XXIV - Malignant Lesions of the Esophagus
Editors: Shields, Thomas W.; LoCicero, Joseph; Ponn, Ronald B.; Rusch, Valerie W.
Title: General Thoracic Surgery, 6th Edition
Copyright 2005 Lippincott Williams & Wilkins
> Table of Contents > Volume II > The Mediastinum > Section XXIX - Primary Mediastinal Tumors and Syndromes Associated with Mediastinal Lesions > Chapter 175 - Pathophysiology of Myasthenia Gravis
Chapter 175
Pathophysiology of Myasthenia Gravis
Jun Li
Robert P. Lisak
Myasthenia gravis (MG) is considered the prototypic autoimmune disease. Salient features of the disease are muscle weakness, decremental motor response to repetitive electrical stimulation, symptomatic improvement with drugs that inhibit acetylcholine esterase, and the presence of anti-acetylcholine receptor (anti-AChR) autoantibodies in most patients with generalized MG.
Historically, the first description of MG is controversial. A clear systematic description of the disease did not occur until 1900, when Campbell and Bramwell reported 60 cases. Subsequent electrophysiologic studies by Harvey and Masland (1941) provided important diagnostic features of the disease. However, successful analysis of MG at the cellular and molecular level only became possible after the isolation of a toxin that specifically binds to AChR, -bungarotoxin (BuTx), from krait snakes, reported by Chang and Lee (1966) and Changeux and associates (1970), and purified AChR from the electric organ of Torpedo by Miledi and colleagues in 1971. An autoimmune mechanism was proved by a serendipitous observation in 1973. Rabbits immunized with Torpedo AChR became paralyzed while experimenters were attempting to produce antibodies to study AChR. Anti-AChR antibodies were demonstrated by Patrick and Lindstrom (1973) to cause the blocking of neuromuscular junction transmission. The causative role of autoimmune mechanism in human MG was firmly established when anti-AChR antibodies were demonstrated in serum of MG patients and MG was reproduced in animals with passive transfer of immunoglobulin G (IgG) from human MG serum or with AChR-specific monoclonal antibodies by numerous investigators, including Patrick and Lindstrom (1973) as well as Lennon (1975), Fambrough (1973), Toyka (1975), Lindstrom (1976a, 1976b), Engel (1976), and Richman (1980) and their colleagues. In this chapter, we review the molecular structure and physiology of AChR, neuromuscular junction signal transmission, and pathophysiologic mechanism of MG.
NEUROMUSCULAR JUNCTION TRANSMISSION
Axons of motor neurons communicate with muscle cells through a specialized structure called neuromuscular synapse (Fig. 175-1). The synapse consists of three parts: (a) a presynaptic terminal, an enlarged part of distal axon, which synthesizes, stores and releases neurotransmitters, such as acetylcholine (ACh); (b) a synaptic cleft, a microscopic space between the presynaptic and postsynaptic terminals; and (c) a postsynaptic terminal, also called the end plate, where the receptors for neurotransmitter (such as AChR) are located. During development, when the motor nerve fiber approaches the muscle cell, it helps induce a specialized indentation of muscle cell surface, the end plate. Upon the activation of presynaptic motor axons by propagated electrical activities, there is concurrent calcium influx to the presynaptic axon. These events activate coordinated intraaxonal molecular processes, which drive the neurotransmitter vesicles containing ACh to the presynaptic terminal membrane. Fusion of vesicles with the presynaptic terminal membrane takes place, and the ACh is released in a quantal fashion. ACh diffuses rapidly across the synaptic cleft, binds to AChR, and directly opens the ion channel of AChR (see Fig. 175-1). An individual ACh ion channel behaves in all-or-none pattern and produces a fixed amplitude of current (2.7 pA) when it opens, as described by Sakmann and colleagues (1980). A normal end plate current reflects the sum of 200,000 ACh-channel currents and generates an end plate potential (EPP) of about 70 mV. The amplitudes of end plate current or potential are large, but the current is not regenerative, unlike the current through voltage-gated ion channels. The depolarizing current through a voltage-gated ion channel, such as a sodium channel, may cause additional sodium channels to open. Thus, the current from many sodium channels may sum up, reach the threshold, and produce action potentials. In contrast, the ACh-induced EPP is unable to activate additional AChR ion channels. EPP must recruit and activate the sodium channels in the
P.2625
vicinity of the postsynapses, leading to the production of action potentials. Thus, the number of available postsynaptic AChRs is critical to the successful postsynaptic signal production and propagation.
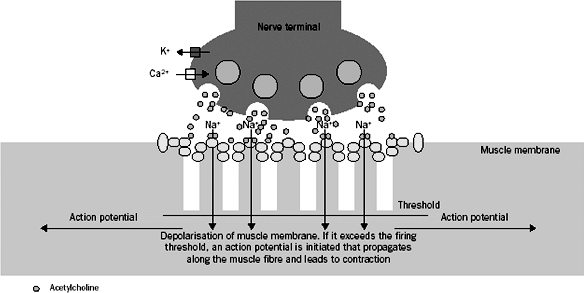 |
Fig. 175-1. Presynaptic terminal is depicted on the top of the picture. Acetylcholine (ACh) vesicles (pink circles) fuse with the terminal membrane after the presynaptic depolarization. The ACh is released, diffuses across the synaptic cleft, and binds to the acetylcholine receptor (AChR) (green circles). Once the AChR is activated, sodium influx occurs through the AChR ion channels, leading to the production of end-plate potential (EPP). From Vincent A, Palace J, Hilton-Jones D: Myasthenia gravis. Lancet 357:2122, 2001. With permission. |
To secure the neuromuscular transmission, a normal neuromuscular junction usually has abundant reservoir of necessary biological machineries, including an excess of AchR as well as voltage-gated sodium ion channels. This is called the safety factor (SF). One may quantitatively define, as noted by Kaminski and associates (1997), the SF by the following formula: SF = EPP/(action potential threshold membrane potential). In MG, the SF is impaired because of the reduction of postsynaptic AChR by the antibodies to AChR, diminishing the amplitude of EPP. Impairment of the SF may cause the failure of neuromuscular transmission. Excitation of postsynaptic muscle cell does not occur, leading to weakness.
MOLECULAR STRUCTURE OF ACETYLCHOLINE RECEPTOR
Acetylcholine receptor (AchR) is a ligand-gated ion channel that, in mature innervated muscles, is composed of several homologous subunits (Fig. 175-2A), including two alpha ( 1, 2), one beta ( ), one delta ( ), and an epsilon ( ) subunits. In fetal or denervated muscles, a subunit replaces the subunit. Five subunits line up like a barrel around the ACh ion channel. The ACh binds at the junctions between and or subunits and between and subunits. Each subunit has four transmembrane domains, M1 to M4 (see Fig. 175-2B). Variations of amino acid sequence between the different subunits are generally in the large cytoplasmic domains. Amino acid sequences for the transmembrane domains are generally conserved. Sequences of the M1 and M2 of each subunit form the lining of AChR ion channel. Saedi and coinvestigators (1990) have shown that amino acids from the -subunit sequence 66 76 are the critical autogenic region for development of pathogenic AChR antibodies. Interestingly, antibodies bound to this region in humans generally do not impair the ACh binding to AChR; rather, they can fix complement and initiate the destruction of postsynaptic membranes. In addition, AChR bound to the antibodies may have an increased rate of internalization and degradation resulting in reduced available AChR, as reported by Kao and Drachman (1977) as well as by Appel and associates 1977).
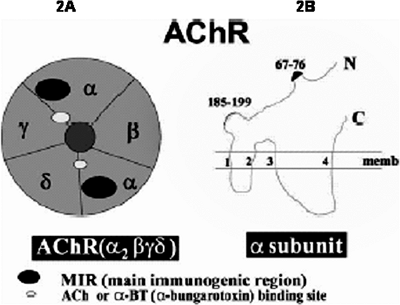 |
Fig. 175-2. A. Acetylcholine receptor (AChR) is a pentameric transmembrane protein composed of five subunits in a stoichiometry of 2, , , and . B. Amino acids from the -subunit sequence 66 76 are the critical region of immunogenesis for the AChR antibodies, called the main immunogenic region (MIR). The acetylcholine binding site (185 199) is also shown. From De Baets M, Stassen MHW: The role of antibodies in myasthenia gravis. J Neurol Sci 202:5, 2002. With permission. |
When compared with voltage-gated ion-selective channels, the channel in the center of AChR is less selective among particular cations, including sodium, potassium, and calcium. However, the channel does possess a relative selectivity to the positivity of ions, which is mainly controlled by the charged side chains of amino acids within the pore of the ACh ion channel, as discussed by Dani (1993). Once ACh binds to the channel, the influx of sodium and efflux of potassium occur, but the net current is positive influx and depolarizes the muscular membrane in the vicinity of postsynapses. The depolarization may reach the threshold of action potential and propagate to other areas of the muscle.
P.2626
PATHOGENESIS OF MYASTHENIA GRAVIS
The Pathologic Locus of Myasthenia Gravis is at the Neuromuscular Junction
A series of early important electrophysiologic studies by Harvey and Masland (1941) accurately localized the pathologic locus of MG to the neuromuscular junction. Action potentials were recorded from human muscles and evoked with a train of electrical stimuli. The amplitudes of action potentials were stable in normal subjects. However, a rapid reduction of the amplitudes was observed in patients with MG, which imitated the changes induced by D-tubocurarine, which blocks the muscle side of the neuromuscular junction. Moreover, neostigmine, a cholinesterase inhibitor, restored the amplitudes of action potentials. These findings suggested that an impairment of the neuromuscular junction transmission in patients with MG contributed to the muscle weakness.
The snake venom -bungarotoxin was discovered to bind specifically to AChR by Chang and Lee (1966) and Changeux and colleagues (1970). By using the radioactive labeled toxin, Fambrough and associates (1973) demonstrated that there were fewer AChRs in the muscular end plate in patients with MG as compared with those in normal subject. These important findings corroborated the pathologic site of MG to the muscle side of the neuromuscular junction.
Myasthenia Gravis Is an Autoimmune Disorder
To study AChR, Patrick and Lindstrom (1973) purified AChR by using the -bungarotoxin from eel electroplax, a natural resource enriched with AChR. They injected the AChR into rabbits and intended to develop AChR antibodies to test the parity of the AChR. They did induce and detect the antibodies. More importantly, the animals showed myasthenialike symptoms and the cholinesterase inhibitors, neostigmine or edrophonium, were able to reverse the symptoms. These phenomena were also observed in other species, such as mice, rats, and monkeys. Moreover, detailed electrophysiologic studies were able to duplicate all characteristic features of human MG in these animals. Repetitive stimulation evoked the decremental response of compound motor action potential (CMAP) in animals immunized with AChR. Microelectrode recording from the end plate areas showed the reduction of miniature end plate potentials as noted by Patrick and Lindstrom (1973) as well as by Lennon (1975), Fambrough (1973), Toyka (1975), Lindstrom (1976a, 1976b), Engel (1976), and Richman (1980) and their colleagues. Under electrical microscope (EM), the convoluted postsynaptic membrane was seen to be simplified with significant reduction of AChR in a study by Engel and co-workers (1976). These data confirmed the pathologic localization of MG and strongly suggested that antibodies against AChR can produce MG.
An immediate question would be whether the same mechanism accounts for human MG. To clarify this issue, serum from patients with MG was studied, and antibodies against AChR were found in the investigations of Newsom-Davis and associates (1978). B lymphocytes from MG patients were cultured by Vincent and Newsom-Davis (1980) and produced antibodies against AChR. Serum immunoglobin from patient with MG was passively transferred to mice and induced symptoms of MG in these animals by Toyka (1975) and Lindstrom (1976a, 1976b) and their colleagues. These data established the concept that MG is an autoimmune disorder. MG was subsequently reproduced in animals with the passive transfer of monoclonal antibodies against AChR by Richman and co-workers (1980), which further strengthened the concept.
The aforementioned evidence demonstrated that MG is an antibody-mediated disease, and the main consequence of the antibody-mediated process is the reduction of AChR. Further studies have shown that there are multiple mechanisms through which anti-AChR antibodies are able to reduce the number of AChRs. Using the radioactive isotope labeled -bungarotoxin, the fate of AChR can be traced, and the rate of the receptor degradation can be estimated. Serum from patients with MG caused an escalated degradation rate of AChR up to twofold to threefold that of normal controls. Endocytosis of the AChR was also accelerated, as noted by Kao and Drachman (1977) and Saedi and coinvestigators (1990). As mentioned, EM studies by Engel and associates (1976) had revealed that the postsynaptic membranes were simplified with flattening of the usually convoluted folds of the postsynaptic membranes in MG. Immunocytochemical techniques used by Engel and Arahata (1987) had detected the membrane attack complex of complement at the neuromuscular junction, supporting the role of complement-mediated destruction of neuromuscular junction in MG. Interestingly, as reported by Lindstrom and co-workers (1976b), the serum concentration of AChR-binding antibodies in patients with MG did not correlate with the severity of the disease. The observation was explained by the variation of antibody functions and different targets of epitopes on the AChR subunits. Indeed, according to Drachman and colleagues (1982), the functional activities of the AChR antibodies in accelerating degradation of the receptors were correlated closely with the severity of weakness in MG.
Although we have learned much about the effect of antibodies in MG, we still do not understand what initiates the autoimmune process. However, Sommer and associates (1990) and Ragheb and one of us (RPL) (1998) have noted that the accumulated immunologic evidence suggests that both B cells and T cells are involved in the autoimmune process of MG.
P.2627
MYASTHENIA GRAVIS IS A HETEROGENOUS DISORDER
Myasthenia Gravis Without Serum Negative Anti-AChR Antibodies
It has been long recognized that about 10% to 15% of patients with acquired generalized MG have no detectable AChR antibodies, as described by Soliven associates (1988). However, these patients clearly have antibody-mediated disease. Passive transfer of their immunoglobulin to animals reproduced the important features of MG and immunologic therapy, including plasma exchange, is effective in anti-AChR seronegative patients. Moreover, immunoglobulin from patients without detectable anti-AChR antibodies accelerated the degradation of AChR of cultured muscle cells.
A recent study has demonstrated that a different autoantibody is present in the serum of patients with seronegative MG. Hoch and colleagues (2001) showed that 70% of MG patients without anti-AChR antibodies have serum autoantibodies against the muscle-specific receptor tyrosine kinase (MuSK). MuSK plays an important role in the agrin-induced clustering of AChR during synapse formation and is also expressed at the mature neuromuscular junction. Hoch and co-workers (2001) reported that the anti-MuSK antibodies inhibited MuSK function. Anti-MuSK antibodies might also damage the neuromuscular junction by a complement-mediated process. This study clearly confirmed that the MG is a heterogeneous disorder even at the neuromuscular junction.
HEREDITARY FORMS OF MYASTHENIA GRAVIS AND MYASTHENIC SYNDROMES
With the advances in molecular biology and genetics, the etiology and pathogenesis of many congenital myasthenic syndromes have been elucidated. The diseases, as noted by Engel and coinvestigators (1999), are caused by mutations in different subunits of AChR, acetylcholinesterase, and other synaptic components. Details of hereditary MG are beyond the scope of discussion of this chapter. However, the discovery of specific genetic abnormalities extends the boundary of pathogenic mechanisms of MG. The mutations in different components of neuromuscular junction reveal the important physiologic functions of these components of the neuromuscular junction.
SUMMARY
MG is an antibody-mediated autoimmune disorder. The synthesis of these antibodies is T cell dependent. In most patients with generalized MG, serum autoantibodies against AChR are detectable. The antibodies bind to the AChR, leading to the acceleration of AChR degradation and endocytosis. They also recruit and activate other immunoreactive complement to damage the postsynaptic membranes. The main consequence of these pathologic processes is the reduction of available AChR and simplification of postsynaptic folds, leading to the impairment of transmission at the neuromuscular junction. Although the initial events in the induction of autoimmunity are as yet unknown, evidence suggests that abnormalities of both T cells and B cells are important in the development of autoimmune process of MG.
REFERENCES
Appel SH, et al: Accelerated degradation of acetylcholine receptor from cultured rat myotubes with myasthenia gravis sera and globulins. Proc Natl Acad Sci U S A 74:2130, 1977.
Campbell H, Bramwell E: Myasthenia gravis. Brain 23:277, 1900.
Chang CC, Lee CY: Electrophysiological study of neuromuscular blocking with cobra neurotoxin. Br J Pharmacol 28:172, 1966.
Changeux JP, Kasai M, Lee CY: Use of a snake venom toxin to characterize the cholinergic receptor protein. Proc Natl Acad Sci USA 67:1241, 1970.
Dani JA: Structure, diversity, and ionic permeability of neuronal and muscle acetylcholine receptors. EXS 66:47, 1993.
De Baets M, Stassen MHW: The role of antibodies in myasthenia gravis. J Neurol Sci 202:5, 2002.
Drachman DB, et al: Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med 23:769, 1982.
Engel AG, Arahata K: The membrane attack complex of complement at the endplate in myasthenia gravis. Ann N Y Acad Sci 505:326, 1987.
Engel AG, Ohno K, Sine SM: Congenital myasthenic syndromes: recent advances. Arch Neurol 56:163, 1999.
Engel AG, et al: The motor end plate in myasthenia gravis and in experimental autoimmune myasthenia gravis. A quantitative ultrastructural study. Ann N Y Acad Sci 274:60, 1976.
Fambrough DM, Drachman DB, Satyamurti S: Neuromuscular junction in myasthenia gravis: decreased acetylcholine receptors. Science 19:293, 1973.
Harvey AM, Masland RL: The electromyogram in myasthenia gravis. Bull Johns Hopkins Hosp 65:1, 1941.
Hoch W, et al: Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 7:365, 2001.
Kaminski HJ, Suarez JI, Ruff RL: Neuromuscular junction physiology in myasthenia gravis: Isoforms of the acetylcholine receptor in extraocular muscle and the contribution of sodium channels to the safety factor. Neurology 49:S8, 1997.
Kao I, Drachman DB: Myasthenic immunoglobulin accelerates acetylcholine receptor degradation. Science 29:527, 1977.
Lennon VA, Lindstrom JM, Seybold ME: Experimental autoimmune myasthenia: a model of myasthenia gravis in rats and guinea pigs. J Exp Med 141:1365, 1975.
Lindstrom JM, et al: Pathological mechanisms in experimental autoimmune myasthenia gravis. II. Passive transfer of experimental autoimmune myasthenia gravis in rats with anti-acetylcholine receptor antibodies. J Exp Med 144:739, 1976a.
Lindstrom JM, et al: Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology 26:1054, 1976b.
Miledi R, Molinoff P, Potter LT: Isolation of the cholinergic receptor protein of Torpedo electric tissue. Nature 229:554, 1971.
Newsom-Davis J, et al: Function of circulating antibody to acetylcholine receptor in myasthenia gravis: investigation by plasma exchange. Neurology 28:266, 1978.
P.2628
Patrick J, Lindstrom J: Autoimmune response to acetylcholine receptor. Science 180:871, 1973.
Ragheb S, Lisak RP: Immune regulation and myasthenia gravis. Ann N Y Acad Sci 841:210, 1998.
Richman DP, et al: Monoclonal anti-acetylcholine receptor antibodies can cause experimental myasthenia. Nature 286:738, 1980.
Saedi MS, et al: Determination of amino acids critical to the main immunogenic region of intact acetylcholine receptors by in vitro mutagenesis. FEBS Lett 267:55, 1990.
Sakmann B, Patlak J, Neher E: Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature 286:71, 1980.
Soliven BC, et al: Seronegative myasthenia gravis. Neurology 38:514, 1988.
Sommer N, et al: Myasthenic thymus and thymoma are selectively enriched in acetylcholine receptor-reactive T cells. Ann Neurol 28:312, 1990.
Toyka KV, et al: Myasthenia gravis: passive transfer from man to mouse. Science 190:397, 1975.
Vincent A, Newsom Davis J: Anti-acetylcholine receptor antibodies. J Neurol Neurosurg Psychiatry 43:590, 1980.
Vincent A, Palace J, Hilton-Jones D: Myasthenia gravis. Lancet 357:2122, 2001.
EAN: 2147483647
Pages: 203