10 - Oculoplastics
Editors: Tasman, William; Jaeger, Edward A.
Title: Wills Eye Hospital Atlas of Clinical Ophthalmology , The, 2nd Edition
Copyright 2001 Lippincott Williams & Wilkins
> Table of Contents > Chapter 10 - Oculoplastics
function show_scrollbar() {}
Chapter 10
Oculoplastics
Joseph C. Flanagan
Robert A. Mazzoli
Edward H. Bedrossian Jr.
Robert B. Penne
Mary A. Stefanyszyn
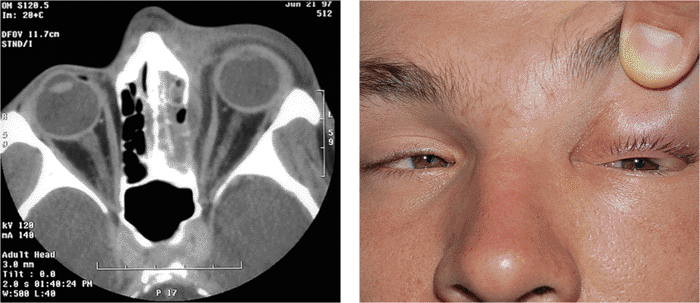 |
| Orbital cellulitis in a 23-year-old male. Findings include left-sided proptosis, chemosis, orbital congestion, decreased ocular motility, and superficial inflammation. Computed tomography demonstrates ethmoid sinus opacification, with a medial subperiosteal abscess. Sinoorbital drainage is required. |
P.366
Anatomic Considerations
The orbicularis oculi muscle is responsible for eyelid closure. This circular muscle is innervated by the seventh cranial nerve. The muscle is divided into three portions (Fig. 10.1A). The orbital portion is the thickest and is responsible for forcible lid closure. It inserts on the medial canthal tendon. The preseptal portion as its name implies overlies the orbital septum. It is important in eyelid closure, involuntary blinking, and the function of the lacrimal pump, aided by multiple insertions medially into the medial canthal tendon and around the lacrimal sac. These insertions contribute to the complicated lacrimal pump mechanism. Laterally, the muscle inserts into the lateral canthal tendon. The pretarsal orbicularis is the smallest portion and acts mainly in involuntary blinking and the lacrimal pump mechanism. Lateral and medial insertions are similar to those of the preseptal muscles.
The frontalis muscle supports the eyebrow and along with the procerus and glabellar muscles is responsible for its animation (Fig. 10.1B). It is also innervated by the seventh cranial nerve. The orbital septum originates from the orbital rim by a fibrous band called the arcus marginalis. Superiorly, the septum fuses with the levator aponeurosis variably above the superior tarsal border. This syncytial structure defines the entrance to the orbit proper. Anterior to the septum lies the preseptal orbicularis. Immediately posterior is the preaponeurotic fat, an important surgical landmark. It is convenient to consider this fat to be contained in two fat pockets superiorly, the nasal and central pads, while in the lower lid the fat is typically divided into three pockets: the nasal, central and lateral pockets. The inferior oblique muscle separates the nasal and central fat compartments in the lower lid, a critical anatomic relationship in lower lid and orbital surgery. The lacrimal gland occupies the lateral pocket in the upper lid, another key anatomic consideration. The levator complex lies immediately beneath the aponeurotic fat. The levator muscle originates posteriorly from the periorbita above the annulus of Zinn and extends anteriorly along the orbital roof, traveling above the superior rectus muscle. At the level of the equator of the globe, the muscle changes to aponeurosis and extends inferiorly over Whitnall's ligament to insert along the tarsus and medial and lateral rims (the levator horns). Attachments along the tarsus are complex. The main attachment is to the inferior two thirds of the anterior border of the tarsus, which effects opening the eyelid. In occidental lids, lesser attachments are made superficially through the orbicularis muscle to the skin near the superior border of the tarsus to form the upper eyelid crease. These attachments are significantly
P.367
more diverse in Asian and oriental lids. The aponeurotic complex is innervated by the third cranial nerve. Not labeled in Figure 10.1 is M ller's muscle, which lies posterior to the levator aponeurosis. This is a sympathetically innervated muscle that attaches to the superior aspect of the tarsal plate and acts to elevate the lid. The tarsal plate is made of dense fibrous tissue and forms the structural framework of the eyelids. The upper tarsal plate is 25 mm long and 10 mm wide. The tarsal plates are attached to the lateral and medial orbital rims by the lateral and medial canthal tendons. The lid margin is rather flat anatomically, making critical landmarks relatively easily recognizable. Anteriorly, the lashes number approximately 100 in the upper lid and 50 in the lower. Posteriorly, 20 to 25 meibomian glands mark the area where the margin blends into the palpebral conjunctiva at the mucocutaneous border. Between the two sits the gray line, a superficial reflection of the muscle of Riolan (a small portion of the pretarsal orbicularis) visible in most patients.
 |
Figure 10.1. The musculature of the eyelid. A: The orbicularis oculi. B: The eyelid in cross section. (From Bosniak SL. Cosmetic blepharoplasty. New York: Raven Press; 1991:40, with permission.) |
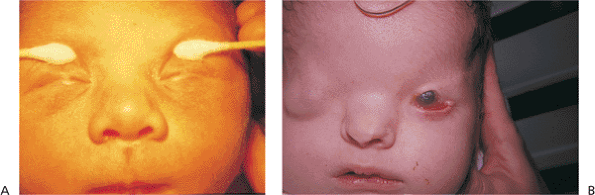 |
Figure 10.2. A: Ankyloblepharon. (Courtesy of Joseph Calhoun, M.D., Philadelphia, PA.) B: Cryptophthalmos. Patient with multiple developmental anomalies. The right eye demonstrates cryptophthalmos, and the left eye shows a large central lid coloboma. |
Congenital Lid Abnormalities
Ankyloblepharon and Cryptophthalmos
Embryologically, the eyelids develop from extensions of the frontonasal and maxillary processes, growing vertically over the developing globe during approximately the seventh week of gestation. The folds fuse along their horizontal margins and remain fused while the lid structures develop, until the fifth to seventh month, when the developed lids gradually separate. Ankyloblepharon (i.e., ankyloblepharon filiforme adnatum) describes a partial failure of the fused lids to separate, while the more severe condition cryptophthalmos is a complete failure of the lids to develop or separate (Fig. 10.2). The latter condition is almost uniformly associated with severe malformations of the globe, orbit, and adnexa, but ankyloblepharon may be an isolated condition. If the fusion is sufficiently extensive, amblyopia may develop, and medial fusion may obstruct the punctum.
P.368
Management
Ankyloblepharon is treated with simple lysis of the adhesive bands and reconstruction (e.g., punctoplasty), as needed. The adhesions do not generally recur. Treatment of cryptophthalmos can require extensive multidisciplinary reconstructive efforts.
Blepharophimosis
Blepharophimosis refers to an abnormally small palpebral fissure that is shortened both vertically and horizontally, but in which the lids themselves are normally developed (Fig. 10.3). Although it can be an associated finding in a variety of syndromes, as an isolated entity the condition is rare, transmitted as an autosomal-dominant trait. Characteristic features include severe congenital ptosis with poor levator function, epicanthus inversus, telecanthus, and an absolute shortage of skin in all four lids. Lateral ectropion, strabismus, motility disturbances, and nystagmus can also be associated features, as can flattening of the nasal bridge, and hyperteleorbitism. Though various degrees of mental deficiency have been reported, mental capacity is generally unaffected. Menstrual irregularities and infertility have also been reported in female patients. Differentiation from simple epicanthus, bilateral congenital ptosis, medial ankyloblepharon, and telecanthus requires close examination. The manifestations can vary from mild to severe.
Management
Patients should undergo a complete family history and physical examination to look for associated developmental anomalies. Radiographic images are obtained before surgical correction to rule out teleorbitism. Surgery is staged, beginning at 3 to 5 years of age, unless the ptosis is severe enough to warrant earlier intervention.
Epicanthus, Telecanthus, and Teleorbitism
Epicanthus (i.e., epicanthal folds) refers to prominent folds of skin in the inner canthus and is most prominent along the upper lid. Although it is a normal finding in those of Asian ancestry, it is abnormal in non-Asians (Fig. 10.4A). Epicanthus inversus exists if the fold extends predominantly from the lower lid to the upper lid; it is almost always an abnormal finding in all races and is associated with significant ptosis and poor levator function (Fig. 10.4B). If the fold is equally divided between the two lids, the condition is called epicanthus palpebralis (Fig. 10.4C). Although epicanthus commonly exists as an isolated finding, it can also be a component of several congenital conditions, such as blepharophimosis and Down's syndrome. The condition presents no particular ocular hazard. However, it often causes parental concern because of the appearance of or, more commonly, the semblance of pseudoesotropia. In the latter case, cover and cross-cover testing reveals orthophoria, and gentle distraction of the soft tissues of the nasal bridge reduces the webbed appearance of the medial canthi to reveal normal ocular alignment.
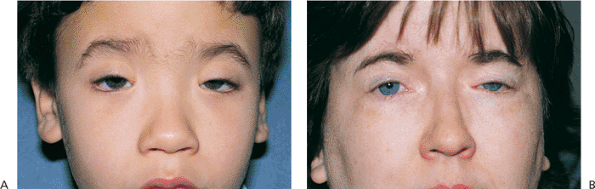 |
Figure 10.3. Blepharophimosis. A: Patient demonstrates typical findings of the condition, including significant congenital ptosis, lid phimosis, and epicanthus inversus. A wide, flat nasal bridge with telecanthus and pseudoesotropia are also present (Fig. 10.4B,C). The condition was inherited as a dominant trait. B: Mother of the patient in (A) after surgical correction as a child. Two of her brothers, her father, and maternal grandmother were also affected. |
Telecanthus is a soft tissue abnormality defined as an abnormally widened separation of the medial canthi. As an isolated condition the interpupillary distance remains normal (Fig. 10.5). It is most often seen as a result of severe nasoorbital-ethmoid trauma in adults, but can occur congenitally, generally in conjunction with other ocular and facial anomalies. Teleorbitism (hypertelorism, or hyperteleorbitism) is a bony abnormality of the orbit wherein the medial walls are too widely separated (Fig. 10.6). In this condition, the interpupillary distance is abnormally widened. Soft tissue telecanthus is a secondary finding. The condition is almost invariably associated with other maldevelopments and is most often encountered in craniofacial dysmorphisms. Hypoteleorbitism describes abnormally close orbital walls.
Management
Rarely is surgery recommended for simple epicanthus. However, a variety of sliding flap techniques have been described for correction when performed for reconstruction of other abnormalities (e.g., blepharophimosis). Simple epicanthus normally resolves spontaneously as
P.369
P.370
the nasal bridge and midface develop. Pseudostrabismus resolves concomitantly.
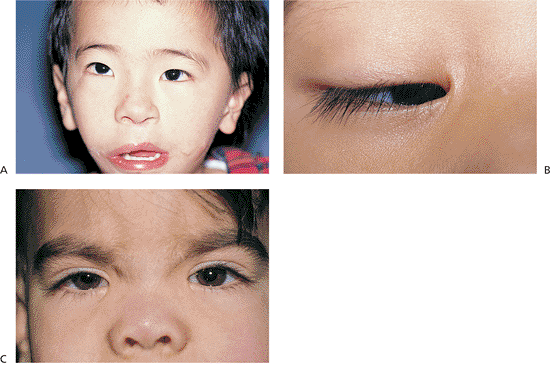 |
Figure 10.4. A: Epicanthus. Patient with Goldenhar's syndrome. The predominant fold extends from the upper lid. Telecanthus is also present (Fig. 10.5). B: Epicanthus inversus. The predominant fold is in the lower lid in this patient with blepharophimosis (Fig. 10.3A). C: Epicanthus palpebralis and pseudoesotropia. Patient with Cornelia de Lange syndrome. The lid fold is equally divided along the upper and lower lid. Pseudoesotropia is also apparent. However, the corneal light reflex demonstrates normal ocular alignment. |
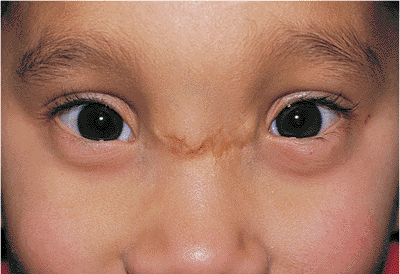 |
Figure 10.5. Telecanthus in a patient after surgical repair of frontal-nasal encephalocele. The lid malposition was congenital. The intercanthal distance is widened. However, the interpupillary distance is normal, indicating that the orbits themselves are not malpositioned. |
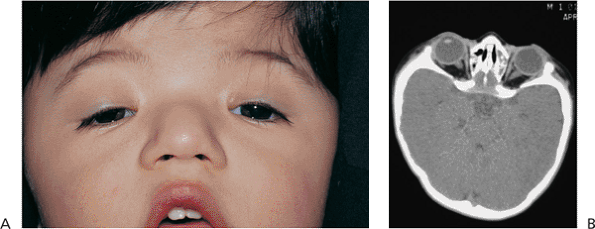 |
Figure 10.6. Teleorbitism. A: Patient with Crouzon's syndrome. The interpupillary distance is abnormally wide. B: Computed tomography demonstrates the widely separated medial walls and orbits. |
Surgical correction of telecanthus necessitates resuspension of the medial canthal tendon, often requiring transnasal wiring. If teleorbitism coexists, as in the craniosynostoses, the bony medial walls must be narrowed as well. These procedures are best performed with the assistance of trained craniofacial surgeons.
Euryblepharon, Congenital Ectropion, and Lid Coloboma
Euryblepharon (i.e., megaloblepharon) is a rare congenital condition (Fig. 10.7). Contrasted to blepharophimosis in which the lid apertures are too small (Fig. 10.3) euryblepharon is characterized by palpebral fissures that are too wide. The eyelids are normally developed; however, the insertions of the canthal tendons appear to be too widely or anteriorly placed. This leads to poor apposition of the lid to the globe (lid stand-off) that is most marked laterally. Congenital ectropion may also occur. There may also be a downward dystopia of the canthus. The condition is most often isolated, but it may be inherited as an autosomal-dominant trait or may be associated with other congenital abnormalities. Microphthalmos or enophthalmos may give the appearance of euryblepharon but can be differentiated by a thorough examination.
Lid colobomas occur when the embryonic lids fail to fuse horizontally, producing a cleft in the lid margin (Fig. 10.8). Amniotic bands have been implicated, but colobomas are known to occur in conjunction with other anomalies of the head and face, particularly of the eyes. They occur commonly in the craniofacial clefting syndromes. Dermoids and dermolipomas are commonly found in proximity to the coloboma, most often at the apex. Large colobomas may affect the entire lid, simulating ablepharon. Lateral coloboma of the lower lid is a frequent finding in mandibulofacial dysostoses such as the Treacher Collins syndrome.
 |
Figure 10.7. Euryblepharon with congenital ectropion. Notice the large lids, with downward and lateral dystopia of the lateral canthus, and poor globe apposition. There are other congenital anomalies. |
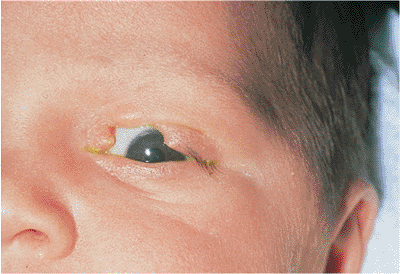 |
Figure 10.8. Upper lid coloboma was unassociated with other abnormalities. A dermoid is seen at the apex of the cleft. |
Management
If severe euryblepharon causes epiphora or exposure, resuspension of the lateral canthus using lateral tarsal strip may be contemplated. Small colobomas may be excised directly, with reconstruction of the affected lid through standard techniques. Larger colobomas require earlier intervention with more extensive reconstructive efforts to prevent corneal complications.
Epiblepharon, Distichiasis, and Congenital Entropion
True congenital entropion is a rare condition, occurring secondary to the absence, hypoplasia, or hypertrophy of the tarsal plate and pretarsal orbicularis (Fig. 10.9). In extreme cases, the tarsus may be bent permanently inward by amniotic bands, causing a tarsal kink that requires prompt surgical correction. More common, but still rare, are the conditions
P.371
of epiblepharon (Fig. 10.10), in which an extra fold of skin exists at the lid margin, occasionally causing a mechanical entropion, and distichiasis (Fig. 10.11), wherein an extra row of lashes grows toward the cornea from metaplastic meibomian gland orifices. This condition may be inherited, and often causes annoying corneal irritation, precipitating intervention.
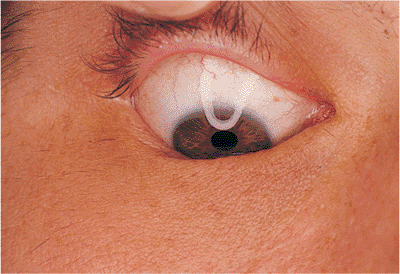 |
Figure 10.9. Congenital entropion. Adult patient with congenital, familial absence of lower lashes. Hypoplasia of the tarsal plate causes the lid margin to roll inward, especially during downgaze. |
 |
Figure 10.10. Epiblepharon. An extra roll of skin below the lashes can cause the lashes to roll inward, resulting in corneal irritation. |
Management
The conditions are generally isolated and well tolerated, but they may cause symptomatic corneal irritation. Treatment is supportive, with lubricants, unless keratitis exists, in which case surgery may be contemplated. Epiblepharon usually resolves with maturation and growth of the midface, although an elliptical skin and muscle excision is sometimes indicated to resolve the keratitis. Electrolysis or cryotherapy can be used to ablate distichiatic lashes. Rarely is a lid-splitting procedure required for this condition. True congenital entropion is treated by marginal rotation techniques.
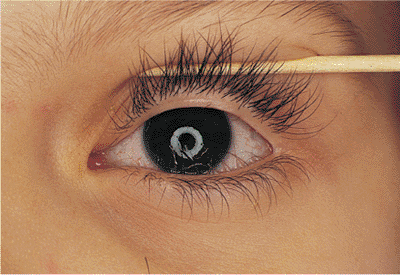 |
Figure 10.11. Distichiasis. A familial condition and congenital disorder in which extra lashes grow from the openings of metaplastic meibomian glands. The position of the lid margin is normal. |
Acquired Lid Malpositions
Entropion
Involutional entropion is a common clinical finding. Laxity of the skin, orbicularis, and other supporting structures contribute to a lack of posterior lid support and tarsal stability, leading to inturning of the lid margin and lashes with resultant keratitis, conjunctivitis, and discomfort (Fig. 10.12A). The tissues can be lax at the lateral canthus, at the medial canthus, or in the horizontal length of the lid. Loss of the normal relation between the lower lid retractors and the tarsal base (i.e., lower lid retractor disinsertion) allows the preseptal orbicularis to override the lid margin, causing the lid to invert. Microvascular insufficiency may be a contributing factor. Involutional entropion is almost exclusively a phenomenon of the lower lid; upper lid entropion is more likely to be secondary to cicatricial changes. Although most cases are idiopathic, factors such as chronic irritation and eye rubbing (e.g., blepharitis, allergy), smoking, and familial tendencies toward skin laxity are undoubtedly contributing factors. The direction of the lashes at the lash border is usually anatomically normal, unlike the condition of trichiasis, in which the lid margin is typically stable but abnormal lashes are misdirected posteriorly (Fig. 10.12B). The two conditions may coexist.
Acute spastic entropion may result from various inflammations and irritations, or from trauma, such as cataract or lid surgery. Cicatricial entropion can occur as a result of surgery (e.g., tumor resection), reaction to medication, or chronic infectious or inflammatory diseases such as trachoma, pemphigoid, or Stevens-Johnson syndrome (Fig. 10.13). It may involve the upper or lower lids.
Management
Treatment is directed toward correcting the anatomic abnormality and removing inciting factors. Blepharoconjunctivitis is treated medically with warm compresses, antibiotic ointments, and lubricants. Canthal tendon laxity is corrected with plication procedures or resuspension (e.g., lateral tarsal strip). Horizontal laxity can be corrected with full-thickness wedge resection. Procedures to correct horizontally tight lids include excision of a base-down triangle from the posterior lamella, placement of fornix sutures (i.e., Quickert-Rathbun sutures), and suture plication of the orbital septum. Temporary measures for comfort may include taping the eyelid with lateral traction or placement of Quickert-Rathbun sutures at the bedside. Cicatricial changes of the posterior lamella often require grafting of spacer materials. Autogenous mucus membrane such as buccal mucosa or hard palate are excellent replacements. However, diseases such as Stevens-Johnson and pemphigoid, which affect the oral mucosa and the conjunctiva, may limit the feasibility of the mouth as a donor site. Other potential graft materials include fascia lata, processed human dermis, and banked sclera, although these materials may be especially irritating on the upper lid because of the
P.372
lack of mucosal surface. Marginal rotation and lash electrolysis may be required.
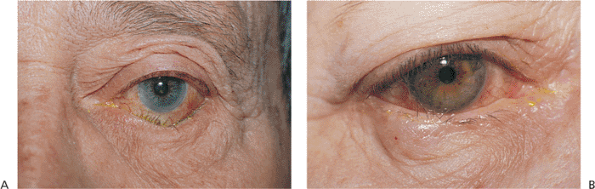 |
Figure 10.12. A: Involutional entropion. Significant lower lid laxity and detachment of the lower lid retractors allow the lid margin to rotate inward. Lashes can create chronic keratoconjunctivitis. When the lid margin is in a normal, everted position, the lash direction is anatomically normal. B: Trichiasis. Unlike entropion, the lid margin position is anatomically stable, but the lashes are misdirected. Corneal touch leads to chronic keratitis. |
Involutional Ectropion
Involutional ectropion, like entropion, is common. It results from poor lid tone and horizontal laxity, allowing gravity and lid weight to pull the lower lid margin away from the globe (Fig. 10.14A). When the lid is pulled away from the globe, it fails to snap back into position as it does in younger patients. Symptoms include irritation and blepharoconjunctivitis. In severe cases, particularly if the lower lid retractors have become disinserted, frank tarsal eversion with severe keratinization of the conjunctiva can occur (Fig. 10.14B). Dysfunction of the lacrimal pump and punctal ectropion lead to symptomatic epiphora. Horizontal laxity is often exacerbated by excess skin and fat, called dermatochalasis. Occasionally, extreme dermatochalasis can produce festoons of the lower lid, also known as malar bags, secondary bags, and bags-on-bags.
Management
Treatment of involutional ectropion involves tightening of the lid sling (e.g., lateral tarsal strip, wedge resection) and, if necessary, removal of redundant skin and fat with a modified Kuhnt-Szymanowski or lower lid blepharoplasty. Punctal eversion is corrected by a variety of punctoplasty techniques. Treatment of floppy eyelid syndrome requires lubrication and horizontal tightening of the upper and lower lids (Fig. 10.15). Aggravating conditions are treated concomitantly.
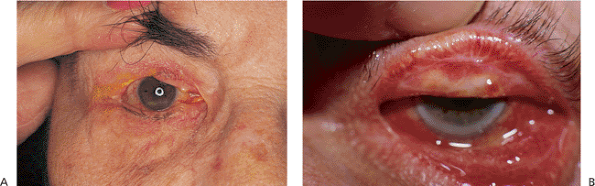 |
Figure 10.13. Cicatricial entropion. A: Multiple tumor resections of the upper lid resulted in cicatricial entropion and corneal irritation. B: Chronic glaucoma medication use led to cicatricial changes of the posterior lamella and entropion of both upper and lower lids. |
Paralytic and Mechanical Ectropion
Paralytic ectropion is a consequence of palsy or paralysis of the facial nerve (Fig. 10.16). Lid hypotonicity leads to marked ectropion, particularly in older adults, in whom involutional changes and laxity exacerbate the paralytic component. The palsy may be permanent or temporary, but it can lead to severe exposure keratopathy in either instance. Loss of normal lid position and function also leads to significant epiphora.
Mechanical ectropion can result from cicatricial changes of the anterior or middle lamella or tumor mass (Fig. 10.17). Any condition leading to scarring, including inflammation
P.373
P.374
(e.g., atopic dermatitis), trauma (e.g., tumor resection, cosmetic surgery, laceration), and chemical or thermal burns, can cause abnormal foreshortening of the skin and muscle layer (Figs. 10.18, 10.19A). Contracture of the middle lamella and septum can result from orbital surgery (Fig. 10.19B). Large tumors of the lid and significant periocular edema can physically push the lid away from the globe.
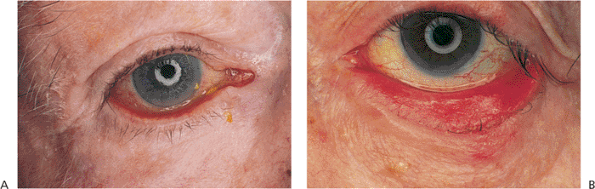 |
Figure 10.14. Involutional ectropion. A: Excess laxity and poor horizontal lid tone lead to sagging and eversion of the lid margin. Secondary mucus discharge and epiphora are not uncommon. B: In this severe case with retractor disinsertion and frank tarsal eversion, chronic exposure has led to secondary keratinization of the tarsal conjunctiva. Exposure changes usually resolve after correction of the ectropion. |
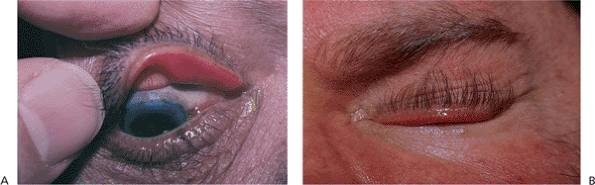 |
Figure 10.15. Floppy eyelid syndrome. A: The upper tarsus has begun to lose its natural rigidity and can be folded and everted easily. There is also significant laxity of the canthal ligaments. The velvety appearance of the conjunctiva is secondary to chronic papillary conjunctivitis. B: Aggravating conditions of floppy eyelid syndrome include overriding eyelids, which may contribute to chronic internal irritation. Associated physical conditions include obesity and obstructive sleep apnea. |
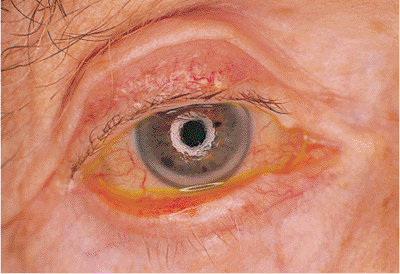 |
Figure 10.16. Paralytic ectropion. Patient after radical parotidectomy for mucoepidermoid carcinoma with subsequent facial palsy. Paralysis produces lagophthalmos and significant ectropion, with marked exposure keratopathy. Previous attempts at correction included implantation of auricular cartilage spacer in the lower lid and a gold weight in the upper lid, which is beginning to extrude at the inferonasal corner. |
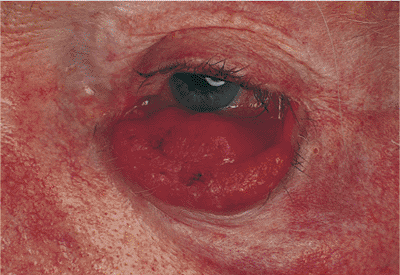 |
Figure 10.17. Mechanical ectropion: tumor. In a patient with metastatic prostate cancer, a mass of the lower lid physically pushes the lid margin away from globe. |
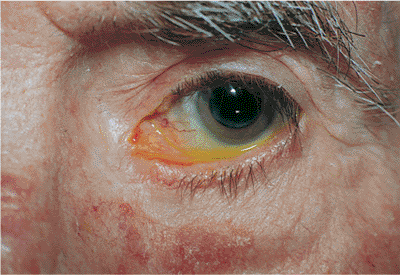 |
Figure 10.18. Mechanical ectropion: cicatrix. A patient with a cicatricial lower lid ectropion, most marked nasally, secondary to previous excision of skin cancer below the punctum. |
Management
In the case of paralytic ectropion, the cause of the facial palsy must be investigated. Conditions such as herpes zoster infection (e.g., Ramsay Hunt syndrome), tumor (e.g., cranial nerve VII or VIII involvement), and trauma must be evaluated. Bell's palsy is an idiopathic palsy that resolves spontaneously and requires little more than supportive treatment. Other conditions, however, are best managed in conjunction with an otorhinolaryngologist. Ocular manifestations are treated with lower lid resuspension procedures such as the lateral tarsal strip operation. If exposure keratitis is severe, lateral or medial tarsorrhaphies may be needed, on either a temporary or permanent basis. Techniques for enhancing lid closure include insertion of a gold weight into the upper lid, wire springs and silicone slings, and nerve transplantation. The eye is kept well lubricated throughout the duration of the palsy.
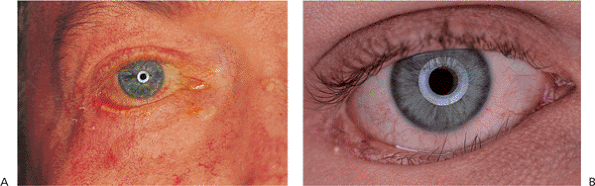 |
Figure 10.19. Mechanical ectropion: cicatrix. A: A patient with a cicatricial shortening of the anterior lamella secondary to chronic atopic dermatitis. B: An orbital trauma patient, status post orbital repair, with cicatricial contracture of the middle lamella and septum, leading to lower lid retraction. A previous unsuccessful attempt at correction included full-thickness wedge excision. |
If mechanical ectropion is caused by a lid mass, it is biopsied and managed as required. Cicatricial ectropion often requires lysis and revision of scar tissue, with placement of anterior lamellar skin grafts or middle lamellar spacer grafts as needed. Every attempt should be made to control inflammatory conditions such as dermatitis before any surgical intervention.
Ptosis
Congenital and Acquired Ptosis
Congenital ptosis occurs most commonly as an isolated finding but also occurs in association with a myriad of other abnormalities and syndromes, such as blepharophimosis and Down's syndrome (Fig. 10.20). Strabismus may also be found. The ptosis is most often unilateral, but it may be bilateral in as many as 25% of cases. A familial inheritance pattern is not unusual. Unlike acquired ptosis, most cases of congenital ptosis are myogenic in origin and are often associated with poor levator and superior rectus muscle function (Fig. 10.21).
Pathologic studies have shown poor muscular development of the levator-superior rectus complex, with fibrous tissue replacing the normal musculature, and a paucity of muscle fibers. The degree of ptosis generally corresponds inversely to the degree of levator function; mild ptosis is generally associated with good levator function, and severe ptosis is indicative of poor levator function. An ill-defined or absent upper lid crease, downwardly directed lashes, and difficulty with lid eversion also imply poor levator development. The hallmark of congenital ptosis is lagophthalmos in downgaze, when the ptotic lid fails to fully follow the globe downward and assumes a higher position than the fellow lid (Fig. 10.22). Reduced ocular motility especially in
P.375
P.376
upgaze and poor Bell's phenomenon can occur in 10% to 15% of patients, and 2% to 5% of patients may manifest the Marcus-Gunn jaw-winking phenomenon.
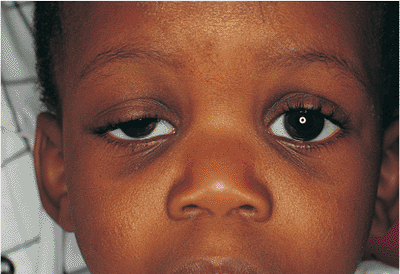 |
Figure 10.20. Significant congenital ptosis of the right upper lid encroaching into the visual axis. Such significant ptosis can lead to amblyopia, and should be repaired. |
 |
Figure 10.21. Congenital ptosis. A: Patient with incontinentia pigmenti. Ptosis is severe enough to cause her to assume a chin-up posture. This head position significantly compromises the inferior visual field, causing some patients to trip over objects on the floor. B: Elevating the lids manually shows good underlying ocular alignment. |
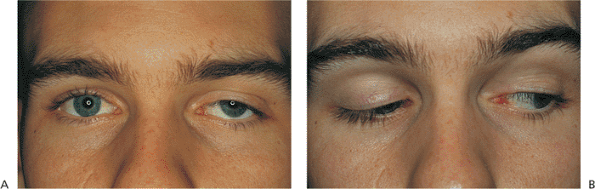 |
Figure 10.22. Congenital ptosis. A: Adult patient with mild left ptosis present since birth. B: Downgaze reveals characteristic lagophthalmos. |
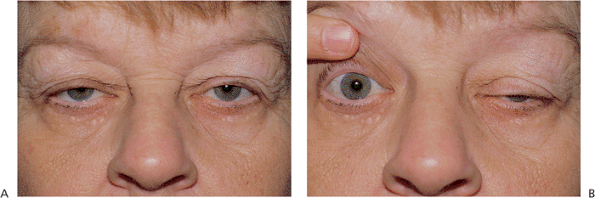 |
Figure 10.23. Acquired ptosis: aponeurotic. A: The patient shows several hallmarks of involutional ptosis, including moderate ptosis. The levator function was characteristically good. B: Manual elevation of one lid to an anatomically normal position causes the fellow eye to become more pronouncedly ptotic, a vivid demonstration of Hering's law of equal innervation. The same occurred when the left lid was manually lifted. |
Acquired ptosis is most commonly secondary to pathology of the levator aponeurosis. Aponeurotic ptosis can occur with dehiscence, fatty degeneration, or frank disinsertion of the tissues (Fig. 10.23). Involutional atrophy is the most common underlying cause, although inflammation, repeated edema, and ocular surgery can also result in separation of the aponeurosis from its tarsal attachments. Chronic use of contact lenses and some topical medications have also been implicated. The hallmarks of aponeurotic pathology include mild-to-moderate ptosis with preservation of good levator function and upward migration of the normal lid crease (Fig. 10.24A). In contrast to the lagophthalmos in downgaze, which is characteristic of congenital ptosis, the lid with an aponeurotic defect often shows greater than normal depression on downgaze. The tissues are sometimes so attenuated that the underlying iris is almost visible through the thin lid skin (Fig. 10.24B). Most cases occur in older adults, but the condition can occur in younger patients with underlying susceptibilities, such as atopy or fatty degeneration. Because the levator subnucleus is a single subnucleus of CN III, equal innervation is sent to both levators. Consequently, elevation of a ptotic lid may result in worsening or unmasking of ptosis in the fellow lid, a demonstration of Hering's law (Fig. 10.23).
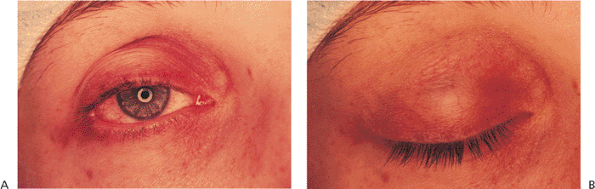 |
Figure 10.24. Acquired ptosis: aponeurotic. A: The patient has a high lid crease and moderate lid droop. B: The iris is visible through the thin lid skin above the tarsal base. |
Traumatic ptosis can occur with any damage to the levator or its aponeurosis (Fig. 10.25). Lid lacerations should be evaluated for levator involvement. Subperiosteal hematomas and blow-out fractures of the roof may induce transient or permanent ptosis. Mechanical ptosis (Fig. 10.26) can also result from trauma (e.g., lid edema, hematoma, emphysema, cicatrix), infection (e.g., preseptal or orbital cellulitis), tumor, and dermatochalasis.
Management
The evaluation of congenital or acquired ptosis is directed to identifying the cause. Neurologic and myogenic causes must be eliminated before surgical correction, and they may require referral to appropriate subspecialists. The height of the palpebral fissures and position
P.377
of lid margins should be inspected to determine if a true ptosis exists instead of secondary ptosis or pseudoptosis. In cases of true ptosis, evaluation of levator function is critical in planning appropriate surgical correction. Minimal ptosis can be corrected by internal (e.g., conjunctival mullerectomy, tarsal-conjunctival mullerectomy) or external (e.g., external levator resection) approaches. Moderate ptosis often requires levator aponeurotic reconstruction, but severe ptosis usually demands frontalis fixation or ptosis crutches.
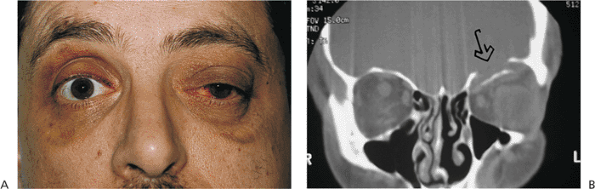 |
Figure 10.25. Acquired ptosis: traumatic. A: The patient suffered a left frontal skull fracture involving the orbital roof secondary to a motor vehicle accident. Other findings included numbness of the forehead (cranial nerve V1), restriction of elevation, and pulsatile exophthalmos. B: Computed tomography shows a depressed roof fracture (arrow) and subperiosteal hematoma. Open reduction through a craniotomy resolved all findings. |
Dermatochalasis, Steatoblepharon, and Blepharochalasis
Dermatochalasis is a common aging change characterized by excess skin of the lids. The baggy eyelid skin may be sufficient to cause a secondary or mechanical ptosis of the upper lids or ectropion of the lower lids (Fig. 10.26). Herniating orbital fat (steatoblepharon) can magnify the problem. Blepharochalasis is characterized by recurrent bouts of bilateral angioneurotic edema, which secondarily distends the overlying skin tissues. The lid becomes thinned and takes on a crenulated or cigarette-paper texture. Women are affected more commonly than are men. The condition may last for several years, but it appears to be self-limited. The condition is benign, but the skin changes may remain (Fig. 10.27).
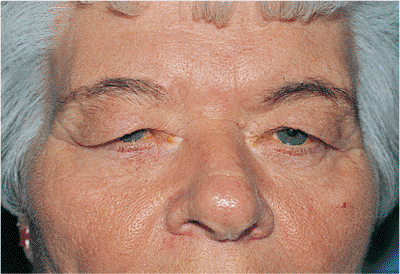 |
Figure 10.26. Dermatochalasis: secondary ptosis. The patient has excessive upper lid skin, creating a mechanical ptosis that interferes with the upper visual field (both eyes). |
Management
The conditions are benign. If the excess skin is sufficient to create secondary ptosis of the upper lid and compromise visual function, or to create ectropion of the lower lid, blepharoplasty can be considered. A variety of techniques have been suggested to manage festoons, including extended blepharoplasty, orbicularis suspension, and direct excision.
Pseudoptosis
Pseudoptosis is the appearance of ptosis although true ptosis does not exist. It can be caused by enophthalmos of any cause including anophthalmos, socket contracture, traumatic orbital blow-out fracture, phthisis, and involutional
P.378
atrophy (Fig. 10.28). Contralateral lid retraction, overcorrection of contralateral ptosis, and contralateral proptosis can also create the appearance of lid droop (Fig. 10.29).
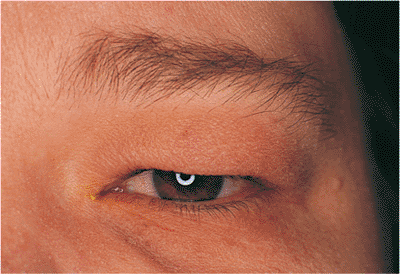 |
Figure 10.27. Blepharochalasis. The patient had recurrent bouts of upper lid swelling. Chronic changes of distended and thinned cigarette-paper skin remain. |
 |
Figure 10.28. Pseudoptosis: enophthalmos secondary to an orbital blow-out fracture (right eye). |
Management
Enophthalmos can occur after trauma, erosion of the orbit by tumors or chronic sinusitis, and atrophy of the orbital tissues because of trauma, involutional atrophy, nutritional deficits, or cachexia. Evaluation includes appropriate history taking, testing for infraorbital dysesthesia, and radiologic evaluation as required. Contralateral lid retraction (e.g., thyroid retraction) should always be looked for and appropriate laboratory tests ordered. Management is directed at correcting the identified defect. For example, enophthalmos secondary to recent orbital trauma is repaired by reduction of the orbital fracture. Thyroid eye disease is treated with topical lubricants, and surgical lowering of the retracted eyelids if needed. Cicatricial and mechanical retraction is treated by releasing the cicatricial bands and placement of spacer grafts to the skin or posterior lamella, as necessary.
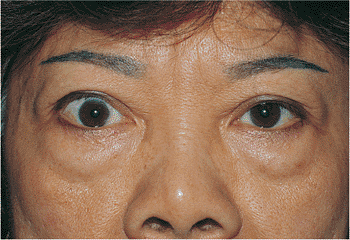 |
Figure 10.29. Pseudoptosis: thyroid eye disease. A 50-year-old patient complained of a drooping left upper lid. Examination revealed proptosis and lid retraction of the right eye. Laboratory studies revealed the presence of thyroid disease. |
Neurogenic and Myogenic Ptosis and Myasthenia Gravis
The neurogenic causes of ptosis include oculomotor palsy and Horner's syndrome.
Cranial nerve III palsy also affects ocular motility and may also affect the pupil (Fig. 10.30). The underlying causes include tumors, infection, inflammation, and vascular and metabolic disorders. This paralytic ptosis is generally severe, with little or no levator function. Aberrant regeneration may cause abnormal lid elevation with attempted ocular movements.
Horner's syndrome results from sympathetic denervation to the lid and orbit. Classic findings include mild ptosis with excellent levator function and miotic anisocoria, which is more apparent in darkness (Fig. 10.31). Lower lid elevation (i.e., reverse ptosis) is often present, resulting in narrowing of the palpebral fissure and apparent enophthalmos. Ipsilateral anhidrosis results from involvement of the sudomotor fibers. Heterochromia is a sign of congenital or infantile onset. The causes include trauma, vascular insult, tumors (particularly those of the pulmonary apex), and intracavernous lesions. The mild ptosis reverses with instillation of Neo-Synephrine.
Acquired myogenic ptosis results from several degenerative conditions affecting the levator and oculomotor muscles. Chronic progressive external ophthalmoplegia (CPEO) encompasses a spectrum of mitochondrially inherited muscular dystrophies that also affect ocular motility and levator function (Fig. 10.32). Patients slowly develop progressively severe ptosis with poor levator function, little or no ocular movement, and poor Bell's phenomenon. A characteristic exotropia is manifested in advanced stages, although patients seldom complain of diplopia. Larger and more distal structures away from the head and neck can be variably affected,
P.379
depending on the specific disorder. In oropharyngeal dystrophy, chewing and swallowing are affected, and in myotonic dystrophy, muscles and genitalia are involved. Ocular manifestations are protean, ranging from pigmentary retinopathy (e.g., Kearns-Sayre syndrome [KSS] or CPEO plus ; Fig. 10.33), to polychromatophilic cataract (e.g., myotonic dystrophy). Cardiac conduction delays (KSS) and peripheral nerve disorders can also occur in mitochondrial encephalopathies. Biopsy of skeletal muscle often shows ragged red fibers that reflect an abnormal number of mitochondria. Although the pigmentary retinopathy of KSS is often referred to as pseudo-retinitis pigmentosa, or RP-like, the visual course and prognosis of the two conditions is not similar. These conditions are often diagnosed clinically, but may require extensive chromosomal and neurologic testing as well as muscle biopsy.
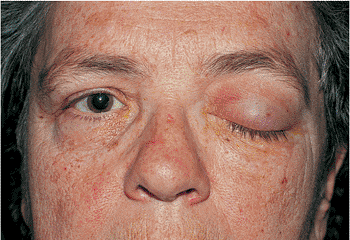 |
Figure 10.30. Neurogenic ptosis: oculomotor palsy. Metastatic paratracheal adenocarcinoma produced a complete orbital apex syndrome. Oculomotor palsy and ptosis were complete. Note the downwardly directed lashes and the loss of the natural lid crease. Other findings include proptosis and significant orbital congestion. |
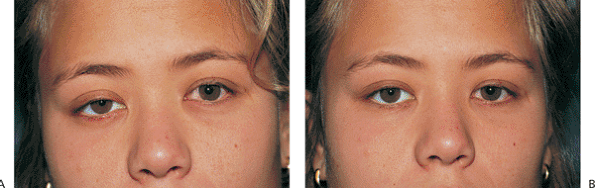 |
Figure 10.31. Horner's syndrome. A: Before Neo-Synephrine. A 17-year-old patient developed Horner's syndrome after resection of a lymphangioma from the right pulmonary apex. Findings include mild ptosis and miosis. B: Ptosis resolves after instillation of Neo-Synephrine. |
Myasthenia gravis is a disorder of adults affecting the neuromuscular endplate, altering the synaptic transmission of acetylcholine. Findings can be multiple, depending on the muscle complex affected, but ocular findings, including intermittent diplopia and ptosis, often arise primarily (Fig. 10.34). The ptosis characteristically varies through the day, depending on the fatigue of the levator. There is a well-recognized association with thyroid dysfunction and thymoma. The condition may be confirmed via edrophonium injection (Tensilon) and ice-pack tests, which can quickly and temporarily reverse ptosis, and by antibody testing. However, both false-positive and false-negative results are known to occur, making the diagnosis still a clinical one.
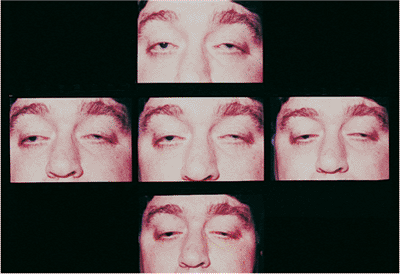 |
Figure 10.32. Myogenic ptosis: chronic progressive external ophthalmoplegia. The composite photograph shows the decrease in ocular motility characteristic of myopathic processes. |
Management
Patients with new-onset ptosis thought to be of neurogenic or myogenic origin or with Horner's syndrome should undergo full evaluation, including laboratory analysis and radiologic and pharmacologic testing, to determine the cause. Patients suspected of having CPEO should undergo routine electrocardiogram testing. Referral to a neuroophthalmologist may be warranted. Treatment of any
P.380
associated ptosis should be approached with caution and depends on the severity of the condition, the degree of visual impairment, and the amount of levator function. Treatment options include conjunctival mullerectomy, levator resection, frontalis suspension, and ptosis crutches. These patients are at risk for postoperative exposure keratitis.
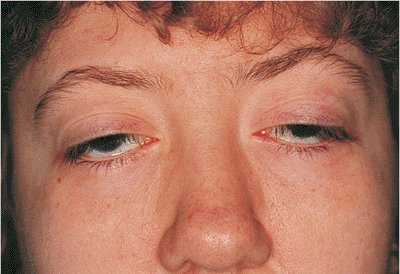 |
Figure 10.33. Myogenic ptosis: chronic progressive external ophthalmoplegia (Kearns-Sayre syndrome). The patient manifests severe ptosis with characteristic facies of longstanding external ophthalmoplegia. The ocular motility is severely limited, and levator function is extremely poor. The patient also has significant visual field defects secondary to pigmentary retinopathy but has not developed cardiac conduction delays. The patient complained of progressive ptosis that interfered with daily functions, requiring her to manually elevate her lids. |
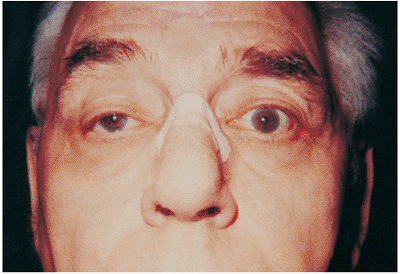 |
Figure 10.34. Myasthenia gravis. The patient noticed gradual worsening of moderate ptosis (right eye). He also complained particularly of having to use his forehead to lift his eyes. Questioning disclosed a history of intermittent diplopia. |
Marcus-Gunn Jaw-Winking
In 1833, Marcus-Gunn described a patient with unilateral congenital ptosis that elevated with jaw movements. The Marcus-Gunn jaw-winking phenomenon is thought to result from an abnormal synkinetic connection between cranial nerves V and III. Branches from the motor root of cranial nerve V destined for the ipsilateral lateral pterygoid muscle (or less commonly, the inner pterygoid) innervate the ipsilateral levator muscle such that jaw movements, especially those to the opposite side, cause elevation of the lid (Fig. 10.35). Jaw-winking may occur in 2% to 5% of patients with congenital ptosis. Familial patterns are not uncommon, and rare bilateral cases have been described. The severity of the disorder appears to lessen with time and age, but this may reflect adaptation to the condition and the ability to suppress the motions that trigger obvious lid retraction. The condition can be confused with aberrant regeneration of cranial nerve VII (i.e., winking jaw of Wartenberg; Fig. 10.36). In this latter, acquired condition, motor fibers originally innervating the orofacial musculature are redirected abnormally to the orbicularis fibers, causing synkinetic closure of the lid with ipsilateral facial movements. It is common after trauma to cranial nerve VII.
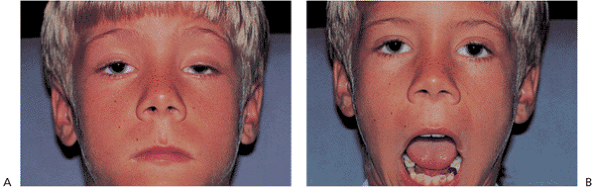 |
Figure 10.35. Marcus-Gunn jaw-winking. A: Significant ptosis of the left eye at rest. B: The lid elevates with jaw movement, reflecting the synkinesis between the pterygoid and levator muscles (cranial nerves V and III). |
Management
The Marcus-Gunn jaw-winking phenomenon is an isolated neurologic finding, and further workup is not indicated. If the ptosis is mild, and the jaw-winking is of little cosmetic concern, the condition may be observed indefinitely. If surgery is contemplated, levator disinsertion with subsequent frontalis suspension is generally recommended. Some ophthalmologists recommend the procedure be done bilaterally to avoid postoperative asymmetry.
Blepharospasm, Hemifacial Spasm, Myokymia, and Aberrant Regeneration
Hyperkinetic dystonias of cranial nerve VII are characterized by involuntary contractures of the muscles subserved by the facial nerve. In blepharospasm, the spasms are bilateral and most often involve the eyelids and brows, but they sometimes extend to the lower face and neck (Fig. 10.37). The spasms can be severe enough to impair daily activities. The disorder begins between 40 and 60 years of age and affects women three times more often than men. Patients often learn tricks that defeat the spasms, such as yawning, whistling, or other facial movements. The spasms can be stress-related but are characteristically absent during sleep. Essential blepharospasm is generally idiopathic, but many factors may cause reflex blepharospasm.
Hemifacial spasm is often caused by compression and irritation of the facial nerve root by an intracranial tumor or
P.381
aberrant vessel, or after facial nerve palsy of any cause (Fig. 10.38). Most cases, however, are idiopathic. The spasms affect only one side of the face, are unaffected by tricks, and persist through sleep, often awakening those so afflicted. The age of onset is later than in blepharospasm.
 |
Figure 10.36. Aberrant regeneration of cranial nerve VII. A: Patient after extensive right parotidectomy for malignant oncocytic carcinoma with secondary right facial palsy. The lid is in a normal position while at rest. B: Aberrantly directed nerve fibers from the lower face now innervate the orbicularis and cause the lid to close with facial movement. |
Aberrant regeneration typically occurs after traumatic injury of the seventh nerve. Unlike blepharospasm and hemifacial spasm, these facial contractures are not involuntary spasms but are coincident with facial movements. Involuntary eyelid closure with smiling, chewing, or grinning results from misdirected nerve fibers originally destined for lower facial musculature regenerating into the orbicularis. Consequently, the eyelid spasms occur only with facial expression. Similarly, upper fibers initially innervating the eyelids can reinnervate the lower oral musculature, resulting in oral spasms on blinking. Salivary fibers may reinnervate the lacrimal gland, resulting in gustatory epiphora, or crocodile tears.
Intermittent, rapid-frequency, fine-motor fasciculations are known as myokymia. The condition is benign, is more transitory than any of the other conditions, and is often related to stress.
 |
Figure 10.37. Essential blepharospasm. Severe involuntary spasms of the eyelids can significantly impede daily functions such as driving, essentially incapacitating the patient. |
Management
Patients with facial hyperkinesis should be thoroughly questioned for a history of facial nerve disorders, previous trauma, or palsy. Dizziness, vertigo, decreased hearing and tinnitus suggest eighth cranial nerve pathology, which may contiguously induce hemifacial spasm. An audible bruit may indicate vascular compression of the nerve root. All patients with hemifacial spasm should undergo radiologic imaging of the cerebellopontine angle to rule out intracranial pathology. Medical management of these disorders is generally ineffective. Anxiolytics, antidepressants, antiparkinsonians, and neuroleptics have all been tried without much success. Local chemodenervation with Clostridium botulinum toxin type A (BoTox) and other medications has proven to be successful in controlling the spasms, but the effects are temporary, and retreatment is often required. The length of effect is generally longer for
P.382
hemifacial spasm than for blepharospasm. Denervation is less widely used because of its poor record of success. Some patients may benefit from wide orbicularis myectomy, which requires meticulous extirpation of the involved muscles. Neurosurgical decompression of the facial canal with interposition of a sponge between the nerve root and an encroaching vessel can be successful in selected patients.
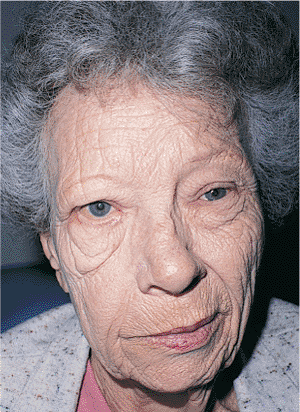 |
Figure 10.38. Patient with left hemifacial spasm. The spasms extend into the neck, as evidenced by the significant platysmal band. The neurologic evaluation was negative. |
Tumors
Xanthelasma, Histiocytoses, and Juvenile Xanthogranuloma
Xanthelasmas are the most common form of cutaneous xanthoma, typically arising in middle-aged and older women. They appear as flat, plaque-like, creamy yellow lesions and are the result of abnormal lipid deposition within the dermis (Fig. 10.39). The medial aspect of the lids is most commonly affected. Although there is no firm association with hyperlipidemia, they can occur in diabetes and in hyperlipidemic or hereditary conditions, particularly in younger patients. The lesion is benign, but it can be cosmetically disfiguring.
The common xanthelasma is not associated with the more uncommon conditions of histiocytosis (e.g., Hand-Sch ller-Christian disease, Letterer-Siwe disease, or eosinophilic granuloma), in which the lesions usually occur in childhood, are more nodular, involve deeper structures, and produce lytic bony lesions. These latter conditions are often associated with significant multisystem disease and can be profoundly morbid or fatal.
Juvenile xanthogranuloma is a benign condition that arises in childhood. The typical lesion is a yellow-orange, solid nodule, but the mass can also take on a deeper color (Fig. 10.40). Although the lesion is typically located on the skin, it can occur intraocularly or, more uncommonly, in the orbit. Spontaneous hyphema has been reported from rupture of the vessels in iris lesions.
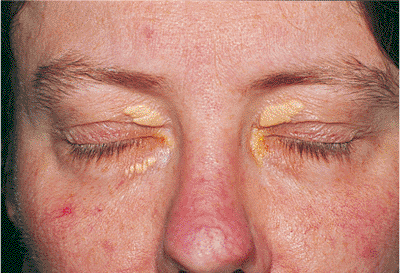 |
Figure 10.39. This otherwise healthy woman has creamy yellow plaques in the dermis of all four lids, which is typical of xanthelasma. |
Management
Xanthelasmas are benign growths and are best managed conservatively. Although the association with hyperlipidemia is rare, if the lesion appears in a young patient, lipid evaluation is warranted. Otherwise, no workup is needed. If the lesion is significantly large or cosmetically disfiguring, simple excision is curative. However, the lesion may recur. Some lesions may be amenable to treatment with cutaneous laser.
Patients suffering from the histiocytoses should be referred for further evaluation and care. Eosinophilic granuloma can be excised locally or treated with irradiation. The lesion sometimes involutes spontaneously. Juvenile xanthogranuloma is a benign condition. Lesions typically resolve spontaneously. The eyes should be thoroughly examined to rule out intraocular involvement.
Hidrocystoma and Syringoma
Hidrocystomas (i.e., sudoriferous cysts, sweat gland cysts, eccrine hydrocystomas) are apocrine or eccrine in origin. They often arise on the lid margin as single or multiple translucent cystic nodules. On incision, a clear or milky fluid is expressed from a smooth-walled cavity (Fig. 10.41). Cystic basal cell carcinoma is often initially misdiagnosed as hidrocystoma.
Syringoma are solid nodules that resemble milia. They are more common in young female patients and typically arise on the lower lids as multiple, irregular nodules, although they may also arise on the cheeks and forehead (Fig. 10.42). The nodule may contain keratin. The masses are probably eccrine in origin.
Management
These lesions are benign and require no further workup. Simple excision of the mass, including the cyst wall, is generally curative. However, because rare malignant counterparts have been reported, large or recurrent lesions should be sent for biopsy.
Papilloma, Keratoacanthoma, Inverted Follicular Keratosis, and Seborrheic Keratosis
Several benign epithelial tumors of the lid are common. Squamous papillomas are by far the most common. They may be single or multiple and tend to involve the lid margin. They are typically sessile or pedunculated, with many finger-like projections containing fibrovascular cores (Fig. 10.43).
Keratoacanthoma is a rapidly growing, dome-shaped lesion with elevated, rolled edges and a central keratin-filled crater (Fig. 10.44). These lesions typically grow over a period of 2 months or less, are generally painless, and often regress spontaneously but may leave a scar. The lesion is benign but is sometimes confused clinically and histologically with squamous cell carcinoma.
Inverted follicular keratosis often presents as a cutaneous
P.383
P.384
horn at the lid margin (Fig. 10.45). It tends to grow rapidly, often in less than 3 months. Most physicians think the lesion represents an irritated seborrheic keratosis.
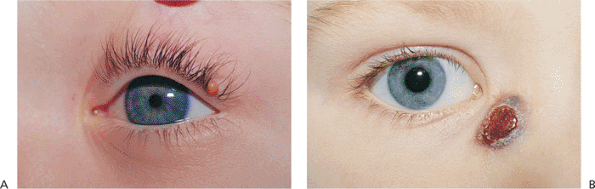 |
Figure 10.40. Juvenile xanthogranuloma (JXG). A: Characteristic yellow-orange, solid tissue nodule arose on the lid margin of this 2-year-old boy. This was the sole tumor found on the patient. The ocular examination was normal. B: Another patient with a JXG lesion on the side of the nose. Its color is somewhat darker than that of the tumor in A. Biopsy specimens of each patient showed characteristic Touton giant cells and foamy histiocytes. |
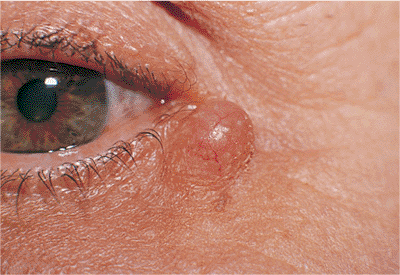 |
Figure 10.41. Hidrocystoma is a nontender cyst of the lid. The lesion is well circumscribed and translucent. |
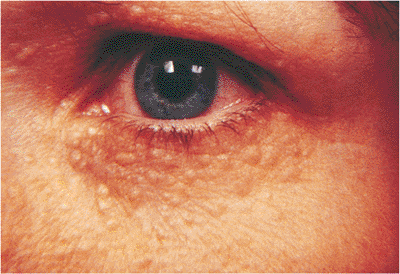 |
Figure 10.42. Syringoma. These are small solid nodules that resemble milia and are more common in young women. |
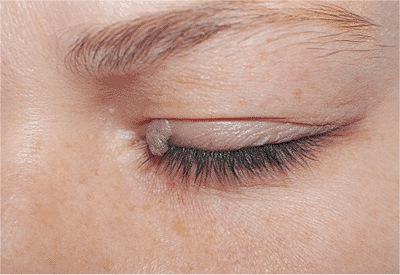 |
Figure 10.43. Squamous papilloma. A sessile, fleshy nodule with finger-like projections and fibrovascular cores. |
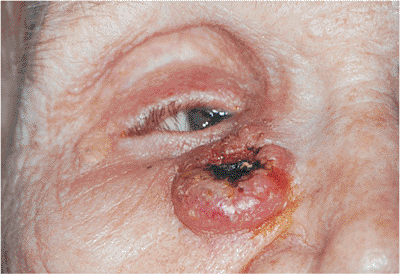 |
Figure 10.44. Keratoacanthoma. This dome-shaped lesion arose over a matter of months. A central, keratin-filled crater can be seen. |
Seborrheic keratosis is common in older persons. It appears as a flatly elevated, verrucous growth of variable pigmentation. It is typically well demarcated, appears stuck on the skin, and may have a friable surface (Fig. 10.46).
Management
These benign skin lesions can be managed by single excision. Because of their similarity to cancerous lesions, submission for pathologic examination should be considered.
Capillary Hemangioma
Capillary hemangioma (i.e., strawberry nevus, benign hemangioendothelioma) is the most common primary orbital and cutaneous tumor of childhood. Only rarely is the lesion seen at birth; it usually arises shortly thereafter as a red, irregularly dimpled, raised lesion, called a strawberry nevus (Fig. 10.47). The lesion characteristically goes through a period of rapid growth over the first 6 to 12 months, with gradual spontaneous involution. Most regress by 3 years of age. Before involution, hemangiomas may be tender to touch and may swell with crying (without significantly changing color), but with regression, the mass becomes less tender, much smaller, more fibrotic, and less vascular, making surgical excision less hemorrhagic and possibly less difficult. The overlying skin remains abnormal, with mild atrophic changes and pallor and developing a crepe paper texture. Unlike the port wine stain angioma of Sturge-Weber, capillary hemangiomas are raised and irregular, are redder than port wine nevi, and blanch with pressure. Feeder vessels can often be identified. If the tumor is primarily orbital or deeper in the dermis, the cutaneous appearance takes on a more bluish hue (Fig. 10.48).
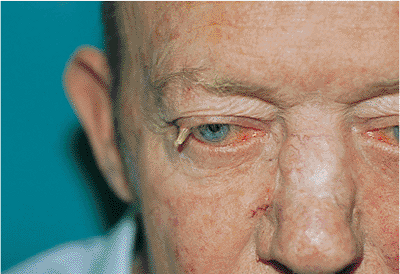 |
Figure 10.45. Inverted follicular keratosis. The lesion at the lid margin has a cutaneous horn. |
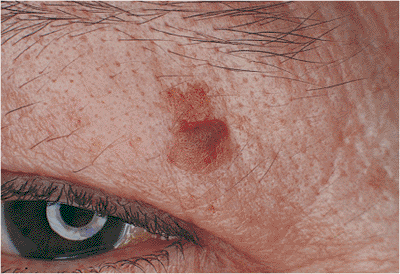 |
Figure 10.46. Seborrheic keratosis. Typical appearance of a flat, elevated plaque that appears stuck on. The lesion does not have deep extensions through the skin. |
The lesion is benign with no tendency for malignant transformation. However, a generalized bleeding disorder secondary to profound thrombocytopenia has been associated in newborns with giant hemangiomas (i.e., Kasabach-Merritt syndrome). If a periorbital mass is large enough, it may induce a mechanical ptosis or astigmatism, resulting in amblyopia. Most lesions are solitary, but as many as 20% of patients may have multiple foci. The head and neck region is most commonly affected, but visceral, central nervous system (CNS),
P.385
and lymphatic hemangiomas have also been described (Fig. 10.48C). Emotional and cosmetic considerations are often the overwhelming parental concerns.
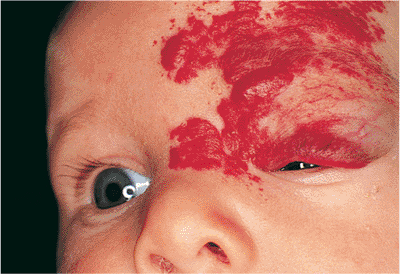 |
Figure 10.47. Capillary hemangioma. A large, diffuse lesion of the left face. The surface is raised and irregular, with a bright red color characteristic of superficial hemangiomas. Significant ptosis can produce occlusion amblyopia. |
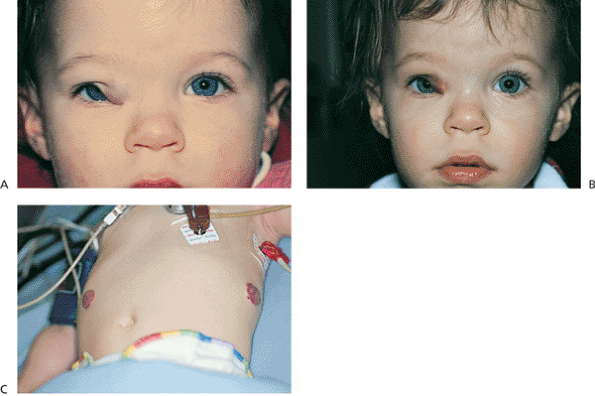 |
Figure 10.48. Capillary hemangioma. A: The lesion of the upper lid is large enough to partially occlude the visual axis and induced a significant astigmatic error and strong visual preference for the left eye, requiring occlusion therapy. Cycloplegic refraction: OD, +5.50 030 degrees; OS, +1.75 sph. Because it is located in the deeper tissue layers, this hemangioma appears more bluish than more superficially located lesions. B: Three days after intralesional steroid injection, the hemangioma has shrunk out of the visual axis. C: The patient also had more peripheral lesions on the trunk. |
Management
The desire for intervention should be tempered with knowledge of the natural history of spontaneous involution. Patients must be closely followed for development of ptosis or amblyopia. If the lesion is small and inconsequential, observation is recommended. The parents should be queried about the presence of other lesions. Although the precise mechanism of action is not well understood, intralesional corticosteroid injection is the most common treatment. Complications have included systemic corticosteroid absorption, eyelid necrosis, and central retinal artery occlusion. The fundus should be examined immediately after injection. Systemic and topical steroids, irradiation, cryotherapy, and injectable sclerosing agents have also been used with variable success. However, each carries its own risks. If surgical excision is contemplated, it should be postponed until the lesion has involuted. Because of the vascularity of the tumor, laser scalpels may be useful.
Nevus Flammeus
Nevus flammeus (i.e., port wine stain, claret stain, Sturge-Weber syndrome, encephalotrigeminal syndrome) is a large, flat, violaceous, vascular malformation characterized by diffusely ectatic, cavernous vascular spaces and channels in the upper layers of the dermis. Unlike capillary hemangioma, which usually arises just after birth, nevus flammeus is well circumscribed and grows with the patient throughout life. The tumor typically follows the distribution along the branches of the trigeminal nerve (Fig. 10.49), but it commonly crosses the midline or is bilateral. It often occurs away from the head and neck region. Unlike capillary hemangioma, it neither regresses nor involutes with age, nor does it blanch with pressure. It is more violaceous (i.e., port wine) than strawberry red, which is perhaps a reflection of the blood moving slower through the tumor, and the color often deepens with crying or a Valsalva's maneuver. Feeder vessels are uncommon. If the tumor is nodular, it is softer and more compressible than a hemangioma, feeling somewhat spongier than the firmer counterpart. With time, the mass often becomes more nodular and fibrotic (Fig. 10.50).
The tumor may be associated with other vascular malformations, such as the Klippel-Tr naunay-Weber syndrome, in which venous and lymphatic malformations coexist on the trunk and extremities, and the Sturge-Weber syndrome. The latter condition, one of the phakomatoses, is the most well-known abnormality associated with facial nevus flammeus. Associated findings include diffuse choroidal hemangioma, retinal detachment, and glaucoma. Ocular involvement is more frequent if the malformation affects the upper trigeminal distributions (i.e., V1 and V2). Central anomalies include vascular malformations of the ipsilateral leptomeninges,
P.386
with calcification of the underlying cortex and consequent risk of seizure disorders, and various degrees of mental retardation.
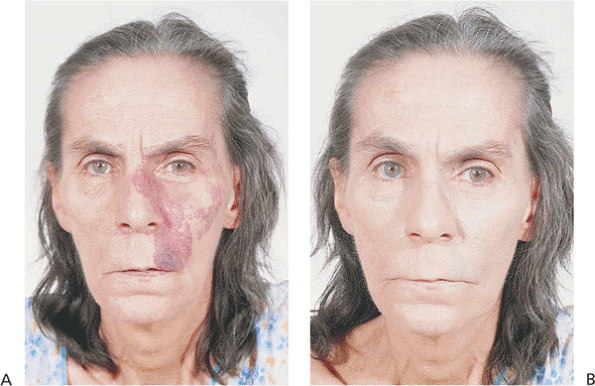 |
Figure 10.49. Nevus flammeus. A: Without makeup. The lesion is much darker than a capillary hemangioma. B: Many patients are able to disguise the lesion well by using heavy makeup. |
Management
Because of the risk of glaucoma and retinal detachment, patients require full examinations at routine intervals, especially if the lid, forehead, and globe are involved. Glaucoma may develop at any age and is difficult to treat. Patients with lesions of the head and neck should also be referred for neurologic evaluation and imaging, because seizures and mental retardation are not uncommon. Treatment of the lesion itself is often frustrating, because the overlying soft tissue and underlying skeleton are often malformed. Facial stains may be covered conservatively with heavy makeup. Although surgery may be considered for older patients whose lesions have become nodular, advances with dermatologic lasers have made photocoagulation a popular alternative in all ages.
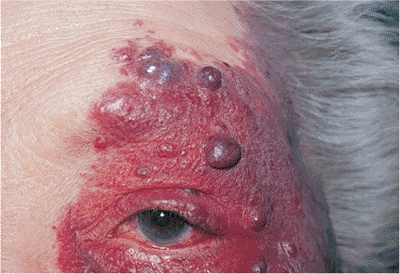 |
Figure 10.50. Chronic Sturge-Weber syndrome. The lesion shows the typical nodularity and pedunculation of chronicity. As suspected from the eyelid and conjunctival involvement, the patient also suffers from unilateral glaucoma. |
Neurofibromatosis
Plexiform neurofibromas are a hallmark of neurofibromatosis, also called von Recklinghausen's syndrome. They occur along peripheral nerves of the lid and orbit but are infiltrative rather than isolated or encapsulated. Arising in the first decade, they present a typical appearance that diffusely involves all tissues of the lid and can extend into the orbit (Fig. 10.51). The normal architecture is distorted, often with the loss of most soft tissue landmarks. Diffuse soft tissue involvement typically creates a secondary ptosis, often worse laterally, which produces
P.387
a lazy S configuration. The infiltrative nature of these benign tumors can massively enlarge the lid, giving a bag of worms sensation. The lid and lash margins are often abnormal, and ocular function can be compromised.
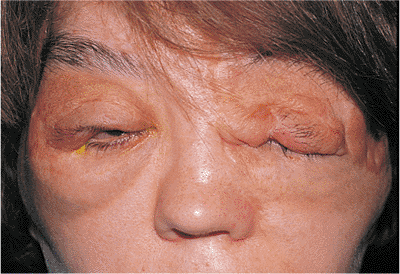 |
Figure 10.51. Neurofibromatosis. The upper lids are diffusely involved with plexiform neurofibromas. The patient is anophthalmic (left eye), having lost the eye to massive proptosis and exposure secondary to an optic nerve glioma. She has had multiple debulking procedures. |
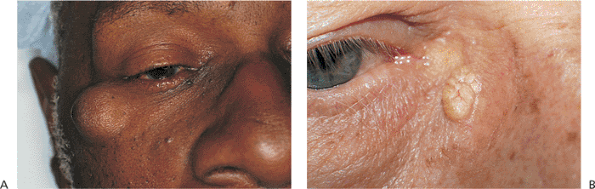 |
Figure 10.52. Epidermal inclusion cyst. A: A slowly growing mass over the right zygoma with normal overlying skin. The mass is nontender and is attached to the overlying skin but has no deep attachments. B: A more superficial lesion shows the typical characteristics of yellow color and telangiectasis, which can lead to an erroneous diagnosis of sebaceous cyst. |
Patients with neurofibromatosis (especially Type II) are at increased risk of CNS tumors such as optic nerve glioma and acoustic neuroma, as well as congenital glaucoma.
Management
Because plexiform neurofibromas are pathognomonic for neurofibromatosis, the patient should undergo a thorough ocular and physical examination, including radiographs of the head. Large, unsightly lid masses may be debulked for cosmetic reasons, with reconstruction dictated by the extent of excision. Because of the infiltrative nature of the tumor, the lesions typically recur, necessitating multiple procedures.
Epidermal Inclusion Cysts and Milia
Inspissation of the hair follicle, with subcutaneous entrapment of keratin produces a freely movable dermal or subcutaneous nodule known as an epidermal inclusion cyst or a sebaceous cyst (Fig. 10.52A). The color of the mass depends on the location within the dermis. Deep lesions are covered by normal skin, and more superficial lesions may appear yellow or fatty, with overlying telangiectatic vessels, leading to the common misnomer of sebaceous cyst (Fig. 10.52B). The mass is often attached to the skin but is not anchored to deep structures. The cyst may rupture, leading to secondary inflammation and scarring. Epidermal inclusion cysts can occur anywhere on the body, including upper and lower lids and the brows. If allowed to enlarge, upper lid lesions can create a mechanical ptosis.
Milia are very small superficial keratin cysts, approximately 1 to 4 mm in diameter, that can arise in large numbers. Their creamy yellow or white color is characteristic of their location in the superficial dermis (Fig. 10.53). They can occur spontaneously or secondary to lid trauma and can be thought of as miniature epidermal inclusion cysts. They are benign.
Management
Epidermal inclusion cysts can be effectively managed by excision of the mass and entire cyst wall. This can be done through a skin incision over the lesion, with subcutaneous dissection of the cyst, or by simple excision and closure of the entire mass.
Milia can be treated by decapitating the mass or incising the overlying skin with a hypodermic tip or razor blade scalpel. The keratin cyst is expressed with gentle pressure.
Hair Follicle Tumors
Hair follicle tumors, including trichoepithelioma, tricholemmoma, trichofolliculoma, and pilomatrixoma, are benign, with little malignant potential. Most occur as solitary masses or nodules that have a marked predilection for the face, head, and neck. Many simulate basal cell carcinoma, intradermal nevus, neurofibroma, and other adnexal tumors, and they may be difficult to differentiate clinically. Most arise in adulthood.
 |
Figure 10.53. Milia are multiple, small, yellow lesions of the superficial skin. The patient had similar lesions on all eyelids. |
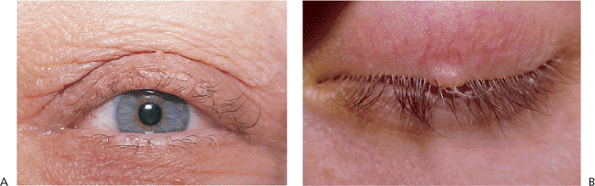 |
Figure 10.54. A: Trichoepithelioma. A 65-year-old woman was observed incidentally to have nodular growths on all eyelids. The lesions were painless and had been present for an unknown length of time. Biopsy specimens revealed them to be trichoepithelioma. B: Trichofolliculoma. Fine white hairs growing from a central pore typify trichofolliculoma. A painless mass was present for several months in this 25-year-old man. |
P.388
Trichoepithelioma presents as a solitary, flesh-colored, dome-shaped papule (Fig. 10.54A). It may be particularly difficult to differentiate from basal cell carcinoma clinically and histologically. An autosomal-dominant form (i.e., Brooke's tumor) begins in adolescence with multiple tumors, associated with multiple cylindromas and syringomas. Tricholemmoma can also present as multiple lesions but most commonly appears as a solitary, flesh-colored growth in later adulthood. However, the lesion is sometimes pearl gray, leading to confusion with basal cell carcinoma. Multiple tricholemmomas of the face, head, and hands are associated with an increased risk of breast, thyroid, and gastrointestinal cancers and with other fibrous hamartomas (e.g., Cowden disease). Trichofolliculoma is the most highly differentiated hair follicle tumor. It is characterized by fine white hairs growing from a central pore in a flesh-colored papule. (Fig. 10.54B)
Unlike the previous entities, which typically arise in adulthood, pilomatrixoma (i.e., calcifying epithelioma of Malherbe) arises primarily in childhood and adolescence. The typical lesion is a solitary, firm, subcutaneous nodule, which is mobile under the skin. Telangiectatic vessels are common, as is a pink or violaceous color.
Management
The lesions are benign and have little malignant potential. However, they can be cosmetically disfiguring. Because of the similarity in appearance with other skin malignancies, biopsy should be considered to rule out more significant pathology. Treatment is by simple excision. Dermabrasion and laser vaporization may also be effective in treating lesions away from the eyes.
Nevocellular Nevi
The skin contains a large number of melanocytes, dendritic cells of neural crest origin that produce melanin. Nevus cells are thought to be a specialized form of melanocyte that possesses the ability to form nests and aggregations within the skin. Nevocellular or melanocytic nevi are categorized according to the histologic location of the cells within the dermis. Junctional nevi occur at the junction of the dermis and epidermis. They appear most commonly in childhood as round or ovoid, flat macules. Pigmentation varies from light to dark brown. They often enlarge and become darker during childhood and adolescence, becoming raised, pigmented nodules, called compound nevi (Fig. 10.55). Hair growth is common. These lesions have characteristics of junctional and intradermal nevi. Malignant transformation can occur with junctional and compound nevi. As the name suggests, intradermal nevi are primarily located purely within the dermis. They are raised nodules and may be minimally pigmented (Fig. 10.56). Malignant transformation is unusual.
Management
Nevi are benign but may be cosmetically unsightly. Excision is usually curative, although the lesion may recur if incompletely removed. Subsequent differentiation from melanoma may be difficult. Any excised pigmented lesion should be sent for pathologic examination.
P.389
Other methods of destruction, such as cryotherapy or electrodesiccation, should not be used.
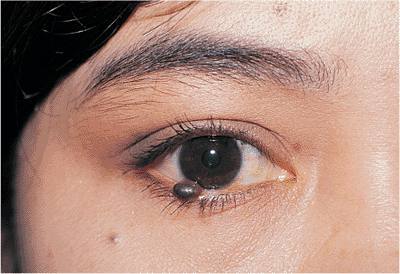 |
Figure 10.55. This compound nevus grew slowly over years from a previously flat mole that had been present since childhood. |
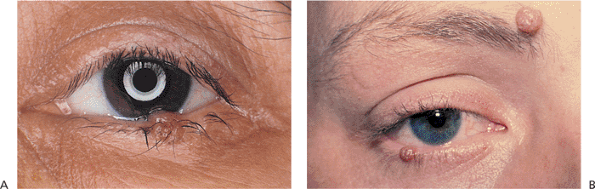 |
Figure 10.56. Intradermal nevus. A: Verrucous appearance of a flesh-colored, lightly pigmented, elevated mass that had been present for longer than 4 years. B: Multiple intradermal nevi. |
Basal Cell Carcinoma
Basal cell carcinoma is the most common skin malignancy. It is more common in the lower lid than the upper, but it can occur anywhere on the ocular adnexal skin (Fig. 10.57). Although it has low metastatic potential and is generally well controlled by local excision, significant morbidity can occur through neglect, recurrence, or inadequate surgical treatment. Chronic sun exposure is known to be a risk factor, as are age and fair skin. Skin cancers of all types are uncommon in darkly pigmented populations.
The tumor starts in the epithelium but can extend into the deeper tissues to involve the orbit or lacrimal drainage system. Several clinical types are recognized, though the distinction is often made at the pathologist's microscope. Although the nodular and sclerosing types of tumor are generally well circumscribed, the morphea type may be poorly demarcated, with multilevel finger-like projections and extensions. Basal cell carcinoma may also take on a cystic or nevoid appearance, sometimes mimicking hidrocystoma or pigmented lesions such as melanoma. Basosquamous carcinoma may be more aggressive than the nodular type. Clinically, the lesions appear as firm masses on the epithelium with variable, deep extensions. The edges of the lesion are typically raised, with the rolled edges forming a central crater of pearl gray tissue (Fig. 10.58). Inherent vascularity and easy excoriation may cause chronic ulceration and bleeding. A lesion at the lid margin often causes lash loss. The most common lesion is found in older adults. However, multiple tumors can arise in the second decade as part of the basal cell nevus syndrome. In this autosomal-dominant condition, multiple basal cells of the skin arise from ordinary-looking nevi, predominantly on the face and eyelids. The lesions, especially those around the nose and eyes, behave more aggressively than most. Other associated systemic findings include prognathism with mandibular bone cysts,
P.390
skull and skeletal anomalies, teleorbitism, partial agenesis of the corpus callosum, and medulloblastoma.
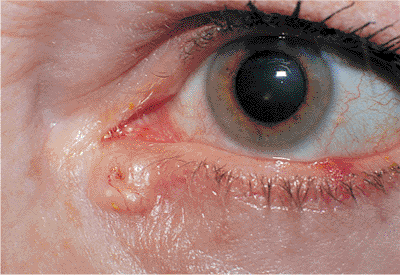 |
Figure 10.57. Basal cell carcinoma. Well-circumscribed, pearly gray tumor of the epithelium, with raised, rolled edges and central ulceration. An independent vascular pattern is also visible. Lesions of the medial canthal area can easily involve the lacrimal drainage system and orbit. |
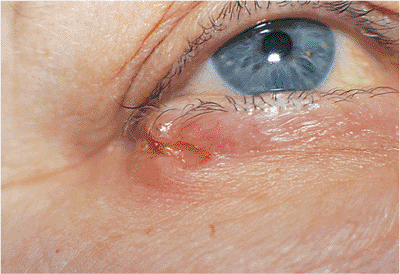 |
Figure 10.58. Basal cell carcinoma. A well-defined, firm mass of the lateral canthus with a deep central crater and markedly rolled edges, although the mass is not characteristically pearly gray. A biopsy confirmed basal cell carcinoma. Like lesions of the medial canthus, the tumors of the lateral canthal region can give rise to deep orbital extension. |
Basal cell carcinoma must be differentiated from chronic chalazion, keratoacanthoma, dermal nevus, and other skin malignancies.
Management
Evaluation should include inspection and palpation of the deeper tissues and orbit. If basal cell carcinoma is suspected, an incisional biopsy in the office can provide the diagnosis. Particular attention should be paid to lesions of the medial and lateral canthi, as these sites offer easy entrance of tumor cells into the deep orbit. After the diagnosis is established, complete excision by Mohs' micrographic technique or frozen-section control and reconstruction is curative, because the lesion has little metastatic potential. However, the patient must be continually monitored for recurrences and the development of new tumors. Although cryotherapy is often used on other cutaneous sites, it should not routinely be a primary treatment for tumors of the lid and adnexa because of the inability to guarantee complete excision. Lesions of the medial and lateral canthal regions require careful attention because of the proximity of the lacrimal drainage system, the increased tendency to deep orbital extension, and the difficulty in reconstruction of these areas. Patients should be advised of the need for sun protection.
Squamous Cell Carcinoma
Squamous cell carcinoma occurs much more commonly on the conjunctiva, cornea, and mucus membranes than on the lid. Like squamous cell carcinoma elsewhere, lid lesions are much more aggressive than the more common basal cell carcinoma. They have a much greater potential for local extension and regional and widespread metastasis, although the overall incidence of metastasis is still low and the risk of death rare. Primary lesions of the lid are less likely to metastasize than those that arise in other locations. The tumor occurs most commonly in older patients. However, patients with underlying skin diseases such as xeroderma pigmentosum, discoid lupus, and lupus vulgaris are commonly affected at an earlier age. Actinic keratosis is the most common precursor lesion. However, the tumor may arise in other areas of chronic irritation and trauma, such as ulcers, scars, burns, previous irradiation sites, and keloids. While a lesion of the upper lid is more likely to be a squamous cell carcinoma than a basal cell carcinoma, the lower lid is still more commonly involved than the upper. But both cancers can occur anywhere along the adnexa. The squamous cell lesion tends to be more infiltrative and less nodular than basal cell carcinoma, with more keratinization and less distinct margins (Fig. 10.59). Lash loss is common if the lid margin is involved. The lesion grows less rapidly than keratoacanthoma with which it shares some pathologic features but it grows more rapidly than basal cell carcinoma. The potential for deep invasion, orbital extension, and metastasis is higher than for basal cell carcinoma. Squamous cell carcinoma must be included in the differential diagnosis of any form of nontender lid lesion.
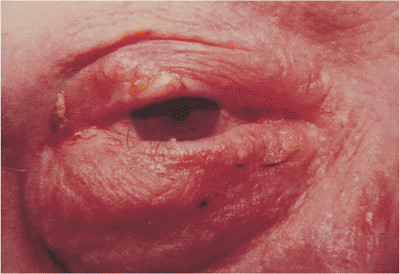 |
Figure 10.59. Squamous cell carcinoma. A firm mass of the upper lid with marked lash loss and destruction of the normal lid margin. Incisional biopsy showed squamous cell carcinoma, which was subsequently excised with clear frozen-section margins. |
Management
Incisional biopsy of any suspicious or recurrent lesion is diagnostic. After the diagnosis is made, wide, full-thickness excision with surgical margin control is recommended. Close follow-up is mandatory because of the risk for deep or orbital invasion and metastasis. Orbital exenteration may be required if orbital extension or deep perineural involvement is confirmed.
Sebaceous Cell Carcinoma
This highly malignant tumor may arise from any of the sebaceous components of the lid, including the meibomian glands and the glands of Zeis of the lashes, brow, and caruncle. Although it is an uncommon tumor, it is certainly not rare, accounting for more than 3% of eyelid malignancies. Some reports show it to be the second most common skin cancer of the lid. Like most epithelial tumors, it occurs primarily in older adults, but can also occur in younger patients, especially those who have received prior orbital radiotherapy. Sebaceous carcinomas of the ocular adnexa are more common and more aggressive than those arising elsewhere on the body. The mass may be subcutaneous and therefore be confused with chalazion, although sebaceous cell carcinoma tends to be firmer, yellow, and involves the lid margin more frequently than the tarsal surface, leading to lash loss (Fig. 10.60). The tumor often spreads superficially along the epithelium (i.e., pagetoid spread), with crusting, ulceration, and the appearance of chronic, unilateral blepharoconjunctivitis that is unresponsive to medical therapy (i.e., masquerade syndrome). The tumor is very often multicentric, with skip areas that may involve other areas of the lid, the conjunctiva, or the cornea before the tumor becomes clinically evident. These characteristics often lead to unfortunate delays in diagnosis and treatment.
P.391
Although the long-term survival rate has dramatically increased, the overall mortality rate of the disease still approaches 20%, with local recurrences common and lymphatic metastasis occurring in roughly 25% of patients. The lung and liver are often involved.
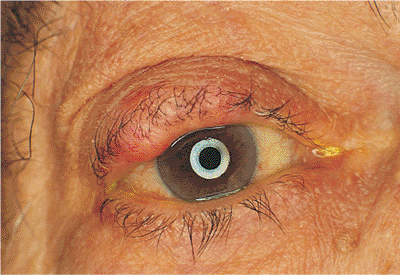 |
Figure 10.60. Sebaceous cell carcinoma. A large, firm, painless mass of the right upper lid. Although this lesion does not show the loss of lashes associated with carcinoma, the mass is still suspected of being malignant. As an irregular mass of the lid border with suggestions of a firm, nodular mass or as a diffusely indurated and thickened lid, the tumor typically grows slowly, with a minimal amount of epithelial involvement, but it may invade deep tissues and orbital structures early, requiring aggressive surgical excision. |
Management
The effective treatment of sebaceous cell carcinoma rests with early detection and wide excision, coupled with close follow-up for local, regional, and distant metastasis. A high index of suspicion is needed when evaluating a solid mass in the elderly, because a delay of even 6 months may prove fatal. Chalazia that recur after excision, any solid tissue tumor, or a mass that is in any way atypical, particularly if associated with lash loss, should be biopsied for examination. Wide excision with frozen-section control is required for the primary lesion. However, because of multicentric growth, negative frozen section margins are not definitive. Consequently, map biopsies should also be submitted from other areas of the lids, conjunctiva, and possibly the globe. Orbital exenteration may be required in cases of orbital or diffuse eyelid involvement. Regional lymph nodes should be palpated before surgery, and if enlarged, should be biopsied as well. The role of sentinel lymph node dissection in this tumor is not yet clear. Patients must be followed closely for the development of lymphatic metastases. Adjunctive radiotherapy may be of some benefit for diffuse disease.
Malignant Melanoma, Eccrine Sweat Gland Carcinoma, and Merkel Cell Carcinoma
Like malignant melanoma elsewhere, lesions arising on the lids are rare but potentially lethal, with a great tendency for widespread hematogenous and lymphatic spread. Although the tumor accounts for less than 1% of all skin tumors, it is responsible for more than 60% of all skin cancer deaths. The incidence of melanoma has increased dramatically over the last 2 decades. There may be a hereditary component. The tumor tends to arise in older individuals, is more common in the fair-skinned and less common in the darker-pigmented population, and may arise from preexisting nevi that have undergone malignant change. Although solar exposure is thought to increase the risk of melanoma, absence of actinic history is not protective against the tumor, particularly the superficial, spreading variety. Most melanomas appear as flat pigmented lesions of various colors, but they may be nodular and melanotic (Fig. 10.61). Prognosis is correlated to depth of invasion. Because of the thin skin of the lids, conventional staging schemes such as the Clark and Breslow systems are not accurate predictors of prognosis. Patients having one lesion are at risk of developing other lesions and require lifetime follow-up.
Several malignant tumors arise in the deeper subcutaneous tissues of the lid, without the epithelial surface changes that are characteristic of the more common skin cancers. Typical of these tumors, the uncommon incidence is inversely related to their ease of diagnosis and malignant potential. Benign tumors of eccrine sweat glands of the lids are rather common, including syringoma, hidrocystoma, spiradenoma, acrospiroma, and chondroid syringoma. Eccrine sweat gland carcinoma is unusual but often very highly malignant, with a potential for local recurrence, deep invasion, and regional or distant lymphatic and visceral metastasis. The tumor typically arises predominantly in middle-aged to elderly men as a discrete, firm, nodular mass or as a diffusely indurated and thickened lid (Fig. 10.62). The tumor typically grows slowly, with a minimal amount of epithelial involvement, but it may invade deep tissues and
P.392
orbital structures early, requiring aggressive surgical excision.
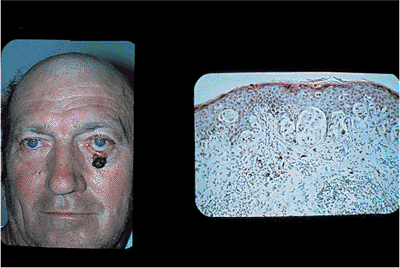 |
Figure 10.61. Malignant melanoma. This darkly pigmented lobular lesion with irregular borders was diagnosed on biopsy to be a malignant melanoma. Two thirds of the lower lid was resected and reconstruction was accomplished with a two-stage Hughes procedure including a full-thickness skin graft. Low magnification photomicrograph (right) showing junctional nests of tumor cells with extension into the dermis (hematoxylin and eosin 10). |
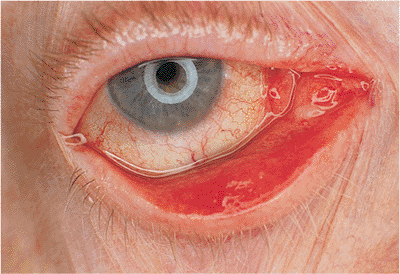 |
Figure 10.62. Eccrine sweat gland carcinoma. The patient was referred for evaluation of mechanical ectropion secondary to possible basal cell carcinoma. Inspection revealed the epithelium to be normal. However, a large, firm mass was palpable subcutaneously. It did not respect the tarsus and had deep extensions medially in the area of the medial canthus. Biopsy revealed sweat gland carcinoma, requiring eventual orbital exenteration. |
Merkel cells are neuroepithelial cells in the deep dermis that contribute to sensation. Their potential for malignant transformation is low, but when malignancy occurs, the tumor is exceptionally aggressive, with a capacity for lymphatic spread to regional and distant sites, particularly the liver and spleen. The tumor is potentially highly lethal, just as malignant melanoma. It is most common in older adults, and although an actinic history may be found, it is not a well established risk factor. Because of the rarity of the tumor, delay in diagnosis is common. As a rapidly growing, violaceous, painless mass, it is most commonly mistaken for chalazion. Unlike many other skin malignancies, alopecia is not a common finding. The mass is firm but not nontender and often shows inherent vascularity and telangiectasia (Fig. 10.63). Without a biopsy, the diagnosis is invariably missed, but even histologic analysis presents difficulty, because it is commonly confused with other small cell tumors such as lymphoma and metastatic oat cell lung cancer. Special neuroendocrine stains are needed for accurate diagnosis.
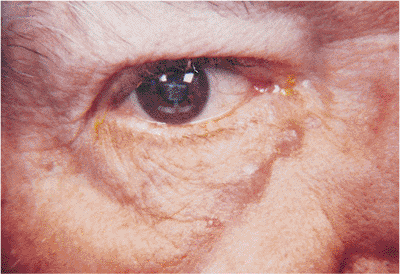 |
Figure 10.63. Merkel cell carcinoma. The lesion was initially treated as a chalazion. Subsequently, biopsy disclosed a Merkel cell carcinoma. This is an aggressive tumor with a high rate of metastasis. |
Management
Because of the very highly malignant and lethal potential, a high index of suspicion is necessary when evaluating pigmented or subcutaneous tumors. Pigmented tumors arising from longstanding skin lesions, particularly those demonstrating any change in color, size, or shape, should be considered for biopsy. Any subcutaneous mass, whether typical for chalazion or not, should also be regarded suspiciously, particularly if the lid shows any evidence of telangiectasia, if the mass is composed of solid tissue rather than typical lipogranulomatous material on biopsy, or if the mass recurs after initial excision. Aggressive, early treatment by wide resection is recommended. Unfortunately, because of the aggressive nature of these tumors, frozen section control is not definitive, though it is still recommended. The role of sentinel node dissection and biopsy in these tumors is being investigated, and may play a part in advancing survival. Adjunctive radiotherapy may be of benefit, and lifetime follow-up is required. The prognosis is guarded, both for the eye and for life.
Other Malignant Tumors of the Skin
Several malignancies are commonly found in the lid. These tumors tend to affect the deeper tissues of the lids rather than the epithelium and clinically appear as subcutaneous or deep nodules with variable mobility. Most masses are well circumscribed and painless, and they arise over the course of weeks to months, with gradual enlargement. They are often misdiagnosed as chalazion or epidermal inclusion cysts before biopsy.
Although metastases are more common to the orbit than the lid, the adnexal tissues can also be affected. The most common primary sites are breast, lung, colon, and prostate, although many other primary tumors have been known to metastasize. Although most patients have a known history of primary cancer, a lid mass may be the presenting symptom.
Hemangiopericytoma is a potentially malignant tumor that more commonly affects the orbit than the lid. It arises primarily in adults in the fourth and fifth decades, but it has been known to occur in younger and older patients. The tumor is most often benign, but it has a well-known potential for local recurrence, malignant transformation, widespread metastasis, and death.
Fibrous histiocytoma can arise anywhere on the body. In the ocular adnexa, it is most commonly found in the orbit, but it may arise within the lid tissues as well. The tumor's behavior in either site is similar; it varies from frankly benign, to locally aggressive, to frankly malignant. The tumor arises as a well-circumscribed, painless mass and may be confused with several neural tumors, including hemangiopericytoma,
P.393
neurilemmoma, and fibrosarcoma. The tumor may recur locally, usually as a result of incomplete excision, but it can metastasize with lethal results. Local extension of aggressive tumors is common.
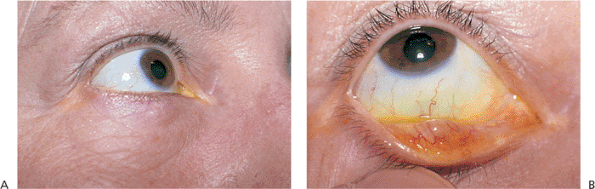 |
Figure 10.64. Chalazion. A: Painless mass of the tarsus, unresponsive to warm compresses. B: A conjunctival component is visible on lid eversion. |
Management
Because of the potential for oversight and misdiagnosis, any discrete lid mass unresponsive to conventional, conservative measures should be considered for excisional biopsy. Simple incision and curettage should be used cautiously, opting instead for pathologic examination of any suspicious lesion. Before excisional biopsy, a photograph or drawing of the lesion should be made to facilitate relocation of the mass if further excision is required. Masses should be excised completely, with adequate surrounding tissue removed to ensure clear margins.
Inflammations
Blepharitis, Chalazion, Hordeolum, and Molluscum Contagiosum
Blepharitis is a nonspecific inflammation of the eyelids, generally involving the lid margin and typically arising secondary to staphylococcal colonization. Seborrhea, acne rosacea, and Demodex folliculorum infection are additional etiologic agents. The classic findings include redness of the lid margin, collarette formation, scurf accumulation, irregularity of the lid margin, and inspissation of the meibomian gland orifices. Chalazion occurs if the meibomian gland duct is obstructed, with subsequent extrusion of the lipid into the adjacent soft tissues, stimulating lipogranulomatous inflammation and fibrosis (Fig. 10.64A). The lesion is localized to the tarsus and often is tender initially but resolves to become nontender. A conjunctival component is typically evident on lid eversion (Fig. 10.64B). Incision and drainage of the lesion produces a characteristic yellow gelatinous material (lipogranuloma) surrounded by dense fibrous tissue (Fig. 10.65). Hordeolum is an acute infection of any gland of the lid, e.g., meibomian, Zeis, or Moll (Fig. 10.66). Styes are painful, tender masses that arise abruptly and that may incite a surrounding cellulitis. Incision and drainage produces a purulent discharge. Molluscum contagiosum characteristically produces painless, pearly nodules with central umbilications (Fig. 10.67). An accompanying follicular conjunctivitis attests to the viral etiology.
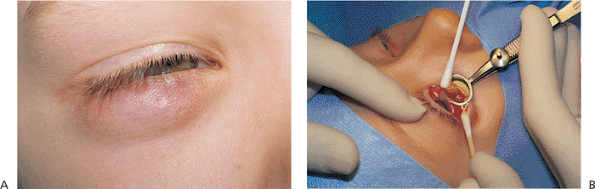 |
Figure 10.65. Chalazion. A: Large, painless mass that grew slowly over 4 months affected the entire lower lid of this 7-year-old boy. B: Incision and drainage revealed a lipogranulomatous material characteristic of chalazion. |
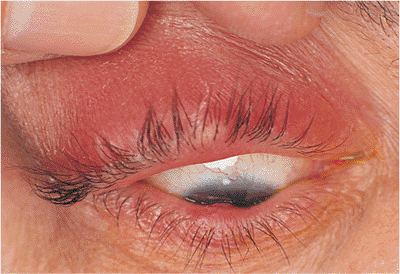 |
Figure 10.66. Hordeolum. A tender mass of the right upper lid is seen with surrounding cellulitis. A chalazion of the central lower lid is also present. |
P.394
Management
Because of the ubiquity of staphylococcal lid disease and the commonplace nature of blepharitis, typical chalazion and hordeolum pose little diagnostic or therapeutic challenge. However, clinicians should be suspicious of atypical characteristics that would warrant further evaluation. Asymmetry of blepharitis involvement or unilateral conjunctivitis especially if unresponsive to routine therapeutic measures should call to mind alternative diagnoses such as a masquerading condition like sebaceous cell or Merkel cell tumor, particularly if there is lid thickening, induration, and lash loss. Atypical locations of chalazia and hordeola, such as the base of the tarsus or exclusively at the lash margin especially if there is no identifiable internal component may prompt the clinician to send any incision and drainage specimens for biopsy, particularly if the chalazion has recurred after prior excision or if the characteristic lipogranulomatous debris is not expressed. Rare tumors with grave prognoses, such as sebaceous cell and Merkel cell carcinomas, are often initially misdiagnosed as chalazion.
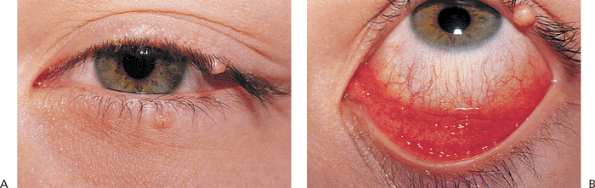 |
Figure 10.67. Molluscum contagiosum. A: A 13-year-old boy was referred for evaluation of lid masses of the left eye. Central umbilication of these pearly nodules is characteristic of molluscum contagiosum. B: Examination also revealed unilateral follicular conjunctivitis. |
Vitiligo, Poliosis, Madarosis, and Trichotillomania
Vitiligo is characterized by progressive, patchy loss of skin pigmentation. It is uncommon but can be associated with other autoimmune disorders, such as alopecia areata, thyroid dysfunction, Vogt-Koyanagi-Harada syndrome, and sympathetic ophthalmia. It has also occurred secondary to the use of topical medications, steroid injections, and as postinflammatory or posttraumatic sequela. Poliosis (progressive whitening of the lashes) and madarosis (loss of lashes) occur with regularity in patients with alopecia areata and dysthyroidism (Fig. 10.68A,B). Madarosis in the face of a lid mass should provoke a search for malignancy, such as squamous cell, sebaceous cell, or Merkel cell carcinoma. However, not all malignancies will cause lash loss.
Trichotillomania is the act of intentionally pulling out one's lashes. It may be the result of nervous tic, habit, or excess anxiety (Fig. 10.68C). When asked, most patients will admit to voluntarily pulling their lashes.
Management
Because of the strong association with other autoimmune disorders, patients with poliosis, madarosis, or vitiligo should undergo thorough ocular and medical evaluation, including thyroid function testing. Referral to specialists in retina-vitreous-uveitis, rheumatology, and dermatology may be appropriate. Alopecia areata is often treated with intralesional corticosteroid. However, involvement of the brows and eyelashes portend poor long-term prognosis for hair regrowth. Systemic steroids are rarely used.
Sarcoidosis
Sarcoidosis is a multisystem granulomatous disorder predominantly affecting young adults. Ocular involvement can be found in as many as 40% of persons of African descent and 20% of Caucasians with the condition. The most common
P.395
ocular finding is granulomatous anterior uveitis. Various skin and lid lesions can be found in approximately 30% of patients with chronic disease. Erythema nodosum, millet seed subcutaneous nodules, and confluent violaceous nodules of the midface (lupus pernio) are all seen. The most common skin findings are slightly raised, granulomatous plaques and nodules that are brown to yellow (Fig. 10.69).
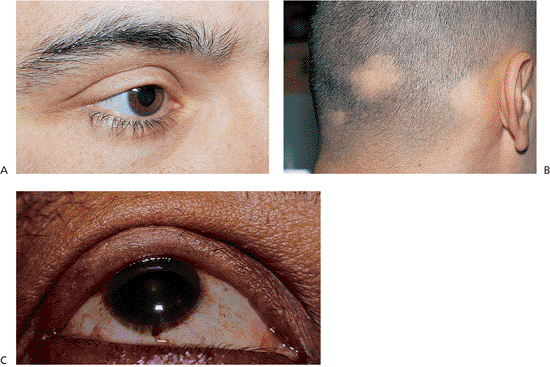 |
Figure 10.68. Madarosis, poliosis, alopecia areata, and trichotillomania. A: The patient complained of a loss of lashes from the right upper lid over 2 years. There was no history of intraocular inflammation. There was a loss of approximately 50% of lashes from the lid, with poliosis of the remaining lashes. B: The patient's scalp shows areas of patchy alopecia and whitening of the remaining hair. The patient had previously undergone treatments for alopecia areata with intralesional corticosteroid injections without success. Metabolic evaluation revealed undiagnosed primary hypothyroidism. C: This patient was asymptomatic, and admitted to intentionally plucking her eyelashes. |
Most patients with sarcoidosis present with intraocular or other orbital findings, such as a lacrimal gland mass. Occasionally, superficial manifestations may be the presenting problem. Ocular evaluation should include examination of the lids, lacrimal glands, and salivary glands. If the diagnosis has not been confirmed, biopsy of a superficial lesion can be performed quickly. Patients with known histories of sarcoidosis should be biopsied to rule out other processes and malignancies.
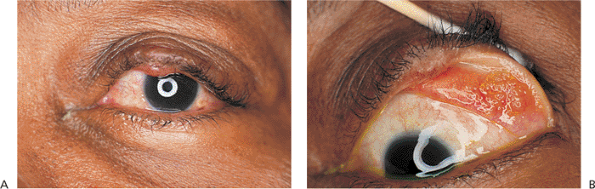 |
Figure 10.69. Sarcoid granuloma. A: A patient with known pulmonary sarcoid noticed irregular lid masses of both eyes that enlarged over 2 years. B: Eversion shows large, yellow granulomas of the tarsal conjunctiva. There were similar lesions on the right eye. The patient had no other ocular or orbital findings, except dry eyes. |
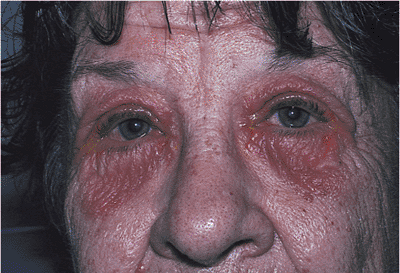 |
Figure 10.70. Allergic dermatitis. The patient had acute onset of redness, swelling, and itching of all lids. The patient had been using neomycin ophthalmic ointment. After discontinuing the ointment, she was treated with cool compresses and a mild topical steroid ointment, producing prompt resolution of her symptoms. |
P.396
Allergic and Atopic Dermatitis
A variety of agents can incite allergic dermatitis of the eyelids. Seasonal allergens and pollens, fumes, cosmetics, and medications can trigger allergic and toxic reactions. The condition is characterized by marked edema, erythema, intense itching, and excoriation of the lids (Fig. 10.70). The remainder of the skin, face, and hands may be unaffected. Atopic dermatitis also manifests with pruritus, swelling, and erythema, but the onset is earlier in childhood, and there is generally a family history of other atopic conditions such as hay fever, asthma, or atopic dermatitis. With repeated insults and chronic inflammation, the skin becomes thickened and excoriated, causing induration of all layers of the skin. Cicatricial changes can lead to lid retraction and ectropion.
Management
Avoidance and discontinuation of known aggravating factors is key in the management of both conditions. Antihistamines may help seasonal factors. Topical steroids may help control the inflammatory component. Surgery may be needed if cicatricial changes occur.
Lacrimal Drainage Dysfunction
Congenital Dacryostenosis, Dacryocystocele, and Dacryocystitis
Although as many as 73% of term stillborn fetuses have been shown to have lacrimal obstruction, only 6% of children have symptomatic congenital dacryostenosis. The obstruction most commonly occurs at the distal end of the tear duct, in the area of the valve of Hasner, but can occur anywhere along the lacrimal drainage system. Symptoms include epiphora and chronic crusting and mattering of the eyelids (Fig. 10.71). Pressure over the nasolacrimal sac may produce reflux of mucopurulent matter. The incidence of dacryostenosis is significantly higher in the presence of craniofacial abnormalities such as cleft lip and palate, and the clefting and synostotic syndromes. Congenital dacryocystocele presents at birth or shortly thereafter as a nontender, soft, bluish mass below the medial canthal tendon (Fig. 10.72). In addition to distal obstruction, proximal obstruction in the area of the common internal punctum prevents outward reflux and decompression, and pressure over the sac is therefore nonproductive. When aspirated or expressed, the contents are generally thick and mucoid, but they may be xanthochromic and viscous. The contents are most often sterile, but if bacteria are sequestered, the condition can develop into frank dacryocystitis. Infantile dacryocystitis is uncommon, but signs and symptoms are similar to those in adults, with painful, tender swelling of the inferior medial canthal area and surrounding cellulitis (Fig. 10.73).
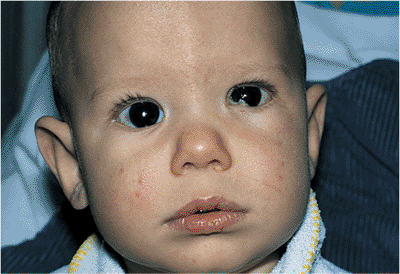 |
Figure 10.71. Congenital dacryostenosis. The child's parents complained of epiphora of both eyes since the child's birth. The right eye cleared spontaneously, but the left eye continued to tear, with morning crusting and heavy mattering. |
Management
Because epiphora and photophobia are often presenting symptoms of congenital glaucoma, patients should be completely examined to rule out this grave entity. After eliminating the possibility of glaucoma, the treatment of dacryostenosis includes warm compresses and antibiotic drops and ointments. Gentle downward massage over the nasolacrimal sac may be added. Most symptomatic obstructions clear spontaneously. If epiphora persists until the child
P.397
is 9 to 12 months of age, probing and irrigation are recommended. Some clinicians advocate early probing in the office, using light sedation. Additional measures include medialization of the inferior nasal turbinate, balloon catheter dilation of the duct, and silicone intubation under general anesthesia.
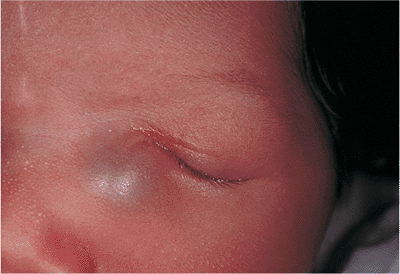 |
Figure 10.72. Congenital dacryocystocele. The painless, uninflamed bluish mass is located below the medial canthal tendon. |
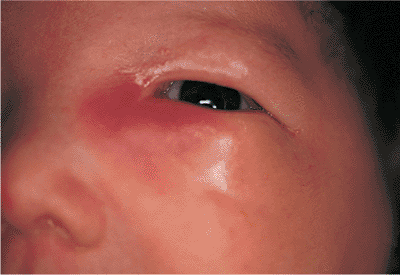 |
Figure 10.73. Infantile dacryocystitis is a painful, tender swelling of the inferior medial canthal area, with surrounding cellulitis. At birth, the child's parents had noticed a nontender, bluish mass in this area, which probably represents an underlying dacryocystocele that had become infected. |
Spontaneous resolution of dacryocystocele is uncommon. Early treatment with probing, irrigation, and decompression is recommended. Percutaneous aspiration provides temporary relief but does nothing to correct the causal obstructions. Dacryocystitis should be treated aggressively with topical and systemic antibiotics, with particular attention given to adequate coverage of Haemophilus influenzae. Hospitalization is usually required. Incision and drainage are sometimes necessary. After the infection is controlled, probing and irrigation should be performed promptly. Occasionally, dacryocystorhinostomy may be required for recurrent or anomalous cases.
Supernumerary Puncta, Lacrimal System Atresia, and Lacrimal Fistula
Congenital anomalies of the lacrimal system are uncommon. Supernumerary puncta are multiple openings into a single canaliculus. However, not all puncta are always patent. The punctal promontory may be poorly developed or atretic or may be occluded by imperforate membranes. Although imperforate puncta generally overlie otherwise intact drainage systems, severe punctal atresia often indicates significant underlying pathology, such as canalicular atresia (Fig. 10.74). This represents a complete failure of the canalicular system to develop and may be associated with other significant lacrimal drainage anomalies. Complete atresia of the lacrimal sac and duct is rare and represents agenesis or malformation of the distal aspect of the lacrimal anlage. This condition can be an associated finding in malformations of the midface.Congenital fistulae of the lacrimal sac are epithelial tracts that extend from the sac to the skin surface (Fig. 10.75). They may be unilateral or bilateral and may be completely patent or end as a blind sac. If the fistula is patent, tears or mucopurulent drainage can be found at the skin ostium, usually located just inferior and lateral to the inferior punctum.
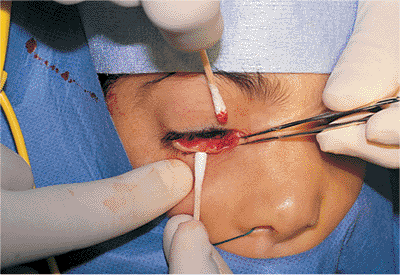 |
Figure 10.74. Punctal and canalicular atresia. A 3-year-old child with Nager's syndrome, a variation of Treacher Collins syndrome, presented with tearing of the right eye since birth. Examination with the patient under anesthesia revealed an absence of upper and lower puncta in the right eye. Cut-down of the lower lid revealed complete absence of the canaliculus. The nasolacrimal sac and duct were also found to be poorly developed. |
Management
As with congenital dacryostenosis, epiphora is the most common presenting complaint. If a fistula is present, parents may complain of tears or discharge on the skin or along the nose. The lacrimal drainage system should be inspected thoroughly in the office to identify atretic or imperforate structures. Atretic or imperforate puncta can be opened easily with a simple incision, after which the canaliculus is probed and irrigated to ensure distal patency. Atretic canaliculi require canalicular reconstruction
P.398
or conjunctivodacryocystorhinostomy. Unlike acquired lacrimal fistulae, which heal spontaneously, congenital lacrimal fistulae must be excised and closed surgically.
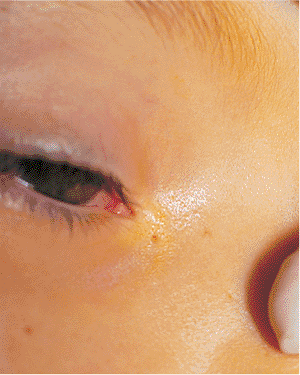 |
Figure 10.75. Congenital lacrimal fistula inferior to the medial canthus. There was a positive paternal family history of similar fistulas. The fistula did not connect to the sac and was excised completely. |
Acquired Dacryostenosis
Obstruction of the tear drainage system can occur at any age. Punctal and canalicular stenosis can develop secondary to chronic irritation and inflammation, autoimmune disorders such as Stevens-Johnson syndrome or pemphigoid, viral infections, or trauma, and as a sequela of topical and systemic medications such as antivirals and antineoplastics (Fig. 10.76). Canaliculitis, an acute infection of the canaliculi, can be caused by a host of organisms, but Actinomyces israelii is the most common pathogen (Fig. 10.77). Canalicular stones and abscesses can form, requiring incision and curettage removal. Acquired dacryostenosis (i.e., obstruction of the lacrimal sac and nasolacrimal duct) occurs most commonly as an idiopathic entity, although biopsy specimens may show chronic inflammatory changes consistent with chronic, indolent, or subacute dacryocystitis. Fungal hyphae may also form dacryoliths, stones, and casts of the sac. Midfacial trauma and prior sinus surgery can cause secondary compression and scarring of the duct. Occult tumors of the sac or sinuses are rare but must always be considered. Patients with dacryostenosis complain of epiphora, often of sufficient magnitude to require keeping a tissue (Fig. 10.78). Chronic mattering, morning crusting, and mucopurulent discharge with chronic blepharoconjunctivitis can also occur. Evaluation may require diagnostic imaging.
Patients complaining of epiphora should be evaluated for reflex hypersecretion (e.g., keratitis sicca) and underlying lid position abnormalities (e.g., entropion, ectropion). Particular attention should be given to the medial canthal drainage structures. Punctal stenosis may be treated with one- or two-snip punctoplasty. If associated with canalicular stenosis secondary to chronic inflammation or medications, canalicular reconstruction may be necessary, including management with warm compresses and topical antibiotics. Occasionally, canaliculotomy must be performed to remove stones or drain abscesses. Dacryostenosis is most commonly diagnosed by office probing and irrigation (Fig. 10.79A). The cannula should not be advanced beyond the lacrimal sac, because this area is difficult to anesthetize adequately. The area of the lacrimal sac should be palpated for masses. Imaging may be useful in cases of trauma or if underlying stones or tumors are suspected (Fig. 10.79B,C). Dacryocystorhinostomy can successfully treat acquired dacryostenosis.
 |
Figure 10.76. Punctal stenosis. An adult patient previously treated with 5-fluorouracil developed epiphora secondary to punctal and canalicular stenosis. The punctum had completely closed and was reopened with an incision. |
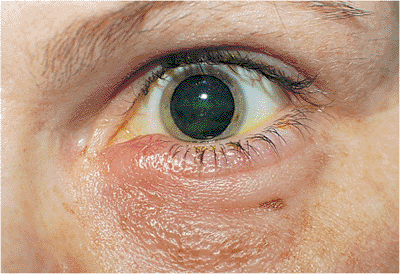 |
Figure 10.77. Canaliculitis. A 32-year-old woman had an acute onset of painful swelling of the inner canthal region. Inspection revealed painful induration and erythema of the punctum and canaliculus. |
Dacryocystitis
Acute dacryocystitis can occur at any age. However, it is more common as an acquired condition among middle-aged or older adults. A variety of conditions can lead to dacryostenosis, which predisposes to stagnation of tears and bacterial overgrowth, which can then fulminate into an acute infection. Midfacial trauma, sinus surgery, oral-maxillofacial surgery, dacryoliths, tumors, and chronic indolent dacryocystitis have been associated with acute dacryocystitis. Often, no underlying condition is identified. The patients typically have a history of epiphora or chronic blepharoconjunctivitis, with mucopurulent discharge and crusting (Fig. 10.80). A history of bloody tears or a mass above the medial canthus should heighten suspicion of a lacrimal sac tumor. Patients present with acute onset of a painful mass in the medial canthal area, which is swollen, tender, erythematous, and warm to the touch, with a surrounding cellulitis. Often, an abscess is located medially below the medial canthal tendon. With spontaneous drainage, a fistula may form to the sac (Fig. 10.81).
Management
Acute dacryocystitis requires aggressive treatment with intravenous and oral antibiotics. Abscesses, if not draining spontaneously, must be incised and drained.
P.399
P.400
Although most clinically significant dacryostenosis can be diagnosed on the basis of clinical history and simple irrigation, a computed tomography (CT) scan is often performed to identify tumors, bony erosion and landmarks, or stones. Irrigation should not be performed in the setting of acute dacryocystitis. Definitive therapy consists of dacryocystorhinostomy. Submission of tissue for pathologic examination is highly recommended to look for unsuspected pathology.
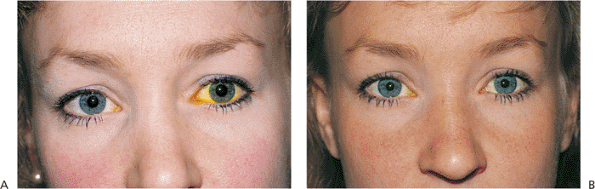 |
Figure 10.78. Acquired dacryostenosis. A: A 24-year-old patient presented with constant epiphora of the left eye which caused her to keep tissues on hand. The fluorescein dye disappearance test is markedly delayed. Irrigation revealed 100% obstruction of the nasolacrimal duct. B: Six months after dacryocystorhinostomy with silicone intubation, the patient is asymptomatic and has a normal result with the dye disappearance test. |
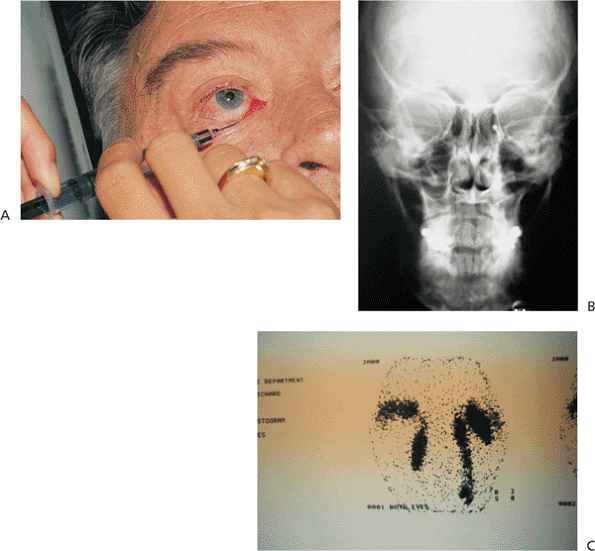 |
Figure 10.79. Diagnosis of dacryo-stenosis. A: Reflux on irrigation of the lacrimal system can indicate significant obstruction. Bloody reflux is highly suggestive of tumor. Palpation of the nasolacrimal sac can disclose tumor masses. B: Irrigation with contrast dye can disclose physical obstructions and irregularities of the lumen. In this plain film dacryocystography (DCG), a dacryolith is visible as an irregular outline in the left nasolacrimal system. DCG can also be performed with a computed tomography scan. C: With dacryoscintigraphy, the instillation of a radioisotope (technetium 99) can be useful in evaluating tear flow physiology and demonstrating occlusions. This scan reveals distal obstruction of the right duct. |
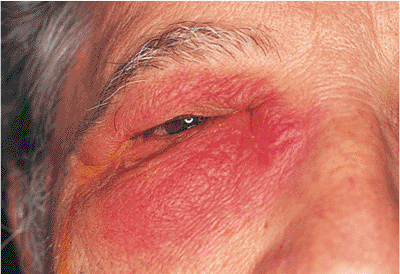 |
Figure 10.80. Acute dacryocystitis is evidenced by a large, tender, swollen, fluctuant erythematous mass arising in the medial canthal area. |
Lacrimal Sac Tumors
Tumors of the lacrimal sac are rare. Approximately 25% are inflammatory lesions, including granulomatous disease. The remainder are true neoplasms, most of which are malignancies of epithelial origin (e.g., squamous cell). Nonepithelial tumors (e.g., lymphoma, fibrous histiocytoma) have been described as well (Fig. 10.82). The clinical presentation varies, but the presence of bloody tears or a painless, noncompressible mass above the medial canthus is highly suspicious for tumor. Although benign lesions may have slower growth, with longer symptoms of epiphora or recurrent dacryocystitis than malignancies, such histories do not rule out malignancies. A history of pain, rapid growth, and bloody discharge suggests a tumor. A history of chronic sinusitis, sinus surgery, or midfacial trauma may indicate mucocele formation.
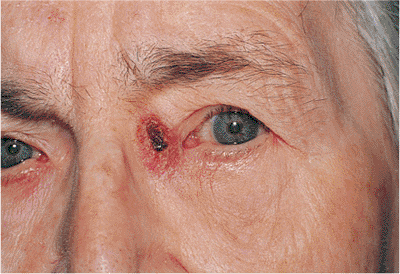 |
Figure 10.81. Acute dacryocystitis, the spontaneous drainage of a sac abscess, may lead to fistula formation. Unlike congenital fistulas of the sac, acquired fistulas generally close spontaneously. |
Palpation generally reveals a firm, irreducible mass in the area of the medial canthus. Irrigation may not be obstructed (Fig. 10.83). The CT scan may show a mass in the area of the lacrimal sac, bony erosion, local extension into the orbit, or diffuse infiltration, depending on the nature of the tumor. Many epithelial tumors (e.g., squamous, inverted squamous, transitional papillomas) may arise primarily or concurrently in the sinuses. Such tumors may be prone to recurrence. Tumors may extend into and protrude through the canaliculi (Fig. 10.84).
Management
Patients complaining of epiphora should be questioned closely about a history of bloody tears and prior sinus surgery or disease, as well as of systemic diseases such as Wegener's granulomatosis, sarcoid, and lymphoma. Office irrigation may reveal partial or complete obstruction and may produce bloody reflux. Palpation of the medial canthus may disclose a firm, irreducible mass. Inspection and palpation of regional lymph nodes are recommended. If
P.401
there is a strong suspicion of tumor or stone, radiologic imaging is recommended. CT is preferred to evaluate bony integrity. Dacryocystography can be performed concurrently. Surgical management depends on the history and CT findings and can include incisional biopsy, excisional biopsy, dacryocystectomy, or wide excision. Extensive surgery should be based on pathology. Combined procedures with an ear, nose, and throat surgeon may be necessary. Because of the small possibility of occult tumors of the sac, some surgeons routinely send biopsy specimens from routine dacryocystorhinostomies for histologic evaluation.
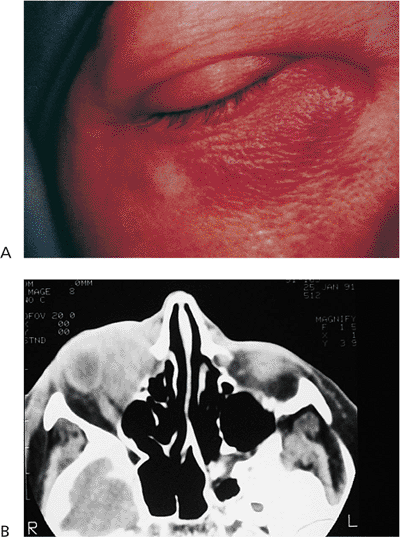 |
Figure 10.82. Lymphoma. A: The patient presented with acute dacryocystitis and a 4-month history of tearing. B: Computed tomography shows a diffuse, infiltrative mass conforming to the orbital walls without bony erosion. The globe is markedly indented. The area of the sac is also infiltrated. Biopsy confirmed the clinical suspicion of orbital lymphoma. |
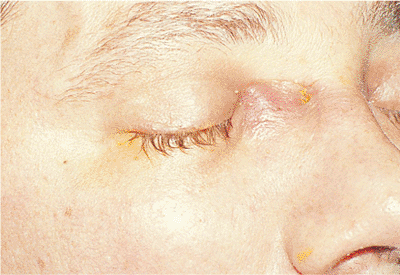 |
Figure 10.83. A firm mass above the medial canthus was of recent onset. Computed tomography showed underlying bony erosion and a mass in the area. Biopsy revealed squamous cell carcinoma. |
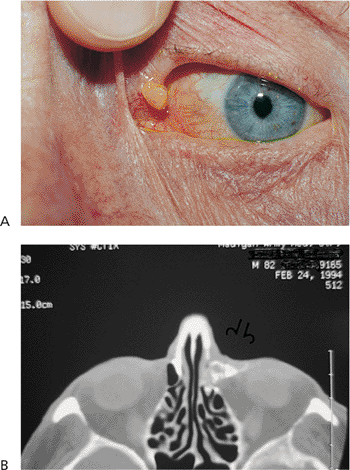 |
Figure 10.84. Squamous papilloma. A: The patient presented with epiphora of a 5-year duration and a papillomatous growth from the upper canaliculus. Biopsy revealed squamous papilloma. B: Computed tomography dacryocystogram shows involvement in the sac (arrow). |
Oculoplastic Trauma
Eyelid Trauma
Blunt Trauma
Blunt trauma or contusion to the eyelids is a nonpenetrating injury by an external force that does not break or lacerate the epidermis or dermis (Fig. 10.85).
Management
Patients deserve careful history taking and an examination, including slit lamp and dilated ophthalmoscopy, to rule out associated intraocular involvement. Care should be taken to ensure that the globe has not been ruptured. Plain radiographs and CT scans are often necessary to rule out orbital fractures. Ten-minute ice compresses every 2 hours can reduce posttraumatic edema if applied within the first 2 days of injury.
Penetrating Trauma
Lacerations Not Involving the Lid Margin
These lacerations penetrate the skin and deeper structures but do not involve the eyelid margin (Fig 10.86). They can be caused by blunt or sharp objects, including human fingernails or animal paws.
Management
A complete history and physical examination should be performed to rule out intraocular involvement. Surgical repair is performed, using an anatomic approach
P.402
to correct any associated levator muscle involvement. Imbedded foreign bodies should be removed, and necrotic tissue should be debrided. The use of 6-0 absorbable or nonabsorbable sutures, eversion of the wound edges, and early suture removal can reduce posttraumatic scarring.
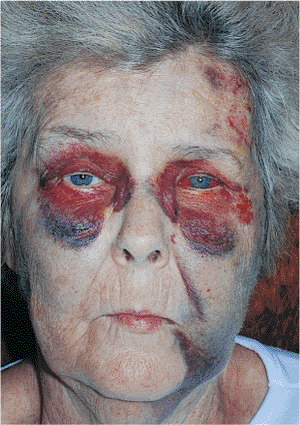 |
Figure 10.85. Ecchymosis and edema are the most common presenting signs of blunt trauma. |
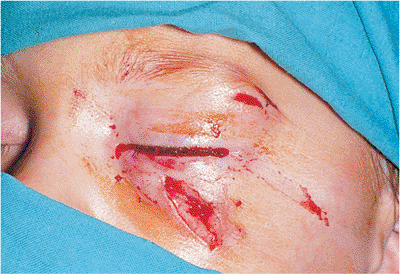 |
Figure 10.86. This 3-year-old boy was hit on the left side of his face by a dog's paw. The superficial lacerations were debrided, irrigated, and examined for foreign bodies. They were then closed in an anatomic fashion using 6-0 plain gut sutures. |
Lacerations Involving the Lid Margin
Full-thickness lacerations extend through the eyelid margin and may be regular or irregular (Fig. 10.87).
Management
The eyelid margin is closed using a three-suture technique. Suture no. 1 is placed through the tarsus of one wound edge and then through the tarsus of the other wound edge, avoiding the palpebral conjunctiva. The suture is secured and cut long. Suture no. 2 is passed in the eyelash line through both wound edges. It is secured and left long. With the eyelid held on stretch by pulling on the first two sutures, suture no. 3 (Fig. 10.88) is then passed through the gray line of one wound edge and then through the gray line of the other wound edge. This suture is then secured and cut long. Associated lacerations of the eyelid are then closed in a layered fashion. The tarsus is closed with 5-0 chromic or Vicryl sutures without passing through the conjunctiva. The skin and orbicularis are then closed with a 6-0 silk or nylon suture. The long ends of the three eyelid margin sutures are incorporated in the superior-most skin suture to prevent corneal irritation by the margin sutures.
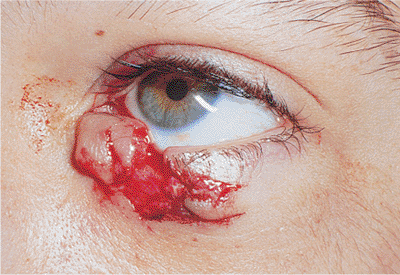 |
Figure 10.87. This 11-year-old boy fell on a shelf bracket, suffering an irregular, full-thickness lid laceration. The eye was not involved. |
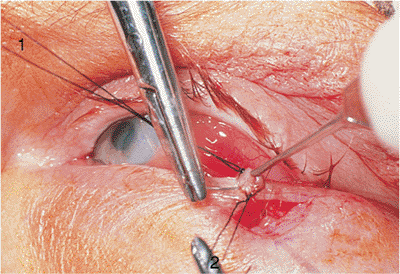 |
Figure 10.88. The tarsal suture (suture no. 1) and lash line (suture no. 2) have been placed. Suture no. 3 is being placed through the gray line. These three sutures together were then incorporated in the superior-most skin suture to prevent corneal irritation. |
Lacerations Involving the Medial Canthal Tendon
The superficial heads of the pretarsal orbicularis oculi muscle unite to form the medial canthal tendon. Often, transmarginal lacerations that involve the canaliculus also sever the superficial head of the medial canthal tendon.
Management
Lacerations in the medial canthal angle (Fig. 10.89) demand evaluation of the upper and lower
P.403
canaliculus, the nasolacrimal sac and duct, and the medial canthal tendon. Involvement is usually confirmed by inspection. The integrity of the inferior and superior limbs of the medial canthal tendon can be assessed by grasping each lid with forceps or fingers and tugging the lid laterally. Disinsertion can be palpated with the finger from the other hand or can be visualized by temporal displacement of the punctum beyond the nasal limbus.
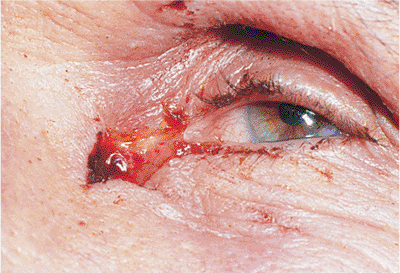 |
Figure 10.89. This 59-year-old man was hit in the left medial canthus with a meat hook. The skin, orbicularis oculi muscle, and anterior head of the inferior horn of the medial canthal tendon were severed. The canalicular system was intact. The inferior horn of the medial canthal tendon was reattached to its stump at the periosteum using 5-0 chromic sutures. The overlying orbicularis oculi muscle was closed with 5-0 chromic sutures. The skin was closed with 6-0 silk sutures. |
Treatment of medial canthal tendon evulsion depends on the nature of the evulsion. If the upper and lower anterior limbs are evulsed but the posterior attachment of the tendon is intact, the evulsed limbs may be sutured to its stump or to the periosteum overlying the anterior lacrimal crest with 5-0 chromic cat gut. The overlying skin is closed with interrupted 6-0 silk or nylon sutures. If the posterior limb of the medial canthal tendon is evulsed, then transnasal wiring may be necessary.
Lacerations of the Lateral Canthal Tendon
Isolated lacerations of the lateral canthal tendon are relatively rare and less complicated than those involving the medial canthal tendon. Although the medial canthal angle is slightly rounded, the lateral canthal angle is sharp. Recognizing this difference helps to achieve good postoperative cosmetic and functional results.
Management
The integrity of the inferior and superior limbs of the lateral canthal tendon can be assessed by grasping each lid and pulling it medially (Fig. 10.90). Disinsertion of the tendon can be seen by an almond-shaped deformity or rounding of the lateral canthal angle. The cut end of the lateral canthal tendon should be reattached to the lateral canthal tendon stump in the area of the lateral orbital tubercle. This is performed with 5-0 double-arm absorbable or nonabsorbable sutures. The lid margin is then closed using the three-suture technique previously described.
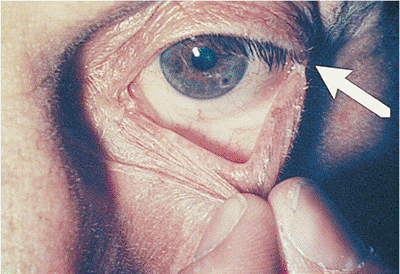 |
Figure 10.90. Disinsertion of the lateral canthal tendon is demonstrated by rounding of the lateral canthal angle as the lid is pulled medially. Medial canthal tendon laxity is also demonstrated by lateral displacement of the punctum tangent to the medial limbus. |
Burns
Patients who sustain burns of the eyelid are often critically ill or injured. Injury may be in the form of thermal (Fig. 10.91), chemical, or radiation burns. Protection of the eyeball to avoid corneal exposure, ulceration, and infection must be of primary consideration. Burns are caused by the application of heat to the body through direct contact or indirectly by radiant heat. The resultant burns vary only in degree, depending on the intensity and duration of application. Burns are classified as first, second, and third degree.
First-degree burns are characterized by erythema of the skin and only microscopic destruction of superficial layers of the epidermis. This is typical of a mild sunburn or when the head of a match strikes the eyelid skin.
Second-degree burns demonstrate greater tissue destruction in the epidermis and superficial layers of the adjacent dermis. Because regeneration occurs from remaining epithelial elements, second-degree burns are described as partial-thickness burns. These burns can be caused by boiling water or direct contact with a hot piece of metal or flame. This results in an erythematous, weeping, painful lesion associated with blisters and bullae. Superficial layers of the skin can be readily wiped away. The remaining skin appears waxy, white, and dry.
Third-degree burns are characterized by total, irreversible destruction of the epidermis and dermis. Because spontaneous regeneration of the epithelium is not possible, third-degree burns are known as full-thickness burns. Clinically, the skin appears dry, hard, and inelastic. These burns are caused by direct flame contact, by immersion in scalding water, or by direct chemical or electrical contact.
Thermal Burns
A common mild thermal burn is caused by the head of a match or cigarette ashes that strike the skin of the eyelids.
P.404
Although it is quickly cooled, it results in local destruction and coagulation of proteins, usually in the epidermis. The local pink or white discoloration usually resolves as the epidermis regenerates.
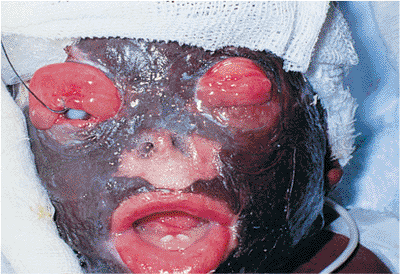 |
Figure 10.91. An 8-month-old boy, who was a victim of a domestic fire, received third-degree burns to his entire face, resulting in complete ectropion of both upper lids. Conservative treatment with moist chambers and lubrication was initiated while he was being stabilized medically. The patient died before definitive surgical repair could be initiated. |
More severe thermal burns (Fig. 10.91) are caused by prolonged contact with a high temperature heat source or fire. In some situations, total destruction is the final result. However, some portion of the eye or eyelids is usually salvaged.
Management
Severe burns of the eyelid are treated conservatively by keeping the exposed area free from contamination. If destruction of the tissues is so extensive that the cornea is exposed, immediate steps are taken to protect the eye. These include lubricating drops, antibiotic drops and ointments, moist chambers, and temporary tarsorrhaphies. After cicatricial changes occur in the eyelids, there is often a rapid deterioration of the patient's ocular status. With the progression of cicatrization, more aggressive treatment may be necessary. Permanent tarsorrhaphies should be more extensive than seem necessary. In the past, skin grafting was usually delayed until cicatricial changes stabilized. However, the use of full-thickness skin grafts from the opposite eyelid or split-thickness skin grafts from a hairless portion of the extremities may be necessary early in the course to reduce ocular morbidity.
Late treatment of thermal burns involves excision of the cicatrix with the use of split- or full-thickness skin grafts. The recipient lid should be placed on stretch to increase the area to be grafted. The use of tissue expanders may also be helpful.
Chemical Burns
The severity of chemical burns of the eyelids is determined by the substance, its concentration, and the duration of contact. Acid burns (Fig. 10.92) are usually self-limited and not as destructive as alkali burns (Fig. 10.93). Clinical signs of mild-to-moderate chemical burns include mild eyelid edema and hyperemia of the periocular skin. Ocular signs include corneal epithelial defects, focal areas of conjunctival chemosis, hyperemia, and a mild anterior chamber reaction. Evidence of more severe chemical burns includes pronounced chemosis, perilimbal blanching, corneal edema, moderate-to-severe anterior chamber reaction, increased intraocular pressure, and local necrotic retinopathy.
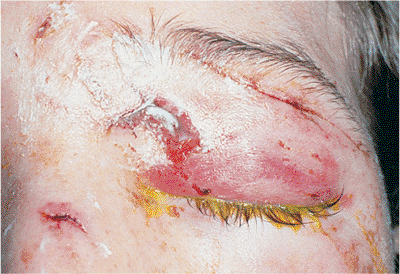 |
Figure 10.92. A 19-year-old car mechanic had a battery explode in his face. Mild hyperemia and edema of the eyelids are associated with areas of necrotic skin. Fortunately, the eye was not involved. The lids were irrigated, and necrotic tissue was debrided. He healed with minimal local scar formation. |
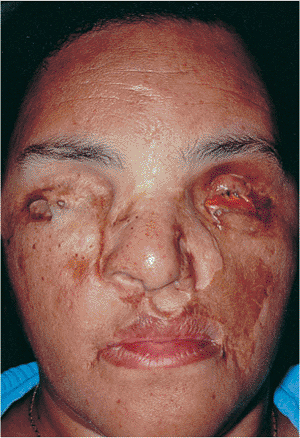 |
Figure 10.93. A 50-year-old woman had received lye burns to her face at 3 years of age. She lost function of both her eyes. Notice the residual ectropion of her left lower lid despite numerous skin grafts. |
Management
An accurate history is necessary to determine the time of injury, the nature of the chemical, and the duration of exposure before irrigation. Emergent treatment includes copious irrigation of the eyelids, preferably with saline or Ringer's lactated solution for at least 30 minutes. If neither of these is available, nonsterile water should be used. The lower and upper fornices should be irrigated if possible. Manual use of intravenous tubing connected to an irrigation solution facilitates the irrigation process. Five minutes after ceasing irrigation, litmus paper should be touched to the inferior fornix. If the pH is not neutral (pH = 7), irrigation should be continued. After irrigation, the fornices should be further examined, and any sequestered particles of caustic material and necrotic conjunctiva should be removed. Similarly, the periocular skin should be cleaned. Sequestered particles should be removed and necrotic skin debrided. Silvadene cream or similar antibiotics can be applied to the skin. Therapy for uveitis (e.g., 0.25% scopolamine), along with mild topical antibiotic ointments (e.g., erythromycin) and oral hypotensive medication (e.g., acetazolamide) and oral pain medication are used as needed. Management of more severe ocular burns may require hospitalization for close monitoring of intraocular pressure and corneal healing. Lysis of early conjunctival adhesions using
P.405
a glass rod may be necessary. If symblepharon forms despite attempted lysis, the use of a scleral shell, therapeutic contact lens, or ring conformer may be necessary.
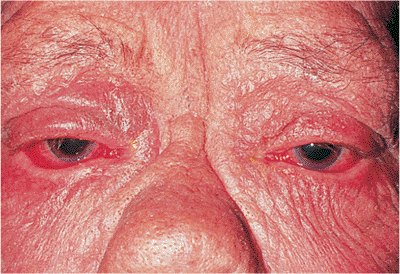 |
Figure 10.94. A 70-year-old man underwent 4 weeks of radiotherapy for a malignant ethmoid tumor. The point source of radiation was located over the bridge of his nose. Notice the local erythema, dry scaly skin, and bilateral lower lid ectropion. |
Late surgical treatment of chemical burns involves various procedures. A mucous membrane graft or conjunctival graft may be necessary to reconstruct the fornices and repair symblepharon. Full-thickness or split-thickness skin grafting may be necessary to correct eyelid ectropion resulting from loss of the anterior lamella of the eyelids.
Radiation Burns
Unlike thermal burns, the burns from an ionizing radiation source (Fig. 10.94) do not initially appear to be severe. If the lids are exposed to high does of radiation, erythema is the only early sign. Later, the skin may atrophy or undergo chronic ulceration. Massive doses cause areas of necrosis. Fortunately, the tarsus is resistant to this type of damage, but it may become distorted by superficial cicatrization. Corneal compromise or cataracts may develop months or years after irradiation. Basal cell carcinoma or squamous cell carcinoma may develop in areas of intense irradiation. This presents an unfavorable situation for surgical revision and repair.
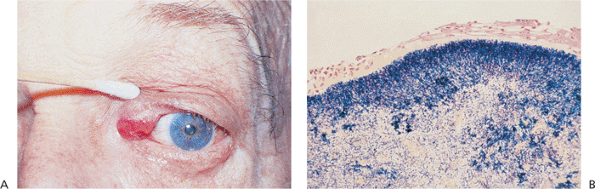 |
Figure 10.95. A: Canaliculitis with local edema and erythema of the left upper canaliculus. B: High-power, hematoxylin-and-eosin preparation of a canalicular stone shows filamentous, gram-positive rods. |
Management
Early management is similar to that of thermal burns with respect to protection of the eye. Mild radiation burns of the skin can be treated conservatively with mild antibiotic ointments, digital massage, and topical antibiotic ointments, if necessary. Delayed surgical treatment for ectropion and cicatrization follow the same principles of lid reconstruction discussed for thermal burns.
Lacrimal System Trauma
Canalicular trauma can be in the form of burns, toxic medication, autoimmune disease, infections, or mechanical injury. Chemical, thermal, or radiation burns can damage the lacrimal system. Toxic medications, including antiviral agents (e.g., adenine arabinoside), antineoplastic agents (5-fluorouracil), cholinergic drugs (e.g., pilocarpine), anticholinergic drugs (e.g., eserine, phospholine iodide), and adrenergic drugs (e.g., epinephrine), can cause punctal or canalicular stenosis. Autoimmune diseases, including ocular pemphigoid and Stevens-Johnson syndrome, can cause punctal occlusion. Fungal, viral, bacterial, and chlamydial infections can cause canaliculitis. Mechanical injury includes repeat canalicular and nasolacrimal probings, ocular or adnexal surgery involving the canaliculus, and canalicular lacerations.
Canaliculitis
The patient with canaliculitis presents with epiphora and localized pain over the canaliculus. There is an associated fish mouth or dilated punctum (Fig. 10.95A). The most common cause of canaliculitis is A. israelii (Streptothrix). This bacterium is a filamentous, gram-positive rod (Fig. 10.95B).
Management
Medical treatment consists of warm compresses; expression of concretions, which may have a cottage cheese texture; and topical antibiotics such as penicillin
P.406
(100,000 U/mL, one drop qid for 2 weeks). Sulfonamides or erythromycin are acceptable alternatives. Chronic canaliculitis requires curettage alone or in combination with canaliculotomy (Fig. 10.96) to remove canaliculoliths. Dacryocystorhinostomy may be required.
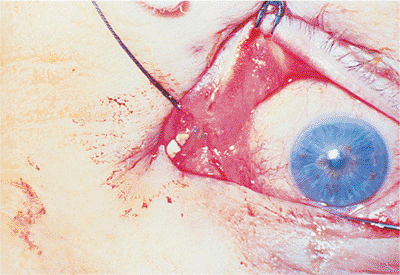 |
Figure 10.96. Canaliculotomy with canalicular stones. |
Canalicular Lacerations
Periocular trauma may involve one canaliculus (i.e., single canalicular laceration) or the upper and the lower canaliculi (i.e., bicanalicular lacerations). The diagnosis is made by careful inspection, with diagnostic probing and irrigation when necessary. Primary repair is desired within 24 to 48 hours of injury.
Management
The choice of general or local anesthesia depends on the age of the patient, associated ocular trauma, lid lacerations, medial canthal tendon avulsions, and nasolacrimal fractures (Fig. 10.97). General anesthesia is preferred for children, for anxious adults, and for placement of a bicanalicular stent. Identification of the severed ends of the canaliculi is facilitated by using the operating microscope.
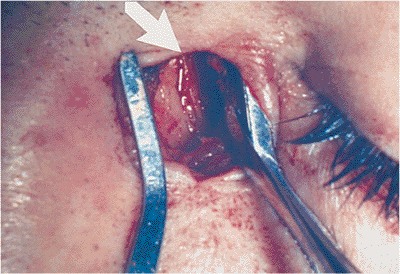 |
Figure 10.97. An 18-year-old boy had persistent epiphora 3 months after the repair of a canalicular laceration. Lacrimal irrigation confirmed a nasolacrimal duct obstruction. A fracture of the nasolacrimal bone was confirmed by a computed tomography scan. A fracture is seen through the left nasolacrimal bone during the dacryocystorhinostomy. |
Anastomosis of the cut ends of the canaliculi, with two or three preplaced 9-0 nylon sutures and two or three preplaced 5-0 chromic paracanalicular sutures, is performed over a stent (Fig. 10.98A). The chromic sutures are secured before the nylon sutures to reduce tension on the wound. Numerous stent materials exist, including Veirs rods, Johnson rods, or silicone tubing. Silicone is preferred because it is soft, flexible, and comfortable. It is less likely to cause corneal irritation and may be left in place for 6 months or longer, if necessary.
Monocanalicular stents are preferred for monocanalicular lacerations, avoiding damage to the normal canaliculus. One end of the silicone tube is allowed to coil in the nasolacrimal sac while the other end is passed through the punctum and secured to the eyelid (Fig. 10.98B). Newer monocanalicular tubes have attached punctal plugs to allow stabilization of the tube in the punctum (Figs. 10.98C,D).
Bicanalicular intubation is necessary for repair of bicanalicular lacerations. The loose ends can be tied under the inferior turbinate in the nose. Alternatively, a Beyer pigtail probe can be used cautiously to pass the loose ends of the silicone tubing into the palpebral fissures; the ends are then tied near the caruncle. Associated medial canthal tendon lacerations, skin lacerations, or lid margin lacerations are then closed appropriately.
Orbital Trauma
Retrobulbar Hemorrhage
A retrobulbar hemorrhage (Fig. 10.99) is a hemorrhage into the orbital cavity, anteriorly bound by the orbital septum and canthal tendons. Limited increases in orbital volume result in anterior displacement of the septum and the globe. A retrobulbar hemorrhage also can result in increased intraorbital and intraocular pressure. The most common causes of retrobulbar hemorrhage are blunt trauma and penetrating injury by the entry of foreign bodies. Retrobulbar hemorrhage from surgery of the eyelids and orbit has occurred after retrobulbar injection, blepharoplasty, dacryocystorhinostomy, palpebral dacryoadenectomy, repair of orbital and facial fractures, and sinus surgery.
Idiopathic orbital hemorrhages are less common. Spontaneous retrobulbar hemorrhages have occurred with orbital lymphangiomas, cavernous hemangiomas, and aneurysms of the ophthalmic artery. Blood disorders associated with spontaneous orbital hemorrhage include hemophilia, leukemia, vitamin K deficiency, anemia, hypertension, scurvy, and von Willebrand's disease.
Clinical Features
Sudden orbital and ocular pain and decreased vision associated with a recent history of trauma to the eye or orbit are frequent complaints. Lid ecchymosis, chemosis, proptosis with resistance to retropulsion, diffuse subconjunctival hemorrhage with nonvisible sclera posterior to the subconjunctival hemorrhage, limited motility in any or all fields of gaze, and elevated intraocular pressure are
P.407
clinical signs. Progressive deterioration of vision and the visual fields, dyschromatopsia, and an afferent pupillary defect signify optic nerve dysfunction. Disc edema and choroidal folds may be seen.
 |
Figure 10.98. A: Preplaced 9-0 nylon canalicular sutures and preplaced 5-0 chromic paracanalicular sutures are shown. The Silastic intubation tube is coiled in the nasolacrimal sac. B: The completed monocanalicular repair. The Silastic tube exiting from the punctum is secured to the eyelid with a full-thickness lid suture on a bolster. C: Monocanalicular intubation tube with attached punctal plug (mini-minoka tube) is seen in right upper punctum of this 11-year-old boy who is 6 months status post repair of canalicular laceration. D: The mini-minoka tube is easily removed from the patient in Figure 10.98C with jeweler's forceps. |
 |
Figure 10.99. A 59-year-old man was hit in the left eye with a wrench and suffered a retrobulbar hemorrhage. His decreased vision, increased intraocular pressure, lid edema, ecchymosis, proptosis, and a subconjunctival hemorrhage improved with conservative treatment. |
The differential diagnosis includes orbital cellulitis; orbital fractures; ruptured globe; subperiosteal hemorrhage; and optic nerve sheath hemorrhage.
Management
A complete ophthalmic examination should check for signs of threatened vision, such as decreased visual acuity, an afferent pupillary defect, loss of color vision, elevated or decreased intraocular pressure, central artery pulsations, and choroidal folds.
Treatment of retrobulbar hemorrhages is reserved for patients with visual loss or marked increases in intraocular pressure. If vision is threatened, CT scans of the orbit, including axial and coronal views, should be delayed until initial treatment has been instituted. Initial measures for the treatment of increased intraocular pressure associated with threatened vision include carbonic anhydrase inhibitors (500 mg of acetazolamide, taken orally), topical -blockers (0.5% timolol, every 30 minutes for 2 doses), hyperosmotic agents (20% mannitol, 1 to 2 g/kg, administered intravenously for 45 minutes), and a lateral canthotomy with superior and inferior cantholysis (Fig. 10.100). Patients with mildly elevated intraocular pressure or mild proptosis who do not have visual deficits may be observed, because hemorrhages
P.408
usually resolve in several weeks. If vision remains threatened after the treatment, emergent orbital decompression may be required if the optic nerve becomes compromised. The retrobulbar hemorrhage may be drained directly through a lateral or medial orbitotomy, depending on the location of the hemorrhage. A subperiosteal hemorrhage (Fig. 10.101) is drained after the periosteum is elevated with a Freer elevator and the clot evacuated. Bone removal in either situation is usually unnecessary.
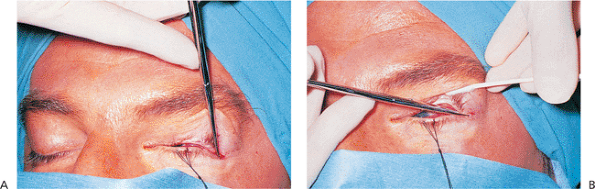 |
Figure 10.100. A: Lateral canthotomy. After a hemostat is placed horizontally over the lateral canthus for 1 minute, it is released, and sterile scissors are used to make a horizontal incision 1 cm toward the lateral orbital rim. B: Cantholysis of the inferior arm of the lateral canthal tendon. The scissors are redirected from 45 degrees to 90 degrees, and the skin and conjunctiva of the initial incision are separated so the scissors can be placed with the posterior blade anterior to the conjunctiva and the anterior blade posterior to the skin, engaging only the inferior arm of the lateral canthal tendon. Release of tension is felt when the inferior arm of the lateral canthal tendon is cut. |
Patients who fail to respond to these measures may require a two-wall orbital decompression to remove the orbital floor and medial wall. Visual loss secondary to hemorrhage within the intraorbital portion of the optic nerve sheath should be treated with optic nerve sheath decompression.
Pulse therapy with corticosteroid therapy is often used concomitantly. A regimen of 250 mg of methylprednisolone, administered intravenously every 6 hours for 12 doses, is followed by 80 mg taken orally daily for 1 week.
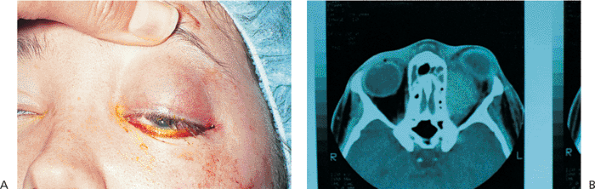 |
Figure 10.101. A: This 19-year-old woman suddenly developed proptosis and no light perception in her left eye after 2 weeks of preseptal cellulitis and ethmoid sinusitis. B: Computed tomography demonstrates a large subperiosteal hemorrhage along the medial orbital wall, compressing the optic nerve and distorting the globe. Blood was surgically drained through a supranasal orbitotomy. Six weeks after surgery, her vision returned to 20/25. |
Intraorbital Foreign Bodies
Intraorbital foreign bodies are composed of inorganic, organic, or mixed materials. Inorganic foreign bodies include stone, plastic, glass, iron, lead, steel, aluminum, and most other metals (Fig. 10.102). These are usually well tolerated in the absence of infection. Pure copper foreign bodies may create a noninfectious, suppurative response requiring removal. Copper alloys with less than 85% copper (i.e., brass, bronze) are fairly well tolerated and usually do not generate such a response. Organic material such as wood and vegetable matter typically produce an acute suppurative infection, subacute infection, or a delayed granulomatous reaction,
P.409
mimicking an expanding orbital mass. Intraorbital foreign bodies should be removed if they are composed of vegetable matter, if they are anterior in the orbit, or if they have sharp edges. Intraorbital foreign bodies may be safely observed without surgery if they are inert, have smooth edges, and are located in the posterior orbit and not compromising the optic nerve.
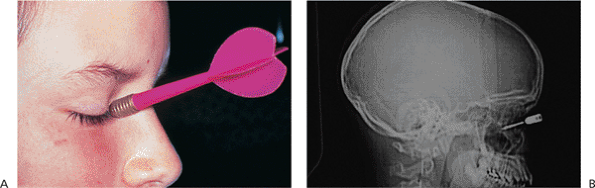 |
Figure 10.102. A: This 12-year-old boy was struck in the right orbit with a dart. The entry site on the right lower lid was inferior to the canaliculus. The results of his eye examination were normal. B: Sagittal computed tomography scan of the patient. |
Clinical Features
Symptoms vary from none to decreased vision, pain, lid edema, and diplopia. There may be no signs of an inert orbital foreign body, or there may be a palpable orbital mass, limitation of ocular motility, proptosis, edema, ecchymosis of the eyelids or conjunctiva, and the presence of an entry site (Fig. 10.103A). An afferent pupillary defect may be present in the case of traumatic optic neuropathy.
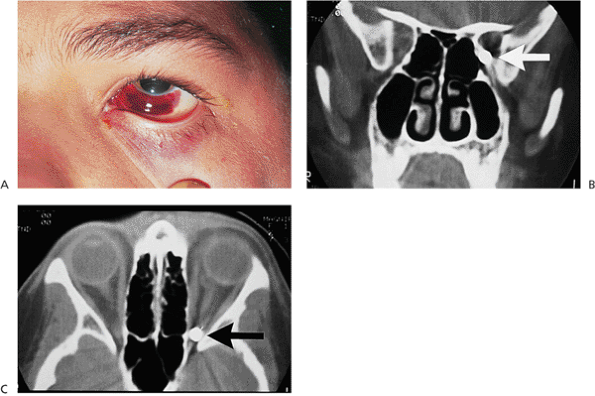 |
Figure 10.103. A: This 10-year-old boy suffered a BB injury to the left eye and orbit. The entry site is below the inferior canaliculus and extends through the caruncle. B: A coronal computed tomography (CT) scan shows the BB in the left orbital apex, inferomedial to the optic nerve. C: An axial CT scan shows the intraorbital BB at the orbital apex, inferior to the optic nerve. This patient was managed conservatively. His visual acuity, optic nerve, color vision, and pupillary reflexes have remained stable 4 years after the trauma. |
Management
A detailed history should determine the composition of the foreign body, the circumstances of the injury, the period since the original injury, and the degree of symptoms. Ocular injury, optic nerve injury, and CNS involvement must be ruled out.
CT scanning is the most useful modality for imaging metallic intraorbital foreign bodies. Magnetic resonance
P.410
imaging (MRI) is useful for imaging inorganic foreign bodies and glass without lead. Axial and coronal views (Fig. 10.103B,C) should be ordered for foreign body localization and to rule out a ruptured globe, optic nerve, CNS involvement, or associated sinus involvement. MRI is contraindicated if a metallic foreign body is suspected. A-scan ultrasonography is helpful for localizing intraocular foreign bodies, but it is less helpful in detecting or localizing small intraorbital foreign bodies.
Treatment of intraorbital foreign bodies involves culturing the wound or object from which the foreign body arose and tetanus prophylaxis. If the patient is febrile, hospitalization and intravenous antibiotics must be considered. A perforation or a ruptured globe should be repaired. Secondary enucleation is indicated if there is no useful vision or a risk of sympathetic ophthalmia exists. Organic foreign bodies (Fig. 10.104A,B) should be removed in toto. The risk of residual wood foreign bodies exists, and additional surgery may be required (Fig. 10.104C,D). Surgical removal along the entry tract is most desirable. The use of a small-diameter endoscope may improve visualization. A close working relationship with an otolaryngologist, neurosurgeon, and neuroradiologist is necessary when adjacent structures are involved.
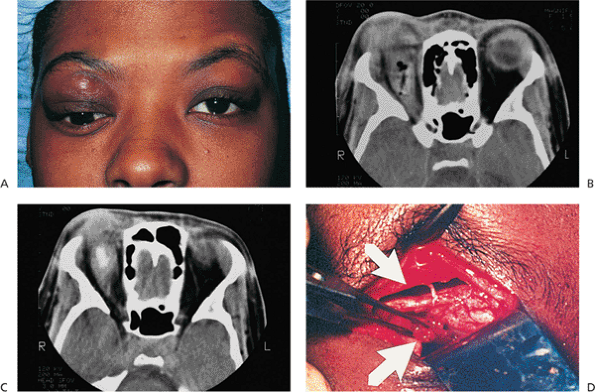 |
Figure 10.104. A: This 29-year-old woman was hit with a tree branch in the right orbit 8 months before this photograph. At that time, her right orbit was explored, and a small wooden foreign body was removed. She was lost to follow-up but returned 8 months later with an inferiorly displaced right globe, proptosis, and a tense right upper lid, as seen here. B: Computed tomography (CT) at the time of initial injury shows a soft tissue density and air in the superior aspect of the right orbit. No obvious foreign body is seen. C: The repeat CT scan of the patient 8 months later shows fibrous encapsulation of a residual piece of wood in the superior aspect of the right orbit. D: A superior orbitotomy was performed. The wooden foreign body was retrieved from the peripheral surgical space (bottom arrow). The subperiosteal space was void of any foreign body (top arrow). |
Fractures
Orbital Floor Fractures
Orbital floor fractures are classified as direct fractures or indirect fractures. Direct fractures (Fig. 10.105) are posterior extensions of inferior orbital rim fractures. Direct fractures are caused by a compressive force at the inferior rim that leads to buckling of the orbital floor. Indirect fractures (Fig. 10.106) of the orbital floor are not associated with an inferior orbital rim fracture, and are commonly known as blow-out fractures. The term blow-out fracture was developed in 1957 by Smith and Regan when they proposed that indirect fractures resulted from a sudden increase in intraorbital
P.411
pressure caused by the force created by an object that was greater than the diameter of the orbital entrance. The orbital contents are compressed, and the intraorbital bones break at their weakest points, the medial wall (0.25 mm thick) and the posterior medial aspect of the orbital floor (0.50 mm thick). The degree of increased intraorbital pressure determines whether or not orbital tissue herniates through these fractures to become entrapped. Most isolated medial wall fractures are asymptomatic unless the patient has orbital or eyelid emphysema.
 |
Figure 10.105. A direct fracture of the orbital floor extends from the infraorbital rim. |
Clinical Features
Patients with a recent history of orbital trauma, commonly with objects larger than the orbital entrance such as a fist, a soft ball, or a dash board injury, may complain of pain on attempted vertical eye movements, local point tenderness at the infraorbital rim, binocular diplopia, or lid swelling, particularly after nose blowing. Signs include ecchymosis and edema of the eyelids, limited vertical movement of the globe, orbital emphysema, subcutaneous emphysema, or subconjunctival emphysema (Fig. 10.107), hypesthesia in the distribution of the ipsilateral infraorbital nerve, enophthalmos, and globe ptosis.
Management
A thorough ophthalmologic examination should include visual acuity, pupillary reactions, adnexal evaluation, orbital palpation, extraocular motility (Fig. 10.108A), slit lamp examination, ophthalmoscopy, and a traction or forced duction test. A neurologic evaluation is indicated if there is a history of loss of consciousness. Severe intraocular damage, such as a ruptured globe, is rarely associated with blow-out fractures. Traumatic iritis, hyphema, and retinal edema are more common associated findings. Loss of vision may result from an associated injury to the optic nerve.
 |
Figure 10.106. Indirect or blow-out fracture of the orbital floor. The entrapped orbital contents have been released. |
 |
Figure 10.107. Subconjunctival emphysema in a patient with a medial orbital wall fracture who blew his nose. |
Orbital floor fractures are radiographically well demonstrated by CT scans of the orbit with coronal (Fig. 10.108B) and sagittal views. They detail the extent of the fracture and associated prolapse of orbital contents. Although plain films of the orbit, specifically a Waters' view, are useful as a screening test, CT scans more accurately demonstrate the fracture size and involvement of the extraocular muscles, which help the surgeon with management decisions.
Small floor fractures with no significant entrapment of orbital tissues may be managed conservatively. This includes nasal decongestants (e.g., Afrin nasal spray, twice daily) for 10 to 14 days, broad-spectrum oral antibiotics (e.g., 250 to 500 mg of cephalexin, taken orally four times daily or 250 to 500 mg of erythromycin, taken orally four times daily) for 10 to 14 days, avoidance of nose blowing, and application of ice compresses to the orbit every 2 hours for the first 48 hours. Some surgeons use prednisone (60 to 80 mg, taken daily for 5 days), beginning during the first 48 hours after injury. Diplopia caused by edema of the muscles resolves more quickly with steroids, but diplopia associated with muscle entrapment does not improve. The use of steroids may allow the surgeon to make an earlier decision concerning the need for surgery. Enophthalmos that was not evident initially may become more apparent as orbital edema subsides.
Surgical indications are controversial but include entrapment of the orbital contents, causing diplopia within 30 degrees of the primary position, confirmed by a positive forced duction test and radiologic evidence of entrapment; cosmetically unacceptable enophthalmos; and fractures involving
P.412
one half of the orbital floor particularly when associated with large medial wall fractures (Fig. 10.109), which may lead to subsequent fibrosis and contracture of the prolapsed tissue.
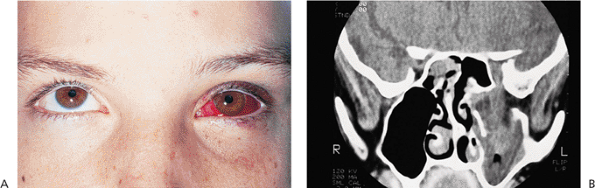 |
Figure 10.108. A: A 17-year-old boy with limited upgaze of the left eye 10 days after being hit with a lacrosse stick. B: The computed tomography scan shows a large orbital floor fracture with a free-floating bone fragment in the maxillary sinus. |
Surgical repair of blow-out fractures is usually delayed 10 to 14 days, a period that permits subsidence of orbital edema and reevaluation of the clinical findings. Commonly, a transconjunctival (i.e., inferior fornix) incision is combined with a lateral cantholysis, or an infraciliary incision is combined with a skin-muscle flap to expose the infraorbital rim. After prolapsed soft tissue is released from the fracture site, a synthetic or homoplastic orbital floor implant is usually placed.
Potential complications of fracture surgery include persistent diplopia, infraorbital nerve dysfunction, enophthalmos, exophthalmos, eyelid malposition, infection, implant extrusion (Fig. 10.110), and, rarely, visual loss.
Tripod Fractures
A tripod fracture is a fracture involving the zygomatic bone. As the name implies, it classically involves fractures at three sites: the zygomaticofrontal suture, the zygomaticomaxillary suture, and the zygomatic arch. With displaced zygomatic fractures, there is usually a fourth or fifth fracture site: the orbital floor or the anterior wall of the maxillary sinus. The name tripod may be a misnomer. The zygomatic bone is responsible for the prominence of the cheek and is an important feature in the facial skeleton. It helps to protect orbital contents from traumatic forces. The lateral canthal tendon and Lockwood's ligament are attached to the zygomatic bone at the lateral orbital tubercle. Patients with fractures and displacement of the zygomatic bone may present with cosmetic deformities involving malar flattening, inferior displacement of the lateral canthal angle, and globe ptosis. The anatomic relations of the zygomatic bone (Fig. 10.111), the zygomatic arch, the mandible, and the temporalis and masseter muscles govern normal movement of the mandible. Tripod fractures can disturb these structural associations and interfere with normal mastication.
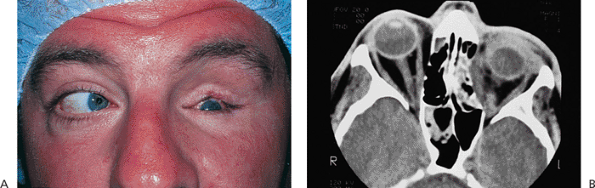 |
Figure 10.109. A: This 35-year-old man was kicked in the left orbit. He has ptosis of his left upper eyelid, 4 mm of left globe ptosis, and limited abduction of the left eye. B: The computed tomography scan shows a large medial wall fracture that has entrapped the medial rectus muscle. |
Tripod fractures are classified according to the type of displacement of the zygomatic bone. The direction of displacement depends on the direction of the traumatic forces.
P.413
Forces from above can cause downward displacement of the zygoma, creating a gap deformity at the lateral orbital rim, inferior displacement of the lateral canthal angle (Fig. 10.112), retraction of the lower lid, and a step deformity of the inferior orbital rim. Forces from below can cause superior displacement and telescoping of bone fragments of the lateral orbital rim. An inwardly displaced fracture of the zygomatic arch may impinge on the coronoid process, limiting movement of the mandible to cause pain or jaw movement.
 |
Figure 10.110. A 37-year-old man with an extruding alloplastic orbital floor implant. |
Clinical Features
Patients frequently complain of numbness of the cheek, nasolabial fold, upper lip, and gingiva around the upper teeth on the injured side. The hypesthesia results from trauma to the infraorbital and alveolar nerves. They may also have pain on movement of the jaw. In addition to the signs seen with orbital floor fractures, patients with tripod fractures may also demonstrate malar flattening, inferior displacement of the lateral canthal angle and telescoping, or gap deformities at the lateral orbital rim.
Management
Life-threatening injuries may be associated with facial trauma. Systemic and neurologic evaluation takes precedence over ophthalmologic assessment. Because the zygomatic bone absorbs the impact of trauma and protects the globe, the frequency and severity of ophthalmic injuries are somewhat less in tripod fractures than with orbital floor fractures. Nevertheless, ocular injury is associated with facial trauma in 11% to 40% of cases. A complete ophthalmologic evaluation is necessary and should include assessment of visual acuity, pupillary reaction, extraocular motility, sensory nerves, and dentition with orbital palpation, slit lamp examination, ophthalmoscopy, forced duction testing, exophthalmometry, and tonometry. Visual loss may be associated with a contusion or transection of the optic nerve.
 |
Figure 10.111. This skull demonstrates the close relation of the coronoid process (arrow) of the mandible with the zygomatic arch. Inward displacement of the zygomatic arch impinges on the coronoid process of the mandible, resulting in pain on movement of the jaw. |
Tripod features are radiographically well demonstrated on a Waters' view radiograph. The zygomatic arch can be evaluated with the submental-vertex view. CT scans of the orbit with coronal (Fig. 10.113) and axial views provide the most detail of the zygomatic bone and other orbital fractures.
Nondisplaced tripod fractures with no significant entrapment of orbital contents, malar flattening, or impingement of the coronoid process of the mandible can be managed conservatively. This includes the use of nasal decongestants, broad-spectrum oral antibiotics, avoidance of nose blowing, application of ice compresses, and the use of systemic prednisone. Surgical indications include persistent pain on mastication, inferior displacement of the lateral canthal tendon and Lockwood's ligament, persistent diplopia with muscle entrapment, and cosmetically unacceptable malar flattening.
Surgical repair can be done within 24 to 48 hours if the deformities are pronounced. In less dramatic cases, surgical repair of tripod fractures can be delayed 7 to 10 days to allow orbital edema to decrease. The transconjunctival approach combined with a lateral cantholysis allows good exposure of the zygomatic bone, the orbital rim, and the orbital floor. Optimal results are obtained with open reduction and internal fixation of bone fragments using interosseous wires or microplates (Fig. 10.114). If necessary,
P.414
an orbital floor implant is placed to prevent recurrent prolapse of the orbital contents.
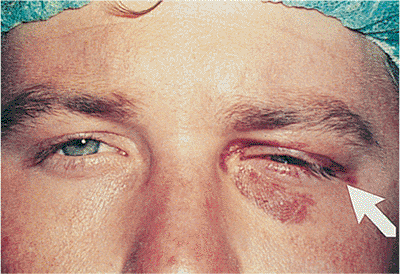 |
Figure 10.112. Inferior displacement of the left lateral canthal angle is seen in this 37-year-old man with a left lateral wall fracture and inferior displacement of the zygomatic bone. |
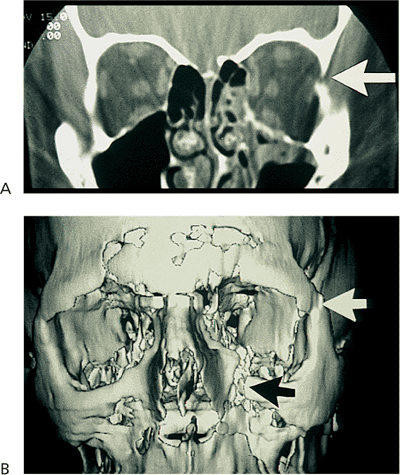 |
Figure 10.113. A: The coronal computed tomography (CT) scan shows a nondisplaced floor fracture and disarticulation of the zygomaticofrontal suture (arrow). B: This three-dimensional CT scan of a tripod fracture from another patient demonstrates disarticulation of the zygomaticofrontal suture (white arrow) and the zygomaticomaxillary suture (black arrow), with inferior displacement of the zygomatic bone. |
Proptosis
Orbital Cellulitis
Orbital cellulitis is a bacterial infection of the orbit most often associated with sinus disease. Patients present with a 1- to 2-day history of progressive redness and swelling of the periorbital areas.
 |
Figure 10.114. Open reduction of the lateral orbital rim and internal fixation with microplates and screws. |
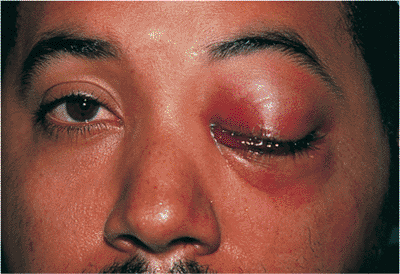 |
Figure 10.115. Orbital cellulitis presenting with massive swelling, chemosis, erythema, and poor ocular motility. |
Clinical Features
Preseptal and orbital cellulitis present with swollen, red, and warm lids, possibly with a fever and an elevated leukocyte count. Orbital cellulitis is differentiated from preseptal cellulitis by orbital signs that may include chemosis, limited motility, proptosis, orbital resistance to retropulsion, an afferent pupillary defect, and decreased vision (Fig. 10.115). The onset of infection may have been preceded by an upper respiratory infection, especially in children. Fever and leukocytosis are more common in children but not always present. Associated sinus disease is thought to cause 85% of cases. Other causes include external wounds or bites, recent dental work, and dacryocystitis. A CT scan is required for patients with orbital cellulitis to rule out an orbital abscess or mass and to assess the paranasal sinuses (Fig. 10.116).
Gram-positive organisms are the most common cause of infection, with Staphylococcus aureus and Streptococcus species often identified as the offending organism. In children younger than 5 years of age, H. influenzae must be considered. Blood cultures are often negative, especially in adults, and the exact organism often remains unidentified.
If a patient is not responding to appropriate antibiotic coverage, an orbital abscess must be suspected. This can be identified with repeat CT scans and is most commonly seen as a subperiosteal collection (Fig. 10.117). The identified abscess requires immediate surgical drainage.
Cavernous sinus thrombosis is the feared complication of orbital cellulitis. The infection travels either by extension or through venous drainage into the cavernous sinus, setting up a potentially fatal intracranial infection. Clinically, this is seen by the sudden onset of bilateral chemosis, pain, proptosis, lid injection, fever, malaise, severe headache, and optic nerve, retinal, and choroidal swelling. This serious condition carries a significant risk of death.
Orbital pseudotumor is a condition that may be difficult to differentiate from orbital cellulitis. Orbital pseudotumor is more commonly associated with pain or pain with eye
P.415
movement. Pseudotumors can have an acute presentation, but they commonly occur subacutely, becoming more noticeable over several days. A fever or leukocytosis points toward orbital cellulitis but, especially in children, can be associated with orbital pseudotumor.
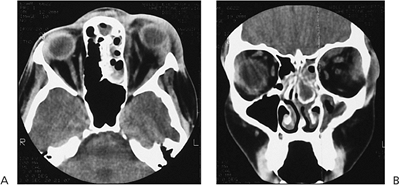 |
Figure 10.116. Axial (A) and coronal (B) computed tomography scans show pansinusitis and diffuse orbital swelling of left side consistent with orbital cellulitis. |
Management
Differentiation between preseptal cellulitis and orbital cellulitis is key in the management of this condition. Preseptal cellulitis in adults can be treated with a broad-spectrum oral antibiotic, with follow-up examination the next day. If the condition continues to worsen over 48 hours or develops signs of orbital cellulitis, admission to the hospital for intravenous antibiotics and a CT scan is required. Management of children with preseptal cellulitis is somewhat controversial. Some institutions admit all children with preseptal cellulitis for intravenous antibiotics. Children who can adequately be examined and have no evidence of orbital involvement or signs of systemic toxicity can be placed on broad-spectrum oral antibiotics and be seen the next day.
Children and adults with orbital cellulitis need immediate broad-spectrum intravenous antibiotics, an orbital CT scan, and a complete blood count and differential. Antibiotics should be instituted quickly and not delayed while the patient is admitted and tested. Once on intravenous antibiotics, there should be some noticeable improvement within 48 hours. Any significant associated sinus disease needs to be evaluated, with possible drainage if the patient is not improving. If no improvement is seen, an orbital abscess should be suspected, and a repeat CT scan may be required.
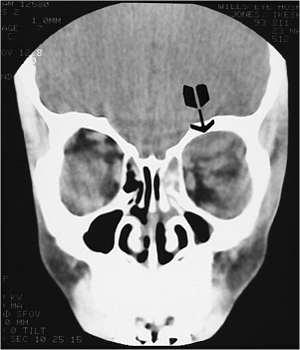 |
Figure 10.117. Coronal computed tomography scan showing subperiosteal abscess formation (arrow) in the left superior nasal orbit. |
Orbital cellulitis generally requires 5 to 7 days of intravenous antibiotics, depending on the clinical response. This is usually followed by 10 to 14 days of oral treatment.
Fungal Infections
Mucormycosis
This aggressive, opportunistic fungal infection is generally an infection of uncontrolled diabetics or immunocompromised patients. Patients present soon after the onset of deep, boring pain, which is followed by progressive proptosis, sudden visual loss, and orbital apex syndrome. Successful treatment depends on early recognition of mucormycosis and an aggressive medical and surgical approach.
Clinical Features
The fungal pathogen, Phycomycetes mucorlais, occurs naturally in soil, air, skin, and food. The organism gains entrance through the nasopharynx or sinuses and, in a person with normal cellular defenses, the organism is controlled by the cellular immune system. In an immunocompromised patient, the organism proliferates in the sinus or nasopharynx. The orbit is invaded secondarily, with the organism causing vascular necrosis and infarction as it progressively invades blood vessels. Early signs are deep, boring pain and sinusitis. Mucormycosis is often initially treated as a bacterial sinusitis, but it must be suspected in any immunocompromised patient with sinusitis and orbital disease. Later in the progression, cellulitis, proptosis, apical neuropathies, and intracranial spread develops. The classic black scar usually develops late in the disease. Without prompt recognition and treatment, the disease is often fatal.
The CT scan usually shows sinus disease with or without bony destruction and adjacent orbital involvement. The CT
P.416
findings can be subtle in early disease and can cause a delay in diagnosis (Fig. 10.118).
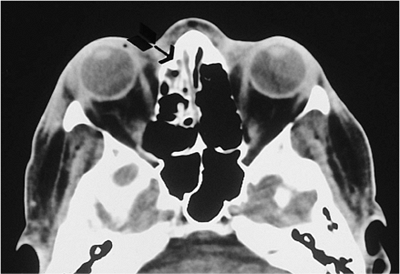 |
Figure 10.118. The axial computed tomography scan of patient with mucormycosis shows clouding and mucosal thickening in the left ethmoid sinus (arrow). |
Management
The key to treatment is early recognition. There is significant potential for a fatal outcome with this disease in these often very sick patients. Once suspected and confirmed with a tissue diagnosis, treatment must be aggressively aimed at controlling the systemic disease, using systemic amphotericin B and surgical excision of involved tissues. This must be done in a hospital with a well-coordinated team effort. The role of hyperbaric oxygen in this disease continues to be debated.
Aspergillosis
Aspergillus is ubiquitous and occurs as an opportunistic infection. The orbital disease occurs in two forms. One form occurs in debilitated patients, and the second occurs in healthy persons who often have a history of sinus disease, polyps, or living in hot, humid areas.
Clinical Features
The diffuse form is often seen in patients with some immune compromise. A history of chronic sinus disease is common. Aspergillus organisms invade the orbital vessels, causing arteritis, thrombosis, and occlusion of the vessels. This leads to a progressive destruction of the orbital bones and infiltration of the optic nerve and orbital contents. This condition usually presents as slowly progressive, unilateral proptosis, ocular pain, and visual loss. Late signs include lid edema, chemosis, fever, and leukocytosis. This infection may even lead to an endophthalmitis and death.
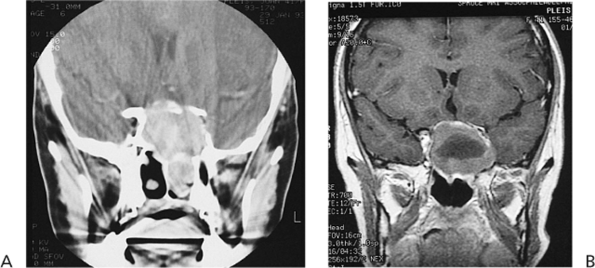 |
Figure 10.119. Allergic noninvasive form of aspergillosis shown by (A) computed tomography and (B) magnetic resonance imaging (MRI). MRI demonstrates the central hypodense fungal ball surrounded by mucin. |
The localized form develops as a slow-growing sclerotic mass that generally begins in the sinuses and extends secondarily into the orbit (Fig. 10.119). There may be an associated allergic component with an IgE-mediated antibody-antigen reaction to specific fungal antigens. This is seen in healthy people who live in hot, humid climates. The symptoms depend on the location of the slow-growing mass. More interior masses present as proptosis and globe displacement, while posterior masses present with visual loss and dysmotility.
Management
Once suspected and confirmed with orbital imaging, the diagnosis must be made pathologically. The primary treatment is then complete drainage and debridement of all areas involved, if possible. The diffuse form is more often fatal but if drained early, the localized form often does well. Amphotericin B and other antifungal agents may be helpful in treating the diffuse form.
Orbital Inflammation
Orbital Pseudotumor
Orbital pseudotumor comprises a wide spectrum of inflammatory syndromes. These vary in their location in the orbit and in onset. They all present with various degrees of pain, proptosis, swelling, and injection of the orbit or globe. Once diagnosed, they respond to corticosteroid treatment.
Clinical Features
This group of disorders is marked by the acute or subacute onset of orbital inflammatory disease. Histologically, these include infiltration by a polymorphous group of inflammatory cells, including neutrophils, lymphocytes, plasma cells, macrophages, and fibrocytes. Clinically, the onset is associated with pain, swelling, erythema, and orbital dysfunction (Fig. 10.120). The exact symptoms depend on the location of the process. The inflammation
P.417
may be anterior, diffusely involving the orbit, apical, lacrimal, a myositis, or any combination. Anterior processes present with pain, redness, and swelling of the lids and anterior orbit. If the process is apical, there may be few external signs but intense pain with loss of vision and motility. Myositis includes pain, proptosis, swelling, and loss of function of the affected muscle. Lacrimal gland involvement presents as swelling and pain of the lacrimal gland and upper lid. This can sometimes be subacute or even chronic, with less pain and a mass effect.
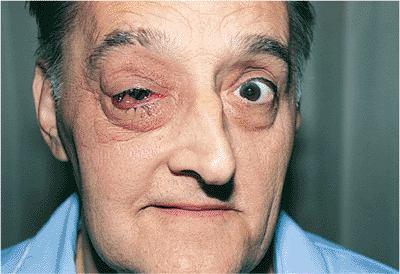 |
Figure 10.120. A patient with orbital pseudotumor presented with swelling, redness, pain, and limited motility. |
Possible ocular findings include uveitis, scleritis, papillitis, and exudative retinal detachment. In children, the disease is more likely to be bilateral and have ocular involvement, and there may be an elevated sedimentation rate and CSF pleocytosis. Children may also have a fever associated with orbital pseudotumor.
CT scanning usually shows diffuse thickening of the involved structures without any bony changes (Figs. 10.121,10.122,10.123). The combination of acute onset of pain along with this CT picture usually make the diagnosis straightforward.
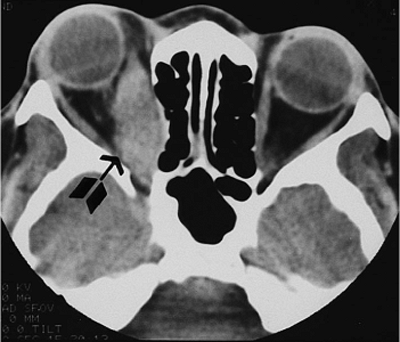 |
Figure 10.121. The axial computed tomography scan of orbital myositis demonstrates an enlarged right medial rectus muscle (arrow). |
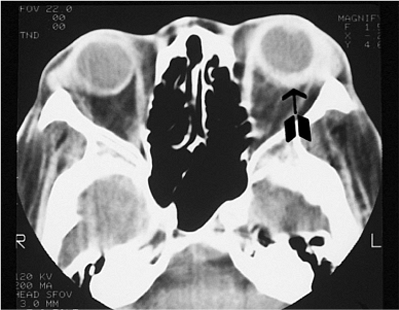 |
Figure 10.122. An axial computed tomography scan shows diffuse scleral thickening (arrow) and anterior orbital inflammation that are consistent with orbital scleritis. |
Management
The diagnosis is based on clinical examination and orbital imaging, and pseudotumor is treated with systemic steroids. Depending on the patient's medical condition, the usual adult dose is 60 to 80 mg of oral prednisone per day. Most patients have a noticeable reduction in pain and swelling in 24 to 48 hours. If the condition has been more chronic, it may take longer to notice a response. After the condition has improved, the steroids can be tapered over 4 to 5 weeks. If there is a poor response to prednisone or a flare condition on tapering the medication, the clinical diagnosis must be suspected. In these cases, an orbital biopsy is done to establish or confirm the diagnosis, because some tumors can mimic pseudotumor.
Inflammation primarily involving the lacrimal gland is more likely to be associated with other conditions such as viral or bacterial dacryoadenitis, sarcoidosis, lymphoid proliferation, and Sj gren's syndrome. Some clinicians suggest routine biopsy of any process primarily involving the lacrimal glands. Lacrimal gland processes must be biopsied if there is not a rapid response to steroids.
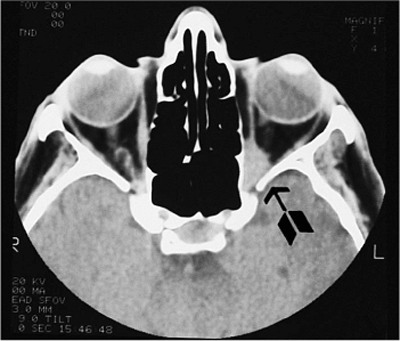 |
Figure 10.123. An axial computed tomography scan shows localized orbital inflammation at the left globe orbital apex, which resulted in decreased vision and dysmotility, with minimal external signs of inflammation. |
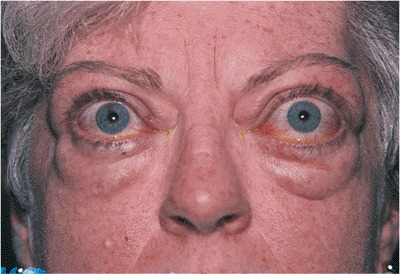 |
Figure 10.124. Thyroid-related ophthalmopathy with proptosis, lid retraction, and limited motility. |
P.418
Thyroid-Related Ophthalmopathy
Thyroid-related ophthalmopathy is an immune-mediated process that results in orbital inflammation and scarring. It occurs four to five times more commonly in women than men and is most common in middle age. The results may be mild swelling of the orbit, or it can result in severe proptosis, lid retraction, corneal exposure, motility disturbances, or visual loss secondary to optic nerve compression. Ninety percent of patients have or have had a systemic thyroid abnormality.
Clinical Features
Thyroid-related ophthalmopathy is characterized by chronic orbital inflammation and scarring. Usually, this is a bilateral process, but it can be asymmetric. This autoimmune process results in infiltration of extraocular muscles and orbital fat by inflammatory cells in the short term and by mucopolysaccharide and collagen in the long term. The disease process is marked by early inflammation and long-term fibrosis and scarring.
Early manifestations of the disease can often be subtle, with nonspecific redness, irritation, chemosis, and eyelid swelling. The presence of a systemic thyroid imbalance is helpful in establishing the diagnosis but not necessary to make the diagnosis, because 10% of the patients are euthyroid. Early signs of lid retraction, lid lag, or evidence of any systemic thyroid imbalance help confirm the diagnosis. As the disease process progresses, eyelid retraction and lid lag become more obvious, and corneal exposure must be watched for. Proptosis and restriction of motility are also later signs (Fig. 10.124). The inferior rectus muscle is the most commonly involved muscle, followed by the medial rectus, making restricted upgaze and lateral gaze the most common motility disturbances. The CT scan shows involvement of the muscle bellies and sparing of the tendons (Fig. 10.125). If extraocular muscle involvement progresses, the optic nerve may become compressed at the orbital apex by these swollen muscles. Visual acuity, color vision, visual fields, and presence of an afferent pupillary defect must all be monitored in these patients to rule out optic nerve compression.
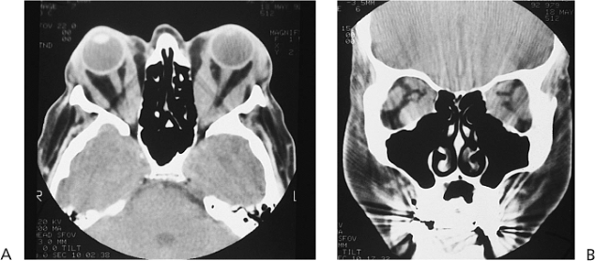 |
Figure 10.125. Axial (A) and coronal (B) computed tomography scans show enlargement of all extraocular muscles in a patient with thyroid-related ophthalmopathy. |
A patient typically goes through a period of 6 to 24 months of active disease. During this period, there is active inflammation, and the ocular manifestations are changing. After this phase is completed, the eye typically remains stable, despite the residual scarring of the muscles and orbit.
Management
Management of thyroid-related ophthalmology is complicated by the wide variability of disease manifestations and by a lack of consensus about the best treatment. Initial assessment of the patient requires determination of disease activity, which is important in making decisions about management. All patients require an evaluation and monitoring of their thyroid status. Patients may be euthyroid on presentation, but their thyroid status must be followed over time. Careful evaluation for optic nerve compression or significant corneal exposure must be done. All patients will benefit from lubrication of the eyes. Good patient education about thyroid-related ophthalmopathy and what the patient can expect is invaluable.
Patients with active disease are candidates for orbital irradiation. Irradiation appears to stop progression of thyroid-related ophthalmopathy in a significant percentage of patients. Irradiation does not reverse the scarring already present from the disease. Irradiation takes up to 6 months to have full effect, so any acute problem such as optic nerve compression needs to be treated with other modalities, often in conjunction with irradiation. Patients with active disease
P.419
will require an orbital CT scan. This helps assess muscle size, activity, and may later assess response.
Patients with inactive disease are without active inflammation and are unlikely to benefit from irradiation. Treatment includes observation, lubrication, and possible surgery to correct changes caused by thyroid-related ophthalmopathy. These patients do not require imaging unless the diagnosis is in question or decompressive surgery is being considered.
Systemic steroids are indicated for short-term management of the inflammation from thyroid-related ophthalmopathy. The exact mechanism of action is unclear but steroids are effective in decreasing inflammation. This is especially helpful in optic nerve compression or severe corneal exposure. Irradiation itself takes weeks to work, so the addition of steroids gives rapid, short-term treatment. Long-term use of steroids is limited by side effects.
Surgical management, which often involves multiple stages and treatments over months to years, may include orbital decompression, eye muscle surgery, and eyelid surgery. The need for acute surgical management of severe disease has decreased with the use of irradiation. There still are the rare patients who will require acute orbital decompression for optic nerve compression. More often, surgery is required to try and reverse the results of the chronic orbital scarring form thyroid-related ophthalmology.
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis, formerly known as histiocytosis X, is a rare collection of disorders of the mononuclear phagocytic system. The former terms eosinophilic granuloma of the bone, Letterer-Siwe disease, and Hand-Sch ller-Christian disease are being replaced by the terms unifocal and multifocal eosinophilic granuloma of bone, and diffuse soft-tissue histiocytosis.
Clinical Features
The nature of involvement and long-term prognosis of this disease appears to be related to the patient's age and the extent of disease. Children younger than 2 years of age have a mortality rate of 55% to 60%, compared with 15% for older children. The extent of the disease is usually apparent early in its course. The diffuse soft tissue disease tends to produce fulminant systemic involvement in very young children. The unifocal disease tends to be in older children and has the best prognosis.
Systemic involvement can vary greatly but may include fever, localized infections, skin lesions, hepatosplenomegaly, lymphadenopathy, and possibly bone marrow involvement.
Orbital disease characteristically involves the superotemporal area. Bony involvement can be quite limited or extensive. Soft tissue involvement alone is rare and is usually secondary to extension from the surrounding bone. This pattern of disease results in a mass effect on the globe or orbital structures. The bony lesions may be sclerotic or have a lytic appearance, with secondary expansion to surrounding tissues seen on CT scans. This bony involvement can lead to the described triad of diabetes insipidus, exophthalmos, and bony lesions.
Management
Suspected isolated orbital lesions must be biopsied. If the biopsy confirms the diagnosis, local curettage is often curative. However, low-dose irradiation may be considered if growth continues. Evidence of multifocal bony involvement or systemic involvement should be looked for in these patients. Diffuse systemic involvement or multifocal involvement requires aggressive treatment by a pediatric oncologist. This may include irradiation, chemotherapeutic agents, and steroids.
Orbital Tumors
Lymphoid Tumors
Lymphoid tumors comprise a spectrum from benign to highly malignant lesions. The presentation of these lesions can vary greatly. They are the most common primary orbital malignancy in adulthood. Lymphoid orbital tumors respond very well to radiation therapy. A tumor in the orbit should alert the clinician to the possibility of lymphoma elsewhere in the body.
Clinical Features
Patients with lymphoproliferative disease of the orbit classically present with a painless mass and its secondary mass effect. Inflammatory signs are rarely present and the onset of lymphoid inflammation is insidious. Lymphoid lesions may present subconjunctivally (Fig. 10.126) with a visible, fleshy mass, in the orbit with globe displacement and dysfunction, or with an enlarged lacrimal gland. The anterior, subconjunctival lesions are more likely to be benign than the more posterior tumors. The orbital involvement is usually extraconal. Patients may have a history of lymphoid disease elsewhere in the body.
CT scans reveal a mass that usually molds around the globe, orbit, and orbital structures rather than causing direct displacement (Fig. 10.127). Bony involvement is rare, and as many as 25% of cases may have bilateral involvement of the orbits.
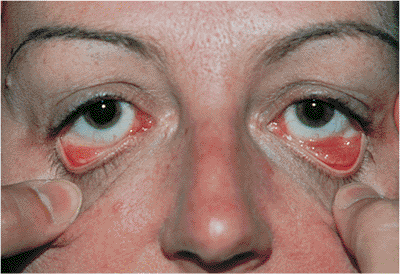 |
Figure 10.126. Bilateral lymphoid infiltrates of the inferior conjunctival fornix. |
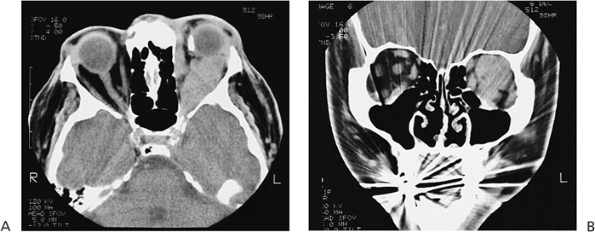 |
Figure 10.127. Diffuse infiltration by a lymphoma of the left orbit. Lymphoid tumors tend to mold around orbital structures rather than displacing them. A: Axial view. B: Coronal view. |
P.420
Management
Management generally involves a team approach of pathology, oncology, and radiation oncology specialists along with the ophthalmologist. After an orbital mass is identified and lymphoma is suspected, tissue is needed to identify the lesion as lymphoid and stage its degree of malignancy. An aggressive systemic workup is also needed to look for any evidence of systemic lymphoid disease. The extent of systemic disease and the histopathologic diagnosis need to be taken into account when deciding upon treatment for each patient. Cases that are localized to the orbit are often treated with orbital irradiation, but systemic involvement by malignant lymphoma usually requires some form of chemotherapy. Even benign lymphoid hyperplasia localized to the orbit implies the patient is at risk of developing lymphoma elsewhere in the body in the future, and long-term follow-up is necessary.
Dermoid and Epidermoid Cysts
These benign cystic lesions comprise about 5% of orbital lesions. They are choristomas that probably arise from sequestration of epidermal tissue in deeper layers during embryonal development. The superficial lesions typically present in the first few years of life as firm, slow-growing, painless masses, often in the anterior superotemporal orbit. Deeper lesions are less common and typically present later in life.
Clinical Features
Dermoid and epidermoid cysts can be separated into superficial and deep lesions. Both are cystic lesions, but epidermoids are keratin-filled cysts lined by epidermis and without dermal appendages. These lesions are more common in the skin and less common in the orbit. A dermoid cyst contains dermal appendages in its wall and is filled with keratin, but it may also contain hairs. Dermoid cysts typically involve the orbit and are thought to arise from ectodermal tissue pinched off in a bony suture during embryonic development.
The superficial lesions are typically recognized early in childhood, often in the first year. They are classically located in the superotemporal orbit, adjacent to the orbital rim (Fig. 10.128), but they can occur in other locations. They grow slowly, are painless, and may be attached to the orbital rim. CT scans reveal a well-defined mass with a lucent center. There may be some bony fossa formation but not true bony erosion.
Deep orbital lesions typically present later because of their deep location and very slow, painless growth. These lesions can arise from virtually any orbital suture and are typically discovered because of their secondary mass effect within the orbit (Fig. 10.129). These deeper dermoids must be differentiated from the superficial lesions, because the dermoid cyst is much more difficult to remove and can sometimes extend intracranially.
Rarely, if the cyst ruptures from trauma or spontaneously, significant orbital inflammation and scarring can result. An incompletely excised cyst can also result in chronic orbital inflammation and scarring from continued leakage of the cyst's contents.
Management
Treatment of these lesions involves excision. The only debate is when and, in the deep cases, how to approach removal. Superficial dermoids tend to grow slowly, and as the child becomes more active, the risk of rupture
P.421
increases, making excision a consideration. These lesions should be completely excised sometime between the ages of 1 and 5 years. If there is any question during the preoperative examination that the lesion is more than superficial, a CT scan should be done to document the lesion's full extent. Superficial lesions can usually be excised through a lid crease approach. Complete excision of the entire cyst, with attempted destruction of any small tail that is attached to a bony suture, is important. Deeper lesions require more extensive surgery and, rarely, a combined neurosurgical procedure. The key is complete excision of the entire lesion, because any remnants left in the orbit can result in chronic orbital inflammation and scarring.
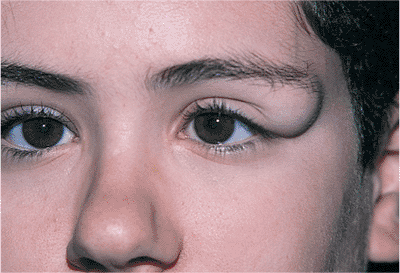 |
Figure 10.128. Superficial dermoid cyst with a classic superotemporal location. |
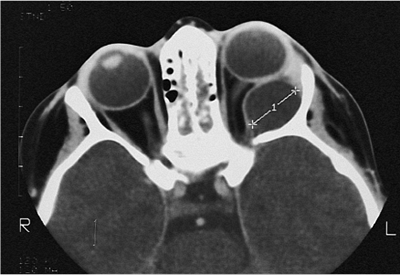 |
Figure 10.129. An axial computed tomography scan of a deep orbital dermoid demonstrates molding of the lateral orbital wall. |
Vascular Tumors
Capillary Hemangioma
Capillary hemangiomas are abnormal proliferations of endothelial cells forming capillary-like vessels. These lesions typically present in the first few months of life. Rapid growth during the first few weeks is followed by spontaneous regression over months to years. Patients often have other hemangiomas elsewhere on their bodies. Amblyopia, because of astigmatism or occlusion and severe proptosis, may necessitate treatment.
Clinical Features
Capillary hemangiomas can be divided into the superficial form that involves skin or conjunctiva and the deep form that is located in the orbit. Patients can also present with a combined form.
Superficial lesions, called strawberry nevi, can be located anywhere on the body. These lesions are located in the dermis and rarely require treatment.
Deep orbital lesions typically present with a mass effect in the orbit, causing proptosis and globe displacement (Fig. 10.130). Orbital imaging shows a variable lesion from well circumscribed to infiltrating. MRI reveals characteristics of blood flow to and within the lesion. This information may be more helpful than CT in treating this lesion.
Combined lesions that are superficial and deep cause the most clinical problems. These lesions can grow and impinge on the visual axis, possibly causing amblyopia. They may press on the globe, causing astigmatic changes. Like the superficial lesions, these tend to grow during the first year of life and then regress with time.
Management
With time, these lesions regress on their own, and if possible, close observation is the therapy of choice. Treatment is required for lesions causing amblyopia or severe proptosis, although the best form of treatment is not clear. Intralesional corticosteroid deposition, systemic corticosteroid therapy, or surgical excision seem to be the most helpful modalities, combined with aggressive treatment of the amblyopia.
Lymphangioma
Lymphangiomas are vascular lesions containing endothelium-lined channels that are isolated from the vascular system of the surrounding structures. These lesions develop in childhood but are often not recognized until early adulthood.
Clinical Features
Lymphangiomas are composed of multiple serous-filled vascular channels, accumulations of lymphocytes, a collagen network, areas of smooth muscle, old hemorrhage, and dysplastic vessels. These lesions are sometimes confused with venous anomalies, but lymphangiomas are isolated from the vascular system.
Lymphangiomas can occur superficially, deep in the orbit, or in a combined form. The superficial lesions present in childhood as clear cystic structures with some blood-filled or partially blood-filled cysts (Fig. 10.131). Deeper lesions often present with a deep orbital hemorrhage and sudden onset of proptosis, pain, and possibly, decreased vision. If the orbital hemorrhage results in visual loss or corneal exposure, drainage of the blood-filled cyst, along with attempted excision of the vascular anomaly causing the bleeding, is required. The surrounding structures are often scarred, and complete excision is usually impossible.
Combined lesions are usually recognized in childhood because of their superficial components. These can be very
P.422
large, cause significant cosmetic deformity, and threaten vision because of optic neuropathy or amblyopia. These lesions may require surgical management on an urgent basis because of a hemorrhage or elective surgery because of the progressive enlargement of the lesion. Because lymphangiomas have an infiltrative growth pattern, complete excision is almost impossible. They are often debulked but continue to grow. It has been suggested that the risk of subsequent hemorrhage is increased after the initial debulking surgery.
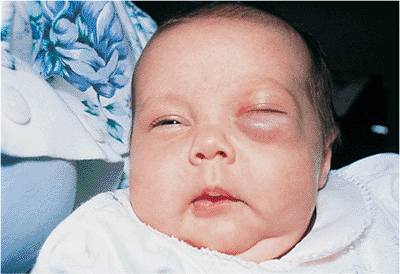 |
Figure 10.130. A child with right subcutaneous capillary hemangioma without the usual strawberry appearance. |
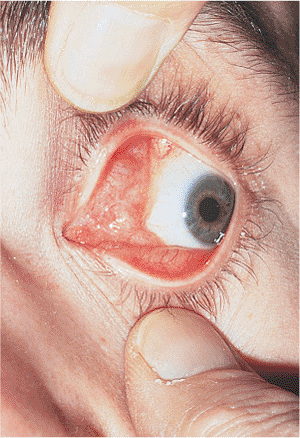 |
Figure 10.131. A patient with lymphangioma of the left orbit presented with a mass involving the medial upper eyelid. Notice the cystic spaces involving the conjunctiva. |
CT scans show a poorly defined, infiltrative lesion with various densities (Fig. 10.132). MRI is often better at identifying these lesions with their cystic areas of blood in patients of different ages.
Management
Management of lymphangiomas is difficult. The slowly progressive course and the infiltrative nature of these lesions make most treatments less than satisfying. After the lesion is identified as a lymphangioma, observation should be the first choice. Some lesions subsequently come to attention because of a spontaneous orbital hemorrhage that may need to be drained if causing visual loss or corneal exposure. Other lesions may need to be debulked in time as they slowly enlarge and infiltrate the orbit. Because of the infiltrative nature of lymphangiomas, complete surgical excision is usually impossible without damaging vital structures. Ultimately, some patients are left with poorly functioning or blind eyes.
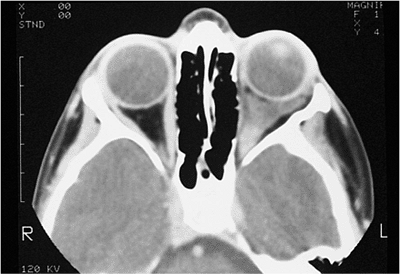 |
Figure 10.132. An axial computed tomography scan shows deep orbital infiltration by a lymphangioma. |
Cavernous Hemangioma
Cavernous hemangiomas are benign, well-circumscribed, intraconal lesions that typically present in adulthood with slowly progressive proptosis. These lesions are successfully treated by surgical excision.
Clinical Features
Cavernous hemangiomas typically present with slowly progressive proptosis that, once noticed, is often identified in old pictures from previous years. These patients present for diagnosis or treatment when they are 20 to 70 years of age. The presenting symptoms depend on the position of the lesion and the size. In addition to proptosis, the symptoms include orbital pain, diplopia, optic nerve compression, and distortion of the globe. The CT scan typically shows a well-circumscribed, intraconal mass that displaces orbital structures (Fig. 10.133). These lesions are low-flow vascular lesions with variable enhancement patterns seen on CT scans.
Management
Management involves surgical excision. Once isolated, these lesions are usually very easily separated from surrounding orbital structures and removed. Bleeding is usually not a problem, because cavernous hemangiomas are low-flow lesions. Cavernous hemangiomas enlarge with time, and once symptomatic should be excised. Asymptomatic lesions may be removed to rule out tumors with malignant potential such as hemangiopericytomas or fibrous histiocytomas.
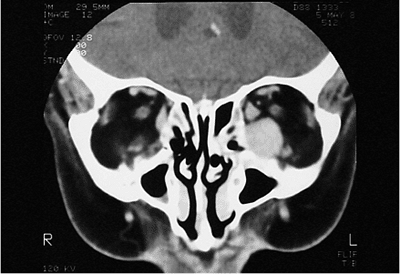 |
Figure 10.133. Computed tomography scan of a cavernous hemangioma, demonstrating a well-encapsulated, contrast-enhanced tumor of the left orbit. |
P.423
Hemangiopericytoma
Hemangiopericytomas are rare vascular orbital tumors that originate from pericytes. The lesion classically presents in the superior orbit as a growing, well-circumscribed mass.
Clinical Features
Hemangiopericytomas present similarly to cavernous hemangioma, with symptoms secondary to mass effect. Unlike cavernous hemangioma, these are more likely to grow in less than 1 year. A CT scan shows a well-circumscribed mass in the retrobulbar orbit. These lesions are often moderately vascular.
Management
Management is complete surgical excision. These lesions are usually well circumscribed, without infiltration, but they may have associated draining vessels. Pathologic evaluation of these lesions is important. The lesions are divided into benign, intermediate, and malignant tumors. The literature indicates that as many as 30% of these lesions recur, and 10% to 15% metastasize. Local recurrence is more common after incomplete or piecemeal excision and by malignant lesions. Metastasis is also more likely for malignant lesions but has been seen in benign tumors. Because local recurrence and metastasis can occur years later, these patients need to be followed on a long-term basis.
Vascular Abnormalities
Arteriovenous Malformation
An arteriovenous malformation is the result of an abnormal connection between an artery and the venous system draining the orbit. Symptoms vary with the amount of arterial flow from mild vascular engorgement to severe pulsatile proptosis, chemosis, and elevated intraocular pressure.
Clinical Features
Arteriovenous malformations are divided into high-flow and low-flow abnormalities. The malformation is an abnormal connection between an artery and venous system, typically the cavernous sinus. The size of the artery that arteriolizes the venous system determines the symptoms associated with this abnormality.
Low-flow malformations typically occur spontaneously and are thought to be related to venous thrombosis within the cavernous sinus. The theory is that, as the sinus recanalizes, small arteries in the wall of the cavernous sinus enlarge to form a shunt into the venous system. Symptoms typically consist of mild swelling of the orbit and engorgement of the veins. Symptoms may wax and wane, and as many as 40% of cases spontaneously resolve.
High-flow malformations are most often related to head trauma resulting in a tear in the cavernous sinus portion of the internal carotid artery. This sets up a high flow of blood into the cavernous sinus and arterialization of the orbital and ocular venous system. Patients often have an audible bruit, pulsatile exophthalmos, chemosis, orbital swelling, episcleral vascular engorgement, and increased intraocular pressure (Fig. 10.134). The CT scan shows diffuse engorgement of orbital structures, enlarged muscles, and an enlarged superior ophthalmic vein. Color Doppler ultrasonography of the orbit shows flow reversal in the venous system.
 |
Figure 10.134. External photograph of a patient with an arteriovenous malformation with dilated, tortuous episcleral vessels of the right eye. |
Management
Treatment involves intravascular embolization of the shunt. Almost all high-flow systems require embolization. Because there is a significant chance the low-flow systems will spontaneously thrombose, observation is indicated unless there is impending visual loss, uncontrollable intraocular pressure, severe pain, swelling, or corneal exposure.
Venous Anomalies
Venous anomalies are composed of segments of dilated venous system. Their manifestations depend on the degree of connection with a functional venous system. Superficially, they appear as distended venous areas; deep lesions can lead to spontaneous orbit hemorrhage.
Clinical Features
These lesions are more understandable if divided into a distensible form and a nondistensible form or varix. Nondistensible venous anomalies are typically isolated from the venous system. When located deep in the orbit, these present with a sudden onset of pain and proptosis associated with an acute hemorrhage. Superficial lesions present with swelling and distortion of the lids and anterior orbital structures that can be quite disfiguring (Fig. 10.135).
 |
Figure 10.135. Extensive varices of the orbit and lids. |
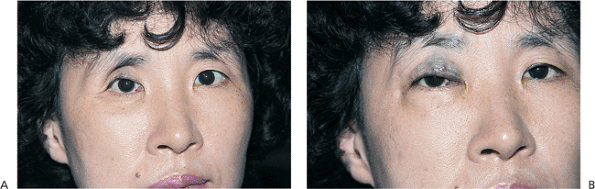 |
Figure 10.136. The patient is shown to have a deep orbital varix with an enophthalmic right eye (A) and proptosis after Valsalva's maneuver (B). |
P.424
The distensible form usually presents in the first decade of life. Typically, these lesions enlarge with increased jugular pressure, because they have a direct connection with the venous system. Patients with deep lesions typically present with enophthalmos secondary to an enlarged orbit and fat atrophy from the chronic, intermittent venous distension (Fig. 10.136). If the lesion is located anteriorly, venous engorgement occurs with increased venous pressure. Valsalva's maneuvers or placing the patient's head in a dependent position can demonstrate the venous engorgement. When comparing the axial and corolla views on CT scans, a significant difference in the lesion size is noticed because of the dependent position of the head during the coronal views (Fig. 10.137).
Management
Observation is generally the rule for these lesions. Severe orbital hemorrhage with severe pain or visual loss may require drainage of the blood. The venous anomaly is usually not able to be identified or removed. The anterior lesions causing extreme disfigurement can be treated with a YAG contact or CO2 laser with some degree of success.
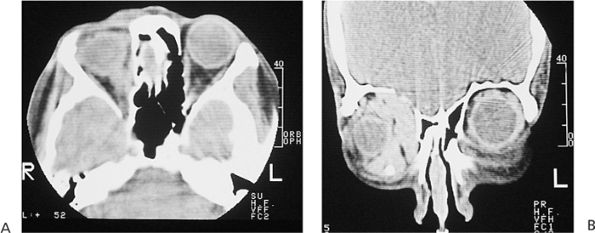 |
Figure 10.137. Axial (A) and coronal (B) computed tomography scans of the patient with an orbital varix (Fig. 10.136). There is a significant increase in the size of the varix when filming the coronal image because the head is in a dependent position. |
Neural Tumors
Optic Nerve Glioma
Clinical Features
Gliomas of the orbital portion of the optic nerve are diagnosed in preschool children with a loss of vision, proptosis, papilledema, optic atrophy, and strabismus. Visual loss is usually the first symptom, which is often picked up on routine screening. Proptosis can be minimal to massive, producing an axial and downward displacement of the globe (Fig. 10.138). Between 25% and 50% of children with optic nerve gliomas have neurofibromatosis. The CT scan demonstrates a fusiform enlargement of the optic nerve and, if the tumor extends intracranially, enlargement of the optic canal (Fig. 10.139). MRI is helpful in delineating the intracranial extent of the tumor. Gliomas are well-circumscribed astrocytic tumors derived from interstitial cells, astroglia, and oligodendroglia. The tumor can be primarily astrocytic with eosinophilic inclusions called Rosenthal fibers, or it can be myxomatous with cystic areas that can expand rapidly.
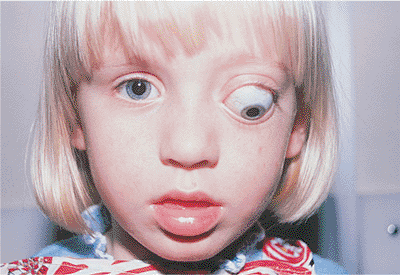 |
Figure 10.138. Child with massive proptosis and downward displacement of the left globe secondary to an optic nerve glioma. |
P.425
Management
Treatment of orbital gliomas is controversial, ranging from observation to aggressive intracranial and orbital surgery. If the tumor is restricted to the orbit, vision is good, and proptosis is minimal, careful follow-up with repeated assessment of visual acuity, color vision, proptosis, visual fields, visual evoked response, and repeat CT or MRI scans is indicated. If vision is compromised, tumor growth documented, and the chiasm or other side are not involved, neurosurgical resection of the optic nerve from the chiasm to its insertion into the globe is the treatment of choice. In bilateral cases or those involving the chiasm, surgery is restricted to orbital resection for massively proptotic blind eyes or ventricular shunting to relieve hydrocephalus. The efficacy of irradiation or chemotherapy for this tumor is questionable.
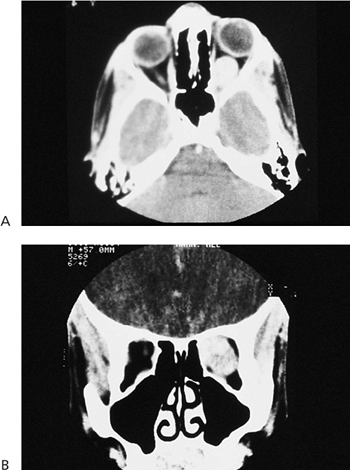 |
Figure 10.139. Axial (A) and coronal (B) computed tomography scans demonstrate fusiform enlargement of the optic nerve caused by a well-circumscribed optic nerve glioma. |
Neurofibroma
Clinical Features
Neurofibromas can occur as solitary tumors or as manifestations of systemic neurofibromatosis. Pathologically, they are composed of intertwining bundles of Schwann cells within the nerve sheaths, endoneural fibroblasts, and axons. In contrast to the solitary neurofibromas that occur in the second decade, the diffuse and plexiform lesions present in the early years. Early plexiform neurofibromas present with ptosis and eyelid thickening; however, the globe itself, including sclera, iris, ciliary body, cornea, and choroid, may be infiltrated, resulting in glaucoma and buphthalmos (Fig. 10.140). Visual loss may occur as a result of occlusion amblyopia, orbital and ocular infiltration, or glaucoma. Neurofibromatosis is associated with caf -au-lait spots, axillary freckling, fibroma molluscum, dysplasia of the orbital walls, congenital glaucoma, iris nodules, optic nerve gliomas, and perioptic meningiomas. Absence of portions of the sphenoid wing can cause pulsating exophthalmos (Fig. 10.141).
Neurofibromatosis is inherited as an autosomal-dominant gene with irregular penetrance. Two forms of the disease, central and peripheral, occur, and they rarely overlap. Peripheral neurofibromatosis is heralded by the development of caf -au-lait spots, associated with visceral neurofibromas, pheochromocytomas, and neurilemomas, and various skeletal abnormalities, including absence of the sphenoid wing. Central neurofibromatosis is characterized by development of tumors in the CNS, including astrocytomas, meningiomas, schwannomas, and ependymomas.
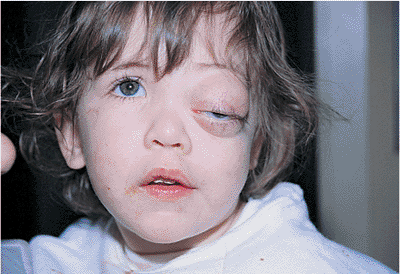 |
Figure 10.140. Neurofibroma of the left orbit results in proptosis, downward displacement of the globe, ptosis, and glaucoma secondary to ocular involvement. |
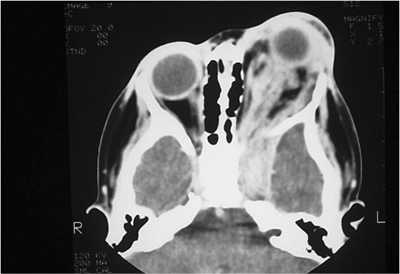 |
Figure 10.141. An axial computed tomography scan shows a diffuse neurofibroma of the left orbit and intracranial extension. |
P.426
Management
Solitary neurofibromas are rare. However, they can be easily excised. Plexiform lesions are difficult to treat, because they infiltrate normal tissues and recurrent debulking procedures may not be cosmetically satisfactory. Some surgeons suggest that debulking procedures are unacceptable and that, in the cases of major orbital involvement, only exenteration can produce an effective result.
Meningiomas
Meningiomas arise from the meningothelial cap cells of the arachnoid villi. They most commonly occur in middle-aged women, and the symptoms depend on the lesion's location. In addition to meningiomas of the optic nerve, intracranial meningiomas can affect orbital and visual structures, especially suprasellar, sphenoid ridge, and olfactory groove tumors.
Clinical Features
Optic nerve sheath meningiomas initially cause visual disturbances that are usually associated with minimal proptosis. Color vision abnormalities and visual field changes such as enlargement of the blind spot or a contracted visual field precede the loss of visual acuity. The optic nerve undergoes progressive edema, with engorgement of papillary and peripapillary vessels, development of optociliary shunt vessels, and eventual optic atrophy. CT scans and MRI studies are helpful in the diagnosis and follow-up (Fig. 10.142). The intracranial extent of the tumor can be best visualized on gadolinium-enhanced MRI scans. The tumor can appear as a tubular, fusiform enlargement or as a globular thickening. Psammomatous meningiomas are calcified and can demonstrate by a railroad track sign on the CT scan, with a lucent optic nerve surrounded by high-density tumor.
The sphenoid wing meningioma is the most common intracranial meningioma to present with orbital findings. This tumor arises from the dura of the sphenoid bone, grows in a plaque pattern, is infiltrative, and causes hyperostosis, making total excision difficult and recurrence common. Vision is often not affected, and early findings can be subtle. Unilateral eyelid edema, temporal fullness, and proptosis may be evident in the absence of cranial nerve palsies and visual deficits (Fig. 10.143). Vision is eventually threatened from optic nerve compression by the tumor or hyperostosis. Visual loss can slowly progress, and similar to the findings in optic nerve sheath tumors, color vision and field loss can precede changes in visual acuity. A combination of CT scans and MRI studies can delineate the extent of the tumor. Hyperostosis and psammomatous calcifications are easily seen on CT scans, and MRI with gadolinium enhancement better delineates the soft tissue extent of the tumor (Figs. 10.144, 10.145).
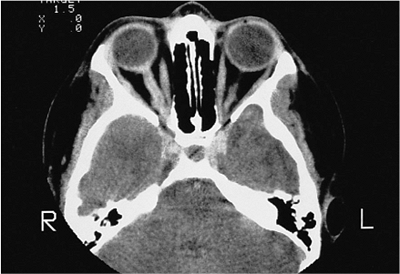 |
Figure 10.142. An axial scan of an optic nerve sheath meningioma of the right optic nerve. Notice the railroad-track sign of a central lucent optic nerve surrounded by tumor. |
Management
Patients with good vision and slowly progressive lesional growth can be observed with repeated CT or MRI studies to document the progression of optic nerve sheath meningiomas. Aggressive tumors or those with intracranial spread require surgical excision. After the tumor is suspected of entering the optic canal, a combined neurosurgical orbital approach is indicated to ensure complete excision and prevent recurrence and spread to the chiasm. Radiotherapy is reserved for certain bilateral cases, and its efficacy is controversial. Chemotherapy is ineffective.
 |
Figure 10.143. The T1-weighted magnetic resonance image demonstrates the intracranial extension of an optic nerve sheath meningioma. |
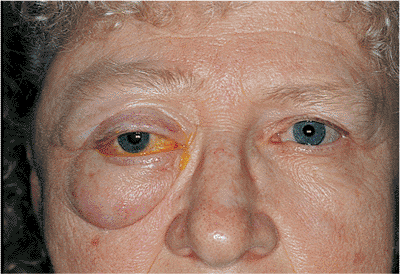 |
Figure 10.144. A patient with a sphenoid wing meningioma presented with massive proptosis, swelling of the eyelids, and fullness in the right temporalis fossa. Vision and globe motility were intact. |
P.427
Observation or surgical excision are the primary options in the treatment of sphenoid wing meningiomas. Slowly progressive lesions in older women can be observed for long periods, until cosmesis is unacceptable and vision is deteriorating. Bony invasion and involvement of vital structures may prevent complete excision; however, major debulking with removal of tumor and hyperostotic bone can decrease proptosis and decrease compressive symptoms, with some reversal of vision. Although radiotherapy is recommended after incomplete excision, its efficacy is questionable.
Mesenchymal Tumors
Rhabdomyosarcoma
Clinical Features
Rhabdomyosarcoma is the most common primary orbital malignancy of childhood. The tumor arises from the pluripotential mesenchyme that normally differentiates into striated muscle. The well-differentiated form is rare, with the embryonal and alveolar type being most common. Rhabdomyosarcoma presents with rapidly evolving exophthalmos associated with eyelid injection and swelling (Fig. 10.146). The tumor is most commonly found in superior and retrobulbar locations, and it can erode through the orbital bones into the sinuses, causing nasal stuffiness or nosebleeds. On CT scans, the mass can be circumscribed or poorly defined and homogeneous in density; it can destroy bone (Fig. 10.147). The differential diagnosis includes neuroblastoma, chloroma (i.e., granulocytic sarcoma), pseudotumor, lymphangioma, and hemangioma.
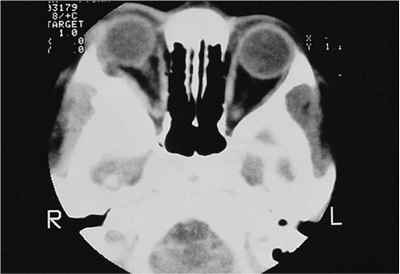 |
Figure 10.145. An axial computed tomography scan of a sphenoid wing meningioma demonstrates massive hyperostosis of the right sphenoid wing. |
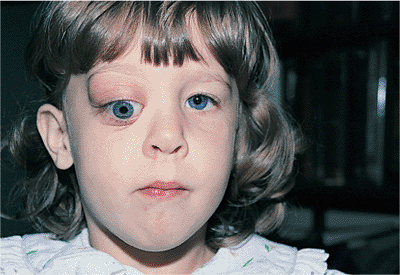 |
Figure 10.146. A child presented with rapidly evolving proptosis, downward displacement of the globe, and erythema of the eyelids. |
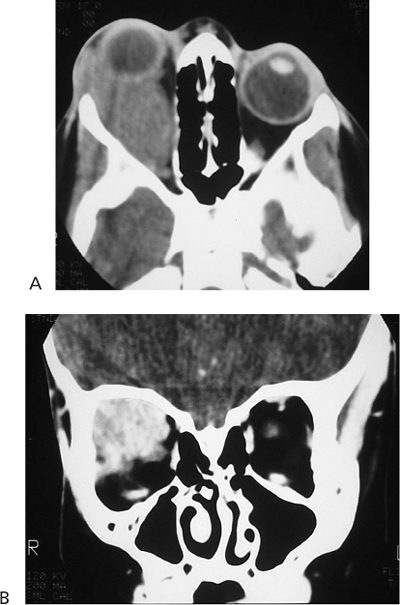 |
Figure 10.147. Axial (A) and coronal (B) computed tomography scans of a rhabdomyosarcoma reveal a large, homogenous mass in the superior aspect of the right orbit. |
P.428
Management
Treatment consists of immediate biopsy and excision, followed by irradiation and chemotherapy. The radiation dose varies from 4000 to 6000 cGy units, depending on the amount of residual tumor and age of the child. After irradiation, visual loss secondary to corneal changes, cataracts, and retinal problems are very common. Enophthalmos, ptosis, lacrimal duct stenosis, facial asymmetry, and bone hypoplasia can also occur. Chemotherapy consists of vincristine, actinomycin, and cyclophosphamide. Some centers are now advocating excision followed by chemotherapy without irradiation, especially in younger children. Exenteration is reserved for the rare radioresistant and recurrent tumor. The overall 5-year survival rate is 95% for cases of orbital rhabdomyosarcoma, much better than for rhabdomyosarcomas elsewhere in the body. All children should be referred to a pediatric oncology center, where a complete workup for metastases and staging of the disease is done before commencing appropriate treatment.
Fibrous Histiocytoma
Clinical Features
The most common mesenchymal orbital tumor of adults is the fibrous histiocytoma. The tumor consists of fibrous-appearing histiocytic cells and usually presents in middle life, occurs equally in men and women, and has a variable growth pattern. Proptosis and visual disturbances secondary to a mass effect are the most common findings. On CT scans, the mass is usually well circumscribed and can resemble other tumors such as a cavernous hemangioma and hemangiopericytoma.
Management
Complete tumor excision is the treatment of choice. However, if the lesion is recurrent and aggressive, a limited exenteration may be necessary to eliminate the disease. Radiotherapy has not been effective.
Lacrimal Gland Tumors
Benign Mixed Tumor or Pleomorphic Adenoma
Clinical Features
The pleomorphic adenoma presents in the fourth and fifth decades with slowly progressive proptosis and displacement of the globe downward and medially. At the time of presentation, the symptoms have usually existed for a few or more months. The tumor is usually not painful unless it has undergone malignant change or corneal exposure has occurred. On CT scans, it appears as a globular, well-circumscribed mass, and pressure may have expanded the lacrimal fossa (Fig. 10.148).
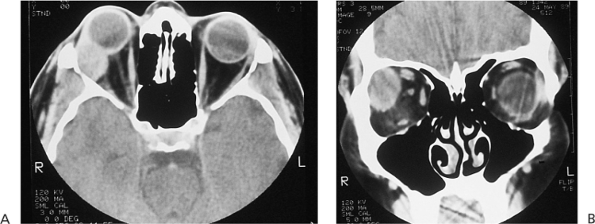 |
Figure 10.148. Axial (A) and coronal (B) computed tomography scans demonstrate a globular, well-circumscribed mass in the right lacrimal fossa, which is typical of a benign mixed tumor. |
Management
Complete excision without biopsy is the treatment of choice. If the tumor is not completely excised, it can recur and rarely undergoes malignant change. Similarly, longstanding pleomorphic adenomas that are not removed can undergo malignant change, heralded by increasing symptoms, especially pain.
Adenoid Cystic Carcinoma
Clinical Features
Adenoid cystic carcinoma is the most common epithelial malignancy of the lacrimal gland. Its incidence peaks in the fourth and second decades. Misdiagnosis is most common in the younger age group, because the tumor is not suspected. The tumor usually occurs as a rapidly developing mass in the lacrimal fossa. Because of its propensity for perineural spread, it can be associated with pain or paresthesia. On CT scans, the tumor can be well circumscribed. However, bony destruction, calcification, or extension toward the orbital apex are more often diagnostic (Fig. 10.149). The course of the disease can vary from rapid progression to a more indolent course, with long periods of remission followed by recurrent disease locally or metastasis to the lungs. The 5-year survival rate is approximately 50%, with a continuing decline to less than 20% in 15 years. The relentlessly fatal course despite long disease-free intervals necessitates aggressive long-term follow-up.
Management
Exenteration followed by radical radiotherapy was regarded as the treatment of choice for adenoid cystic carcinoma. Wide resection followed by radiotherapy does not seem to negatively alter the prognosis and is the current treatment of choice. The dismal prognosis may be related to the escape of the tumor past the surgical margins of resection by early perineural or bony spread before diagnosis.
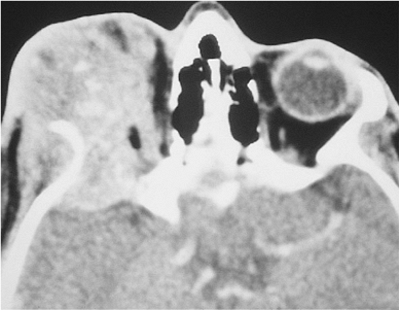 |
Figure 10.149. An axial computed tomography scan shows adenoid cystic carcinoma of the lacrimal gland with intracranial invasion. Notice the bone destruction and calcification. |
P.429
Secondary Tumors
Metastatic Tumors
Metastases to the orbit can be categorized as those appearing in the pediatric or the adult group. In children, the most common metastases include neuroblastoma, leukemia, and Ewing's sarcoma. Breast, lung, genitourinary, gastrointestinal, prostate, thyroid, carcinoid, and renal metastases occur in the adult population. Adult metastatic disease can present with inflammatory findings, diplopia, and pain. The development of enophthalmos rather than proptosis secondary to tumor-induced cicatrization is most commonly seen in sclerosing breast carcinoma. However, it can occur in lung, prostate, and gastrointestinal carcinomas.
Neuroblastoma
Clinical Features
Neuroblastoma arises from the primitive neuroblasts of the sympathetic system, most commonly from the retroperitoneal abdominal region near the adrenal gland, followed by the thorax, cervix, and pelvis. Orbital involvement occurs late in the disease and presents with abrupt ecchymotic unilateral or bilateral proptosis, ptosis, and periorbital swelling. Other ocular manifestations include opsoclonus-myoclonus and Horner's syndrome secondary to mediastinal involvement. CT scans reveal involvement of the superolateral orbit and zygoma with areas of bony destruction and lucency secondary to necrosis.
Management
Treatment consists of aggressive chemotherapy with or without local irradiation. Disease-free survival after 2 years is less than 10%. Total-body irradiation followed by bone marrow rescue has had encouraging results.
Breast Carcinoma
Clinical Features
Breast carcinoma is the most common orbital metastasis in women. It usually occurs a few years after mastectomy, but it can be the presenting symptom or occur some 20 years later. The tumor causes a firm infiltration of the orbit and has a predilection for extraocular muscles, resulting in diplopia. The sclerosing cicatricial breast carcinoma results in enophthalmos, ptosis, and restricted motility (Fig. 10.150). CT scans reveal a diffuse, infiltrative, reticulated pattern and involvement of the muscles. On MRI, the lesions often are hyperdense on the T2-weighted image (Fig. 10.151).
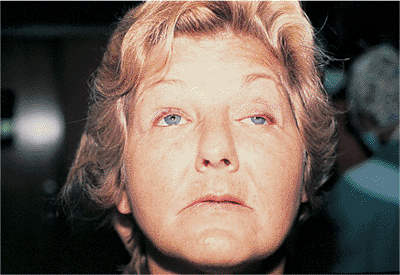 |
Figure 10.150. The patient has breast metastases to the left superior orbit, resulting in ptosis, decreased elevation of the left globe, and enophthalmos. |
Management
The treatment of orbital breast carcinoma is primarily radiation therapy, with adjunctive hormonal manipulation if indicated. The indication for chemotherapy is a rapidly progressing systemic disease, especially in an estrogen receptor negative tumor. The purpose of the treatment is palliative, because even the most aggressive treatment does not usually increase survival after the disease is metastatic.
Mucocele
Clinical Features
Mucoceles are cystic lesions arising most commonly from the frontal and ethmoidal sinuses. Obstruction of the normal sinus ostia results in entrapment of secretory epithelium, expansion of the sinus, thinning of the bony walls, and eventual erosion into the orbit. Patients with frontoethmoidal mucoceles present with a downward and lateral displacement of the globe. Maxillary mucoceles can cause an upward or downward displacement of the globe, depending on the destruction of the orbital floor and
P.430
size of the cyst. Mucoceles of the sphenoid and posterior ethmoid sinus can cause functional visual problems such as a loss of vision and diplopia secondary to compression of the optic nerve, apical structures, and cavernous sinus. The diagnosis is easily made on CT scans, which demonstrate an expanded, opacified sinus filled with a soft tissue density mass with well-defined margins and with erosion and sclerosis of bone (Fig. 10.152).
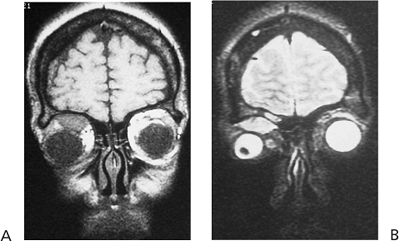 |
Figure 10.151. T1-weighted (A) and T2-weighted (B) magnetic resonance images of a breast carcinoma metastasis in the right superior orbit. Notice the hyperdense lesion on the T2-weighted scan. |
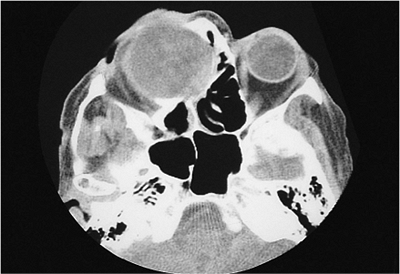 |
Figure 10.152. Computed tomography scan of a large, globular frontoethmoidal mucocele. |
Management
Treatment is complete removal of the mass and secretory epithelium with reestablishment of normal sinus drainage or obliteration of the sinus with a fat implant. External and endoscopic surgical approaches are used.
Bibliography
Abramson DH, Ellsworth RM, Tretter P, et al. The treatment of orbital rhabdomyosarcoma with radiation and chemotherapy. Ophthalmology 1979;86:1330.
Abramson DH, Jereb B. Radiation treatment of malignant ophthalmic neoplasms. In: Smith BC. Ophthalmic plastic and reconstructive surgery, vol 2. St. Louis: CV Mosby; 1987:1165.
Alper MG. Management of primary optic nerve sheath meningiomas. Current status-therapy in controversy. J Clin Neuroophthalmol 1981;1:101.
Arndt KA. Manual of dermatologic therapeutics. Boston: Little Brown & Co; 1978.
Arthurs B, Silverstone P, Della Rocca RC. Medial wall fractures. In: Bosniak SL, Smith BC. Advances in ophthalmic plastic and reconstructive surgery, vol 6. New York: Pergamon Press; 1987:393.
Baghdassarian SA, Shammas HF. Eosinophilic granuloma of the orbit. Ann Ophthalmol 1977;9:1247.
Ballen PH. Thermal chemical and radiation burns. In: Steward WB. American Academy of Ophthalmology manual on ophthalmic plastic and reconstructive surgery. San Francisco: American Academy of Ophthalmology; 1984:280.
Beard C. Ptosis. 3rd ed. St. Louis: CV Mosby; 1981.
Bedrossian EH Jr. Eyelid trauma repair. In: Albert DM. Ophthalmic surgery: Principles and techniques. Blackwell Science; 1999:1155.
Bedrossian EH Jr. Evaluation of orbital injuries. In: Bosniak SL, Smith BC. Advances in ophthalmic plastic and reconstructive surgery. New York: Pergamon Press; 1987:37.
Bedrossian EH Jr. Banked fascia lata as an orbital floor implant. Ophthal Plast Reconstr Surg 1993;1:66.
Berlin AJ. Acute adnexal trauma. In: Stewart WB. Ophthalmic plastic and reconstructive surgery. Academy manuals program. San Francisco: American Academy of Ophthalmology; 1984:273.
Bosniak SL. Ectropion. In: Smith BC. Ophthalmic plastic and reconstructive surgery. St. Louis: CV Mosby; 1987:562.
Boyd MJ, Collin O Jr. Capillary hemangiomas: an approach to their management. Br J Ophthalmol 1991;75:298.
Bullock JD, Warwar RE, Bartley GB, et al. Unusual orbital foreign bodies. Ophthal Plast Reconstr Surg 1999;15:44-51.
Bullock JD, Yanes B. Ophthalmic manifestations of metastatic breast cancer. Ophthalmology 1980;87:961.
Caronni EP. Craniofacial surgery. Boston: Little Brown & Co; 1985.
Cassady JV. Developmental anatomy of the nasolacrimal duct. Arch Ophthalmol 1952;47:141.
Castronuovo S, Prokhel GB. Orbital hemorrhage. In: Linberg JV. Oculoplastic and orbital emergencies. East Norwalk, CT: Appleton & Lange; 1990:145.
Char DH. Thyroid eye disease. 2nd ed. New York: Churchill-Livingstone; 1990.
Collin JR. Entropion and trichiasis. In: Stewart WB. Ophthalmic plastic and reconstructive surgery. San Francisco: American Academy of Ophthalmology; 1984:131.
Crawford JS, Pashby RC, Apt RK. Embryology and congenital anomalies. In: Stewart WB. Ophthalmic plastic and reconstructive surgery. San Francisco: American Academy of Ophthalmology; 1984:90.
Della Rocca RC, Bedrossian EH Jr. Orbital floor fractures. In: Clayman HM. Atlas of contemporary ophthalmic surgery. St. Louis: CV Mosby; 1980;899.
Della Rocca RC, Bedrossian EH Jr. Zygomaticomaxillary fractures. In: Clayman HM. Atlas of contemporary ophthalmic surgery. St. Louis: CV Mosby; 1980:909.
Demant E, Hurwitz JJ. Canaliculitis: review of twelve cases. Can J Ophthalmol 1980;15:73.
Dortzbach RK, Segrest DR. Orbital aspergillosis. Ophthal Surg 1983;14:240.
Duke-Elder S. Congenital anomalies of the ocular adnexa. In: Duke-Elder S. System of ophthalmology, vol 3. St. Louis: CV Mosby; 1963:827.
Ellis JH, Banks PM, Campbell J, et al. Lymphoid tumor of the ocular adnexa. Ophthalmology 1985;92:1311.
Engrav LH, Heimbach DM, Walkinshaw MD, et al. Excision of burns of the face. Plast Reconstr Surg 1986;77:744.
Ferry AP, Abedi S. Diagnosis and management of rhino-orbital-cerebral mucormycosis (phycomycosis): a report of 16 personally observed cases. Ophthalmology 1983;90:1096.
Flanagan JC, Mauriello JA. Management of lacrimal sac tumors. Adv Ophthal Plast Reconstr Surg 1984;3:399.
Flanagan JC, Stokes DP. Lacrimal sac tumors. Ophthalmology 1985;85:1282.
Font RF, Ferry AP. Carcinoma metastatic to the eye and orbit: III. A clinicopathologic study of 28 cases metastatic to the orbit. Cancer 1976;38:1326.
Font RL, Gamel JW. Adenoid cystic carcinoma of the lacrimal gland. A clinicopathologic study of 79 cases. In: Nicholson DH. Ocular pathology update. New York: Masson Publishing USA; 1980:277.
Font RL, Gamel JW. Epithelial tumors of the lacrimal gland: an analysis of 265 cases. In: Jakobiec FA. Ocular and adnexal tumors. Birmingham: Aesculapius; 1978:787.
Font RL, Hidayat AA. Fibrous histiocytoma of the orbit: a clinicopathologic study of 150 cases. Hum Pathol 1982;13:199.
Font RL. Eyelids and lacrimal drainage system. In: Spencer WH. Ophthalmic pathology. 3rd ed. Philadelphia: WB Saunders; 1986:2141.
Frank A, Wachtel T. The early treatment and reconstruction of eyelid burns. J Trauma 1983;23:874.
Friedberg MD, Rapuano CJ. Wills Eye Hospital office and emergency room diagnosis and treatment of eye disease. Philadelphia: JB Lippincott; 1990:17.
P.431
Garber PF, MacDonald D, Beyer-Machale CK. Management of trauma to the eyelids. In: Smith BC. Ophthalmic plastic and reconstructive surgery. St. Louis: CV Mosby; 1987:437.
Garber PF. Management of injuries to the lacrimal system. In: Bosniak SL, Smith BC. Advances in ophthalmic plastic and reconstructive surgery, vol 3. New York: Pergamon Press; 1984:1175.
Geeraetes WJ. Ocular syndromes. 3rd ed. Philadelphia: Lea & Febiger; 1976.
Glaser JS. Infranuclear disorders of eye movement. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 2. Philadelphia: JB Lippincott; 1993.
Glassburn JR, Klionsky M, Brady LW. Radiation therapy for metastatic disease involving the orbit. Am J Clin Oncol 1984;7:145.
Griffith DG, Salasche SJ, Clemons DE. Cutaneous abnormalities of the eyelid and face. New York: McGraw-Hill; 1987.
Grove AS, McCord CD. Orbital disorders: diagnosis and management. In: McCord CD, Tannenbaum M. Oculoplastic surgery. New York: Raven Press; 1987.
Grove AS. Giant dermoid cysts of the orbit. Ophthalmology 1979;86:1513.
Haik BG, Carroll GS, Kilmer SL. Periocular hemangiomas. In: Mannis MJ, Macsai MS, Huntley AC. Eye and skin disease. Philadelphia: Lippincott-Raven; 1996.
Haik BG, Karcioglu ZA, Pechous BP. Capillary hemangioma (infantile periocular hemangioma). Surv Ophthalmol 1994;38:399.
Hanneken AM, Miller NR, et al. Treatment of carotid-cavernous sinus fistulas using a detachable balloon catheter through the superior ophthalmic vein. Arch Ophthalmol 1989;107:87.
Harris GJ, Sakol PJ, et al. An analysis of thirty cases of orbital lymphangioma: pathophysiologic considerations and management recommendations. Ophthalmology 1990;97:1583.
Harris GJ, Will BR. Orbital aspergillosis. Ophthal Plast Reconstr Surg 1989;5:207.
Harris GT, DiClementi D. Congenital dacryocystocele. Arch Ophthalmol 1982;100:1763.
Hawes MJ. Canalicular lacerations. In: Linberg JV. Oculoplastic and orbital emergencies. East Norwalk, CT: Appleton & Lange; 1990:15.
Houle TV, Ellis PP. Aspergillosis of the orbit with immunosuppressive therapy. Surv Ophthalmol 1975;20:35.
Hoyt C, Lambert S. Lids. In: Taylor D. Pediatric ophthalmology. Boston: Blackwell Scientific Publications; 1990:141.
Hoyt WF, Baghdassarian SA. Optic glioma of childhood. Natural history and rationale for conservative management. Br J Ophthalmol 1969;53:793.
Iliff CE. Mucoceles in the orbit. Arch Ophthalmol 1973;89:392.
Imes RK, Hoyt WF. Childhood chiasmal gliomas: update on the fate of patients in the 1969 San Francisco study. Br J Ophthalmol 1986;70:179.
Jackson IT, Laws ER, Martin RD. The surgical management of orbital neurofibromatosis. Reconstr Surg 1983;71:751.
Jakobiec FA, Depot MJ, Kennerdell JS, et al. Combined clinical and computed tomographic diagnosis of orbital glioma and meningioma. Ophthalmology 1984;91:137.
Jakobiec FA, Iwamoto T, Patell M, et al. Ocular adnexal monoclonal lymphoid tumors with a favorable prognosis. Ophthalmology 1986;93:1547.
Jakobiec FA, Jones IS. Neurogenic tumors. In: Duane's clinical ophthalmology, vol 2. Philadelphia: JB Lippincott; 1992.
Jakobiec FA, Jones IS. Vascular tumors, malformation, and degenerations. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 2. Philadelphia: JB Lippincott; 1992:6.
Jakobiec FA, Rootman J, Jones IS. Secondary and metastatic tumors of the orbit. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 2. Philadelphia: JB Lippincott; 1992.
Janetta PJ. Hemifacial spasm. In: Samili M, Janetta PJ. The cranial nerves. Berlin: Springer-Verlag; 1981:484.
Jones DB, Steinkuller PG. Microbial preseptal and orbital cellulitis. In: Duane TD. Clinical ophthalmology, vol 4. Philadelphia: JB Lippincott; 1989.
Jones HM. Some orbital complications of nose and throat conditions. J R Med Soc 1981;74:409.
Jones KL. Smith's recognizable patterns of human malformation. 5th ed. Philadelphia: WB Saunders; 1997.
Jordan DR, Patrinely JR, Anderson RL, et al. Essential blepharospasm and related dystonias. Surv Ophthalmol 1989;34:123.
Katowitz JA, Low JE. Lacrimal drainage surgery. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 6. Philadelphia: JB Lippincott; 1993.
Kennerdell JS, Dresner SC. The nonspecific orbital inflammatory syndromes. Surv Ophthalmol 1981;29:93.
Kilmer SL. Adnexal tumors. In: Mannis MJ, Macsai MS, Huntley AC. Eye and skin disease. Philadelphia: Lippincott-Raven; 1996.
Kingston JE, McElwain TJ, Malpas JS. Childhood rhabdomyosarcoma: experience of the children's solid tumor group. Br J Cancer 1983;48:195.
Kivela T, Tarkkanen A. The merkel cell and associated neoplasms in the eyelids and periocular region. Surv Ophthalmol 1990;35:171.
Kobalter AS, Roth A. Benign epithelial neoplasms. In: Mannis MJ, Macsai MS, Huntley AC. Eye and skin disease. Philadelphia: Lippincott-Raven; 1996.
Kobryn JL, Blodi FC, Weingeist TA. Ocular and orbital manifestations of neurofibromatosis. Surv Ophthalmol 1979;24:45.
Kohn R. Hepler Management of limited rhino-orbital mucormycosis without exenteration. Ophthalmology 1985;92:1440.
Kulwin D. Acute eyelid and periocular burns. In: Linberg JV. Oculoplastic and orbital emergencies. East Norwalk, CT: Appleton & Lange; 1990:39.
Kulwin D. Modern management of ophthalmic burns. Highlights Ophthalmol 1990;30:1.
Kulwin D. Treatment of periorbital burns. In: Bosniak SL. Advances in ophthalmic plastic and reconstructive surgery, vol 7. New York: Pergamon Press; 1987:167.
Kuper Smith MJ, Berenstein A, et al. Management of nontraumatic vascular shunts involving the cavernous sinus. Ophthalmology 1988;95:121.
Kushner BJ. Congenital nasolacrimal system obstruction. Arch Ophthalmol 1982;100:597.
Kushner BJ. Intralesional corticosteroid injection for infantile adnexal hemangiomas. Am J Ophthalmol 1982;93:496.
Lee DA, Campbell JR, Waller RR, et al. A clinicopathologic study of primary adenoid cystic carcinoma of the lacrimal gland. Ophthalmology 1985;92:128.
Leone CR, Lloyd WC. Treatment protocol for orbital inflammatory disease. Ophthalmology 1985;92:1325.
Leone CR. The management of ophthalmic Graves' disease. Ophthalmology 1984;91:770.
Leone CR. The management of pediatric lacrimal problems. Ophthal Plast Reconstr Surg 1989;5:34.
Lewis RA, Gerson LP, Axelson KA, et al. Von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology 1984;91:929.
Long JA. A method of monocanalicular silicone intubation. Opthalmic Surg 1988;3:204.
Lowry JC, Bartley GB. Complications of blepharoplasty. Surv Ophthalmol 1994;38:327.
Mansour AM, Cheng KP, et al. Congenital dacryocystocele; a collaborative review. Ophthalmology 1991;98:1744.
Maurer H, Foulkes M, Gehan E. Intergroup Rhabdomyosarcoma Study II, preliminary report. Proc Am Soc Clin Oncol 1983;2:70.
Mauriello JA, Flanagan JC. Management of orbital and ocular adnexal tumors and inflammations. New York: Field and Wood Medical Publishers; 1990:226.
P.432
McCord CD, Tanenbaum M, Dryden RM, et al. Eyelid malpositions. In: McCord CD, Tanenbaum M. Oculoplastic surgery. New York: Raven Press; 1987:279.
McCord CD, Tannenbaum M. Oculoplastic surgery. New York: Raven Press; 1987.
McNab AA, Colin D Jr. Eyelid and canthal lacerations. In: Linberg JV. Oculoplastic and orbital emergencies. East Norwalk, CT: Appleton & Lange; 1990:1.
Moster ML. CPEO, MERRF and MELAS. In: Abstracts from the North American Neuro-ophthalmology Society 18th Annual Meeting; 1992.
Mulliken JB, Young AE. Vascular birthmarks: hemangiomas and malformations. Philadelphia: WB Saunders; 1988.
Musarella MA, Chen HSL, DeBoer G, et al. Ocular involvement in neuroblastoma: prognostic implications. Ophthalmology 1984;91:936.
Nelson CC, Oestriecher J. Eyelid trauma. In: Focal points. San Francisco: American Academy of Ophthalmology; 1991:10.
Nunery WR, Martin RT. Zygoma and complex facial fractures. In: Linberg JV. Oculoplastic and orbital emergencies. East Norwalk, CT: Appleton & Lange; 1990:1167.
Nunery WR, Wilson FM. Suppurative canaliculitis. In: Bosniak SL, Smith BC. Advances in ophthalmic plastic and reconstructive surgery, vol 3. New York: Pergamon Press; 1984:157.
Orcutt JC. Orbital foreign bodies. In: Linberg JV. Oculoplastic and orbital emergencies. East Norwalk, CT: Appleton & Lange; 1990:183.
Osher RH, Schatz NJ, Duane TD. Acquired orbital retraction syndrome. Arch Ophthalmol 1980;98:1798.
Paris GL, Bullock JD, Lauring L. Ectropion. In: Stewart WB. Ophthalmic plastic and reconstructive surgery. San Francisco: American Academy of Ophthalmology; 1984:143.
Peterson RA, Robb RM. The natural course of congenital obstruction of the nasolacrimal duct. J Pediatr Ophthalmol Strabismus 1978;15:246.
Petro J, Tooze FM, Bales CR, et al. Ocular injuries associated with periorbital fracture. J Trauma 1979;19:730.
Reifler DM. Orbital metastases with enophthalmos: a review of the literature. Henry Ford Hosp Med J 1985;33:171.
Rice CD, Kersten RC, Mrak RE. An orbital hemangiopericytoma recurrent after 30 years. Arch Ophthalmol 1989;107:552.
Rootman J, Hay E, Graeb D, Miller R. Orbital-adnexal lymphangiomas. A spectrum of hemodynamically isolated vascular hamartomas. Ophthalmology 1986;93:1558.
Rootman J, Nugent R. The classification and management of acute orbital pseudotumors. Ophthalmology 1982;89:1040.
Rootman J, Robertson WJ. Tumors. In: Rootman J. Diseases of the orbit. Philadelphia: JB Lippincott; 1988.
Rootman J. Diseases of the orbit. Philadelphia: JB Lippincott; 1988:231.
Roy FH. Ocular differential diagnosis. 3rd ed. Philadelphia: Lea & Febiger; 1984.
Ryan SJ, Font RC. Primary epithelial neoplasms of the lacrimal sac. Am J Ophthalmol 1973;76:73.
Sauer GC. Manual of skin diseases. 6th ed. Philadelphia: JB Lippincott; 1991.
Scott AB. Botulinum treatment for blepharospasm. In: Smith BC. Ophthalmic plastic and reconstructive surgery. St. Louis: CV Mosby; 1987:609.
Sergott RC, Grossman RI, Savino PJ, Bosley TM, Schatz NJ. The syndrome of worsening of dural cavernous sinus arteriovenous malformation. Ophthalmology 1987;94:205.
Setzkorn RK, Lee DJ, Iliff NT, Green WR. Hemangiopericytoma of the orbit treated with conservative surgery and radiotherapy. Arch Ophthalmol 1987;105:1103.
Sherman RP, Rootman J, Lapointe JS. Orbital dermoids: clinical presentation and management. Br J Ophthalmol 1984;68:642.
Shore JW, McCord CD, Popham JK. Surgery of the eyelids. In: Duane's clinical ophthalmology, vol 6. Philadelphia: JB Lippincott; 1992.
Silver, Bernard, et al. Ophthalmic plastic surgery: a manual prepared for the use of graduates in medicine. San Francisco: American Academy of Ophthalmology; 1977:242.
Slamovits TL, Glaser JS. The pupils and accommodation. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 2. Philadelphia: JB Lippincott; 1993.
Slamovits TL. External disease and cornea. Basic clinical science course, sect 8. San Francisco: American Academy of Ophthalmology; 1993:87.
Slamovits TL. Neuro-ophthalmology. Basic clinical science course, sect 5. San Francisco: American Academy of Ophthalmology; 1993:119.
Slamovits TL. Orbit, eyelid and lacrimal system. Basic clinical science course, sect 7. San Francisco: American Academy of Ophthalmology; 1993:96.
Smit TJ, Mouritis MP. Monocanalicular lesions: to reconstruct or not. Ophthalmology 1999;106:1310.
Smith B, Regan WF Jr. Blowout fractures of the orbit: mechanism and correction of internal orbital fracture. Am J Ophthalmol 1957;44:733.
Smith BC. Ophthalmic plastic and reconstructive surgery. St. Louis: CV Mosby; 1987.
Smith BC, Nesi FA. Practical techniques in ophthalmic surgery. St. Louis: CV Mosby; 1981:50.
Sutula FC, Glover AT. Eyelid necrosis following intralesional corticosteroid injection for capillary hemangioma. Ophthalmic Surg 1987;24:103.
Tanenbaum M, McCord CD. The lacrimal drainage system. In: Tasman W, Jaeger EA. Duane's clinical ophthalmology, vol 4. Philadelphia: JB Lippincott; 1993.
Vinken PJ, Bruy GW. Handbook of clinical neurology, vol 42, part 1. Amsterdam: North Holland Publishing; 1981:319.
Vogintzks KV. Lymphoid tumors of the orbit and ocular adnexa: a long-term follow-up. Ann Ophthalmol 1984;16:1046.
Weinstein GS, Anderson RL. Diagnosis and treatment of blepharospasm. In: Smith BC. Ophthalmic plastic and reconstructive surgery. St. Louis: CV Mosby; 1987:600.
Weiss A, Friendly D, Eglin K, et al. Bacterial periorbital and orbital cellulitis in childhood. Ophthalmology 1983;90:195.
Wharam M, Beltangady M, Hays D, et al. Localized orbital rhabdomyosarcoma. An interim report of the Intergroup Rhabdomyosarcoma Study Committee. Ophthalmology 1987;94:251.
Wilson ME, Parker PL, Chavis RM. Conservative management of childhood orbital lymphangioma. Ophthalmology 1989;96:484.
Wilson WB, Gordon M, Lehman RAW. Meningiomas confined to the optic canal and foramina. Surg Neurol 1979;12:21.
Woog JJ, Albert DM, Solt LC, et al. Neurofibromatosis of the eyelid and orbit. Int Ophthalmol Clin 1982;22:157.
Wright JE. Orbital vascular anomalies. Trans Am Acad Ophthalmol 1974;78:OP606.
Wright JE, Call NB, Liaricos S. Primary optic nerve meningioma. Br J Ophthalmol 1980;64:553.
Wright JE, McDonald WI, Call NB. Management of optic nerve gliomas. Br J Ophthalmol 1980;64:545.
Wright JE. Factors affecting the survival of patients with lacrimal gland tumors. Can J Ophthalmol 1982;17:3.
Wright JE. Primary malignant neoplasms of the lacrimal gland. Br J Ophthalmol 1992;76:401.
Wright JE. Primary optic nerve meningiomas: clinical presentation and management. Trans Am Acad Ophthalmol Otolaryngol 1977;83:617.
Zimmerman RA, Bilaniuk LT, Metzger RA, et al. Computed tomography of orbital facial neurofibromatosis. Radiology 1983;146: 113.
EAN: 2147483647
Pages: 17