8 - Hematuria, Proteinuria, or Both, and Abnormal Findings on Urinary Microscopy
Editors: Schrier, Robert W.
Title: Manual of Nephrology, 6th Edition
Copyright 2005 Lippincott Williams & Wilkins
> Table of Contents > 8 - The Patient with Hematuria, Proteinuria, or Both, and Abnormal Findings on Urinary Microscopy
8
The Patient with Hematuria, Proteinuria, or Both, and Abnormal Findings on Urinary Microscopy
Sharon G. Adler
Kenneth Fairley
Urine analysis. A urine sample is usually easy to obtain, and it can be studied by very simple techniques on the ward or in the physician's office. Examination of the urine is one of the most rewarding steps in clinical medicine. Not only does it uncover renal parenchymal and urinary tract disease that eludes detection by other methods of investigation, but it frequently points to a specific diagnosis. Almost every patient with parenchymal renal disease shows abnormal findings on urine microscopy, an increase in urine protein, or both. Urine microscopy reveals abnormalities in many patients with renal disease who have no significant proteinuria. Many renal diseases that require treatment are first suspected on the basis of urinary findings in particular, infections, glomerulonephritis, and interstitial nephritis.
Method of collection of urine specimens. The method of collecting a urine sample is of critical importance when the specimen is to be examined microscopically. For dipstick testing, the sample should be examined promptly for accurate results. Urine pH may change with time after collection, and contaminating bacteria multiply and convert nitrate to nitrite, causing a false-positive test for bacteriuria. At low specific gravity (less than 1.010), cells lyse and casts form less readily. Casts also dissolve in alkaline urine.
Midstream urine collection. In collecting urine for microscopical examination, avoiding contamination with bacteria, squamous cells, and leukocytes is important. Contamination is very common in women, in whom contaminating cells and bacteria arise in the vagina and vulval area. The important points in collecting a good midstream urine sample are shown in Table 8-1. Although bacteria are often detected on microscopy, infection is best proved by culture, which also allows the antibiotic sensitivity of infecting organisms to be determined. Special media are required for the culture of some organisms, such as the tubercle bacillus, Mycoplasmas, anaerobic organisms, and yeasts. Culture results are reported as colony-forming units (CFU) per mL of urine; the significance of a count depends on the method of collection (Table 8-2).
Table 8-1. Guidelines for Collecting Midstream Urine Sample
Women When possible use a vaginal tampon. Hold labia well separated during collection of the specimen. Gently cleanse the periurethral area from anterior to posterior with several moistened gauze squares. Men Hold the retracted foreskin back throughout the collection. Clean the urethral meatus with moist gauze. If urethral or prostatic infection is suspected: The first 10 mL of voided urine reflects urethral infection (it also includes bladder contents). The midstream urine collected, as above, reflects the bladder contents. Prostatic fluid, if available, reflects prostatic infection most accurately. The first 10 mL of urine voided after prostatic massage provides information on infection in prostatic secretions (it also includes bladder contents). Note: In both sexes, at least 200 mL should be passed before a midstream urine specimen is collected without interruption of the flow of urine. Table 8-2. Influence of Collection Technique on Accuracy of Detection of Urine Abnormalities
Collection technique Hematuria Pyuria Significant bacteriuria (CFU/mL) Fastidious microorganisms Midstream urine Excellent Faira >100,000 Poor Open-ended catheter Good Very good >1,000 Good Suprapubic aspiration Poor Excellent >1 Excellent CFU, colony-forming units. aDepends on collection technique. A catheter specimen in women is best collected as a midcatheter specimen using a short open-ended catheter. At least 200 mL must pass through the catheter to flush out contaminating urethral contents before the specimen is collected. Conventional side hole catheters push urethral contents into the bladder before urine can run out, and critical bacterial counts are 30-fold higher using a conventional catheter than those obtained on urine collected with an open-ended catheter.
Suprapubic aspiration. A fine lumbar puncture needle with stylet in place is passed through sterilized suprapubic skin directly into a full bladder. Uncontaminated urine can then be aspirated.
Dipstick testing. Dipstick testing of urine provides a rapid determination of urine pH, specific gravity, and the presence of protein, blood (hemoglobin), leukocytes, nitrites, glucose, and bile. Dipsticks are less sensitive in detecting pyuria and bacteriuria.
The method of screening for proteinuria is the examination of a randomly excreted urine sample by dipstick. Tetrabromophenol is buffered to a pH of 3, and this results in a yellow-to-blue color that changes with increasing protein concentration. False-positive dipstick results for proteinuria are
P.117
seen when urine pH is greater than or equal to 8 and when the patient is excreting metabolites of penicillins, aspirin, or oral hypoglycemic agents in the urine. Standard dipsticks for protein fail to detect very small quantities of abnormal albuminuria (e.g., microalbuminuria) and are relatively insensitive to even large amounts of nonalbumin protein. To alleviate the first of these shortcomings, dipsticks capable of detecting microalbuminuria are currently available and are useful, when used properly, as a screening test to identify patients with diabetes mellitus and incipient nephropathy or to detect patients with a high risk of coronary artery and/or cerebrovascular disease. Dipsticks that are more sensitive to nonalbumin proteins are under development. Currently, a 24-hour urine collection is required to detect such proteins.Urine color varies with its concentration and with the presence of drugs such as phenazopyridine (Pyridium), phenindione, and multivitamin tablets, and of blood, bile, porphyrin, melanin, and homogentisic acid. Some of these color changes may be mistaken for hematuria. Dipstick examination of the urine accurately determines the presence of hemoglobin, which depends on the oxidation of orthotolidine by cumene hydroperoxide, although cross-reactivity with myoglobin does occur. Although the microscopic examination of urine for erythrocytes provides far more information about the underlying renal lesion, a dipstick is quick, simple, and provides quantitative information. The Ames N Multistix and Boehringer nephron tests provide sensitive and specific methods of detecting microscopical
P.118
hematuria. Ascorbic acid, a strong reducing agent, prevents the chemical reaction in dipsticks that detects hemoglobin and is the cause of many false-negative tests. In the case of a low-specific-gravity urine (below 1.010), lysis of erythrocytes leads to false-negative microscopy, but the dipsticks provide a positive test.Microscopic analysis. In the United States, urine is characteristically examined using a standard light microscope to make a semiquantitative estimate of the frequency of the formed elements in the urine by counting their number per high-power field. More quantitative measures can be obtained by examining the urine on a coverslipped counting chamber rather than on a plain glass slide. The counting-chamber method is more accurate, largely because it eliminates the error introduced by the variability of the depth of the layer of urine on the slide, which occurs as a function of viscosity. The accuracy of the microscopic examination can also be enhanced by using phase-contrast microscopy, which allows for more sensitivity in the observation of morphologic detail. If a phase-contrast microscope is unavailable, staining the urine sediment with Sternheimer-Malbin stain may also enhance visualization of cellular elements using a standard light microscope.
Hematuria. A large amount of blood present in the urine is obvious to the naked eye. The red blood cell count in such cases is always well above 106 cells per mL. The morphology and quantitation of the urinary erythrocytes are two of the most important investigations in clinical nephrology. They not only provide information about the underlying cause, but also assist in determining the nature of investigations required. Because the recognition that glomerular bleeding can be determined by the appearance of erythrocytes on phase-contrast microscopy, prompt assessment of the number and morphology of urinary erythrocytes in the urine has assumed new significance. In the case of crescentic glomerulonephritis, which can be suspected when red blood cell casts and large numbers of dysmorphic erythrocytes are seen on urine microscopy, the physician is alerted to the urgent need for biopsy confirmation and appropriate treatment.
A high count of glomerular erythrocytes (more than 106 red blood cells per mL) has both prognostic and therapeutic implications, because it suggests the presence of crescents on renal biopsy. If 4+ dipstick testing for heme is present, immediate urine microscopy is indicated to determine the nature and hence the source of erythrocytes, and, if possible, the count per mL. The source of erythrocytes can be determined by measuring their size and by examining their morphology. After centrifuging the urine specimen, if a red pellet results, adequate numbers of erythrocytes are present to measure their volume in an automated counter of the type customarily used for complete blood cell counts (CBCs). If the urinary erythrocyte mean corpuscular volume is less than 72 fL, a glomerular bleeding source is likely.
The best method for assessing erythrocyte morphology is phase-contrast microscopy. If bright-field illumination is used, many erythrocytes will not be correctly identified. In particular, those that have lost their hemoglobin staining are not seen, a common occurrence in an acid urine. The small and distorted erythrocytes characteristic of glomerular bleeding may also be missed. Characteristic dysmorphic erythrocytes, which indicate that hematuria is arising in glomeruli, are illustrated in Fig. 8-1. These vary considerably in size, shape, and hemoglobin content, and many bizarre forms are present. Normal urine contains up to 8,000 glomerular erythrocytes per mL in a centrifuged specimen and 13,000 per mL in uncentrifuged urine. Erythrocytes arising from a nonglomerular source are uniform in size and shape but may show some ghost cells, cells that are losing their hemoglobin (Fig. 8-2). The latter change occurs particularly in an acid urine.
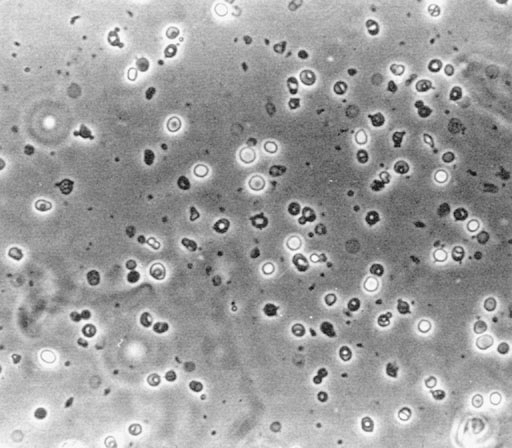
Figure 8-1. Erythrocytes, showing the wide variation in size, shape, and hemoglobin content, in the urine of a patient with glomerulonephritis (phase-contrast microscopy). (From Fairley KF. Urinalysis. In: Schrier RW, Gottschalk CW, eds. Diseases of the kidney, 4th ed. Boston: Little, Brown, 1988. Reprinted with permission.)
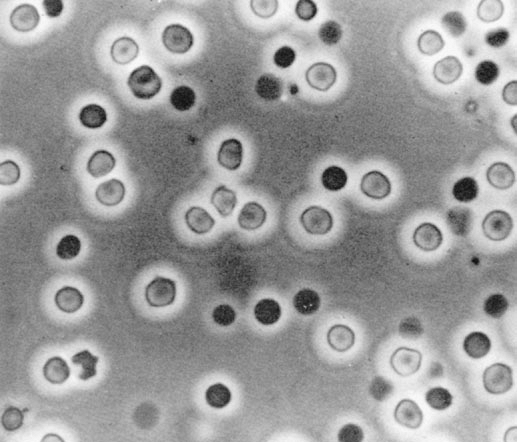
Figure 8-2. Nonglomerular bleeding, showing two populations of cells (phase-contrast microscopy). (From Fairley KF. Urinalysis. In: Schrier RW, Gottschalk CW, eds. Diseases of the kidney, 4th ed. Boston: Little, Brown, 1988. Reprinted with permission.)
Urinary casts. Casts are formed from Tamm-Horsfall glycoprotein, which is synthesized and secreted in the ascending limb of the loop of Henle and distal convoluted tubules.
Physiologic casts. Hyaline casts are transparent and cylindrical and are seen in the urine of normal subjects. Their presence does not indicate renal disease. Granular casts are semitransparent cylinders with refractile granules of uncertain origin. These, too, are present in individuals without renal disease. The number of hyaline and granular casts in urine may be increased by fever, exercise, and volume depletion.
Pathologic casts. Casts may contain cellular material (erythrocytes, leukocytes, tubular cells, bacteria, or fungi), fibrin, lipids, bile, and crystals.
The most important cellular cast is that composed of erythrocytes (Fig. 8-3). This indicates glomerular bleeding, as do erythrophagocytic cells. Large numbers of erythrocyte casts also increase the likelihood of crescents in glomeruli. Casts composed of polymorphonuclear leukocytes usually indicate renal parenchymal infection. These may also be seen in acute interstitial nephritis and occasionally in glomerular lesions, where numerous polymorphonuclear leukocytes are in glomeruli. Casts may also contain all varieties of mononuclear leukocytes
P.120
P.121
and may include tubular epithelial cells from all segments of the nephron. In nephrotic syndrome, all casts usually contain fat particles and some contain oval fat bodies. Because the fat bodies consist of cholesterol esters, they can be easily identified by their Maltese cross birefringence (Figs. 8-4 and 8-5). Many fat bodies, however, are composed of fat particles too small to show crosses, and these appear as a faint glow in polarized light.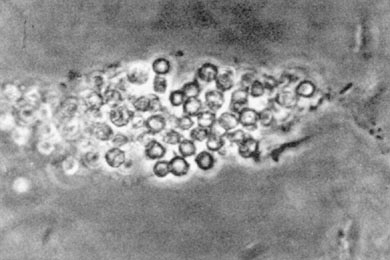
Figure 8-3. An erythrocyte cast in an acid urine, composed of red cells from which much of the hemoglobin has disappeared. (From Fairley KF. Urinalysis. In: Schrier RW, Gottschalk CW, eds. Diseases of the kidney, 4th ed. Boston: Little, Brown, 1988. Reprinted with permission.)
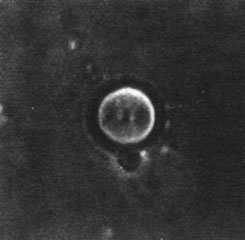
Figure 8-4. A cholesterol ester spherulite approximately the same size as an erythrocyte. (From Fairley KF. Urinalysis. In: Schrier RW, Gottschalk CW, eds. Diseases of the kidney, 4th ed. Boston: Little, Brown, 1988. Reprinted with permission.)
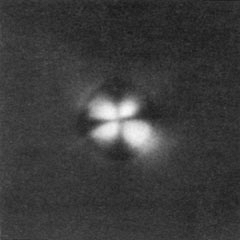
Figure 8-5. The particle in Figure 8-4, when viewed with polarized light, shows the classic Maltese cross. (From Fairley KF. Urinalysis. In: Schrier RW, Gottschalk CW, eds. Diseases of the kidney, 4th ed. Boston: Little, Brown, 1988. Reprinted with permission.)
Crystals in casts are commonly present in patients who are taking triamterene. The crystals disappear when the urine is alkalinized or the drug is discontinued. Crystals may also occasionally be present in casts in patients with hypercalcemia or hyperuricosuria, or as an isolated finding.
P.122
Broad waxy casts signify chronic renal disease. These typically have a very sharp border and are easily seen with bright-field illumination. They do not dissolve in alkaline urine.
Leukocytes and nucleated cells in the urine. Normal midstream urine contains up to 2,000 nucleated cells per mL, and most of these are leukocytes. Normal bladder urine obtained by needle aspiration contains very low counts of leukocytes (mean, 283 per mL, with only 2 of 25 urines containing more than 1,000 per mL). In one author's (KF) laboratory, the mean count in a midstream specimen in the same subjects performed at the same time was 2,018 leukocytes per mL, even when meticulous care was taken to avoid contamination. The additional cells in the midstream sample presumably arise in the urethra.
An increase in the leukocyte count in the urine most commonly implies infection. Infected urines show an increase in leukocytes in more than 90% of cases. When pyuria is present without bacteriuria, three-fourths of patients show an underlying urinary tract abnormality.
Sterile pyuria may be seen in tuberculosis, renal papillary necrosis, acute interstitial nephritis, urate nephropathy, glomerulonephritis, and polycystic disease of the kidney. It may also be present in patients with calculi and in a variety of other urinary tract abnormalities. Before a diagnosis of sterile pyuria is made, fastidious organisms such as ureaplasmas, anaerobic bacteria such as Gardnerella vaginalis, and chlamydia should be sought in an appropriate specimen using special cultural techniques.
When it is difficult to distinguish leukocytes from renal tubular epithelial cells, a drop of acetic acid makes it easier to recognize the lobed nuclei of polymorphonuclear leukocytes. Staining the cells also distinguishes them.
Renal tubular cells. Large numbers of renal tubular cells are found in the urine in acute tubular necrosis and acute interstitial nephritis. Acute interstitial nephritis can be distinguished by a much higher count of leukocytes (more than 15,000 per mL) and a higher total nucleated cell count (more than 75,000 per mL) than are seen in acute tubular necrosis. Eosinophils are present in the urine in most cases of interstitial nephritis, in counts above 5,000 per mL; these are rare in acute tubular necrosis. Eosinophils in the urine are best seen with Hansel's stain, which, unlike Wright's stain, is not pH dependent.
Nucleated cells are also present in the urine in glomerulonephritis, particularly crescentic glomerulonephritis, in which total cell counts, leukocytes, and renal tubular cells are present in higher numbers than in noncrescentic glomerulonephritis. Glomerular epithelial cells identified by monoclonal antibody staining are also present in a much higher percentage of urines from patients with crescentic glomerulonephritis than from those with noncrescentic glomerulonephritis.
Crystals. Although crystals of calcium oxalate and uric acid may be seen in normal urine samples, large, bizarre crystals of any type, including calcium oxalate and uric acid, usually indicate increased urinary excretion and may indicate calculous disease. Cystine crystals are always abnormal and indicate cystinuria.
P.119
Hematuria
Localization and differential diagnosis. An abnormal quantity of erythrocytes in the urine can either be due to a glomerular disorder or to nonglomerular bleeding. Glomerular bleeding should be suspected if any of the following are present: dysmorphic urinary erythrocytes, erythrocytes with mean corpuscular volume less than 72 fL, red blood cell casts, and concomitant proteinuria, especially higher than 1 g per day. In Table 8-3, an asterisk denotes those glomerular disorders that frequently manifest hematuria. Nonglomerular hematuria is characterized by the presence of isomorphic urinary erythrocytes with a mean corpuscular volume of more than 72 fL in the absence of red blood cell casts or significant proteinuria. The further
P.123
localization of nonglomerular bleeding can be suggested by the three-glass test. In this procedure, patients with macroscopic hematuria urinate 10- to 15-mL aliquots of urine each into three containers. A urethral site of bleeding is suggested if the hematuria predominates in the first 10 mL, whereas a bladder origin is implied if the majority of blood is in the final aliquot. Upper-tract bleeding is characterized by hematuria in all three collection vessels. The differential diagnosis of nonglomerular proteinuria isP.124
outlined in Table 8-4. Many of these disorders are either characterized by or occasionally present with hematuria.Table 8-3. Causes of Glomerular Proteinuria
Primary glomerular disorders Minimal change, mesangial proliferative (IgA, IgM*), focal and segmental glomerulosclerosis*, membranous, membranoproliferative*, crescentic* Hereditary Alport syndrome*, Fabry disease*, nail-patella syndrome* Infectious Bacterial*, viral*, fungal*, protozoal*, and helminthic* causes, including bacterial endocarditis*, poststreptococcal glomerulonephritis*, visceral abscesses*, secondary syphilis*, hepatitis B and C*, human immunodeficiency virus*, malaria* Metabolic Diabetes mellitus Immunologic Systemic lupus erythematosus*, mixed connective tissue disease, Sj gren syndrome, Henoch-Sch nlein purpura*, Wegener granulomatosis*, microscopic polyarteritis nodosa*, Goodpasture syndrome*, cryoglobulinemia* Medications Penicillamine, gold- or mercury-containing compounds, lithium, nonsteroidal anti-inflammatory drugs, angiotensin-converting enzyme inhibitors, heroin* Neoplasms Multiple myeloma*; colon, lung, or breast carcinoma; lymphoma; leukemia Miscellaneous Sickle cell disease*, allergies*, immunizations*, cirrhosis*, immunotactoid glomerulopathy*, amyloidosis*, reflux nephropathy*, congenital nephrotic syndrome IgA, immunoglobulin A; IgM, immunoglobulin M. *Denotes glomerular disorders that frequently present with hematuria. Table 8-4. Causes of Tubular Proteinuria
Hereditary Polycystic kidney disease, medullary cystic disease Infectious Pyelonephritis, tuberculosis Metabolic Diabetes mellitus, hyperuricemia, uricosuria, hypercalcemia, hypercalciuria, hypokalemia, oxalosis, cystinosis Immunologic Sj gren syndrome, renal transplant rejection, drug hypersensitivity, sarcoidosis Toxic Analgesic abuse, radiation nephritis, lithium, heavy metals (lead, cadmium, mercury), Balkan nephritis, cyclosporine, cisplatinum, aminoglycosides Anatomic Obstruction, ureterovesical reflux, medullary sponge kidney Miscellaneous Multiple myeloma, amyloidosis, sickle cell disease, medullary sponge kidney Clinical evaluation and treatment. Patients with hematuria may present with quantities of blood visible to the naked eye (macroscopic or gross hematuria) or visible only by microscopic examination (microscopic hematuria). The initial evaluation of the patient with gross or microscopic hematuria should include those steps outlined above to distinguish glomerular from nonglomerular causes. If a glomerular cause is suspected, patients should be further evaluated as described in section III.C.
For patients who have been determined to have nonglomerular hematuria, the differential diagnosis outlined in Table 8-4 may be narrowed by obtaining a relevant history and physical examination. The history and review of systems should include questions regarding passage in the urine of stones, tissue, or blood clots; frequent urinary tract infections (UTIs); episodic gross hematuria; fever; weight loss; medication or drug use, particularly oral contraceptives or analgesics; dysuria; tuberculosis; diabetes mellitus; trauma; coagulation disorders; and family history of renal disease. Pertinent aspects of the physical examination should include auscultation for renal bruits, palpation of the prostate for nodules or prostatitis, and palpation of the abdomen for enlarged kidneys indicative of polycystic kidney disease.
Table 8-5 outlines a suggested diagnostic approach to the patient with nonglomerular hematuria. Initial laboratory studies should include a complete blood count (CBC), prothrombin time, and partial thromboplastin time to rule out coagulopathy and hemoglobinopathy. A serum chemistry
P.125
panel is useful to determine the level of renal function and to rule out the presence of hypercalcemia, hyperuricemia, and hyperglycemia. Urine should be cultured for bacterial and mycobacterial infection and, if clinically suspected, mycoplasma or other more fastidious organisms. Once a UTI has been excluded, an examination of the urinary tract anatomy should be undertaken to define the bleeding site. In the presence of normal or near-normal glomerular filtration rate (GFR), intravenous urography is the most useful diagnostic radiologic procedure available for defining renal anatomy. It should disclose the presence of renal cysts (hereditary and acquired); calculi; papillary necrosis; medullary sponge kidney; renal, pelvic, and ureteral tumors; and ureteral strictures. In the event of a nondiagnostic intravenous urographic study, cystoscopy can be performed to more definitively examine the bladder for the presence of tumors or infectious (inflammatory/interstitial) cystitis and the urethra for the presence of urethritis, strictures, or both. A nondiagnostic cystoscopic examination can be followed, if clinically indicated, by arteriography to pursue the possibility that hematuria is secondary to ureteral varices or aneurysms, arteriovenous malformations, or the loin-pain hematuria syndrome.
The treatment of hematuric states is entirely dependent on the underlying cause of erythrocyturia. The reader is therefore referred to textbooks of internal medicine, nephrology, and urology for detailed coverage of these topics. In this section, however, UTIs are discussed briefly, because they are commonly encountered by the general practitioner as well as by the specialist.
The most common presentation with UTI is dysuria and frequency in a woman of childbearing years. The urine may show macroscopic or heavy nonglomerular microscopic hematuria, as well as pyuria and bacteriuria. Some have recently advocated that even in patients with diabetes, pyuria without clinical signs or symptoms of infection should not be treated with antibiotics. In those in whom treatment is indicated, a single dose of an appropriate antibiotic (e.g., 2 g of amoxicillin or ampicillin) cures the infection in 80% of cases. Antibiotic resistance may be responsible for some treatment failure (e.g., ampicillin-resistant Escherichia coli). However, unresponsiveness to treatment may be an indication for further investigation, because a high percentage of those who do not respond to a single dose show abnormalities on further investigation by imaging techniques, such as intravenous urography. Failure to respond to treatment may also suggest an upper rather than lower UTI.
Urinary tract infection in a child or a man should always be investigated, because of the high incidence of underlying abnormalities. Infection in the presence of an underlying urinary tract abnormality usually requires longer courses of treatment. Eradicating the infection without removing the abnormality may be impossible in some cases, notably with infected stones or staghorn calculi.
Proteinuria
Biochemical evaluation. Although not specific for glomerular disease, an abnormal amount of protein excreted in the urine is a cardinal manifestation of disease in virtually all patients with glomerulonephritis. Fever, exercise, hyperglycemia, and severe hypertension can transiently increase proteinuria.
To precisely quantitate and qualitatively analyze the amount and composition of urinary proteins, an examination of a 24-hour collection of urine is usually required. The collection method involves emptying the bladder and discarding the first urine on the morning of collection, and scrupulously collecting all subsequent urine for the next 24 hours. The final urine at the end of the 24-hour period is also kept as part of the collection. If the urine is refrigerated during the 24-hour period, no preservative is required. If refrigeration is unavailable, a preservative such as acetic acid (or a cup of vinegar) can be added to the collection vessel.
P.126
Quantification of the 24-hour urine creatinine should be performed to ensure that a complete collection was submitted. In female patients under steady-state conditions of renal function, the 24-hour urinary excretion of creatinine should equal approximately 15 to 20 mg per kg of ideal body weight; in male patients, the excretion should be 18 to 25 mg per kg. Accurate quantitative methods of protein excretion by precipitation include the sulfosalicylic acid precipitation test, the micro-Kjeldahl method, Esbach's picric acid citric acid reagent, and the biuret method. Results are expressed either as grams excreted in 24 hours or as a ratio of protein to creatinine excretion.
In following a patient with heavy proteinuria (e.g., when assessing responsiveness to therapy), a urinary protein creatinine concentration ratio is useful as a substitute for repeated full 24-hour urine collections. The normal 24-hour urine protein excretion in the adult ranges from 30 to 130 mg. Children and adolescents may excrete as much as twice this amount. Normal spot urinary protein creatinine ratios on random samples generally fall below 0.2. Values higher than 3 suggest the presence of nephrotic-range proteinuria.
Qualitative assessments of urinary protein composition are often a valuable addition to the quantitative examination. Urine protein electrophoresis (UPEP) separates urinary proteins on the basis of molecular weights into five peaks: albumin and 1, 2, , and globulins. Normally, urinary proteins are composed of filtered proteins from plasma (50%) and proteins that are secreted into the urine from urinary tract cells (50%). Of the filtered proteins, albumin is the most abundant, representing approximately 15% of the total urinary protein. This is followed by immunoglobulins (5%), light chains (5%), 2 microglobulin (<0.2%), and other plasma proteins (25%). Of the secreted proteins, Tamm-Horsfall protein enters the urine after synthesis in the tubular cells of the ascending limb of the loop of Henle. It is the single most abundant protein in normal urine, accounting for 50% of the total urinary protein.
UPEP and immunoelectrophoresis (IEP) can be helpful in identifying the nature of proteins present in the urine. Immunofixation may be more sensitive than either of the two latter methods. Examining the urine for Bence-Jones proteinuria by seeking urinary precipitates at 45 to 55 C that redissolve at a higher temperature is a less sensitive method than are UPEP and IEP for identifying overflow proteinuria.
Pathophysiologic classification of proteinuria. Given the diverse sources of urinary proteins and the implication of source for the site of pathology, proteinuria is classified into three major categories reflecting pathogenesis.
Overflow proteinuria is due to the filtration by the normal glomerulus of an abnormally large amount of small-molecular-weight protein present in serum whose filtration exceeds the capacity of normal tubules for reabsorption. This occurs in monoclonal gammopathies (such as multiple myeloma), in intravascular hemolysis (hemoglobinuria), and in rhabdomyolysis (myoglobinuria). Overflow proteinuria can be identified by an abnormal peak or spike on the UPEP. For example, a spike occurring in the region (or, less commonly, in the 2 or region) suggests the presence of a monoclonal gammopathy. Further identification of the protein can be accomplished by performing IEP.
Tubular proteinuria is found in both acute and chronic injuries involving the renal tubulointerstitial region. Usually quantitatively less than 2 g per day, tubular proteinuria is derived from three sources. First, injured tubules fail to completely reabsorb small-molecular-weight proteins filtered by the glomerulus, such as 2 microglobulin and amylase. Second, injured tubules secrete brush border components and cellular enzymes, such as n-acetylglucosamine and lysozyme, into the urine. Finally, with tubulointerstitial injury, Tamm-Horsfall protein may be secreted into the
P.127
urine in greater amounts by the tubular cells of the ascending limb of the loop of Henle and the distal nephron. UPEP and IEP may also aid in distinguishing glomerular from tubular proteinuria. The overwhelming predominance of albumin rather than globulins on a UPEP suggests the presence of glomerular proteinuria. A quantitative comparison of the urinary albumin and 2 microglobulin levels performed by IEP or other immunologic techniques (including immunoprecipitation, immunodiffusion, or radioimmunoassay) may also be helpful in this regard. A urinary albumin to 2 microglobulin ratio of 10 to 1 suggests the presence of tubular proteinuria, whereas in glomerular proteinuria, this ratio usually exceeds 1,000 to 1. In normal urine, the albumin to 2 microglobulin ratio ranges from 50 to 1 to 200 to 1.Glomerular proteinuria occurs when injury to the glomerulus results in an ultrafiltrate characterized by a fractional increase in the clearance of serum proteins. In some forms of glomerulonephritis, this is due to changes in the size-selective properties of the glomerular capillary wall that allow the passage of larger-molecular-weight proteins or even of cells (e.g., in crescentic glomerulonephritis). In other forms, this is due to changes in the charge-selective properties of the glomerular capillary wall that enhance the ultrafiltration of negatively charged albumin (e.g., minimal change nephropathy). Some glomerular disorders are characterized by changes in both size and charge selectivity (e.g., diabetic nephropathy). Mesangial injury may also induce proteinuria, perhaps by interfering with normal mesangial clearance functions.
Glomerular proteinuria is composed predominantly of albumin and, when quantitatively large (i.e., more than 3.0 to 3.5 g per day, or more than 2 g per day per m2), it is said to be in the nephrotic range. The nephrotic syndrome consists of a pentad of nephrotic-range proteinuria, hypoalbuminemia, hyperlipidemia, lipiduria, and edema. With the exception of minimal-change glomerulopathy, the occurrence of heavy proteinuria in glomerular disorders is associated with a higher risk for progressive renal insufficiency.
Other types of proteinuria. Two forms of proteinuria do not fit easily into the classification described above. Benign orthostatic proteinuria is typically found in tall adolescents and occurs in the lordotic position. Protein is found in the urine collected on retiring and in the morning after the patient has been ambulant, but not in the overnight specimen collected immediately on rising. No abnormality should be present in the urine sediment, and proteinuria should not exceed 1 g per day. In one-half of these patients, proteinuria disappears within 10 years; however, in a small proportion, overt renal disease develops in later life. Finally, functional, transient proteinuria may be associated with such diverse causes as cardiac failure, fever, or heavy exercise. It disappears within hours after cessation of exercise and with resolution of the disease process. Proteinuria after marathon running may be as heavy as 5 g per L of urine.
Clinical evaluation and treatment. The most important first step in the development of an appropriate differential diagnosis and subsequent treatment plan for patients with proteinuria is classification of the urinary protein.
Classification of proteinuria
Overflow proteinuria. The presence of overflow is suggested by a disparity characterized by a small amount of proteinuria demonstrated by dipstick and a disproportionately larger amount measured on a 24-hour collected specimen. This is most often due to excretion of monoclonal light chains, which can be confirmed by IEP. Identification of a monoclonal immunoglobulin in the urine should stimulate diagnostic testing for multiple myeloma, amyloidosis, or a lymphoproliferative disorder. Hemoglobinuria and myoglobinuria can also cause overflow
P.128
proteinuria. However, these are readily identifiable, because in these conditions the dipstick for blood is strongly positive, whereas the microscopical examination of urine demonstrates few or no red blood cells. This finding should stimulate an evaluation for hemolysis and for rhabdomyolysis.Tubular proteinuria. A wide range of conditions can cause tubulointerstitial injury (see Table 8-4). An investigation of tubular proteinuria should begin with a detailed history regarding other affected family members (to rule out polycystic kidney disease); prescribed or over-the-counter medication use (analgesic nephropathy); frequent UTIs (reflux); flank pain; passage of renal stones; skin rash, arthralgias, arthritis (drug hypersensitivity, collagen-vascular disease); dry mouth or eyes (Sj gren's syndrome); occupational or recreational exposure to potential toxins; and manifestations of systemic disease. Physical findings confirmatory of disorders in the differential diagnosis listed in Table 8-4 should be sought. These might include palpably enlarged kidneys (polycystic kidney disease), band keratopathy (hypercalcemia, hyperparathyroidism), skin rash (systemic lupus, drug hypersensitivity), arthritis (gout, lupus), or oral mucosal lead line (lead toxicity). Laboratory tests including CBC with examination of the peripheral smear; measurements of serum creatinine, urea nitrogen, glucose, calcium, phosphorus, uric acid, and potassium; and urine culture are adjunctive information to the history, physical examination, urinalysis, and quantitative urine studies in focusing the differential diagnosis. Positive or negative results in these areas may suggest the need for further tests, which may include renal ultrasound (polycystic kidney disease, renal stones, obstruction); urine, serum, or hemoglobin electrophoresis (monoclonal gammopathy, sickle cell disease or trait); urine culture and sensitivity (pyelonephritis, renal tuberculosis); serum angiotensin-converting enzyme level (sarcoidosis); intravenous urography (medullary sponge kidney); or serum lead levels (lead toxicity). Although some tubulointerstitial disorders have characteristic histomorphologic features (medullary cystic disease, amyloidosis, myeloma kidney, hypokalemia), the microscopic appearance of most tubulointerstitial disorders lacks uniquely distinguishing characteristics. Renal biopsy is therefore used infrequently in the diagnosis of patients with tubulointerstitial disease. Therapy for the tubulointerstitial disorders is dependent on the underlying cause.
Glomerular proteinuria. When proteinuria is characterized by a disproportionate amount of albumin, a glomerular origin is implied. Mild transient proteinuria, especially associated with acute and reversible disease, is usually of little long-term significance. However, heavy and sustained proteinuria suggests a more serious disorder. Because the differential diagnosis is large, and many of the implicated disorders are uncommon, consultation with a nephrologist is recommended during the initial evaluation and treatment.
Patients with constant heavy proteinuria require close diagnostic scrutiny. Glomerular proteinuria in this group is often categorized either as non-nephrotic (less than 3.5 g per day per 1.73 m2 body surface area) or nephrotic (more than 3.5 g per day per 1.73 m2 body surface area). This somewhat arbitrary division of patients into those with lesser and greater amounts of urinary protein is justified by two major observations. First, patients with non-nephrotic proteinuria tend to have a better prognosis with regard to renal function than do patients with heavier proteinuria. As a result, an aggressive diagnostic or therapeutic approach may not be immediately indicated. Thus, after a search for underlying causes discernible by history, physical examination, and serologic assessment (see section III.D.), therapy including the use of antiproteinuric agents such as angiotensin
P.129
converting enzyme inhibitors (ACEi) with or without angiotensin receptor blockers (ARB), and follow-up to observe the course of the renal function and degree of proteinuria may be indicated in individual patients before a consideration of renal biopsy and the use of potentially risky immunosuppressive therapeutic regimens. Second, the morbidity and prognosis of patients with heavier proteinuria is conferred not only by the patient's renal functional outcome, but also by the pathophysiologic consequences of heavy proteinuria (e.g., the nephrotic syndrome).The nephrotic syndrome is defined by the presence of more than 3.5 g of proteinuria per 1.73 m2 of body surface area per day, hypoalbuminemia, hyperlipidemia, lipiduria, and edema. Heavy proteinuria induces enhanced tubular reabsorption and catabolism of proteins leaked into the glomerular ultrafiltrate, thus contributing to hypoproteinemia. Sodium and water retention, with resultant edema, occurs secondary to hypoproteinemia in some patients and as a direct result of glomerular injury in others. Hypoproteinemia and an associated decrement in plasma oncotic pressure may stimulate apolipoprotein synthesis in the liver, causing hyperlipidemia and lipiduria. It is postulated that in nephrotic disorders that persist for many years (e.g., membranous nephropathy), hyperlipidemia may contribute to accelerated atherosclerosis. Heavy proteinuria also predisposes to hypercoagulability; variable losses of antithrombin III, protein S, and protein C have been described in some but not all nephrotic patients. Other urinary losses may variably induce subtle abnormalities of function in some patients with nephrotic syndrome, including losses of immunoglobulin and complement (predisposition to infection), thyroid-binding globulin (low total thyroxine, normal thyroid-stimulating hormone), and vitamin D (hypovitaminosis, hypocalcemia, and secondary hyperparathyroidism). Individuals with heavy proteinuria express the complications of nephrotic syndrome variably, further reflecting differences in urinary protein losses, dietary intake, and genetic predisposition.
The differential diagnosis of heavy proteinuria. Once proteinuria is determined to result from glomerular injury, an underlying disorder should be sought. The very large number of differential diagnoses outlined in Table 8-3 can be narrowed substantially by obtaining a detailed history, physical examination, and appropriate serologic testing. The history should cover the following important details: the presence of diabetes; deafness; similarly affected family members (suggestive of Alport nephropathy or other familial nephritis); ethnicity [immunoglobulin A (IgA) nephropathy is frequent in Asians and infrequent in African Americans]; presence of fever; travel; medication use; transfusions, drug abuse, sexual orientation and partners [human immunodeficiency virus (HIV), hepatitis, syphilis]; arthritis, arthralgias, malar or skin rash, oral ulcers, alopecia [systemic lupus erythematosus (SLE) and other immune or hypersensitivity disorders]; hemoptysis (Goodpasture disease, Wegener granulomatosis); sinusitis, sterile otitis (Wegener granulomatosis); paresthesias, angiokeratomas, dyshidrosis, focal neurologic deficits (Fabry disease); weight loss, cough, breast mass (malignancy and secondary membranous nephropathy); allergies; childhood or adolescent UTI's (focal sclerosis secondary to reflux nephropathy); and episodes of gross or persistent microhematuria (IgA nephropathy, thin basement membrane nephropathy). The physical examination should be directed at establishing whether systemic disease is present and whether the nephrotic syndrome or its complications are present. All adult patients with abnormal proteinuria should also have, at a minimum, a chest x-ray; CBC; serum and urine protein electrophoresis; serum chemistry panel, including tests of renal and hepatic function; and a measurement of serum albumin, total protein, total and high-density lipoprotein cholesterol, triglycerides, glucose, and calcium levels. For patients older than the age of 40, stool guaiac examination in men and women and mammography in women should also
P.130
be performed. For those over the age of 50, a screening colonoscopy should be performed if it has not already been done. Additional serologic testing may be warranted, depending on the presence or absence of hematuria and the results of the aforementioned studies. Such additional tests include, but are not limited to, antinuclear and anti-double stranded DNA antibodies (lupus); antineutrophil cytoplasmic antibody (ANCA), anti-proteinase 3 and anti-myeloperoxidase antibodies (Wegener granulomatosis and other vasculitic syndromes); C3, C4 (may be low in endocarditis, poststreptococcal glomerulonephritis, lupus, membranoproliferative glomerulonephritis, cryoglobulinemia); antihyaluronidase, anti-DNase B, antistreptolysin O (poststreptococcal glomerulonephritis); anti-glomerular basement membrane antibody (Goodpasture disease); rheumatoid factor (endocarditis, cryoglobulinemia, rheumatoid arthritis); serum cryoglobulins; angiotensin-converting enzyme (sarcoidosis); glycosylated hemoglobin; serologic testing for syphilis; hepatitis B antigens and antibody; recombinant immunoblot testing and viral load for hepatitis C; and enzyme-linked immunoabsorbent assay/Western blot for human immunodeficiency virus. In the spirit of cost-consciousness, these tests should not be routinely ordered in all patients with glomerular proteinuria. Rather, a thoughtful synthesis of the history and physical examination should guide the selection of appropriate tests among those listed (and potentially others not mentioned).A renal biopsy should be considered if no underlying disorder emerges as a cause for glomerular proteinuria after a thorough evaluation. In addition, renal biopsy is indicated when a secondary cause has been identified and a histologic examination of the renal tissue would help guide therapy, as is the case for SLE.
Therapy. The treatment of glomerular disorders falls into three categories: treatment of the underlying systemic disease, general management of the nephrotic syndrome, and therapies for specific glomerular diseases.
Treatment of underlying systemic disease. Due to the large number of entities that potentially cause glomerulonephritis, the reader is referred to a textbook of internal medicine or nephrology for their treatment. However, special mention should be made of diabetes mellitus. Data suggest that tight glycemic control and ACEi and/or ARBs are useful in preventing or forestalling the development of overt diabetic nephropathy in patients with diabetes mellitus. These also have been shown to have a favorable impact on serum creatinine levels, progression to end-stage renal disease, and overall mortality in patients with diabetic nephropathy.
General management of the nephrotic syndrome. The treatment of edema due to the nephrotic syndrome begins with sodium restriction and diuretics. Care should be taken to avoid inducing a state of marked prerenal azotemia.
Many strategies have been advocated to decrease heavy proteinuria. Nonsteroidal anti-inflammatory drugs (NSAIDs) reportedly produce a decrease in proteinuria in some patients, along with a smaller decrement in GFR. Despite their utility in a few patients, however, the overall impact of NSAIDs in substantially diminishing proteinuria for the majority of patients with nephrosis is disappointingly small. ACEi and ARBs are also useful to decrease proteinuria, and their efficacy has been tested both in patients with diabetic nephropathy and in patients with the idiopathic nephrotic syndrome. Combinations of these agents appear to be additive in limiting proteinuria. Many months may elapse between ACEi and/or ARB initiation and the achievement of the maximal decrement in proteinuria from that dose, a phenomenon that suggests more than hemodynamic change as its mechanism of action. Proteinuria may also be reduced by lowering patients' mean arterial pressure to levels below 92 mm Hg, independent of the class of antihypertensive agents used to achieve this target. Finally, dietary protein restriction in the range of 0.6 to 0.8 g per kg per day has been suggested, both to slow the rate of loss
P.131
of renal function and as a further means of diminishing proteinuria. The efficacy of angiotensin blockade, the conflicting supporting data for the effcicacy of low-protein diets, and concerns regarding nutritional safety in patients with heavy proteinuria (i.e., more than 10 g per day) has made stringent dietary protein restriction less frequently prescribed in recent years. Nevertheless, patients with heavy proteinuria should be advised to eat a diet close to the recommended daily allowance of protein, which is 0.8 gm protein/kg body weight.The hyperlipidemia of the nephrotic syndrome may contribute to accelerated atherosclerosis. Use of the HMG-CoA (3-hydroxy-3-methylglutaryl coenzyme A) reductase inhibitors or agents such as gemfibrozil often lower serum low-density lipoprotein cholesterol and triglyceride levels substantially in patients with nephrosis. However, they should not be used together because of the increased risk of rhabdomyolysis. For the patient with venous or, less commonly, arterial thrombosis or pulmonary emboli, long-term anticoagulation for the duration of the nephrotic syndrome is recommended.
Therapies for specific glomerular diseases. The so-called primary glomerular disorders fall into essentially seven histologic categories: minimal-change glomerulopathy, IgM mesangial proliferative glomerulonephritis, focal and segmental glomerulosclerosis, IgA nephropathy, membranous nephropathy, membranoproliferative glomerulonephritis (MPGN), and crescentic glomerulonephritis. From a therapeutic standpoint, the first three are often considered as a group, and the latter four are considered separately. Because these disorders are relatively uncommon and therapies often involve the use of potentially toxic drugs, referral to a nephrologist is advised.
Minimal-change glomerulopathy IgM mesangial proliferation glomerulonephritis focal and segmental glomerulosclerosis. If the nephrotic syndrome is present, conservative general care as described in section IV.A.1 may be appropriate as an adjunct to specific therapy. These glomerular disorders are variably responsive to high-dose prednisone (1 mg per kg, maximum, 80 mg). More than 90% of children with minimal-change glomerulopathy have a complete remission of proteinuria within 2 months of starting steroid therapy. In adults, this figure is approximately 80% to 90%. Response rates for the latter two disorders are not as high (approximately 40% to 50% and 20% to 30%, respectively). Extending the duration of high-dose prednisone to 4 to 6 months may accrue as many as 10% to 15% additional complete remissions. Prednisone should then be slowly tapered over approximately 4 months. Relapse, steroid dependence, or both can be treated with the addition of cytotoxic agents (e.g., cyclophosphamide or chlorambucil), cyclosporine A, or possibly mycophenolate mofetil, depending on the severity of the nephrotic syndrome and the level of renal function. Treatment of prednisone-resistant patients is controversial. Genetically inherited forms of focal and segmental glomerulosclerosis have been identified and involve mutations of podocyte proteins. These inherited forms may or may not be steroid-responsive.
IgA nephropathy (Berger's disease). This is the most common form of primary glomerular disease in the world. It is particularly prevalent in Asia and Australia, and rare in African-Americans. Originally thought to be benign, it is now apparent that the condition progresses to end-stage renal disease in 20% to 40% of patients affected. Occasionally, reversible acute renal failure has been noted, particularly in association with gross hematuria. It may, rarely, be complicated by the presence of crescents. Clinically, it may be confused with a benign familial disorder, thin basement membrane nephropathy, because the presenting manifestation of each is predominantly glomerular hematuria. These two entities may be distinguished by a family history of
P.132
hematuria (occasional in IgA nephropathy, frequent in thin basement membrane nephropathy), the presence of abnormal proteinuria (frequent in IgA nephropathy, uncommon in thin basement membrane nephropathy), and renal biopsy (no glomerular immunoglobulins present in thin basement membrane nephropathy, mesangial IgA in Berger's disease). No therapeutic regimen is agreed upon to clearly affect outcome in IgA nephropathy, although there is support in different geographic locations for warfarin and dipyridamole with or without cyclophosphamide, omega-3 fatty acids, ACEi with or without ARBs, long-term steroids, and steroids with cytotoxic agents, the latter particularly for those with progressive renal insufficiency.Membranous nephropathy. Two-thirds of patients with this disorder either have a spontaneous remission or have stable or very slowly progressive renal insufficiency. Thus, the majority do well with conservative therapy (including ACEi and ARBs) directed at minimizing complications from the nephrotic syndrome. For patients at high risk for progression (e.g., those with heavy proteinuria, more than 10 g per day; hypertension; diminished GFR; male gender; or tubulointerstitial fibrosis on renal biopsy), specific therapy may be indicated. In this setting, steroids with a cytotoxic agent (either chlorambucil or cyclophosphamide) appears to be preferable to steroids alone in inducing a timely complete or partial remission. Cyclosporine A has been shown to be of benefit in a randomized trial; the benefit of mycophenolate mofetil remains anecdotal.
MPGN. Hepatitis C is the most common cause of MPGN type 1. Definitive therapy is uncertain, but a combination of interferon-alpha and ribavirin (or pegylated interferon alpha with ribavirin) may be indicated. Side effects due to ribavirin increase when the GFR falls below 50 mL per min. Steroids and cytotoxics may increase viral load, and should be used only for the cryoglobulinemic vasculitis complications of hepatitis C. Some investigators controversially suggest that children with MPGN type I (mesangial and subendothelial deposits) benefit from steroid therapy. Initial enthusiasm for the use of aspirin and dipyridamole has waned. No therapy is effective for type II (dense-deposit disease).
Crescentic glomerulonephritis. When glomerulonephritis is accompanied by crescents, a nephrologic emergency is present, due to the propensity for rapid progression to end-stage renal disease. Early diagnosis and implementation of therapy are essential to preserve renal function. After excluding poststreptococcal glomerulonephritis, three forms of crescentic glomerulonephritis are recognized. The reader is referred to a textbook of nephrology for the treatment of type I (antiglomerular basement membrane nephritis, or Goodpasture syndrome) and type III (pauci-immune glomerulonephritis, Wegener granulomatosis). Type II (immune complex mediated glomerulonephritis) is treated initially with intravenous methylprednisolone sodium succinate (Solu-Medrol ), followed by high-dose oral prednisone with or without cytotoxics. Because of the relative rarity of the syndrome, no randomized controlled trials have proved that additional benefits accrue from the use of cytotoxic agents. However, largely due to the clear-cut benefits of cyclophosphamide in types I and III and plasmapheresis in type I, some investigators have recommended their use in type II.
Suggested Readings
Addis T. The number of formed elements in the urinary sediment of normal individuals. J Clin Invest 1926;2:409.
Birch DF, Fairley KF, Whitworth JA, et al. Urinary erythrocyte morphology in the diagnosis of glomerular hematuria. Clin Nephrol 1983;20:78.
P.133
Fairley KF, Birch DF. Haematuria: a simple method for identifying glomerular bleeding. Kidney Int 1982;21:105.
Gadehold H. Quantitative estimation of cells in the urine. Acta Med Scand 1968;183:309.
Godfrey K, Harding M, Zhanel GG, Nicolle LE, Cheang M, for the Manitoba Diabetes Urinary Tract Infection Study Group. Antimicrobial treatment in diabetic women with asymptomatic bacteriuria. N Engl J Med 2002;347:1576 1583.
Haver MH. Urinary sediment: a textbook atlas. Chicago: American Society of Pathologists, 1983.
Kincaid-Smith P, Whitworth JA, eds. The kidney, 2nd ed. Oxford, UK: Blackwell, 1987.
Murphy BM, Fairley KF, Birch DF, et al. Culture of mid catheter urine collected via an open-ended catheter: a reliable guide to bladder bacteriuria. J Urol 1984;131:19.
Kim M, Corwin H. Urinalysis. In: Schrier RW ed. Diseases of the kidney and urinary tract, 7th ed. Philadelphia: Lippincott Williams & Wilkins: 317 332.
Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination of treatment of angiotensin II receptor blocker and angiotensin-converting enzyme inhibitor in nondiabetic renal disease (COOPERATE): a randomised controlled trial. Lancet 2003;361:117 124.
Ruggenenti P, Gaspari F, Perna A, Remuzzi G. Cross-sectional longitudinal study of spot morning urine protein: creatinine ratio, 24-hour urine protein excretion rate, glomerular filtration rate, and end-stage renal failure in chronic renal disease in patients without diabetes. BMJ (Clinical Research Edition) 1998;316(7130):504.
Scherberich JE. Urinary proteins of tubular origin: basic immunochemical and clinical aspects. Am J Nephrol 1990;10[Suppl 1]:43.
Waller KV, Ward KM, Mahan JD, Wismatt DK. Current concepts in proteinuria. Clin Chem 1989;35:755.