Chapter 5: DNA Computing and Its Frontiers
|
Carlo C. Maley
5.1 Introduction
This is a great age of discovery for the intersection between biology and computation. Most of the work in this intersection has consisted of the application of computational techniques to the analysis of biological data—so-called computational biology. This chapter is about the opposite endeavor, the application of biological techniques and substrates to computation. In November 1994, Adleman stunned the scientific community when he demonstrated that through the simple annealing of Watson-Crick complementary base pairs, strands of DNA could compute a solution to an instance of the Hamiltonian Path Problem (HPP). In retrospect, there were a number of precursors that presaged Adleman's discovery. Work in formal language theory (Head 1987) had examined formalisms based on DNA manipulations. At the wet benches of biology, artificial selection of RNA had engineered solutions to a number of biological problems (Beaudry and Joyce 1992; Sassanfar and Szostak 1993; Lorsch and Szostak 1994; Stemmer 1994). But it was not until Adleman (1994) that the field of DNA computing was launched. Research in DNA computing is moving quickly, so many of the references in this chapter will be out of date by the time of publication. Thus, rather than give a comprehensive explication of the entire field, I have chosen to try to provide the reader with an understanding of the techniques, approaches, and issues in DNA computing and related research. It is my hope that with these tools you may both assess the potential of the field and perhaps begin to contribute to it yourself. An alternative review can be found in Reif (1998). Păun and coworkers (Păun, Rozenberg, and Salomaa 1998) provide a thorough treatment of the various theoretical formalisms for DNA computing. I have intentionally skewed the discussion of results in this field toward experimental rather than theoretical work. There is no longer any doubt as to the theoretical potential of DNA computing. Yet only the crucible of the laboratory can extract its true potential.
Sequence That Launched a Thousand Sequences
Adleman has once again reminded us that computation is independent of its medium. He saw that there is computation in the interaction of molecules. His insight was that the sequence-specific binding properties of DNA allow it to carry out massively parallel computation in a test tube. The Hamiltonian Path Problem is a famous NP-complete problem in computer science. The question is, given a graph, a starting node, and an ending node, is there a path through the graph beginning and ending at the specified nodes such that every node in the graph is visited exactly once? Briefly, Adleman's experiment followed these six steps:
-
Create a unique sequence of 20 nucleotides (a 20-mer) associated with each vertex in the graph. Similarly, create 20-mer sequences to represent the edges in a graph. If two vertex sequences v1 and v2, were each composed of two 10-mer sequences x1y1 and x2y2, then an edge, e1,2 from v1 to v2 was a 20-mer composed of the Watson-Crick complement[1] of y1 and x2 (y1x2). Thus, when put in solution, the edge sequence will bind to the second half of the source vertex and the first half of the destination vertex and serve as a sort of "connector" for binding the vertices together.
-
Generate random paths through the graph by putting many copies of the vertex and edge sequences into a solution and letting them anneal to each other, as shown in figure 5.1. That is, any complementary sequences will bind to each other to form double-stranded DNA. In addition to the simple edge and vertex sequences, two further sequences were added. If vin = xinyin was the starting vertex and vout = xoutyout was the terminating vertex, the sequences xin and yout were added in order to "cap off" sequences starting with vin and ending with vout. The process of annealing resulted in long double strands of DNA with breaks in the "backbone" wherever one vertex (or edge) sequence ended and another began. These breaks were filled in by adding ligase enzyme to the solution, thus forming the covalent bonds between subsequences necessary to construct two coherent single strands of DNA annealed together.
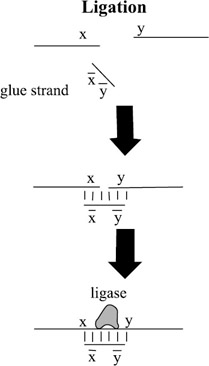
Figure 5.1: In order to connect strand y to strand x a "glue strand" is used to bring the two strands into proximity through annealing. Then a ligase enzyme is added to fuse the two strands together. Here x indicates the Watson-Crick complement of sequence x. -
Separate out all paths starting with vin and ending with vout. This was done not by actually separating the DNA representing good paths from DNA representing bad paths, but by exponentially replicating the good paths using a polymerase chain reaction (PCR). In this way, the proportion of good paths in the solution far outweighed the proportion of bad paths.
-
Separate out all paths that go through exactly n vertices (where n = 7 is the number of vertices in Adleman's graph). This was implemented by separating the strands by their lengths and identifying all 140-mers. Separation by length is typically carried out by gel electrophoresis. Different length strands of DNA move at different rates through an electrical field in a gel.[2] The section of the gel corresponding to 140-mers was identified, the DNA extracted from the gel, and replicated, or amplified, by PCR. This sequence of gel electrophoresis, extraction, and amplification was repeated several times in order to purify the sample.
-
Separate out all paths that go through all n vertices. This was done by first constructing (for each vertex in the graph) a set of magnetic beads attached to the complement sequence of the vertex. The double-stranded DNA from step 3 was "melted" to reduce it to a solution of single-stranded DNA, and then mixed with the magnetized vertex complement sequence for vertex 1. The sequences binding to these beads were magnetically extracted. They should represent paths that include vertex 1. The process was repeated on the results of the previous extraction for each vertex in the graph. At the end, only those paths that include all seven vertices should remain.
-
Detect if there are any DNA sequences in the remaining solution. This was done by first amplifying the results with PCR and then using gel electrophoresis to see if anything of the appropriate length is left.
Elegant in design, clumsy in implementation. This is perhaps appropriate for the first steps of a field. Let us consider the amount of time that was necessary to perform this experiment. The time required to create a desired sequence, such as the sequence representing v1, depends on the enzymes used; however, it is on the scale of hours (Gannon and Powell 1991). There are O(n2) such sequence generation processes in step 0, where n is the number of nodes in the graph. In many cases, subsequences might be taken from a preexisting DNA library or bought from a vendor. Adleman omits a description of how he obtained the sequences representing the vertices, except to say that the sequences were random. The process of annealing (step 1) requires about 30 seconds (Vahey, Wong, and Michael 1995: 20). Each PCR process (step 3, multiple times, and step 5) takes approximately 2 hours (Vahey, Wong, and Michael 1995: 20). The time required for gel electrophoresis (steps 2 and 5) depends on the gel used and the size and charge of the molecules. For the agarose gel that Adleman used, electrophoresis takes about 5 hours. For a polyacrylamide gel, it takes about 70 minutes (Kaplan, Cecci, and Libchaber 1995). It should also be noted that gel electrophoresis currently requires human intervention to visually identify and extract the desired results. Constructing the magnetic beads bound to premade sequences is a simple annealing operation. Finally, separation using the magnetic beads, which Adleman noted was the most labor-intensive step of the algorithm (performed n times in step 4), requires at a minimum approximately 1 hour using standard techniques (L nneborg, Sharma, and Stovgaard 1995: 445). The entire experiment took Adleman 7 days of lab work. Adleman asserts that the time required for an entire computation should grow linearly with the size of the graph. This is true as long as the creation of each edge does not require a separate process. But the limiting factor here is the fact that the volume of DNA needed to compute the path grows exponentially with the size of the graph.
Kaplan and coworkers (Kaplan, Cecci, and Libchaber 1995) rightly criticize Adleman for not using a positive or a negative control. This is a serious issue, in part because the only known attempt to replicate Adleman's experiment produced ambiguous results (Kaplan, Cecci, and Libchaber 1995).
Despite all the practical delays of the biological manipulations, the real excitement over Adleman's work comes from the fact that in step 1, a massively parallel computation of all the possible paths through the graph is carried out in O(1) time. With an estimated upper bound of 1020 "operations" in step 1 (Adleman 1994), the massive parallelism of DNA computing might be able to make the delays in the other steps seem negligible. In fact, Adleman (1994) makes a series of claims for the promise of DNA computing. DNA computation may outstrip modern silicon computers on three counts:
-
Speed. This should fall out from the massive parallism of the approach, amortized across the slow serial operations.
-
Energy efficiency. Because the molecules actually release energy when they anneal together, there is some hope that computations could be carried out using very little energy.
-
Information density. Packing 1020 strands of data into a liter of volume would give us an information density at least five orders of magnitude better than current hard drive technology.
But more immediately, Adleman identified a number of important open questions for the new field:
-
What is the theoretical power of DNA computation?
-
Are there practical algorithms for solving problems with DNA?
-
Can errors be adequately controlled?
-
Gifford (1994) adds: Is there universal computation in naturally occurring systems?
The purpose of this chapter is to introduce the state of the art of DNA computing as well as the related research programs that it has launched. The ships of many proposals have set sail for this new world. Most have not reached the point of implementation in the real world, although some are beginning to establish colonies. It remains to be seen if any will bring back the gold for which they were sent.
Mapping the Known World
The majority of work in DNA computing has taken place in vitro. That is, researchers have sought to manipulate DNA in test tubes and other artificial environments. This is the subject of section 5.2. DNA computing in vitro has no relation to biology or life except that it shares some of the same substrates and tools. A more recent, radical, and less developed approach is to carry out computation within a cell (generally E. coli), as described in section 5.3. In a sense, this in vivo form of computation is the old engineering trick of adding a layer of abstraction to our machines so as to give ourselves more powerful tools. Adleman's techniques are a little like machine code. The researcher must explicitly, and laboriously, drive each operation to change the state of the computation. Computation in a cell might be more like an assembly language. The cell can provide some of the nitty-gritty work to manipulate the computation, while the researcher need only provide an appropriate environment for the cells.
Within the world of in vitro DNA computation, I will discuss in section 5.2 the kinds of manipulations of DNA that have been discussed in the literature. This will provide a sketch of the tools that we have at hand. I will then show how various subsets of these tools have been proposed as the bases for computing with DNA. This is still an area of great ferment, for the choice of computational model will probably determine whether or not DNA computing will ever outperform its silicon brother. The end of section 5.2 reports on some of the daunting practical obstacles that will face any colonists to this new world. Section 5.3 discusses computation in cells. Finally, section 5.4 attempts to summarize the current state of the field and makes some predictions for its future.
[1]The four nucleic acids that make up DNA combine in "Watson-Crick" complementary pairs. Thus Adenine (A) binds to Thymine (T) while Guanine (G) binds to Cytosine (C). Any sequence of nucleotides, such as TAGCC, has a complementary sequence, ATCGG, that will bind to it so as to form a double strand of DNA.
[2]For a strand of length N < 150, biologists have experimentally found that the distance a DNA strand travels is d = a − bln(N), for some constants a and b.
|