitis: a 5-year retrospective study. Am J Kidney Dis 2003;41:776.
Gaskin G et al: Plasmapheresis in antineutrophil cytoplasmic antibody-associated systemic vasculitis. Ther Apher 2001;5:176.
Harper L et al: ANCA-associated renal vasculitis at the end of the twentieth century a disease of older patients. Rheumatology (Oxford) 2005;44:495.
Hudson BG et al: Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med 2003;348:2543.
Jara LJ et al: Pulmonary-renal vasculitic disorders: differential diagnosis and management. Curr Rheumatol Rep 2003;5: 107.
Jennett JC et al: Microscopic polyangiitis (microscopic polyarteritis). Semin Diagn Pathol 2001;18:3.
Levy JB et al: Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med 2001;134:1033.
Madaio MP et al: The diagnosis of glomerular diseases: acute glomerulonephritis and the nephrotic syndrome. Arch Intern Med 2001;161:25.
Vinen CS et al: Acute glomerulonephritis. Postgrad Med J 2003; 79:206.
Nephrotic Syndrome
![]() Essentials of Diagnosis
Essentials of Diagnosis
Urine protein excretion > 3.5 g/1.73 m2 per 24 hours.
Hypoalbuminemia (albumin < 3 g/dL).
Peripheral edema.
General Considerations
In adults, about one-third of patients with nephrotic syndrome have a systemic renal disease such as diabetes mellitus, amyloidosis, or systemic lupus erythematosus. With the current epidemic of type 2 diabetes mellitus, this proportion is slowly increasing. The remainder have idiopathic nephrotic syndrome. The four most common are minimal change disease, focal glomerular sclerosis, membranous nephropathy, and membranoproliferative glomerulonephritis.
Clinical Findings
A. Symptoms and Signs
Peripheral edema is a hallmark of the nephrotic syndrome, occurring when the serum albumin concentration is less than 3 g/dL. Edema is most likely due to sodium retention (from renal disease) rather than arterial underfilling from low plasma oncotic pressure. Initially this presents in the dependent areas of the body such as the lower extremities; however, such edema can become generalized. Patients can experience dyspnea due to pulmonary edema, pleural effusions, and diaphragmatic compromise with ascites. Complaints of abdominal fullness may also be present in patients with ascites.
Patients may show symptoms and signs of infection more frequently than the general population owing to loss of immunoglobulins and certain complement moieties in the urine.
B. Laboratory Findings
1. Urinalysis
Proteinuria occurs as a result of an alteration of the negative charge in the GBM. The screening test for proteinuria is the urinary dipstick analysis; however, this test indicates albumin only. The addition of sulfosalicylic acid to the urine sediment
P.942
allows abnormal paraproteins to be detected. Urine dipstick testing can detect as little as 15 mg/dL of protein, but the results must be interpreted along with the urine specific gravity. Trace protein seen on highly concentrated specimens may be insignificant, while trace protein on dilute specimens may indicate true renal disease.
Table 22-10. Classification and findings in glomerulonephritis: nephrotic syndromes. | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
Microscopically, the urinary sediment has relatively few cellular elements or casts. However, if marked hyperlipidemia is present, patients can have oval fat bodies in the urine. These represent lipid deposits in sloughed renal tubular epithelial cells. They appear as grape clusters under light microscopy and Maltese crosses under polarized light.
2. Blood chemistries
Characteristic blood chemistries include a decreased serum albumin (< 3 g/dL) and total serum protein less than 6 g/dL. Hyperlipidemia occurs in over 50% of those with early nephrotic syndrome. As patients excrete larger amounts of protein per day, the frequency of hyperlipidemia increases. There is increased hepatic production of lipids (cholesterol and apolipoprotein B), owing to a fall in oncotic pressure. There is also decreased clearance of very low-density lipoproteins, causing hypertriglyceridemia. Patients can also have an elevated erythrocyte sedimentation rate as a result of alterations in some plasma components such as increased levels of fibrinogen.
Other less common tests may be necessary depending on the patient's clinical presentation, including complement levels, serum and urine protein electrophoresis, ANA, and serologic tests for hepatitis. Patients may become deficient in vitamin D, zinc, and copper from loss of binding proteins in the urine; they are prone to infection, in part from urinary losses of immunoglobulins.
3. Renal biopsy
Specific classification and findings are shown in Table 22-10. Specimens are examined by light microscopy, with immunofluorescent stains, and by electron microscopy. Renal biopsy is often performed in adults with new-onset idiopathic nephrotic syndrome if a primary renal disease that may require drug therapy (eg, corticosteroids, cytotoxic agents) is suspected. Significantly elevated creatinine levels may indicate irreversible renal disease mitigating the usefulness of renal biopsy. Disease due to amyloid or diabetes mellitus often does not need to be biopsied, since nephrotic range proteinuria in these diseases represents irreversible damage, although bone marrow transplant with high-dose chemotherapy can be considered in some patients with amyloid. The role of biopsy for other systemic renal diseases is debated. However, it can be useful for prognosis and treatment. An occasional unexpected diagnosis is made, such as membranous nephropathy due to lupus erythematosus without serologic evidence of that illness.
P.943
Management of Nephrotic Syndrome
A. Protein Loss
The daily total dietary protein intake should replace the daily urinary protein losses so as to avoid negative nitrogen balance. Protein malnutrition often occurs with urinary protein losses greater than 10 g/d. In the past, protein restriction was suggested for patients with renal insufficiency because experimental animal models had demonstrated a decrease in glomerulosclerosis among those animals fed low-protein diets. The largest human trial to date (the MDRD Study) did not show a significant benefit, but two recent meta-analyses have shown a mild renal benefit. For this reason, the KDOQI recommends protein restriction to 0.6 g/kg/d in patients with a GFR less than 25 mL/min prior to starting dialysis.
B. Edema
Dietary salt restriction is essential for managing edema; most patients also require diuretic therapy. Commonly used diuretics include thiazide and loop diuretics. Both are highly protein bound. With hypoalbuminemia, diuretic delivery to the kidney is reduced, and patients often require large doses. The combination of loop and thiazide diuretics can potentiate the diuretic effect. This may be needed for patients with refractory fluid retention associated with pleural effusions and ascites.
C. Hyperlipidemia
Hypercholesterolemia and hypertriglyceridemia occur as outlined above. Dietary management in patients with nephrotic syndrome is of little value; however, dietary modification and exercise should be advocated. Aggressive pharmacologic treatment should be pursued. This is discussed in Chapter 27.
D. Hypercoagulable State
Patients with serum albumin less than 2 g/dL can become hypercoagulable. Nephrotic patients have urinary losses of antithrombin III, protein C, and protein S and increased platelet activation. Patients are prone to renal vein thrombosis and other venous thromboemboli, particularly with membranous glomerulopathy. Anticoagulation therapy is warranted for at least 3 6 months in patients with evidence of thrombosis. Patients with renal vein thrombosis and recurrent thromboemboli require indefinite anticoagulation.
Nephrotic Disease In Primary Renal Disorders
Minimal Change Disease
Minimal change disease is most commonly seen in children but is occasionally present in adults. In patients over 40 years with primary nephrotic syndrome, the incidence of minimal change disease is 20 25%, with equal distribution between men and women. In younger patients, there is a male predominance. Minimal change disease can be idiopathic but also occurs following viral upper respiratory infections, in association with tumors such as Hodgkin's disease, with drugs (gold and lithium), and with hypersensitivity reactions (especially to NSAIDs and bee stings).
Clinical Findings
A. Symptoms and Signs
Patients can exhibit the manifestations of nephrotic syndrome. They are more susceptible to infection, especially with gram-positive organisms, have a tendency toward thromboembolic events, develop severe hyperlipidemia, and experience protein malnutrition. Minimal change disease can rarely cause acute renal failure due to tubular changes and interstitial edema.
B. Histologic Findings
Glomeruli show no changes on light microscopy or immunofluorescence. On electron microscopy, there is a characteristic fusion of epithelial foot processes. A subgroup of patients also shows mesangial cell proliferation. These people have more hematuria and hypertension and respond poorly to corticosteroid treatment.
Treatment
Treatment is with prednisone, 1 mg/kg/d. In children, the response is excellent, but about 10% of patients become corticosteroid resistant after 4 6 weeks. Adults often require longer therapy. It can take up to 16 weeks to achieve a response to corticosteroids. Treatment should be continued for several weeks after complete remission of proteinuria. A significant number of patients will relapse and require further corticosteroid treatment. Patients with frequent relapses and corticosteroid resistance may need cyclophosphamide or chlorambucil to induce subsequent remissions. Progression to ESRD is rare. Complications most often arise from prolonged corticosteroid use.
Membranous Nephropathy
Membranous nephropathy is the most common cause of primary nephrotic syndrome in adults. It is an immune-mediated disease characterized by immune complex deposition in the subepithelial portion of glomerular capillary walls. The antigens in primary disease are not known. Secondary disease is associated with infections, such as hepatitis B, endocarditis, and syphilis; autoimmune disease, such as systemic lupus erythematosus, mixed connective tissue disease, and thyroiditis; carcinoma; and certain drugs, such as gold, penicillamine, and captopril. Membranous nephropathy occurs most commonly in adults in their fifth and sixth decades and almost always after age 30 years.
P.944
Clinical Findings
A. Symptoms and Signs
Patients exhibit the signs of nephrotic syndrome and have a higher incidence of renal vein thrombosis than most nephrotic patients. A higher incidence of occult neoplasms of lung, stomach, and colon is found in people over 50 years of age. The course of disease is variable, with about 50% of patients progressing to ESRD over 3 10 years. Poorer outcome is associated with concomitant tubulointerstitial fibrosis, male gender, elevated serum creatinine, hypertension, and heavy proteinuria (> 10 g/d).
B. Laboratory Findings
By light microscopy, capillary wall thickness is increased without inflammatory changes or cellular proliferation. When stained with silver methenamine, a spike and dome pattern may be observed owing to projections of excess GBM between the subepithelial deposits. Immunofluorescence shows IgG and C3 uniformly along capillary loops. Electron microscopy shows a discontinuous pattern of dense deposits along the subepithelial surface of the basement membrane.
Treatment
Treatment is controversial. After underlying causes are excluded, treatment depends on the risk of renal disease progression. One algorithm is based on the degree of proteinuria. In patients with proteinuria less than 3.5 g/d, the risk of progression is low. These individuals should be closely monitored with a low-salt diet, strict blood pressure control, and an ACE inhibitor for reduction of proteinuria. Patients with proteinuria of 3.5 8 g/d but normal renal function are at medium risk. They should follow the above suggestions and can elect immunosuppressive regimens with corticosteroids and chlorambucil or cyclophosphamide for 6 months, although 65% of these patients experience partial or complete remission within 3 4 years. Cyclosporine is a second choice. The highest-risk patients those with greater than 8 g/d of proteinuria and possible renal dysfunction might receive corticosteroids with a cytotoxic agent as first-line immunosuppressant therapy, though the choice of cyclosporine is also reasonable. These treatments should be carefully chosen in consultation with a nephrologist. Patients with membranous nephropathy are excellent candidates for transplant.
Focal Segmental Glomerular Sclerosis
This lesion can present as idiopathic disease or secondary to such conditions as heroin use, morbid obesity, and HIV infection. Clinically, patients show evidence of nephrotic syndrome, but they also have more nephritic features than membranous nephropathy or minimal change disease. Eighty percent of patients have microscopic hematuria at presentation, and many are hypertensive. Decreased renal function is present in 25 50% at time of diagnosis. Patients with focal segmental glomerular sclerosis and nephrotic syndrome typically progress to ESRD in 6 8 years.
Diagnosis requires renal biopsy. Light microscopy shows the lesions of focal segmental glomerular sclerosis. It is thought that these lesions occur first in the juxtamedullary glomeruli and are then seen in the superficial renal cortex. IgM and C3 are seen in the sclerotic lesions on immunofluorescence. Electron microscopy shows fusion of epithelial foot processes as seen in minimal change disease (Table 22-10).
Treatment is controversial, though supportive care for nephrotic patients is indicated. Longer courses of corticosteroids are now being used because a higher percentage of patients enter remission. High-dose oral prednisone (1 1.5 mg/kg/d) for 2 3 months followed by a slow taper can induce remission in over half of patients. Most patients achieve remission within 5 9 months. Other cytotoxic drug therapy can be considered but is disappointing (< 20% remission in most series).
Cattran DC: Idiopathic membranous glomerulonephritis. Kidney Int 2001;59:1983.
Chun MJ et al: Focal segmental glomerulosclerosis in nephrotic adults: presentation, prognosis, and response to therapy of the histologic variants. J Am Soc Nephrol 2004; 15:2169.
DeSanto NG et al: Nephrotic edema. Semin Nephrol 2001;21: 262.
Fogo AB: Minimal change disease and focal segmental glomerulosclerosis. Nephrol Dial Transplant 2001;16(Suppl 6):74.
Madaio MP et al: The diagnosis of glomerular diseases: acute glomerulonephritis and the nephrotic syndrome. Arch Intern Med 2001;161:25.
Ponticelli C et al: Treatment of membranous nephropathy. Nephrol Dial Transplant 2001;16(Suppl 5):8.
Schwarz A: New aspects of the treatment of nephrotic syndrome. J Am Soc Nephrol 2001;12(Suppl 17):S44.
Nephrotic Disease From Systemic Disorders
Amyloidosis
Amyloidosis consists of extracellular deposition of the fibrous protein amyloid in one or more sites in the body. The amyloid fibrils are composed of proteins that have formed -pleated sheets, a definitive characteristic. Primary renal amyloidosis (AL amyloidosis) may occur in the absence of systemic disease or associated with multiple myeloma; indeed, both are plasma cell dyscrasias. Secondary amyloidosis (AA amyloidosis) is due to a chronic inflammatory disease, such as
P.945
rheumatoid arthritis, inflammatory bowel disease, or chronic infection. The acute phase reactant serum amyloid A is synthesized in the liver and deposited in the tissues. Primary amyloidosis usually occurs in older age groups and displays a benign urinary sediment; the amyloid is derived from immunoglobulin light chain. The degree of proteinuria is not associated with the extent of renal lesions. Kidneys can be enlarged as a result of amyloid deposition. Pathologically, glomeruli are filled with amorphous deposits that stain positive with Congo red and show green birefringence.
Treatment options are few. Remissions can occur in secondary amyloidosis if the inciting agent is removed. Primary amyloidosis of the kidney progresses to ESRD in an average of 2 3 years. Five-year overall survival is less than 20%, with death occurring from ESRD and heart disease. The use of alkylating agents and corticosteroids eg, melphalan and prednisone can reduce proteinuria and improve renal function in a small percentage of patients. Melphalan and stem cell transplantation are associated with high toxicity (45% mortality) but induce remission in 80% of the remaining patients. Renal transplant is an option in patients with secondary amyloid.
Diabetic Nephropathy
Diabetic nephropathy is the most common cause of ESRD in the United States (about 4000 cases a year). Type 1 diabetes mellitus carries a 30 40% chance of nephropathy after 20 years, whereas type 2 has a 15 20% chance after 20 years. ESRD is much more likely to develop in persons with type 1 diabetes mellitus, probably because of fewer comorbidities and deaths before ESRD ensues. With the current epidemic of type 2 diabetes mellitus, rates of diabetic nephropathy are projected to continue to increase over at least the next 2 decades. Patients at higher risk include males, African Americans, and Native Americans.
The nephrotic syndrome develops in patients at risk for nephropathy. Diabetic retinopathy is often present in these patients. Initial screening of diabetics should always include urine examination for microalbuminuria. Dipstick examination may not be sensitive enough; a 24-hour urine collection is the accepted standard measure. (An albumin excretion of > 30 mg/d is abnormal.) However, an early morning spot urine albumin or albumin-creatinine ratio is adequate. (More than 30 mg of albumin per gram of creatinine is considered abnormal.) In patients prone to nephropathy, microalbuminuria will develop within 10 15 years after onset of diabetes and progress over the next 3 7 years to overt proteinuria. During the onset of subclinical proteinuria, aggressive treatment is necessary. Strict glycemic control and treatment of hypertension have been proven to slow progression of disease. In particular, ACE inhibitors and ARBs lower the rate of progression to clinical proteinuria and slow progression to ESRD. They may reduce intraglomerular pressure as well as treat hypertension. Even in the subset of patients with markedly diminished renal function, ARBs seem to provide renal benefit if patients can tolerate the medication from the perspective of hyperkalemia and the acute decrease in GFR.
The most common lesion in diabetic nephropathy is diffuse glomerulosclerosis, but nodular glomerulosclerosis (Kimmelstiel-Wilson nodules) is pathognomonic. The kidneys in these patients are usually enlarged as a result of cellular hypertrophy and proliferation. At the onset of diabetic nephropathy, glomerular disease will cause an increase in GFR. As the nephropathy progresses, with the development of macroalbuminuria, the GFR returns to normal and continues to decrease.
Patients with diabetes are prone to other renal disease. These include papillary necrosis, chronic interstitial nephritis, and type IV (hyporeninemic hypoaldosteronemic) renal tubular acidosis. Patients are more susceptible to acute renal failure from contrast material and have a poor prognosis once dialysis is begun.
Hiv-Associated Nephropathy
HIV-associated nephropathy can present as the nephrotic syndrome in patients with HIV infection. Most patients are young black men. In these patients, the more common mode of acquisition of HIV is through injection drug use.
Patients can have a nephrotic picture with normal complement levels. Light microscopy shows focal segmental glomerulosclerosis as described above. Lesions can be of the collapsing variety and often exhibit severe tubulointerstitial damage.
Small, uncontrolled studies have shown that highly active antiretroviral therapy (HAART) for a prolonged course can slow progression of disease. Despite this minimal evidence, HAART has been recommended for use in these patients given the therapy's other beneficial effects and reasonable toxicity profile. Corticosteroid treatment has been used with variable success at a dosage of 1 mg/kg/d along with cyclosporine and ACE inhibitors.
Barnett AH et al; Diabetics Exposed to Telmisartan and Enalapril Study Group: Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med 2004;351:1952.
Finne P et al: Incidence of end-stage renal disease in patients with type 1 diabetes. JAMA 2005;294:1782.
Merlini G et al: Molecular mechanisms of amyloidosis. N Engl J Med 2003;349:583.
Remuzzi G et al: Clinical practice. Nephropathy in patients with type 2 diabetes. N Engl J Med 2002;346:1145.
Ruggenenti P et al; Bergamo Nephrologic Diabetes Complications Trial (BENEDICT) Investigators: Preventing microalbuminuria in type 2 diabetes. N Engl J Med 2004;351: 1941.
Strippoli GF et al: Effects of angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists on mortality and renal outcomes in diabetic nephropathy: systematic review. BMJ 2004;329:828.
P.946
Szczech LA et al: The clinical epidemiology and course of the spectrum of renal diseases associated with HIV infection. Kidney Int 2004;66:1145.
Diseases Demonstrating Nephritic & Nephrotic Components
Systemic Lupus Erythematosus
Systemic lupus erythematosus is a systemic autoimmune disease in which renal involvement is common. In various series, clinical renal involvement ranges from 35% to 90%.
Patients present with glomerular syndromes such as nephritic or nephrotic syndromes and asymptomatic renal disease. Nonglomerular syndromes include tubulointerstitial nephritis and vasculitis. All patients with systemic lupus erythematosus should have routine urinalyses to monitor for the appearance of hematuria or proteinuria. If urinary abnormalities are detected, renal biopsy is often performed. The type of glomerular injury depends on the site of immune complex deposition. The World Health Organization (WHO) classifies the renal glomerular lesions as follows: type I, normal; type II, mesangial proliferative; type III, focal and segmental proliferative; type IV, diffuse proliferative; and type V, membranous nephropathy.
Individuals with type I and type II patterns require no treatment. Transformation of these types to a more active lesion is usually accompanied by an increase in lupus serologic activity and evidence of deteriorating renal function (eg, rising serum creatinine, increasing proteinuria). Repeat biopsy to confirm the transformation in these patients is standard. Patients with extensive type III lesions and all type IV lesions should receive aggressive immunosuppressive therapy. Poorest prognostic features in patients with type IV lesions are an elevated serum creatinine, hematocrit less than 26%, and black race. Indications for treatment of type V disease are unclear; however, if superimposed proliferative lesions exist, aggressive therapy should be instituted.
Corticosteroids are the mainstay of treatment (methylprednisolone 1 g intravenously daily for 3 days followed by prednisone, 60 mg orally daily for 4 6 weeks) but are associated with many side effects and may not prevent progression of chronic lesions. Cytotoxic agents, such as cyclophosphamide, are almost always added because they improve long-term renal survival in patients with aggressive type III and type IV nephritis. However, a recent study has shown the potential benefit of mycophenolate mofetil as an alternative form of induction therapy. In a recent randomized noninferiority study, patients using mycophenolate mofetil had similar rates of full remission at 24 weeks in comparison to those using cyclophosphamide. These agents are typically used for 18 24 months (eg, cyclophosphamide intravenously every month for six doses and then every 3 months for six doses). Studies suggest that mycophenolate mofetil is less toxic than cyclophosphamide, does not cause ovarian failure, and may be more acceptable to patients, although long-term follow-up needs to be examined. Ongoing trials of other therapies include cyclosporine and azathioprine as longer-term options, too. The return of serologic measurements to normal can be useful in monitoring treatment. Markers include double-stranded DNA (dsDNA) antibodies, C3, C4, CH50, and serum creatinine. Urinary protein and sediment are also helpful markers. Patients with systemic lupus erythematosus who undergo dialysis have a favorable prospect for long-term survival. Patients with kidney transplants have recurrent renal disease in 8% of cases.
Hepatitis C Virus Infection
Renal disease in the setting of hepatitis C viral infection was not well-recognized until 1993. Now it accounts for approximately 8% of patients with ESRD. Three clinicopathologic glomerular syndromes associated with hepatitis C are secondary membranoproliferative glomerulonephritis, cryoglobulinemic glomerulonephritis, and membranous nephropathy. A type I membranoproliferative glomerulonephritis (MPGN) is the most common lesion in patients requiring renal biopsy. These patients typically have hematuria and proteinuria, hypertension, and anemia. Occasionally, they exhibit the nephrotic syndrome. Many have elevated serum transaminases and an elevated rheumatoid factor. Of patients studied in one series, 50% had hepatomegaly. Hypocomplementemia is very common, with C4 typically more reduced than C3. Cryoglobulinemic disease is discussed above. Membranous glomerulopathy is the least common of the three and presents with a typical nephrotic picture. Neither cryoglobulins nor rheumatoid factor is present.
In patients with MPGN not receiving treatment for liver disease, the question arises whether to initiate therapy for renal disease. The main indications for therapy are poor renal function, nephrotic syndrome, new or worsening hypertension, fibrosis or tubulointerstitial disease on biopsy, and progressive disease. IFN- may result in suppression of viremia and improvement in hepatic function. Renal function rarely improves unless viral suppression occurs; however, renal function often worsens when therapy is abated. Ribavirin is relatively contraindicated in renal disease because of the dose-related hemolysis that occurs with renal dysfunction. Despite this, some case series have shown benefit with combined IFN- and ribavirin in closely monitored settings.
Idiopathic Membranoproliferative Glomerulonephritis
MPGN in its primary form is an idiopathic syndrome that can present with nephritic or nephrotic features. (The secondary form can be seen in the immune complex, paraprotein deposition, and thrombotic microangiopathic glomerulonephritides discussed above.) Most
P.947
patients are under 30 years of age. At least two major subgroups are recognized: type I and type II.
Patients with type I MPGN have a history of recent upper respiratory tract infection about a third of the time. Patients typically have a nephrotic picture, and complement levels are low. Histologically, the GBM is thickened because of immune complex deposition and abnormal mesangial cell proliferation between the GBM and the endothelial cells. This gives a characteristic splitting appearance to the capillary wall. Immunofluorescence shows IgG, IgM, and granular deposits of C3, C1q, and C4 (Table 22-10).
Type II MPGN often presents with a nephritic picture and is less common than type I. Light microscopy is similar to type I. Serologically, type II is associated with C3 nephritic factor, which is a circulating IgG antibody. Electron microscopy shows a characteristic dense deposit of homogeneous material that replaces part of the GBM.
Treatment of this disorder is controversial. After ruling out secondary causes, it consists of corticosteroid therapy (there is no standard dosage for adults) and antiplatelet drugs (aspirin, 500 975 mg/d, plus dipyridamole 225 mg/d). The rationale for antiplatelet therapy is that platelet consumption is increased in MPGN and may play a role in glomerular injury. Fifty percent of patients used to progress to ESRD in 10 years; these rates may now be slightly lower with the introduction of more aggressive therapy. Less favorable prognostic findings include type II disease, early renal insufficiency, hypertension, and persistent nephrotic syndrome. Both types of MPGN will recur after renal transplantation; however, type II recurs more commonly.
Contreras G et al: Sequential therapies for proliferative lupus nephritis. N Engl J Med 2004;350:971.
Fine DM: Pharmacological therapy of lupus nephritis. JAMA 2005;293:3053.
Flanc RS et al: Treatment for lupus nephritis. Cochrane Database Syst Rev 2004;(1):CD002922.
Ginzler EM et al: Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005;353: 2219.
Meyers CM et al: Hepatitis C and renal disease: an update. Am J Kidney Dis 2003;42:631.
Moroni G et al: Antiphospholipid antibodies are associated with an increased risk for chronic renal insufficiency in patients with lupus nephritis. Am J Kidney Dis 2004;43:28.
Nakopoulou L: Membranoproliferative glomerulonephritis. Nephrol Dial Transplant 2001;16(Suppl 6):71.
Tubulointerstitial Diseases
Tubulointerstitial disease may be acute or chronic. Acute disease is most commonly associated with toxins and ischemia. Interstitial edema, infiltration with polymorphonuclear neutrophils, and tubular cell necrosis can be seen. (See Acute Renal Failure, above, and Table 22-11.) Chronic disease is associated with insult from an acute factor or progressive insult without any obvious acute cause. Interstitial fibrosis and tubular atrophy are present, with a mononuclear cell predominance. The chronic disorders are described below.
Table 22-11. Causes of acute tubulointerstitial nephritis. | |
|---|---|
|
Chronic Tubulointerstitial Diseases
![]() Essentials of Diagnosis
Essentials of Diagnosis
Kidney size: small and contracted.
Decreased urinary concentrating ability.
Hyperchloremic metabolic acidosis.
Hyperkalemia.
Reduced GFR.
P.948
General Considerations
There are four main causes of chronic tubulointerstitial disease. These are discussed below. Other causes include multiple myeloma and gout, which are discussed in the section on multisystem disease with variable kidney involvement (below).
A. Obstructive Uropathy
The most common cause of chronic tubulointerstitial disease is prolonged obstruction of the urinary tract. In partial obstruction, urinary output alternates between polyuria (due to vasopressin insensitivity) and oliguria (due to decreased GFR). Azotemia and hypertension (due to increased renin-angiotensin production) are usually present. The major causes are prostatic disease in men; ureteral calculus in a single functioning kidney; bilateral ureteral calculi; carcinoma of the cervix, colon, and bladder; and retroperitoneal tumors or fibrosis.
Abdominal, rectal, and genitourinary examinations are helpful. Urinalysis can show hematuria, pyuria, and bacteriuria but is often benign. Abdominal ultrasound may detect mass lesions, hydroureter, and hydronephrosis. CT scanning and MRI provide more detailed information.
B. Vesicoureteral Reflux
Reflux nephropathy is primarily a disorder of childhood and occurs when urine passes retrograde from the bladder to the kidneys during voiding. It is the second most common cause of chronic tubulointerstitial disease. It occurs as a result of an incompetent vesicoureteral sphincter. Urine can extravasate into the interstitium; an inflammatory response develops, and fibrosis occurs. The inflammatory response is due to either bacteria or normal urinary components.
Patients are adolescents or young adults with hypertension, renal insufficiency, and a history of urinary tract infections as a child. Focal glomerulosclerosis is often seen. This is a cause of substantial proteinuria, unusual in most tubular diseases. Renal ultrasound or IVP can show renal scarring and hydronephrosis. Although most damage occurs before age 5 years, progressive renal deterioration to ESRD continues as a result of the early insults.
C. Analgesics
Analgesic nephropathy is most commonly seen in patients who ingest large quantities of analgesic combinations. The drugs of concern are phenacetin, paracetamol, aspirin, and NSAIDs. Chronic ingestion of 1 g/d for 3 years is the typical amount needed for renal dysfunction. This disorder occurs most frequently in individuals who are using analgesics for chronic headaches, muscular pains, and arthritis. Most patients grossly underestimate their analgesic use.
Tubulointerstitial inflammation and papillary necrosis are seen on pathologic examination. Papillary tip and inner medullary concentrations of some analgesics are tenfold higher than in the renal cortex. Phenacetin once a common cause of this disorder and now rarely available is metabolized in the papillae by the prostaglandin hydroperoxidase pathway to reactive intermediates that bind covalently to interstitial cell macromolecules, causing necrosis. Aspirin and other NSAIDs may worsen the damage by decreasing medullary blood flow (via inhibition of prostaglandin synthesis) and decreasing glutathione levels (which are necessary for detoxification).
Patients can exhibit hematuria, mild proteinuria, polyuria (from tubular damage), anemia (from gastrointestinal bleeding), and sterile pyuria. As a result of papillary necrosis, sloughed papillae can be found in the urine. An IVP may be helpful for detecting these contrast will fill the area of the sloughed papillae, leaving a ring shadow sign at the papillary tip.
D. Heavy Metals
Environmental exposure to heavy metals such as lead and cadmium is seen infrequently now in the United States. Chronic lead exposure can lead to tubulointerstitial disease. Individuals at risk are those with occupational exposure (eg, welders who work with lead-based paint) and drinkers of alcohol distilled in automobile radiators (moonshine users). Lead is filtered by the glomerulus and is transported across the proximal convoluted tubules, where it accumulates and causes cell damage. Fibrosed arterioles and cortical scarring also lead to damaged kidneys. Proximal tubular damage leads to decreased secretion of uric acid, resulting in hyperuricemia and saturnine gout. Patients commonly are hypertensive. Diagnosis is most reliably performed with a calcium disodium edetate (EDTA) chelation test. Urinary excretion of more than 600 mg of lead in 24 hours following 1 g of EDTA indicates excessive lead exposure.
Occupational exposure to cadmium also causes proximal tubular dysfunction. Hypercalciuria and nephrolithiasis can be seen. Other heavy metals that can cause tubulointerstitial disease include mercury and bismuth.
Clinical Findings
A. Symptoms and Signs
Polyuria is common because tubular damage leads to inability to concentrate the urine. Dehydration can also occur as a result of a salt-wasting defect in some individuals.
B. Laboratory Findings
Patients are hyperkalemic because the distal tubules become aldosterone resistant. A hyperchloremic renal tubular acidosis is characteristic. The cause of the renal tubular acidosis is threefold: (1) reduced ammonia production, (2) inability to acidify the distal tubules, and (3) proximal tubular bicarbonate wasting. The urinalysis is nonspecific, as opposed to that seen in acute interstitial nephritis. Proteinuria is typically less than 2 g/d (owing to inability of the proximal tubule to reabsorb freely filterable proteins); a few cells may be seen; and broad waxy casts are often present.
P.949
Treatment
Treatment depends first upon identifying the disorder responsible for renal dysfunction. The degree of interstitial fibrosis that has developed can help predict recovery of renal function. Once there is evidence for loss of parenchyma (small shrunken kidneys or interstitial fibrosis on biopsy), nothing can prevent the progression toward ESRD. Treatment is then directed at medical management. Tubular dysfunction may require potassium and phosphorus restriction and sodium, calcium, or bicarbonate supplements.
If hydronephrosis is present, relief of obstruction should be accomplished promptly. Prolonged obstruction leads to further tubular damage particularly in the distal nephron which may be irreversible despite relief of obstruction. Neither surgical correction of reflux nor medical therapy with antibiotics can prevent deterioration toward ESRD once renal scarring has occurred.
Patients in whom lead nephropathy is suspected should continue chelation therapy with EDTA if there is no evidence of irreversible renal damage (eg, renal scarring or small kidneys). Continued exposure should be avoided.
Treatment of analgesic nephropathy requires withdrawal of all analgesics. Stabilization or improvement of renal function may occur if significant interstitial fibrosis is not present. Hydration during exposure to analgesics may also have some beneficial effects.
Harris DC: Tubulointerstitial renal disease. Curr Opin Nephrol Hypertens 2001;10:303.
Huerta C et al: Nonsteroidal anti-inflammatory drugs and risk of ARF in the general population. Am J Kidney Dis 2005;45: 531.
Kodner CM et al: Diagnosis and management of acute interstitial nephritis. Am Fam Physician 2003;67:2527.
Cystic Diseases of the Kidney
Renal cysts are epithelium-lined cavities filled with fluid or semisolid material. They develop primarily from renal tubular elements. One or more simple cysts are found in 50% of individuals over the age of 50 years. They are rarely symptomatic and have little clinical significance. In contrast, generalized cystic diseases are associated with cysts scattered throughout the cortex and medulla of both kidneys and can progress to ESRD (Table 22-12).
Simple Or Solitary Cysts
Simple cysts account for 65 70% of all renal masses. They are generally found at the outer cortex and contain fluid that is consistent with an ultrafiltrate of plasma. Most are found incidentally on ultrasonographic examination. Simple cysts are typically asymptomatic but can become infected.
The main concern with simple cysts is to differentiate them from malignancy, abscess, or polycystic kidney disease. Renal cystic disease can develop in dialysis patients. These cysts have a potential for progression to malignancy. Ultrasound and CT scanning are the recommended procedures for evaluating these masses. Simple cysts must meet three sonographic criteria to be considered benign: (1) echo free, (2) sharply demarcated mass with smooth walls, and (3) an enhanced back wall (indicating good transmission through the cyst). Complex cysts can have thick walls, calcifications, solid components, and mixed echogenicity. On CT scan, the simple
P.950
cyst should have a smooth thin wall that is sharply demarcated. It should not enhance with contrast media. A renal cell carcinoma will enhance but typically is of lower density than the rest of the parenchyma. Arteriography can also be used to evaluate a mass preoperatively. A renal cell carcinoma is hypervascular in 80%, hypovascular in 15%, and avascular in 5% of cases.
Table 22-12. Clinical features of renal cystic disease. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
If a cyst meets the criteria for being benign, periodic reevaluation is the standard of care. If the lesion is not consistent with a simple cyst, surgical exploration is recommended.
Autosomal Dominant Polycystic Kidney Disease
This disorder is among the most common hereditary diseases in the United States, affecting 500,000 individuals, or 1 in 800 live births. Fifty percent of patients will have ESRD by age 60 years. The disease has variable penetrance but accounts for 10% of dialysis patients in the United States. At least two genes account for this disorder: ADPKD1 on the short arm of chromosome 16 (85 90% of patients) and ADPKD2 on chromosome 4 (10 15%). Patients with the PKD2 mutation have slower progression of disease and longer life expectancy than those with PKD1. Other sporadic cases without these mutations have also been recognized.
Clinical Findings
Abdominal or flank pain and microscopic or gross hematuria are present in most patients. A history of urinary tract infections and nephrolithiasis is common. A family history is positive in 75% of cases, and more than 50% of patients have hypertension (see below) that may antedate the clinical manifestations of the disease. Patients have large kidneys that may be palpable on abdominal examination. The combination of hypertension and an abdominal mass should suggest the disease. Forty to 50 percent have concurrent hepatic cysts. Pancreatic and splenic cysts occur also. Hemoglobin and hematocrit tend to be maintained as a result of erythropoietin production by the cysts. The urinalysis may show hematuria and mild proteinuria. In patients with PKD1, ultrasonography confirms the diagnosis two or more cysts in patients under age 30 years (sensitivity of 88.5%), two or more cysts in each kidney in patients age 30 59 years (sensitivity of 100%), and four or more cysts in each kidney in patients age 60 years or older are diagnostic for autosomal dominant polycystic kidney disease. If sonographic results are unclear, CT scan is recommended and highly sensitive.
Complications & Treatment
A. Pain
Abdominal or flank pain is caused by infection, bleeding into cysts, and nephrolithiasis. Bed rest and analgesics are recommended. Cyst decompression can help with chronic pain.
B. Hematuria
Gross hematuria is most commonly due to rupture of a cyst into the renal pelvis, but it can also be caused by a renal stone or urinary tract infection. Hematuria typically resolves within 7 days with bed rest and hydration. Recurrent bleeding should suggest the possibility of underlying renal cell carcinoma, particularly in men over age 50 years.
C. Renal Infection
An infected renal cyst should be suspected in patients who have flank pain, fever, and leukocytosis. Blood cultures may be positive, and urinalysis may be normal because the cyst does not communicate directly with the urinary tract. CT scans can be helpful because an infected cyst may have an increased wall thickness. Bacterial cyst infections are difficult to treat. Antibiotics with cystic penetration should be used, eg, fluoroquinolones, trimethoprim-sulfamethoxazole, and chloramphenicol. Treatment may require 2 weeks of parenteral therapy followed by long-term oral therapy.
D. Nephrolithiasis
Up to 20% of patients have kidney stones, primarily calcium oxalate. Hydration (2 3 L/d) is recommended.
E. Hypertension
Fifty percent of patients have hypertension at time of presentation, and it will develop in most patients during the course of the disease. Cyst-induced ischemia appears to cause activation of the renin-angiotensin system, and cyst decompression can lower blood pressure temporarily. Hypertension should be treated aggressively, as this may prolong the time to ESRD. (Diuretics should be used cautiously since the effect on renal cyst formation is unknown.)
F. Cerebral Aneurysms
About 10 15% of these patients have arterial aneurysms in the circle of Willis. Screening arteriography is not recommended unless the patient has a family history of aneurysms or is undergoing elective surgery with a high risk of developing hypertension.
G. Other Complications
Vascular problems include mitral valve prolapse in up to 25% of patients, aortic aneurysms, and aortic valve abnormalities. Colonic diverticula are more common in patients with polycystic kidneys.
Prognosis
No medical therapy has been shown to prevent the development of renal failure, though treatment of hypertension and a low-protein diet may slow the progression of disease.
P.951
Medullary Sponge Kidney
This disease is a relatively common and benign disorder that is present at birth and not usually diagnosed until the fourth or fifth decade. It is caused by autosomal dominant mutations in the MCKD1 or MCKD2 genes on chromosomes 1 and 16, respectively. Kidneys have a marked irregular enlargement of the medullary and interpapillary collecting ducts. This is associated with medullary cysts that are diffuse, giving a Swiss cheese appearance in these regions.
Clinical Findings
Medullary sponge kidney presents with gross or microscopic hematuria, recurrent urinary tract infections, or nephrolithiasis. Common abnormalities are a decreased urinary concentrating ability and nephrocalcinosis; less common is incomplete type I distal renal tubular acidosis. The diagnosis is confirmed with IVP, which shows striations in the papillary portions of the kidney produced by the accumulation of contrast in dilated collecting ducts.
Treatment
There is no known therapy. Adequate fluid intake (2 L/d) helps prevent stone formation. If hypercalciuria is present, thiazide diuretics are recommended because they decrease calcium excretion. Alkali therapy is recommended if renal tubular acidosis is present.
Prognosis
Renal function is well maintained unless there are complications from recurrent urinary tract infections and nephrolithiasis.
Juvenile Nephronophthisis-Medullary Cystic Disease
This is a rare disorder associated with almost universal progression to ESRD. The childhood type juvenile nephronophthisis is an autosomal recessive disorder caused by mutations in the NPH1, NPH2, and NPH3 genes; the type appearing in adulthood medullary cystic disease is autosomal dominant. Both types are manifested by multiple small renal cysts at the corticomedullary junction and medulla. The cortex becomes fibrotic, and as the disease progresses, interstitial inflammation and glomerular sclerosis appear.
Clinical Findings
Patients with both forms exhibit polyuria, pallor, and lethargy. Hypertension occurs at the later stages of disease. The juvenile form causes growth retardation and ESRD before age 20 years. Patients require large amounts of salt and water as a result of renal salt wasting. Ultrasound and CT scan show small, scarred kidneys, and an open renal biopsy may be necessary to recover tissue from the corticomedullary junction.
Treatment & Prognosis
There is no current medical therapy that will prevent progression to renal failure. Adequate salt and water intake are essential to replenish renal losses.
Peters DJ et al: Autosomal-dominant polycystic kidney disease: modification of disease progression. Lancet 2001;358:1439.
Rizk D et al: Cystic and inherited kidney diseases. Am J Kidney Dis 2003;42:1305.
Terada N et al: Risk factors for renal cysts. BJU Int 2004;93: 1300.
Torres VE et al: Autosomal dominant polycystic kidney disease. Nefrologia 2003;23(Suppl 1):14.
Wilson PD: Polycystic kidney disease. N Engl J Med 2004;350: 151.
Multisystem Diseases With Variable Kidney Involvement1
Multiple Myeloma
Multiple myeloma is a malignancy of plasma cells (see Chapter 13). Renal involvement occurs in about 25% of all patients. Myeloma kidney is the presence of light chain immunoglobulins (Bence Jones protein) in the urine causing renal toxicity. Bence Jones protein causes direct renal tubular toxicity and results in tubular obstruction by precipitating in the tubules. The earliest tubular damage results in Fanconi's syndrome (a type II proximal renal tubular acidosis). The proteinuria seen with multiple myeloma is primarily due to light chains that are not detected on urine dipstick, which mainly detects albumin. Glomerular amyloidosis can develop in patients with multiple myeloma; in these patients, dipstick protein determinations are positive. Hypercalcemia and hyperuricemia are frequently seen. Other conditions resulting in renal dysfunction include plasma cell infiltration of the renal parenchyma and a hyperviscosity syndrome compromising renal blood flow. Therapy for acute renal failure attributed to multiple myeloma includes correction of hypercalcemia, volume repletion, and chemotherapy for the underlying malignancy. Previously, plasmapheresis had been considered
P.952
appropriate to decrease the burden of existing monoclonal proteins while awaiting chemotherapeutic regimens to take effect. Recently, however, in the largest randomized prospective trial to date, plasmapheresis did not provide any renal benefit to these patients.
Sickle Cell Disease
Renal dysfunction associated with sickle cell disease is most commonly due to sickling of red blood cells in the renal medulla because of low oxygen tension and hypertonicity. Congestion and stasis lead to hemorrhage, interstitial inflammation, and papillary infarcts. Clinically, hematuria is common. Damage to renal capillaries also leads to diminished concentrating ability. Isosthenuria (urine osmolality equal to that of serum) is routine, and patients can easily become dehydrated. Papillary necrosis occurs as well. These abnormalities are commonly encountered in sickle cell trait. Sickle cell glomerulopathy is less common but will inexorably progress to ESRD. Its primary clinical manifestation is proteinuria. Optimal treatment requires adequate hydration and control of the sickle cell disease.
Tuberculosis
The classic renal manifestation of tuberculosis is the presence of microscopic pyuria with a sterile urine culture or sterile pyuria. More often, other bacteria are present in addition. Microscopic hematuria is often present with pyuria. Urine cultures are the gold standard for diagnosis. Three to six first morning midstream specimens should be performed to improve sensitivity. Papillary necrosis and cavitation of the renal parenchyma occur less frequently, as do ureteral strictures and calcifications. Adequate drug therapy can result in resolution of renal involvement.
Gout & The Kidney
The kidney is the primary organ for excretion of uric acid. Patients with proximal tubular dysfunction have decreased excretion of uric acid and are more prone to gouty attacks. Depending on the pH and uric acid concentration, deposition can occur in the tubules, the interstitium, or the urinary tract. The more alkaline pH of the interstitium causes urate salt deposition, whereas the acidic environment of the tubules and urinary tract causes uric acid crystal deposition at high concentrations.
Three disorders are commonly seen: (1) uric acid nephrolithiasis, (2) acute uric acid nephropathy, and (3) chronic urate nephropathy. Renal dysfunction with uric acid nephrolithiasis stems from obstructive nephropathy. Acute uric acid nephropathy presents similarly to acute tubulointerstitial nephritis with direct toxicity from uric acid crystals. Chronic urate nephropathy is caused by deposition of urate crystals in the alkaline medium of the interstitium; this can lead to fibrosis and atrophy.
Treatment between gouty attacks involves avoidance of food and drugs causing hyperuricemia, aggressive hydration, and pharmacotherapy aimed at reducing serum uric acid levels. These disorders are seen in both overproducers and underexcretors of uric acid. The latter situation may seem counterintuitive; however, these patients have hyperacidic urine, which explains the deposition of relatively insoluble uric acid crystals.
The Kidney & Aging
Renal mass declines progressively after the fourth decade. The renal medulla is spared in comparison to the cortex. Renal blood flow decreases with a resultant increase in arteriolar resistance. This allows for an increased filtration fraction and a relative sparing of the GFR. After the age of 40 years, GFR declines at a rate of approximately 0.8 mL/min/1.73 m2/yr (though some older patients show little or no change). Serum creatinine values remain relatively constant because of decreased muscle mass along with the decrease in GFR. GFR impairment is partially due to thickening of the GBM, leading to glomerulosclerosis.
Renal tubular changes include impaired sodium handling, decreased concentration and dilutional ability, and impaired acidification. Thus, older patients are more prone to volume overload, hyponatremia and hypernatremia, and acidosis. Decreased renin synthesis and 1 -hydroxylase activity are also observed. These abnormalities can result in hyperkalemia, hypocalcemia, and elevated PTH activity.
More adverse drug reactions occur in older patients. Three main pharmacokinetic changes occur: (1) altered volume of distribution, (2) altered drug half-life, and (3) altered elimination. The latter two are directly related to impaired renal clearance of drug.
The average age of patients starting dialysis is 61 years; the average age of patients receiving dialysis is 65 years. Both are increasing steadily. Hemodialysis is the modality of choice for those with functional impairment. Peritoneal dialysis is tolerated much better in those with cardiovascular disease. Sudden fluid and electrolyte shifts can cause hypotension, ischemia, and arrhythmias.
Renal transplantation is being offered to older individuals more often as it seems to benefit even those over 65 years. The main complications in this population are infection and cardiovascular disease. A reduced corticosteroid requirement with the introduction of steroid-sparing agents, such as cyclosporine, has diminished infection rates.
Footnote
1Other diseases with variable involvement described elsewhere in this chapter include systemic lupus erythematosus, diabetes mellitus, and the vasculitides such as Wegener's granulomatosis and Goodpasture's disease.
Clark WF et al; Canadian Apheresis Group: Plasma exchange when myeloma presents as acute renal failure: a randomized, controlled trial. Ann Intern Med 2005;143:777.
Corso A et al: Urinary proteins in multiple myeloma: correlation with clinical parameters and diagnostic implications. Ann Hematol 2003;82:487.
Kapoor M et al: Malignancy and renal disease. Crit Care Clin 2001;17:571.
Kramer HJ et al: The association between gout and nephrolithiasis in men: The Health Professionals' Follow-Up Study. Kidney Int 2003;64:1022.
Pandit SR et al: Management of renal dysfunction in multiple myeloma. Curr Treat Options Oncol 2003;4:239.
Scheinman JI: Sickle cell disease and the kidney. Semin Nephrol 2003;23:66.
Wise GJ et al: Genitourinary manifestations of tuberculosis. Urol Clin North Am 2003;30:111.
EAN: 2147483647
Pages: 49