7 - Retinal Tumors
Editors: Tasman, William; Jaeger, Edward A.
Title: Wills Eye Hospital Atlas of Clinical Ophthalmology , The, 2nd Edition
Copyright 2001 Lippincott Williams & Wilkins
> Table of Contents > Chapter 7 - Retinal Tumors
function show_scrollbar() {}
Chapter 7
Retinal Tumors
James J. Augsburger
 |
| Macular retinoblastoma. |
P.264
Retinal tumors comprise a broad spectrum of congenital and acquired, benign and malignant, isolated and syndromic, neoplastic and nonneoplastic intraocular lesions of interest to ophthalmologists. The clinical spectrum of retinal tumors includes retinoblastoma, lesions and conditions simulating retinoblastoma, and miscellaneous other tumors that arise from or involve the neurosensory retina, retinal pigment epithelium (RPE), and pigmented or nonpigmented ciliary epithelium. Several extremely rare tumors that are unlikely to be encountered by even the busiest practitioner (e.g., glioneuroma, adenocarcinoma of the RPE) are not covered. The reader is referred to an encyclopedic text on ophthalmic pathology for information about such lesions.
Retinoblastoma
Retinoblastoma is a primary malignant intraocular neoplasm that arises from immature retinoblasts within the developing retina. It is the most common primary intraocular malignancy of childhood, occurring in about one in 15,000 individuals. Almost all cases occur in children under the age of 6 years. The neoplasm has strong tendencies to invade the brain via the optic nerve and to metastasize widely. Untreated children typically die of their disease within 2 to 4 years of onset of symptoms.
Clinical Features
Laterality and focality
Approximately 60% to 70% of retinoblastoma cases are unilateral, while the remaining 30% to 40% are bilateral. In unilateral cases, only a single tumor is usually present in the affected eye. In bilateral cases, multifocal tumors in both eyes are the rule.
Presenting signs and symptoms
The most common presenting symptom of retinoblastoma is leukokoria, a white pupil, in the tumor-containing eye or eyes (Fig. 7.1). Other common initial manifestations include strabismus (esotropia or exotropia; Fig. 7.2) and symptomatic or asymptomatic visual impairment. Less common presenting symptoms include an abnormal appearance of the eye (Fig. 7.2), such as a red eye, a cloudy cornea, or a change in color of iris, and pain in or around the eye.
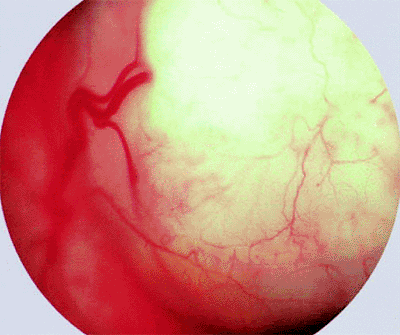
Appearance of tumors. Very small retinoblastoma tumors typically appear as translucent, whitish-pink intraretinal patches (Fig. 7.3), while slightly larger intraretinal tumors appear as well-defined, white, nodular lesions with prominent feeder and drainer retinal blood vessels (Fig. 7.4). Larger tumors commonly produce a nonrhegmatogenous retinal detachment that frequently becomes total and bullous (exophytic growth pattern) (Fig. 7.5). In other cases, tumor cells flake off from the vitreal surface of the mass and infiltrate the gelatinous vitreous (endophytic growth pattern) (Figs. 7.5, 7.6). Many tumors exhibit elements of both endophytic and exophytic growth.
Some eyes with retinoblastoma develop implantation tumors on the iris and accumulation of tumor cells in the aqueous (Fig. 7.7). Most of these eyes have a form of
P.265
P.266
retinoblastoma known as diffuse infiltrating retinoblastoma. Occasional retinoblastoma tumors stop growing spontaneously and either lose their malignant character or never acquire it. Such a tumor is called a retinoma. Tumors of this type (Fig. 7.8) tend to appear virtually identical to active retinoblastomas that have been treated successfully by irradiation. In other cases, massive intraocular retinoblastomas occasionally undergo spontaneous necrosis leading to phthisis bulbi.
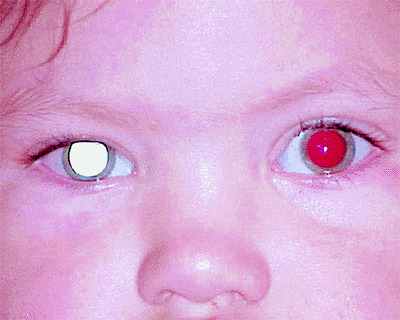 |
Figure 7.1. Leukokoria due to advanced intraocular retinoblastoma of right eye. |
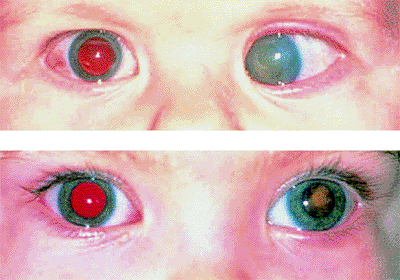 |
Figure 7.2. Strabismus as presenting manifestation of retinoblastoma. Left esotropia plus enlarged corneal diameter, corneal clouding, and loss of red reflex in left eye (top). Left exotropia, plus slightly enlarged corneal diameter and loss of red reflex in left eye (bottom). |
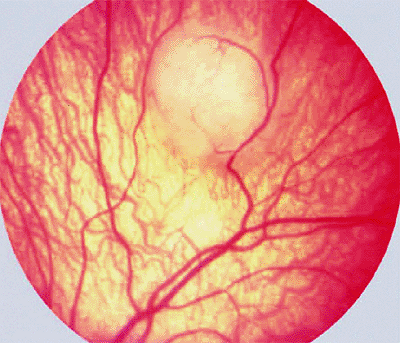 |
Figure 7.3. Small intraretinal retinoblastoma. White tumor is fed and drained by slightly dilated, mildly tortuous retinal blood vessels. |
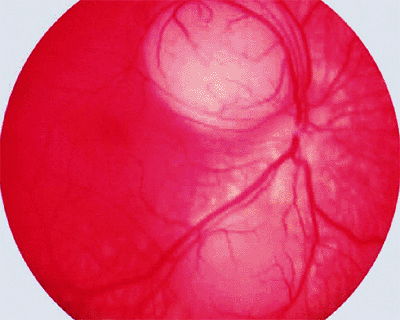 |
Figure 7.4. Slightly larger intraretinal retinoblastoma with prominent feeding and draining retinal blood vessels. |
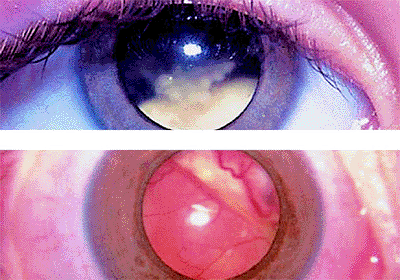 |
Figure 7.5. Endophytic and exophytic forms of retinoblastoma. Advanced endophytic retinoblastoma appears as avascular white mass inferiorly associated with prominent intravitreal tumor seeds (top). Advanced exophytic retinoblastoma appears as ill-defined yellow-white vascularized fundus mass superiorly associated with total bullous retinal detachment (bottom). |
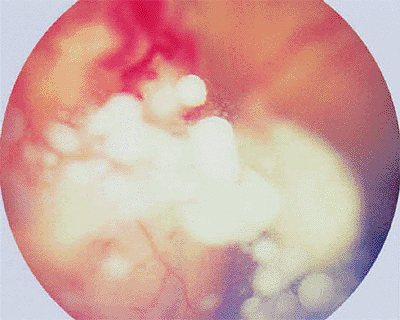 |
Figure 7.6. Prominent vitreous seeds of retinoblastoma overlying large intraretinal vascularized white retinal tumor. |
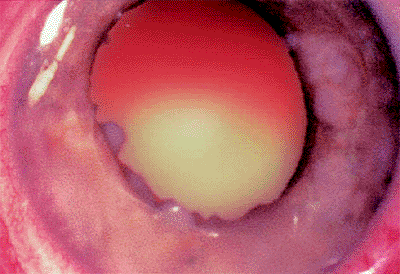 |
Figure 7.7. Eye with retinoblastoma showing implantation tumors on iris. |
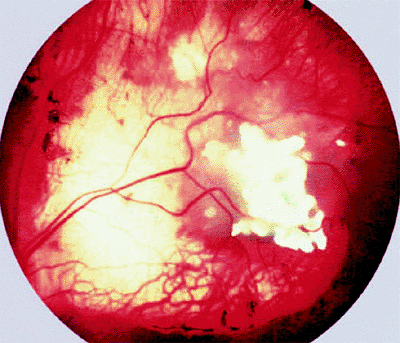 |
Figure 7.8. Retinoma. This spontaneously arrested retinoblastoma appears as a calcific tumor residue centrally surrounded by chorioretinal atrophy. |
Ancillary diagnostic studies. The three diagnostic studies most commonly used today in children with suspected retinoblastoma are ultrasonography, computed tomography (CT), and magnetic resonance imaging (MRI). B-scan ultrasonography generally reveals one or more soft tissue intraocular masses. In about 95% of cases, multifocal particulate intralesional calcification (characteristic of retinoblastoma) can be demonstrated (Fig. 7.9). CT demonstrates intraocular tumors and intralesional calcification better than ultrasonography (Fig. 7.10); however, not all retinoblastoma tumors become calcified. CT is also useful for evaluating the orbital optic nerve and for identifying extraocular tumor extension. MRI is the most useful and informative test currently available for evaluating sellar and parasellar regions of brain (to rule out ectopic intracranial retinoblastoma). At the same time, MRI appears to be less valuable than CT for assessing intraocular tumors because it does not show the intralesional calcification characteristic of this malignancy.
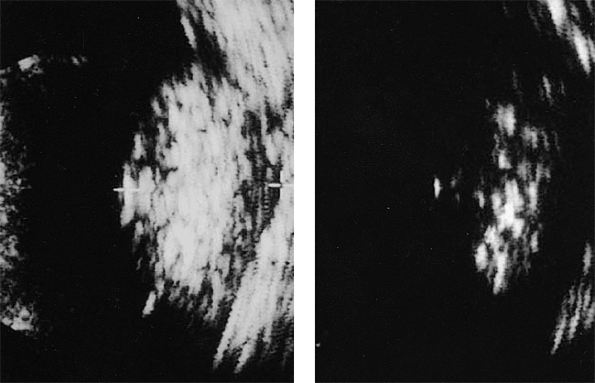 |
Figure 7.9. B-scan ultrasonography of retinoblastoma. Image of tumor obtained at typical gain setting (76 dB) for diagnostic examination of eye (left). Tumor appears generally but nonuniformly bright (hyperreflective). Same tumor at reduced gain setting (60 dB) is ill defined but exhibits multiple persistent strong particulate intralesional echoes (foci of calcification) (right). |
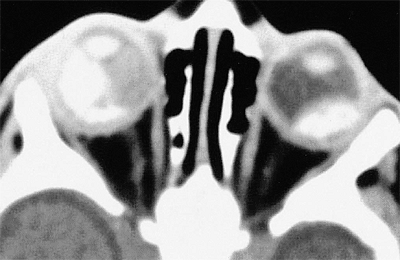 |
Figure 7.10. Computed tomography scan of bilateral retinoblastoma (without contrast enhancement). Intraocular tumors appear bright because of intralesional calcification. |
Genetics of retinoblastoma
Retinoblastoma appears to be due to loss or inactivation of both normal alleles of the retinoblastoma gene, a DNA sequence localized to a small segment of the long arm (the 14q region) of chromosome 13.
P.267
The timing of the loss or inactivation of the two normal alleles determines whether the disease is genetic (i.e., can be inherited by the offspring of an affected person) or somatic (i.e., cannot be inherited by the offspring of an affected person). In genetic retinoblastoma, at least one normal allele must be lost or inactivated prior to the first mitotic division of embryogenesis. This circumstance will arise if either the sperm or the egg contains defective DNA from an affected or carrier parent, or develops that defect by means of spontaneous mutation prior to fertilization. In somatic retinoblastoma, both alleles are present and active beyond the stage of the fertilized egg, but subsequent spontaneous mutation occurs to delete or inactivate both alleles in at least one immature retinal cell (retinoblast).
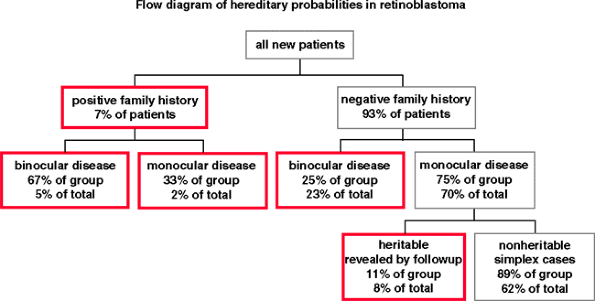 |
Figure 7.11. Flow diagram showing the hereditary probabilities in new cases of retinoblastoma. The boxes with red borders identify the heritable cases. |
The majority of cases of retinoblastoma (about 62%) are sporadic (i.e., diagnosed in patients who have no family history of retinoblastoma and no affected family members on comprehensive familial ophthalmic examination). The hereditary probabilities in a newly diagnosed child with retinoblastoma are shown in Figure 7.11.
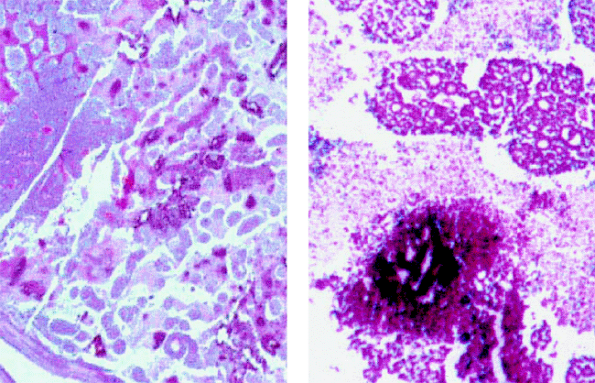 |
Figure 7.12. Histopathology of retinoblastoma. Low-power photomicrograph (left) and higher-power photomicrograph (right) showing cellular necrosis (pale, nonstaining areas) and intralesional calcification (intense reddish-purple foci) surrounding areas of viable retinoblastoma. |
Natural history of retinoblastoma
Left untreated, retinoblastoma is usually relentlessly progressive, leading to death of the affected child from intracranial invasion along the optic nerve, widespread metastasis, or both within about 2 to 3 years of initial symptoms. In occasional cases, however, retinoblastoma undergoes either spontaneous regression, resulting in phthisis bulbi of the involved eye, or shows benign growth arrest, retinoma, which is self-limited and stable in most affected individuals.
Pathology
Retinoblastoma is characterized histopathologically by malignant neuroepithelial cells (retinoblasts) arising within the immature retina. The retinoblasts typically appear to have a large basophilic nucleus and scanty cytoplasm. Cellular necrosis and intralesional calcification (Fig. 7.12) are frequent associations, especially in larger tumors. In some cases, tissue differentiation occurs, often producing Flexner-Wintersteiner rosettes (Fig. 7.13, left) and occasionally resulting in Homer-Wright rosettes (Fig. 7.13, center). In occasional cases, photoreceptor differentiation of individual retinoblasts (fleurettes) can also be observed (Fig. 7.13, right). Retinoblastoma has a strong tendency to invade the optic nerve and choroid and to extend out of the globe via either the optic nerve or the scleral emissarial canals.
Syndromic associations
Some patients with retinoblastoma develop a primary nonretinoblastoma intracranial malignancy, which is usually categorized histopathologically as either a pineoblastoma or an ectopic intracranial retinoblastoma. Presenting features of this tumor include somnolence, headache, or other neurologic symptoms. Central nervous system (CNS) imaging studies show a solid tumor involving the suprasellar or parasellar regions of the brain. Ophthalmoscopy frequently reveals papilledema. Because
P.268
this type of tumor usually occurs in children with genetic retinoblastoma who have bilateral disease, this association is commonly referred to as trilateral retinoblastoma. The intracranial malignancy has a strong tendency to seed the cerebrospinal fluid (CSF) and thereby spawn implantation tumors all along the spinal cord. This malignancy is usually fatal.
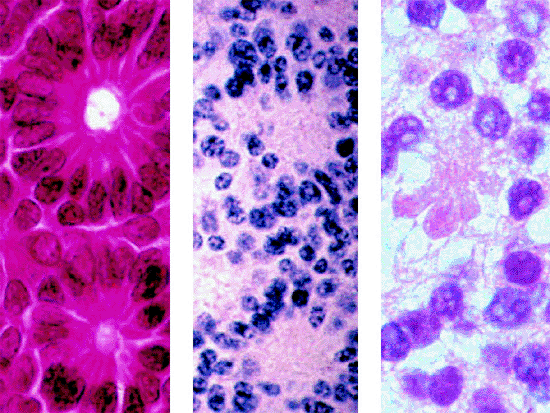 |
Figure 7.13. Tissue and cellular differentiation in retinoblastoma. Left: Flexner-Wintersteiner rosettes. Center: Homer-Wright rosettes. Right: Fleurette. |
Some children who develop retinoblastoma have a syndrome of multiple congenital anomalies attributed to a major deletion in the long arm of chromosome 13 (13q deletion syndrome). In children with this syndrome, the deletion is usually demonstrable by karyotype analysis. All infants with 13q deletion syndrome should be screened ophthalmoscopically for retinoblastoma.
Differential diagnosis
The principal differential diagnosis of retinoblastoma includes advanced Coats' disease (which tends to simulate advanced exophytic retinoblastoma), persistent hyperplastic primary vitreous, nematode endophthalmitis (which can simulate advanced endophytic retinoblastoma), retinopathy of prematurity with retrolental fibroplasia, retinal astrocytoma, intraocular medulloepithelioma, retinal capillary hemangioma (von Hippel tumor), and massive gliosis of the retina.
Management
Chemotherapy is the most commonly employed therapeutic option for children with advanced bilateral retinoblastoma in most parts of the world today. This type of therapy is also used in some children with advanced unilateral disease and in virtually all children with metastatic retinoblastoma. A cyclic, intravenous three-drug regimen consisting of carboplatin, etoposide, and vincristine (or some variation thereof) is currently the preferred combination in most centers. This treatment requires implantation of an indwelling venous port for repeated injection of the chemotherapeutic drugs. In some centers, the chemotherapy is supplemented by cyclosporine A, a drug that is known to reduce resistance of tumor cells to chemotherapeutic agents. The child generally undergoes a chemotherapy cycle every 3 to 4 weeks, depending on his or her tolerance, for six to nine cycles. Because of the potential for drug-related immunosuppression and bone marrow toxicity, children undergoing such treatment require close systemic monitoring by their pediatricians and pediatric oncologists during the course of treatment.
In some children, chemotherapy alone induces complete clinical regression of all treated tumors (Fig. 7.14). More frequently, the chemotherapy shrinks the intraocular tumors, so that the residual lesions can be eradicated by local treatments such as laser therapy (Fig. 7.15), cryotherapy,
P.269
and plaque radiotherapy (Fig. 7.16). This approach is referred to as chemoreduction followed by sequential aggressive local therapy. New tumors frequently arise in the peripheral fundus while the child is on chemotherapy, and residual lesions that regressed initially following the start of chemotherapy often relapse within 1 to 2 months after stopping chemotherapy. Vitreous seeds commonly regress markedly following chemotherapy but virtually always recur following discontinuation of the drug therapy. Because of these likely occurrences, children undergoing chemotherapy must be monitored closely during treatment and for an extended period of time following the completion of chemotherapy.
 |
Figure 7.14. Retinoblastoma treated by chemotherapy only with carboplatin, etoposide, and vincristine. Regressed macular tumor appears as well-defined clump of calcific tissue surrounded by foci of chorioretinal atrophy. |
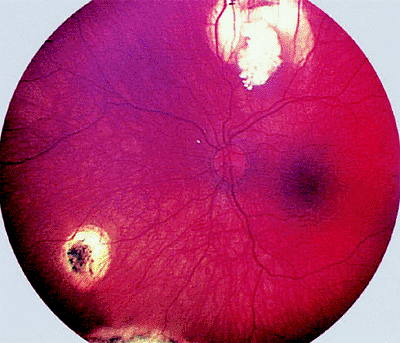 |
Figure 7.15. Retinoblastoma treated by chemotherapy plus laser therapy and cryotherapy. Superior tumor residue is completely calcific and partly surrounded by chorioretinal atrophy following laser therapy. The inferonasal lesion (also treated by laser) is completely atrophic, as is the inferior peripheral lesion (treated by cryotherapy). |
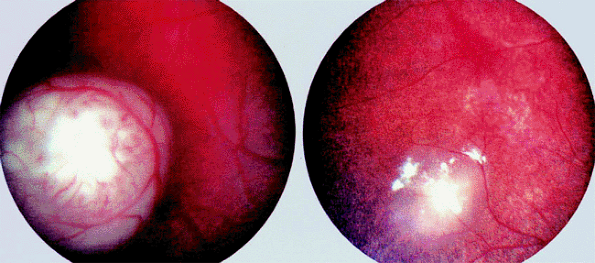 |
Figure 7.16. Retinoblastoma treated by plaque radiotherapy. Left: Tumor prior to treatment. Right: Regressed lesion 3 months following I-125 plaque radiotherapy. Lesion is markedly shrunken and exhibits foci of calcification. |
External beam radiation therapy (typically 4500 cGy delivered through precision lateral or oblique portals in a fractionated dose schedule over 4 to 5 weeks) was the primary treatment of choice for most children with advanced bilateral retinoblastoma until the advent of the chemotherapeutic regimen mentioned above. Although this method of treatment is frequently able to eradicate relatively large intraretinal tumors and multifocal intraretinal lesions (Fig. 7.17), it is often ineffective against vitreous seeding. It usually causes substantial orbital-facial bone growth arrest, induces development of posterior subcapsular radiation-induced cataracts, and causes other side effects. More importantly, long-term follow-up of retinoblastoma survivors who received external beam radiation therapy in childhood (especially prior to 6 months of age) has revealed a substantial increase in the expected frequency of malignant nonretinoblastoma neoplasms in the orbital-facial region (i.e., in the field of radiation). Most of this increased risk appears to be in children irradiated prior to 1 year of age. Because of this, most clinicians who deal with retinoblastoma regularly now try to avoid external beam radiation therapy. If such treatment must be used (e.g., in children who have failed chemotherapy with sequential aggressive local therapy), clinicians currently try to delay the radiotherapy until after the child is 1 year old.
Photocoagulation, the coagulation of tissues by means of intense light energy, is an effective local treatment for small- to medium-sized intraretinal retinoblastoma tumors. Therapeutic photocoagulation of retinal tissue can be produced with either a white light photocoagulator (e.g., xenon arc photocoagulator) or an ophthalmic laser (e.g., argon green laser). In retinal photocoagulation of retinoblastoma, the ophthalmologist creates an intense barrier of overlapping photocoagulator spots or a zone of confluent laser burns around the tumor in an attempt to obliterate its vascular
P.270
supply. Multiple treatment sessions 1 to 3 weeks apart are frequently needed to ensure tumor destruction.
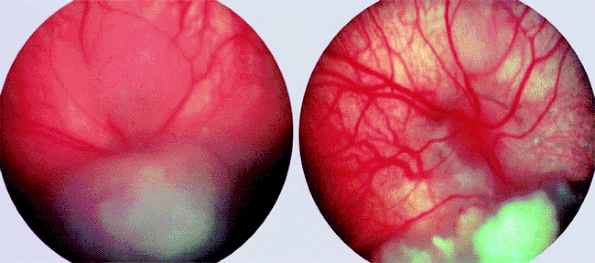 |
Figure 7.17. Retinoblastoma treated by external beam radiation therapy. Left: Multiple tumors prior to treatment. Right: Same tumors 6 months following irradiation (45 Gy over 5 weeks). Superior tumor has almost completely disappeared, and inferior tumor has regressed to a shrunken, calcific nodule. |
Noncoagulative laser therapy using a long wavelength (infrared) ophthalmic laser, relatively low power settings, long exposure times (typically 30 to 90 seconds per exposure), and a large spot size (typically 1 to 3 m in diameter) has recently become popular as an alternative to conventional photocoagulation. This method of treatment has been widely promoted under the term transpupillary thermotherapy (TTT). In this treatment, the ophthalmologist attempts to heat the entire tumor for a prolonged duration without causing frank tissue coagulation. In the operating room, this therapy requires a commercially available adapter to an operating microscope or a specially adapted indirect ophthalmoscope. The end point of treatment is a uniform graying of the treated tissue after a total treatment duration of approximately 15 minutes. Just as with photocoagulation, this type of treatment must be repeated every 1 to 3 weeks until the tumor is totally eradicated (or until the treatment is judged to have failed).
Cryotherapy, the disruption of tissues by repeated freezing and thawing, is applicable to most eyes with one or multiple small tumors, provided that the tumors are peripherally located (i.e., not involving or near the central macula or optic disc) and there is no vitreous seeding. Tumors at and anterior to the ocular equator can usually be treated directly through the overlying conjunctiva, but those posterior to the equator generally require incision of the conjunctiva and dissection of subconjunctival connective tissues down to bare sclera for satisfactory placement of the cryoprobe tip. Cryotherapy can be used as a single treatment in some eyes with one or a few small- to medium-sized peripheral tumors, in conjunction with photocoagulation or noncoagulative laser therapy in eyes with multiple small- to medium-sized anterior and posterior tumors, in conjunction with chemoreduction (as a locally destructive treatment in sequential aggressive local therapy), and as a subsequent treatment in some eyes with local tumor recurrences after plaque radiotherapy or external beam radiation therapy.
Plaque radiotherapy refers to delivery of ionizing radiation to a tumor by means of a temporary interstitial implant (the plaque) containing a radionuclide of specific known activity. The most commonly used radionuclides in eye plaques at this time are I-125 (a low-energy gamma emitter) and Ru-106 (a beta emitter). A plaque of appropriate diameter
P.271
(usually 2 to 4 mm larger than the estimated diameter of the tumor) is sutured to the sclera directly overlying the intraocular tumor. The plaque is left in place long enough to deliver a radiation dose of approximately 45 Gy to the tumor's apex (usually 2 to 4 days). The plaque is then removed at a second surgical procedure. This method of treatment frequently induces profound local regression of the treated tumor (Fig. 7.18).
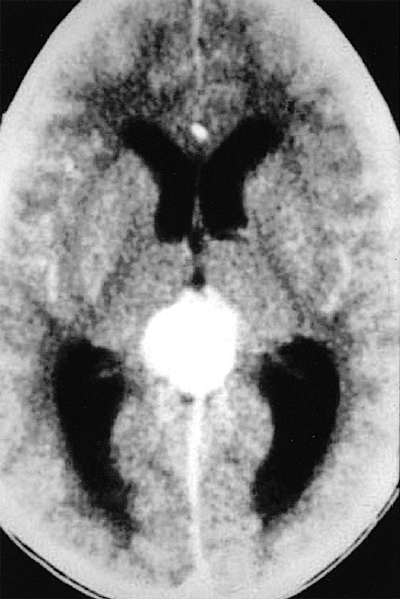 |
Figure 7.18. Ectopic intracranial retinoblastoma in child with bilateral retinoblastoma (trilateral retinoblastoma). Computed tomography scan of brain reveals prominent suprasellar mass that exhibits contrast enhancement. |
Plaque radiotherapy is generally applicable to larger tumors than can be eradicated with cryotherapy, photocoagulation, or noncoagulative laser therapy. Larger tumors typically regress to well-defined calcific residues (type I regression), but medium-sized and smaller tumors frequently shrink down to residual soft tissue masses with a gray fleshy appearance (type II regression). Plaque radiotherapy can be used as a single treatment or as an element in sequential aggressive local therapy. Plaque therapy has the potential to cause delayed radiation retinopathy, radiation-induced optic papillopathy, and radiation-induced cataract but (because of the plaque's shielding) does not appear to increase the risk of second malignant neoplasms in the facial-orbital region the way that external beam radiation therapy does.
Observation without treatment is appropriate for children with a retinoma. However, because such tumors have occasionally been noted to become active later in life, all such children must be monitored on a regular basis throughout life.
Prognosis
Most children with retinoblastoma do not die of this malignancy, at least in developed countries in which affected children present at a relatively early stage of disease. Unfavorable prognostic factors for survival include metastatic disease, orbital extension, optic nerve invasion, and massive choroidal invasion. As long as the tumor is confined within the globe and does not massively involve the choroid, the size of the intraocular tumor and the degree of cellular or tissue differentiation are not important prognostic factors for survival.
Prognosis for survivors
Children with genetic retinoblastoma have a substantially heightened risk of developing an intracranial nonretinoblastoma malignancy within the first decade of life (see above) and of developing bony and soft tissue sarcomas in later years of life. These second malignancies are frequently fatal. All patients who survive their retinoblastoma must be monitored throughout their life for possible development of one or more nonretinoblastoma malignancies.
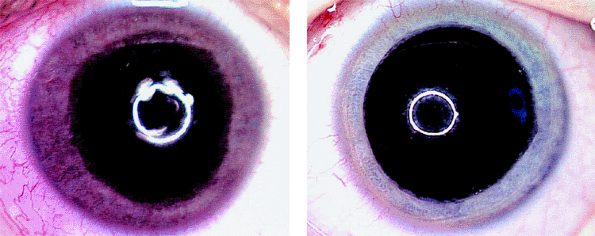 |
Figure 7.19. Heterochromia iridis as presenting manifestation of medulloepithelioma of ciliary body. Right iris (left image) appears substantially darker than the left iris (right image) due to florid iris neovascularization. The child's parents reported that this change of iris color developed abruptly less than 1 week prior to presentation. |
Medulloepithelioma
Intraocular medulloepithelioma is a rare primary intraocular tumor arising from the primitive ocular neuroectoderm. It is an uncommon tumor, but its precise frequency is unknown. It occurs in both benign and malignant varieties. It tends to be locally aggressive but rarely metastasizes. This tumor is usually congenital or infantile, but juvenile and even adult-onset cases have been reported. The average age of the affected individual at diagnosis is about 5 years. Medulloepithelioma affects all races and both sexes equally.
Clinical Features
Laterality and focality
Medulloepithelioma is almost always a unilateral, unifocal tumor. It usually arises in the ciliary body, but occasional lesions of this type have been found in the iris, retina, and optic nerve.
Presenting signs and symptoms
The usual presenting symptoms of medulloepithelioma are a red eye, a change in color of the iris (Fig. 7.19), a visible mass in the iris, and visual problems.
Appearance of tumors
The typical presenting findings are of a white, tan, or pink mass involving the ciliary body (Figs. 7.20, 7.21) and occasionally the root of iris (Figs. 7.22, 7.23),
P.272
often in association with prominent surface cysts (Fig. 7.23). Neovascularization of iris is common in affected eyes. If the tumor was present congenitally, there may be an associated defect in the lens (coloboma; Figs. 7.23, 7.24) due to a deficiency of the zonule in the region of the ciliary body tumor.
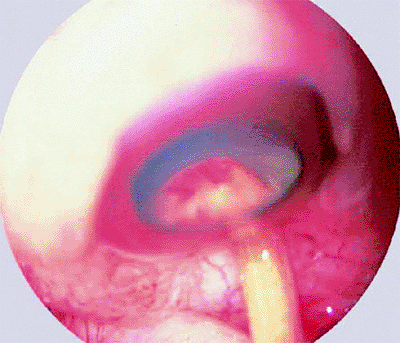 |
Figure 7.20. Medulloepithelioma of ciliary body. Anterior segment photograph taken during scleral depression in the pars plana region reveals pale ciliary body tumor with streaks of blood on its surface. |
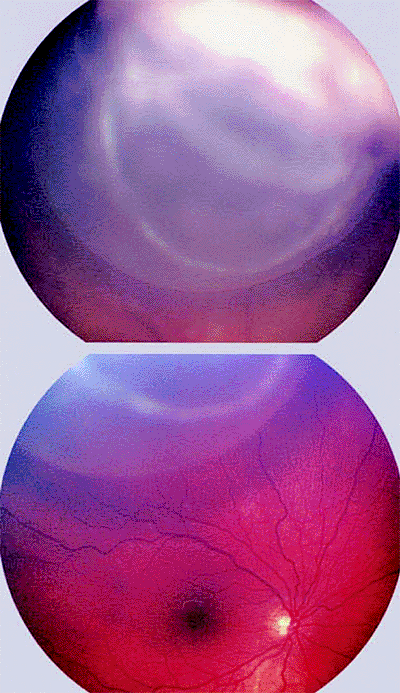 |
Figure 7.21. Cystic medulloepithelioma of ciliary body. Top: Solid portion of tumor appears opaque white while cystic portion appears gray with well-defined vitreal edge. Bottom: Cystic portion of mass is visible above normal optic disc and macula. |
 |
Figure 7.22. Medulloepithelioma of ciliary body with iris involvement presenting as fleshy, vascular peripheral iris tumor (slit lamp view). |
Ancillary diagnostic studies
B-scan ultrasonography is frequently able to image a relatively large ciliary body tumor; however, conventional contact B-scan imaging is usually insufficient to demonstrate a small ciliary body mass. For such a lesion, either a water bath ultrasound or biomicroscopic ultrasonography is required for satisfactory imaging. CT scanning (Fig. 7.25) and MRI are generally capable of imaging ciliary body masses that cannot be demonstrated by conventional contact B-scan ultrasonography. They should be considered if medulloepithelioma of the ciliary
P.273
body is suspected and water bath ultrasonography and biomicroscopic ultrasonography are not available.
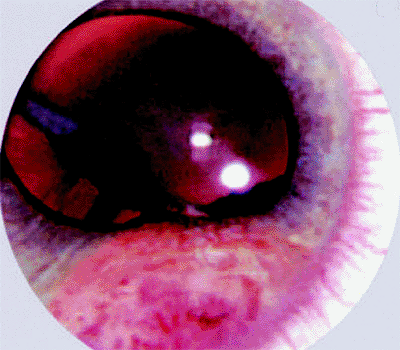 |
Figure 7.23. Fleshy vascularized iris mass and lens coloboma as presenting manifestations of congenital medulloepithelioma of ciliary body with iris involvement. |
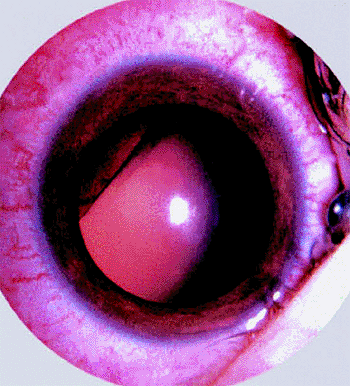 |
Figure 7.24. Sectorial notch in lens (lens coloboma) as sole presenting manifestation of ciliary body medulloepithelioma. |
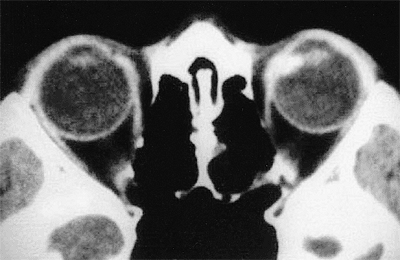 |
Figure 7.25. Computed tomography scan of medulloepithelioma of ciliary body nasally in left eye. |
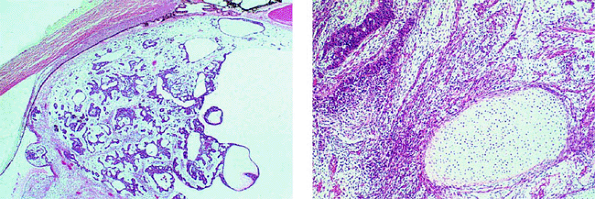 |
Figure 7.26. Histopathology of medulloepithelioma. Left: Low-power photomicrograph of nonteratoid medulloepithelioma showing cords and tubules of tumor cells and prominent associated cysts. Right: High-power photomicrograph of teratoid medulloepithelioma showing intralesional cartilage (oval area of pale tissue at lower right). |
Natural history
The natural history of untreated medulloepitheliomas is essentially unknown. A few lesions of this type that have been observed for prolonged periods of time have remained relatively stable, but others have enlarged progressively over time. Because extraocular extension can occur in some advanced cases, surgical excision of the tumor or enucleation is usually performed shortly after the tumor is recognized.
Pathology
The characteristic histopathological feature of intraocular medulloepitheliomas is a structural arrangement of cells that closely resembles the neural medullary epithelium. The degree of cellular differentiation differs widely from case to case. Many well-differentiated medulloepitheliomas containing prominent rosettes and cystic spaces (Fig. 7.26, left) containing hyaluronic acid are common. Medulloepitheliomas that contain heterotopic elements such as hyaline cartilage (Fig. 7.26, right), striated muscle, or brain are referred to as teratoid medulloepitheliomas. Those that do not contain such elements are referred to as nonteratoid medulloepitheliomas. About two thirds of intraocular medulloepitheliomas are categorized as malignant pathologically, largely on the basis of invasiveness and extraocular extension of the tumor, especially if associated with a prominent degree of undifferentiation and numerous mitotic figures.
Differential diagnosis
The principal differential diagnosis of intraocular medulloepithelioma includes retinoblastoma, nematode granuloma, and juvenile xanthogranuloma.
Management
Enucleation is generally recommended if the eye is blind and painful, if the eye has advanced neovascular glaucoma due to tumor, if the tumor is very large, and if there is clinical evidence of extrascleral extension of tumor. Microsurgical tumor excision is sometimes performed if the intraocular tumor is relatively small, especially if the tumor involves only the peripheral iris and anterior ciliary body only and there is no evidence of neovascular glaucoma or extrascleral extension.
P.274
Astrocytoma of Retina (Astrocytic Hamartoma)
The retinal astrocytoma is a benign glioma arising from the astrocytes of the neurosensory retina. It tends to arise early in life, being detected in many affected persons during childhood or adolescence. It affects all races and both sexes equally. It is extremely rare, but its precise frequency has not been determined. This tumor is regarded by many ophthalmic pathologists as a hamartoma rather than a true neoplasm. In some patients, it occurs as a manifestation of the tuberous sclerosis syndrome.
Clinical Features
Laterality and focality
The retinal astrocytoma occurs in unilateral and bilateral forms. The bilateral form is usually characterized by multifocal tumors in both eyes, and most patients with such lesions have tuberous sclerosis. The unilateral form is usually characterized by a single tumor in the affected eye, and most patients with this form of the disease do not appear to have tuberous sclerosis.
Presenting signs and symptoms
Affected patients usually have no visual symptoms unless one or more tumors involve the macula. In rare instances, a retinal astrocytoma remote from the fovea causes a nonrhegmatogenous retinal detachment that involves the macular retina and abruptly blurs the vision in that eye.
Appearance of tumors
The typical ophthalmoscopic finding is that of one or more whitish superficial retinal tumors ranging from thin translucent patches through somewhat thicker opalescent lesions to well-defined opaque nodules (Figs. 7.27, 7.28, 7.29, 7.30). The retinal vasculature tends to be somewhat irregular at the site of each lesion, but prominent tortuous feeder and drainer retinal blood vessels similar to those in retinoblastoma do not occur.
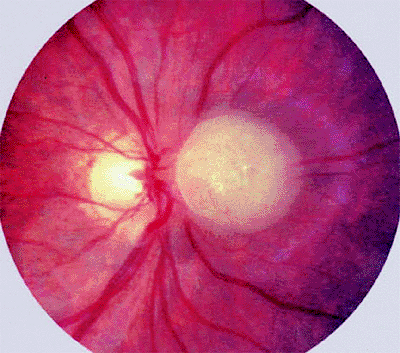 |
Figure 7.27. Astrocytoma (astrocytic hamartoma) of retina. Opaque white intraretinal tumor just nasal to optic disc. The tumor appears solid and has a faintly bumpy surface texture. |
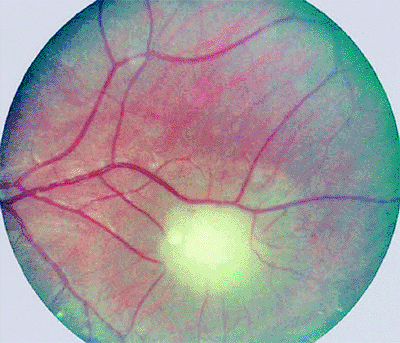 |
Figure 7.28. Astrocytoma (astrocytic hamartoma) of retina. Small, homogeneously white intraretinal tumor in fundus midzone. The tumor appears soft and noncalcified. |
Ancillary diagnostic studies
Fluorescein angiography generally reveals a prominent network of superficial and deep intralesional blood vessels. B-scan ultrasonography shows a solid soft tissue mass that, in some cases, contains focal calcifications. CT scanning is sometimes able to image larger intraocular tumors, but it is more useful for showing associated paraventricular intracranial tumors in patients who have tuberous sclerosis (see below).
Syndromic associations
A substantial but currently unspecified proportion of individuals with one or more retinal astrocytomas is found to have other manifestations of tuberous sclerosis. This syndrome is characterized by cutaneous lesions (adenoma sebaceum of the face, subungual fibromas, ash leaf spots, shagreen patches), CNS lesions (benign paraventricular
P.275
astrocytomas of the brain, retinal astrocytomas), and various visceral lesions (cysts in the lungs, liver, and other organs; benign tumors in the heart; rhabdomyomas), and a host of other abnormalities.
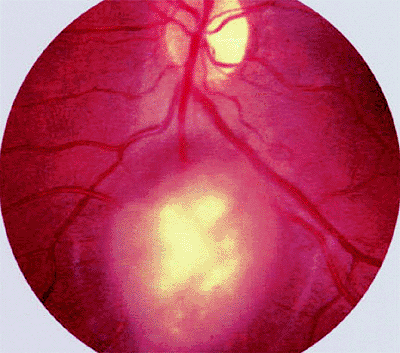 |
Figure 7.29. Astrocytoma (astrocytic hamartoma) of retina. Opaque yellow-white superficial retinal tumor is not associated with dilated, tortuous retinal blood vessels or surrounding subretinal or intraretinal exudates. |
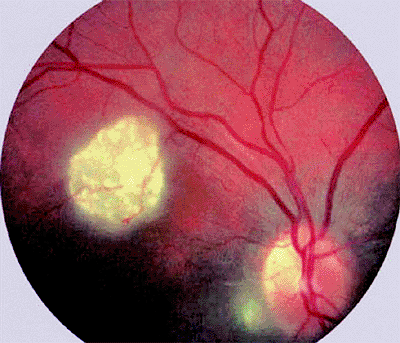 |
Figure 7.30. Multiple retinal astrocytomas (astrocytic hamartomas) in patient with tuberous sclerosis. Larger lesion is located just above center of macula, and small lesion is located just temporal to optic disc. |
Genetics
Tuberous sclerosis is inherited as an autosomal-dominant disease with incomplete penetrance in some families. Several chromosomal loci have been linked with tuberous sclerosis, the most common of which is located on the long arm of chromosome 9 (locus 9q32-34).
Natural history
Retinal astrocytomas appear to have extremely limited malignant potential; however, some tumors of this type have been noted to grow to a rather large size and lead to bullous retinal detachment and blindness in the affected eye.
Pathology
The typical retinal astrocytoma consists of a mass of enlarged fibrous astrocytes containing small oval nuclei and interlacing cytoplasmic processes (Fig. 7.31). Larger lesions frequently contain foci of calcification.
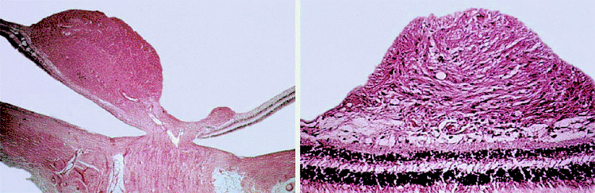 |
Figure 7.31. Histopathology of retinal astrocytomas. Left: Lower-power photomicrograph showing two retinal tumors arising from the inner portion of sensory retina near the optic disc. Right: Higher-power photomicrograph showing tumor composed of benign fibrous astrocytes. |
Differential diagnosis
The principal differential diagnosis of retinal astrocytoma includes intraretinal retinoblastoma, nematode granuloma of the retina, and retinal capillary hemangioma.
Management
In most cases, no treatment is required. For patients whose tumor enlarges progressively and eventuates in a blind, painful eye, enucleation seems to be the only available treatment.
Capillary Hemangioma of Retina (Von Hippel Tumor)
The retinal capillary hemangioma (hemangioblastoma) is a benign retinal vascular tumor that occurs in unifocal and multifocal, monocular and binocular, and hereditary and nonhereditary forms. It is rarely if ever congenital but tends to develop in most patients by the second to fourth decades of life. It affects both sexes equally and has no racial predilection. Its precise frequency is unknown. Untreated tumors commonly cause substantial visual impairment and sometimes lead to total blindness. Most patients with multifocal binocular disease have other features of the von Hippel Lindau syndrome.
Clinical Features
Laterality and focality
The usual patient has a single lesion in one eye, but some patients have multifocal or bilateral tumors or both. The precise relative frequency of unilateral, unifocal lesions versus bilateral, multifocal retinal capillary hemangiomas is unknown.
Presenting signs and symptoms
Blurred vision in the affected eye or loss of visual field are the usual presenting symptoms. In some patients, the affected eye is blind by the time any problem is recognized. In advanced cases, the patient presents with a blind painful eye due to secondary glaucoma. In families known to have members with the von Hippel Lindau syndrome, ophthalmic screening examinations frequently identify asymptomatic small tumors in one or both eyes.
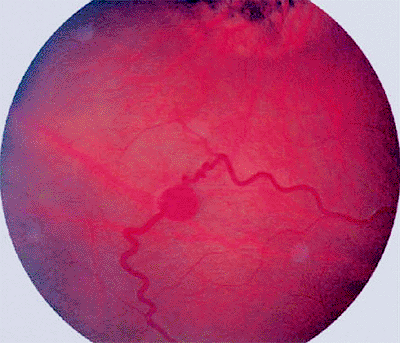 |
Figure 7.32. Capillary hemangioma of retina (von Hippel tumor). Fundus photograph shows small red intraretinal tumor fed and drained by prominent dilated tortuous retinal blood vessels. Superior to the lesion is an area of chorioretinal atrophy subsequent to prior cryotherapy of another retinal capillary hemangioma. |
P.276
Appearance of tumors
The classic retinal capillary hemangioma is a discrete reddish retinal vascular tumor connected to prominent, dilated, tortuous afferent and efferent retinal blood vessels (Figs. 7.32, 7.33, 7.34, 7.35). Intraretinal and subretinal exudates (Figs. 7.33, 7.34) and nonrhegmatogenous retinal detachment with vitreoretinal traction (Fig. 7.35) are frequently associated with such tumors.
Ancillary diagnostic studies
Fluorescein angiography reveals rapid blood flow through the vascular tumor, shows fluorescein leakage through the walls of the vascular lesion, and distinguishes the feeder arteriole from the draining venule. CT scanning and MRI can be used to search for intracranial vascular tumors and abdominal visceral tumors of the von Hippel Lindau syndrome.
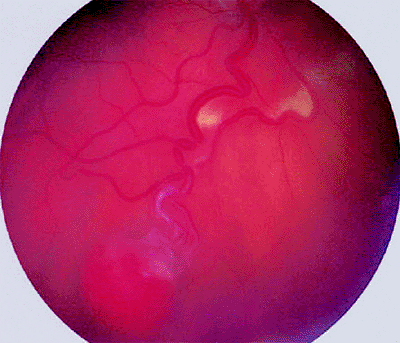 |
Figure 7.33. Capillary hemangioma of retina (von Hippel tumor). Typical globular red retinal lesion with dilated, tortuous afferent and efferent retinal blood vessels. A line of intraretinal exudates is present approximately halfway between the hemangioma and the optic disc. |
 |
Figure 7.34. Capillary hemangioma (von Hippel tumor) involving optic disc. Reddish vascular tumor is surrounded by intraretinal and subretinal exudates. Dilated afferent and efferent retinal blood vessels are not usually observed with tumors at the disc. |
Genetics
The von Hippel Lindau syndrome is an autosomal-dominant disease that has been linked to a defect in the short arm of chromosome 3 (locus 3p25-26). This syndrome is highly penetrant in affected families. Most individuals who have multifocal, bilateral retinal capillary hemangiomas have this syndrome, while most persons with a single tumor in one eye do not.
Natural history
Some untreated retinal capillary hemangiomas remain stable or enlarge minimally over long periods, but most tumors of this type enlarge progressively and lead to exudative, hemorrhagic, and fibrotic complications if followed without treatment.
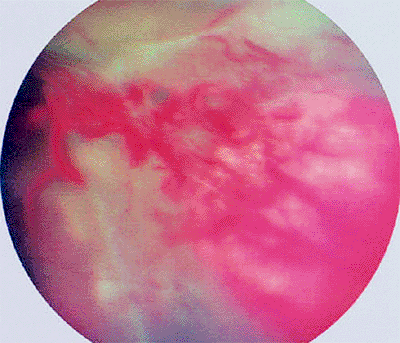 |
Figure 7.35. Large capillary hemangioma of peripheral retina (von Hippel tumor) with an associated exudative-tractional retinal detachment. |
P.277
Pathology
The vascular tumor consists of small capillary-like blood vessels lined by endothelial cells and supported by a delicate tumor stroma that contains vacuolated fibrous astrocytes (Fig. 7.36).
Differential diagnosis
Idiopathic acquired nonfamilial retinal hemangiomatous lesion
Hemangiomatous retinal neovascularization associated with:
Prior scleral buckling surgery for retinal detachment
Retinitis pigmentosa
Retinopathy of prematurity
Familial exudative vitreoretinopathy
Baseline assessment
CT scan or MRI of the CNS and abdomen are generally recommended as part of the patient's baseline systemic evaluation to rule out clinically important extraocular lesions of the von Hippel Lindau syndrome in the CNS (hemangioblastomas) and abdominal viscera (renal cell carcinomas, pheochromocytomas); they are particularly important in patients with a personal or family history consistent with the von Hippel Lindau syndrome and in those with multifocal and/or bilateral retinal hemangiomas.
Management
Photocoagulation is applicable to small tumors that do not involve the optic disc or central macula in eyes that do not have an exudative retinal detachment or vitreous clouding.
Cryotherapy is appropriate for small- to medium-sized tumors of the equatorial or oral zone of fundus and can be used even in the presence of localized exudative retinal detachment.
Diathermy can be used to treat some medium-to-large tumors. This treatment is generally performed as a penetrating treatment using a needle-like diathermy tip inserted into the hemangioma transsclerally via a lamellar scleral bed just external to the lesion. Scleral buckling is usually performed at the same time.
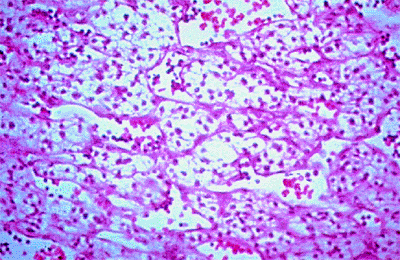 |
Figure 7.36. Histopathology of retinal capillary hemangioma. Tumor consists of small vascular channels lined by mature endothelial cells and a stroma of pale tumor cells. |
Scleral buckling and vitrectomy are sometimes performed in conjunction with endodiathermy, endophotocoagulation, and even endoresection of the tumor in eyes with bullous exudative retinal detachment or tractional retinal detachment.
Enucleation is usually reserved for patients with a blind painful eye due to tumor or for those with an unsightly phthisical eye.
Observation appears to be appropriate management for selected patients who have one or more small tumors not causing visual problems, especially if they are not producing any exudative or hemorrhagic problems.
Cavernous Hemangioma of Retina
The cavernous hemangioma of the retina is a benign retinal vascular tumor having characteristic clinical and pathological architecture. This lesion is very uncommon, but its precise frequency is unknown. It affects both sexes and occurs in all races. Most affected patients have a single lesion in one eye and no evidence of a multi-system syndrome.
Clinical Features
Laterality and focality
The typical patient has a single lesion in one eye; however, some patients have multifocal or bilateral tumors or both. The precise relative frequency of unilateral, unifocal lesions and bilateral, multifocal hemangiomas is unknown.
Presenting signs and symptoms
Most patients with a cavernous hemangioma of the retina are asymptomatic when their tumor is detected on ophthalmic examination. In some cases, vision is blurred due to the macular location of the lesion. In other cases, the patient reports floaters attributable to intravitreal bleeding from the hemangioma.
Appearance of tumors
Ophthalmic examination typically reveals a localized cluster of dark red vascular saccules
P.278
(Figs. 7.37, 7.38) frequently in proximity to a central large-caliber retinal vein. There may be whitish fibrous tissue on the surface of the lesion, and this white tissue can be mistaken on casual viewing for intraretinal exudate. If the component vascular saccules are relatively large, plasma-erythrocyte separation can be seen in some of the saccules. In some affected eyes, one can find intraretinal and preretinal or intravitreal blood arising from the surface of the tumor.
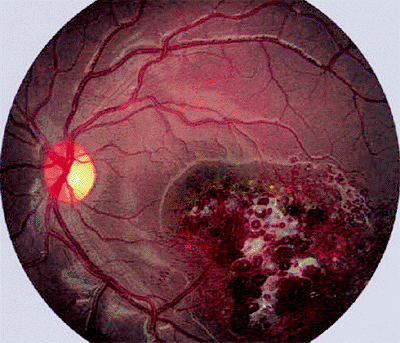 |
Figure 7.37. Cavernous hemangioma of retina. Macular retinal lesion appears as cluster of aneurysmal vascular saccules with some associated white superficial fibrosis (gliosis). |
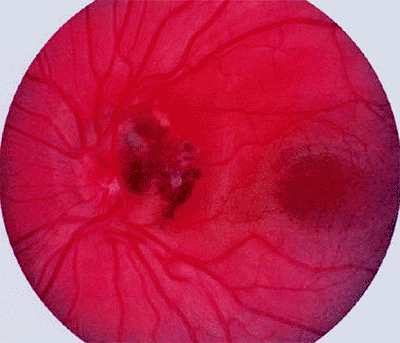 |
Figure 7.38. Cavernous hemangioma of retina involving optic disc. Tumor consists of a collection of small vascular saccules containing dark red blood. |
Ancillary diagnostic studies
Fluorescein angiography reveals slow blood flow through the vascular lesion, accentuates the central vein of the lesion, and confirms plasma-erythrocyte separation in some of the larger vascular saccules.
Genetics
In most patients with retinal cavernous hemangioma, the fundus tumor is a sporadic, nonfamilial, nonsyndromic lesion. However, some patients appear to have an uncommon familial disorder characterized by other benign cavernous or telangiectatic vascular lesions of the skin and CNS.
Natural history
Most retinal cavernous hemangiomas that have been observed for prolonged periods have remained stable in size and clinical appearance. Occasional hemangiomas of this type bleed repeatedly into the vitreous over the course of many years.
Pathology
The retinal capillary hemangioma consists of a cluster of large-caliber, thin-walled intraretinal vascular saccules lined by normal vascular endothelial cells (Fig. 7.39). The tumor thickens and replaces the neurosensory retina at the affected site.
Differential diagnosis
Coats' disease (idiopathic retinal telangiectasis)
Idiopathic perifoveal telangiectasia
Baseline patient assessment
CT scanning or MRI of the CNS is probably appropriate in patients with a personal or family history of retinal or CNS vascular lesions; however, such studies are probably not necessary in patients with a unifocal, unilateral lesion and no symptoms or neurologic signs suggesting an intracranial abnormality.
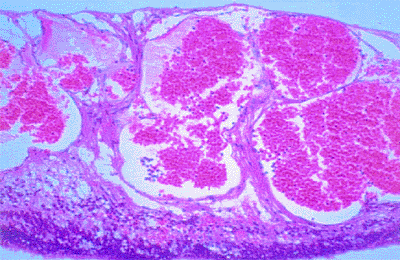 |
Figure 7.39. Histopathology of cavernous hemangioma of retina. Tumor consists of a cluster of large-caliber, thin-walled intraretinal vascular saccules lined by normal vascular endothelial cells. |
Management
Observation is recommended for most lesions of this type because of their benign nature and usual lack of progression.
Coagulation (photocoagulation or cryotherapy) is applicable to small- to medium-sized lesions that bleed repeatedly.
Excision of the lesion via posterior vitrectomy can be performed in eyes with massive or nonclearing intravitreal hemorrhage from the hemangioma.
Metastatic Carcinoma to Retina
Metastatic carcinoma to the retina is an extraocular primary malignancy that has metastasized via the bloodstream to the retina. Although metastatic tumors to the eye are relatively common, most such lesions involve the uveal tract. The retina is an uncommon site for a metastasis to appear. The great majority of retinal metastatic lesions are unifocal and unilateral.
Clinical Features
Laterality and focality
The great majority of retinal metastatic lesions are unilateral and unifocal; however, bilateral and multifocal retinal metastatic tumors have been reported.
Presenting signs and symptoms
Patients with a metastatic retinal tumor tend to have visual symptoms corresponding to the location of the tumor. In some patients, the tumors are detected on routine ophthalmic evaluation of asymptomatic eyes.
Appearance of tumors
The usual metastatic tumor to the retina is a white to tan, placoid intraretinal mass (Fig. 7.40). The most common exception to this appearance is the metastatic melanoma from the skin, which typically appears
P.279
dark brown to almost black. If the tumor involves the optic disc, it appears as an infiltrative lesion, often with finely dispersed tumor cells in the overlying vitreous.
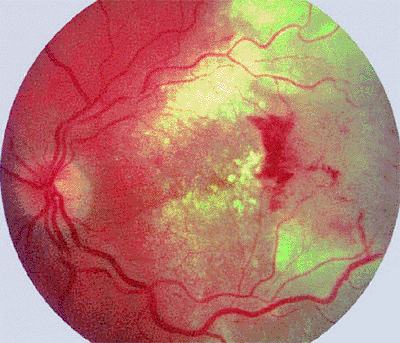 |
Figure 7.40. Metastatic cutaneous malignant melanoma to retina. Tumor is an ill-defined tan infiltration of the retina (at right in photograph) associated with extensive surrounding intraretinal exudates and some localized surface hemorrhage. |
Ancillary diagnostic studies
On fluorescein angiography, a metastatic retinal lesion generally appears hypofluorescent early and somewhat hyperfluorescent late. Leakage of fluorescein from the retinal blood vessels damaged by the lesion is frequently apparent in the late frames.
Systemic implications
The patient who develops a metastatic lesion to the retina is likely to have other bodily sites of metastasis. Consequently, such a patient's prognosis for long-term survival is correspondingly poor.
Natural history
If untreated, most metastatic carcinomas to the retina progress, leading eventually to retinal detachment and marked visual loss.
Pathology
The pathology of metastatic carcinomas to the retina depends largely on the nature of the metastasizing cancer. The retina is thickened and replaced by metastatic tumor cells in the area of involvement.
Differential diagnosis
Microbial infiltrate (e.g., Candida, Nocardia, Cryptococcus, etc.)
Nonmicrobial inflammatory granuloma (e.g., sarcoid granuloma)
Leukemic or lymphomatous infiltrate
Baseline patient assessment
Comprehensive systemic history and physical examination is essential in patients suspected of having a metastatic retinal tumor for identifying other metastatic lesions and, if necessary, finding the primary malignancy. Chest x-ray, mammography, CT scanning, MRI and other studies of these types are needed to evaluate the patient for metastatic lesions in other bodily sites not detectable by physical examination.
Management
External beam radiation therapy is commonly used to treat visually significant metastatic retinal tumors in patients who are not in a terminal phase of their illness.
Chemotherapy hormone therapy, or immunotherapy is appropriate in patients with generalized metastatic disease, assuming that there is a reasonable chance of salvage.
Observation is probably appropriate in most terminal patients, especially in ones who have a healthy fellow eye.
Enucleation of the affected eye is occasionally required for pain relief in patients who develop a blind, painful eye due to their retinal tumor.
Leukemic Infiltration of Retina
Leukemic infiltration of the retina is an extravascular proliferation of leukemic cells within the substance of the retina. Lesions of this type are uncommon, but their precise frequency is unknown. They occur in individuals of any age, but peaks of incidence mirror peaks of incidence of leukemia in general. Retinal involvement is much more common as a manifestation of leukemic relapse than as an initial site of disease. Hemorrhagic retinal lesions are much more common in leukemic patients than are true malignant infiltrates.
Clinical Features
Laterality and focality
Leukemic retinal infiltrates may be unifocal or multifocal and unilateral or bilateral.
Presenting signs and symptoms
The usual symptoms are blurred vision, dim vision, and floaters.
Appearance of tumors
The typical leukemic retinal infiltrate is a fuzzy, flat, white retinal lesion (Fig. 7.41) that is frequently associated with retinal hemorrhages and overlying intravitreal cells (Fig. 7.42).
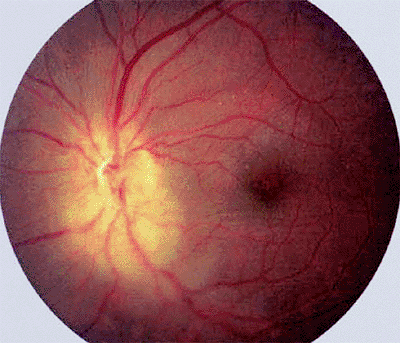 |
Figure 7.41. Leukemic infiltration of retina and optic disc in a patient with chronic myelogenous leukemia. Tumor appears as an ill-defined yellowish infiltration that partly obscures the retina and optic disc. |
 |
Figure 7.42. Leukemic cells in the vitreous of a boy with relapse of acute lymphoblastic leukemia. |
P.280
Ancillary diagnostic studies
All patients with suspected leukemic retinal infiltration should be evaluated thoroughly from a hematologic perspective (i.e., with complete blood count and differential count, bone marrow aspiration or biopsy, complete systemic staging evaluation, and probably lumbar puncture for CSF cytology). In addition, if there is still uncertainty about the ocular diagnosis, the ophthalmologist should consider pars plana vitrectomy or retinal biopsy to confirm the diagnosis.
Systemic implications
Concurrent CNS leukemia is common in patients who develop a retinal leukemic infiltrate. Consequently, CNS imaging and lumbar puncture are especially indicated in any patient suspected of having leukemic relapse in the eye. Not surprisingly, leukemic infiltration of the retina is an unfavorable prognostic factor for survival.
Natural history
Untreated leukemic infiltrates tend to progress at a variable rate and to eventuate in profound visual loss in the affected eye.
Pathology
The infiltrated retina and overlying vitreous contain leukemic cells in various quantities (Fig. 7.43). Leukemic cells are commonly observed in the lumina of retinal blood vessels.
Differential diagnosis
Microbial infiltrate (e.g., Candida, Nocardia, Cryptococcus, etc.)
Nonmicrobial inflammatory granuloma (e.g. sarcoid granuloma)
Lymphomatous infiltrate
Management
In terminal patients, no treatment for the ocular lesions is indicated. In patients who are salvageable, chemotherapy appropriate to the precise type of leukemia is usually indicated. If the infiltration is associated with profound visual loss because of macular or optic nerve involvement, palliative external beam radiation therapy should also be considered.
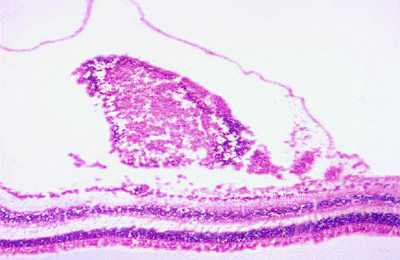 |
Figure 7.43. Histopathology of leukemic retinal infiltrate. This photomicrograph shows an accumulation of leukemic cells under the internal limiting membrane. |
Lymphomatous Infiltration of Retina
Lymphomatous infiltration of the retina is a malignant intraretinal or subretinal collection of atypical lymphoid cells. Such tumors are uncommon, but their precise frequency is unknown. Lymphomatous retinal infiltrates usually occur as a feature of primary lymphoma of the CNS and retina, but they rarely occur as a metastatic tumor in patients with systemic non-Hodgkin's lymphoma. The disease generally affects older adults and occurs somewhat more commonly in women than in men.
Clinical Features
Laterality and focality
Retinal lymphomatous infiltrates can involve one or both eyes, but bilateral involvement is distinctly more common (about 80% of cases). However, asynchronous ocular involvement is often noted in affected patients who develop binocular disease.
Presenting signs and symptoms
The usual symptoms are blurred vision and floaters.
Appearance of tumors
The characteristic retinal lesions are geographic yellowish-white subretinal pigment epithelial lesions (Fig. 7.44) and fuzzy, ill-defined intraretinal patches (Fig. 7.45), both of which are frequently associated with overlying, finely dispersed white cells in the vitreous.
Ancillary diagnostic studies
Patients suspected of having primary retinal lymphoma should undergo CT scanning or MRI of the brain and lumbar puncture for CSF cytology. If primary CNS lymphoma is confirmed by such studies, pathologic confirmation of the intraocular diagnosis by biopsy may be judged unnecessary. However, diagnostic posterior vitrectomy with cytopathologic assessment of intravitreal cells or even transscleral retinal biopsy must be considered when the CNS evaluation is negative for lymphoma and when the intraocular lesions are not absolutely characteristic. The vitreous from eyes with suspected intraocular lymphoma can also be evaluated to determine levels
P.281
of interleukins (IL) 10 and 6. Recent evidence suggests that an IL-10/IL-6 ratio greater than 1 is strongly suggestive of lymphoma.
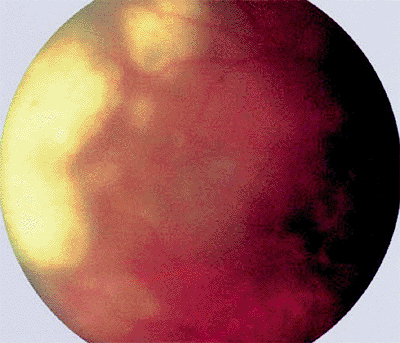 |
Figure 7.44. Lymphomatous infiltration of retina in a patient with primary lymphoma of the retina and central nervous system. Tumor appears as geographic, relatively flat, yellowish-white subretinal pigment epithelial infiltrates (left in photograph) surrounded by less distinct satellite lesions. The lesions appear a bit hazy because of associated diffuse fine intravitreal cells. |
Natural history
Untreated infiltrative lymphoid lesions of the retina tend to progress and result in progressive visual loss in the affected eye or eyes. However, some lesions have been noted to wax and wane without specific intervention.
Pathology
The infiltrative retinal and subretinal pigment epithelial lesions are composed of malignant lymphoid cells that typically have the cytologic features of large cell lymphoma (Fig. 7.46).
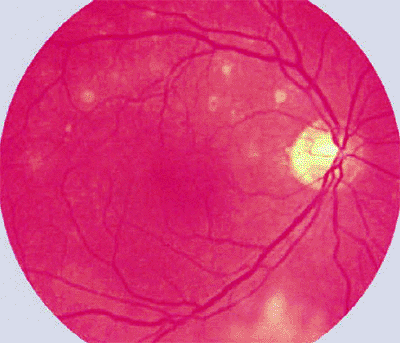 |
Figure 7.45. Primary lymphoma of retina (confirmed pathologically). Multiple small deep retinal or subretinal amelanotic foci are evident above macula, and an ill-defined area of lymphomatous inner retinal whitening is present inferiorly from optic disc. |
 |
Figure 7.46. Cytopathology of subretinal lymphoid infiltrate. High-power photomicrograph showing malignant lymphoid cells, some exhibiting mitotic figures, obtained by fine needle aspiration biopsy. |
Differential diagnosis
Microbial infiltrate (e.g., Candida, Nocardia, Cryptococcus, etc.)
Nonmicrobial inflammatory granuloma (e.g., sarcoid granuloma)
Leukemic infiltrate
Systemic implications
Patients who have primary CNS lymphoma or a visceral non-Hodgkin's lymphoma are at great risk for death due to complications of that disease.
Management
If only the eyes are involved, ocular irradiation by external beam radiation therapy (35 to 45 Gy in fractionated schedule) is generally recommended. If there is concurrent CNS lymphoma, aggressive chemotherapy and sometimes whole brain irradiation are usually recommended. The role of chemotherapy in primary intraocular lymphoma without evidence of CNS lymphoma or visceral lymphoma is controversial. Local intraocular relapses of lymphoma in previously irradiated eyes can sometimes be treated effectively by repeated intravitreal injections of methotrexate.
Hypertrophy of Retinal Pigment Epithelium
Hypertrophy of the RPE is a benign congenital hamartoma of the RPE. The lesion occurs in typical and atypical forms and in unifocal and multifocal patterns. The prevalence of this lesion is unknown, but it appears to be relatively common. There is no apparent racial or gender predilection.
Clinical Features
Laterality and focality
There are three distinct clinical patterns of these lesions:
Unifocal unilateral the typical solitary lesion referred to as congenital hypertrophy of the RPE
Multifocal unilateral this clustering of lesions is commonly referred to as grouped pigmentation of the retina (or bear tracks )
Multifocal bilateral or atypical this clinical pattern is observed most commonly as a marker phenomenon for Gardner's syndrome and several other forms of familial adenomatous polyposis carcinoma of the colon
P.282
Presenting signs and symptoms
These lesions are virtually always asymptomatic and detected on routine ophthalmic examination.
Appearance of tumors
The characteristic tumors are gray to black, flat fundus lesions at the retinal pigment epithelial level (Figs. 7.47, 7.48, 7.49). These lesions typically occur in the midzone to periphery of the fundus, but they occasionally occur in the macular and juxtapapillary regions of the fundus. Flatness of the lesion can usually be confirmed by slit lamp biomicroscopy of the fundus, binocular indirect ophthalmoscopy with scleral depression over the lesion, or, if necessary, B-scan ultrasonography.
Typical unilateral unifocal lesions appear as nummular or geographic lesions with well-defined, smooth margins (Fig. 7.47). These lesions may have one or more central lacunae of depigmentation. Typical unilateral multifocal lesions generally appear less dense in terms of the pigmentation of the individual lesions but are frequently densely clustered in a small area of the retina (Fig. 7.48). The atypical bilateral multifocal lesions tend to be more irregular and angulated in shape and less uniform in pigmentation than are the other two types of lesions (Fig. 7.49). These lesions tend to be scattered widely in the fundus and not localized to one area.
Ancillary diagnostic studies
No diagnostic studies are generally indicated for characterization of these retinal lesions.
Syndromic associations
As noted above, atypical multifocal, bilateral fundus lesions of congenital hypertrophy of the RPE tend to be identifying markers for Gardner's syndrome and some other autosomal-dominant colonic polyposis carcinoma syndromes in affected kindred. Because of this, the ophthalmologist should always review the family history of any individual with multifocal, bilateral, atypical hypertrophy of the RPE and also recommend comprehensive colonic evaluation of all such patients.
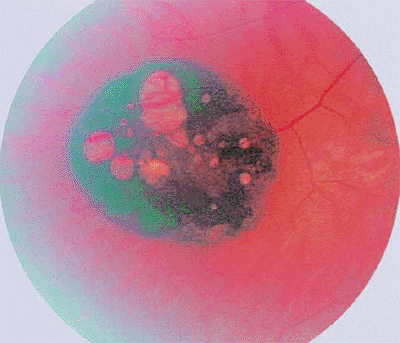 |
Figure 7.47. Hypertrophy of retinal pigment epithelium (RPE), unifocal type. Black lesion appears well circumscribed and nummular, and has several white lacunae of RPE depigmentation. |
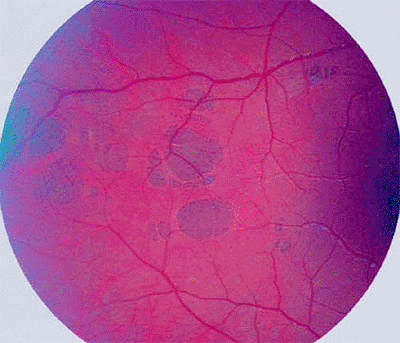 |
Figure 7.48. Hypertrophy of retinal pigment epithelium (RPE), multifocal type. The lesion appears as a cluster of gray to black, well-defined RPE lesions that have been likened to bear tracks on the retina. |
Natural history
Most retinal lesions of this type change minimally if at all over extended periods of follow-up. In rare instances, malignant change (to adenocarcinoma of the RPE) has been documented in lesions of this type. Because of this, periodic life-long monitoring of such lesions is probably advisable.
Pathology
The lesions of hypertrophy of the RPE consist of well-defined foci of taller than normal retinal pigment
P.283
epithelial cells containing an increased number of large oval melanin granules (Fig. 7.50).
 |
Figure 7.49. Hypertrophy of retinal pigment epithelium, atypical-multifocal-bilateral type, associated with colonic adenomatous polyposis carcinoma. |
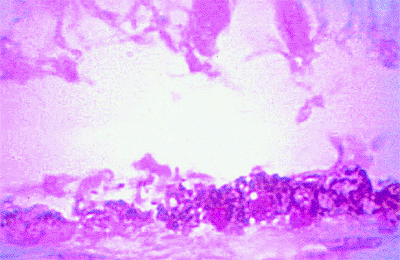 |
Figure 7.50. Histopathology of hypertrophy of retinal pigment epithelium (RPE). High-power photomicrograph showing abrupt transition between normal RPE (on left) and abnormally thickened RPE densely packed with melanin granules (on right). The overlying retina is artifactitiously detached and degenerated. |
Differential diagnosis
Hyperplasia of RPE
Metastatic melanoma to retina
Malignant melanoma of choroids
Management
No treatment is indicated for typical lesions of this type.
Combined Hamartoma of Retina
The combined hamartoma of the retina is a benign tumor comprised of retinal pigment epithelial cells, neurosensory retina, retinal vascular elements, and vitreous. This lesion is usually detected during infancy or early childhood and is probably congenital in a large proportion of affected individuals. Although it can cause profound visual impairment, it has no recognized malignant potential. It sometimes occurs in association with other features of neurofibromatosis type 2.
Clinical Features
Laterality and focality
The combined hamartoma of the retina is almost always unifocal and unilateral. With rare exceptions, it arises in a juxtapapillary or circumpapillary location.
Presenting signs and symptoms
The most common presenting feature is amblyopia in the involved eye due to macular involvement.
Appearance of lesion
The typical lesion is a geographic white gliotic retinal lesion with deep intrinsic gray-black pigmentation, especially at its margins, and marked tortuosity of the retinal blood vessels in the area of the lesion (Figs. 7.51, 7.52). The retinal vascular pattern and chorioretinal appearance peripheral to lesion are normal.
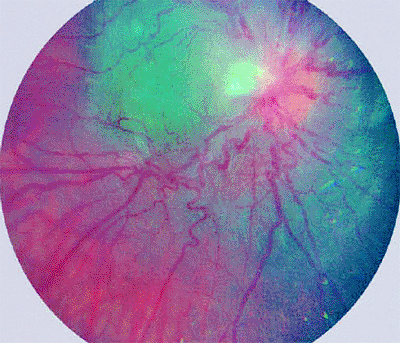 |
Figure 7.51. Combined hamartoma of retina. Typical lesion surrounds optic disc, appears gray in its deeper layers, and exhibits superficial whitish gliosis with pronounced tortuosity and angulation of involved retinal blood vessels. |
Ancillary diagnostic studies
Fluorescein angiography accentuates the prominent tortuous intralesional retinal blood vessels but provides no important differential diagnostic information. B-scan ultrasonography can image thicker lesions but is also not particularly helpful in differential diagnosis. CT scanning and MRI can reveal bilateral acoustic neuromas (the characteristic feature of neurofibromatosis type 2) in patients who have that syndromic association.
Syndromic associations
Although most cases have been sporadic and nonsyndromic, a substantial number of cases have been associated with neurofibromatosis types 2 (central neurofibromatosis, bilateral acoustic neuromas), and an occasional
P.284
case has been linked with neurofibromatosis type 1 (von Recklinghausen's disease). Consequently, neurofibromatosis should always be considered in youngsters found to have a combined hamartoma of the retina.
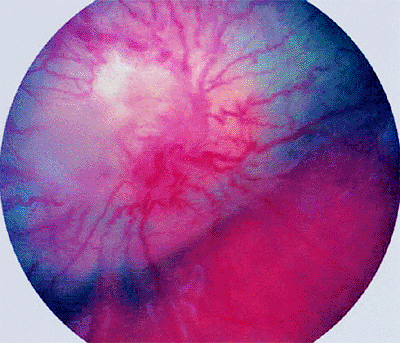 |
Figure 7.52. Combined hamartoma of retina. Lesion surrounds and completely obscures optic disc. Tumor in this case is approximately 3 mm thick and has well-defined margins. It is gray in its deeper layers and appears whitish on its surface due to superficial gliosis. The involved retinal blood vessels are markedly angulated and tortuous. |
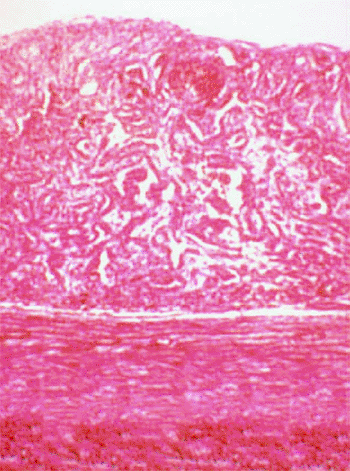 |
Figure 7.53. Histopathology of combined hamartoma of retina. High-power photomicrograph shows tumor to be composed of interlacing cords of retinal pigment epithelial cells and blood vessels within disorganized and thickened neurosensory retina. |
Natural history
Most such lesions are minimally progressive after detection. In those cases that change during follow-up, the apparent progression is usually due to contraction of the vitreoretinal surface of the lesion and not to cellular proliferation of the lesion per se.
Pathology
The few pathologic specimens that have been reviewed have been composed of interlacing cords of retinal pigment epithelial cells and blood vessels within disorganized and thickened neurosensory retina (Fig. 7.53) and a proliferation of benign fibrous cells on its vitreal surface.
Differential diagnosis
Choroidal malignant melanoma (because of the dark intrinsic pigmentation)
Retinoblastoma (because of the whitish appearance of the surface gliosis in many lesions)
Management
No treatment is generally indicated. Posterior vitrectomy has been employed in a few cases to remove the vitreoretinal fibrosis, but such procedures have met with limited visual success.
Massive Gliosis of Retina
Massive gliosis of the retina is a benign proliferation of retinal glial cells in response to some underlying injury or other local process. Recognized causes of such lesions include severe ocular trauma, chronic intraocular inflammation, retinal vascular occlusive diseases, and even congenital malformations. In spite of its name, massive gliosis does not necessarily involve a large portion of fundus; this is because the word massive in the term refers to the mass-like nature of the lesion rather than its size. Massive gliosis is rare, but its precise frequency is unknown. It can arise at any age, but it is rarely identified in childhood.
Clinical Features
Laterality and focality
Massive gliosis of the retina is almost always a unilateral unifocal lesion.
Presenting signs and symptoms
If the lesion develops in childhood, amblyopia is possible. If the lesion develops later in life, profound visual loss usually occurs in the affected eye.
Appearance of tumors
The typical lesion (when it can be observed ophthalmoscopically) is a gray-white to black fundus tumor (Fig. 7.54), depending on the proportion of pigment-laden retinal pigment epithelial cells contained in the lesion. In many patients, no fundus view is possible because of dense cataract, vitreous opacification, or other media abnormalities. Most affected eyes have an extremely limited visual potential.
Ancillary diagnostic studies
B-scan ultrasonography reveals a soft tissue mass that can be difficult if not impossible to distinguish from a posterior uveal malignant melanoma. Fluorescein angiography has rarely been attempted because the optical media of the eye are usually hazy or opaque.
Natural history
Progression of lesions of this type has rarely been observed, in part because enucleation is commonly recommended if the mass is extremely large and in part because the ophthalmoscopic view of the lesion is typically poor to nil in most affected eyes.
Pathology
The tumor is composed of benign spindle-shaped fibrous astrocytes, an abundant fibrous extracellular
P.285
matrix, and numerous dilated thick-walled blood vessels scattered throughout the lesion (Fig. 7.55).
 |
Figure 7.54. Localized form of massive gliosis of retina showing prominent superficial white glial proliferation, marked retinal pigment epithelial hyperplasia with intraretinal pigment migration, and abnormal intralesional blood vessels. |
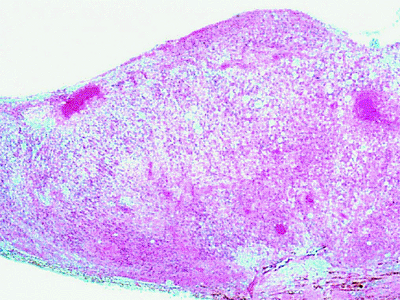 |
Figure 7.55. Histopathology of massive gliosis of retina. Tumor is composed of spindle-shaped fibrous astrocytes, abundant fibrous extracellular matrix, and numerous thick-walled intralesional blood vessels. Retinal pigment epithelial cells are present within basal aspect of mass. |
Differential Diagnosis
Choroidal malignant melanoma
Eccentric disciform lesion
Management
If the lesion is recognized for what it is, simple observation is all that is indicated. In many cases, however, the affected eye comes to enucleation because posterior uveal melanoma cannot be excluded.
Hyperplasia of Retinal Pigment Epithelium
RPE hyperplasia is a benign reactive proliferation of RPE cells that develops in response to traumatic insult of some type, including focal infection and noninfectious inflammation. It is a rather common lesion, but its prevalence in the general population is unknown. It can develop at any age but is rarely congenital.
Clinical Features
Laterality and focality
The lesion is usually unifocal and unilateral.
Presenting signs and symptoms
These lesions are usually asymptomatic unless they involve the macula.
Appearance of lesions
The typical RPE hyperplasia is a focal irregular black retinal lesion, usually with some associated circummarginal chorioretinal atrophy and intraretinal pigment cell migration (Fig. 7.56). On high magnification fundus biomicroscopy, intraretinal migration of the black RPE pigment is usually evident.
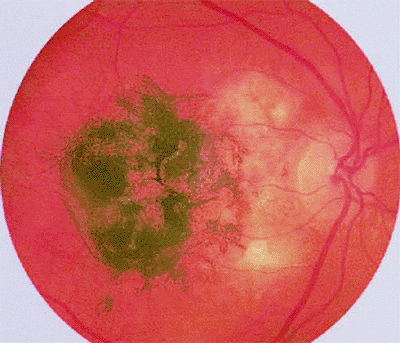 |
Figure 7.56. Hyperplasia of retinal pigment epithelium in macula. |
Ancillary diagnostic studies
No diagnostic studies are generally indicated for lesions of this type.
Natural history
The lesions of RPE hyperplasia, once developed, usually remain dormant or change minimally during long-term follow-up. However, occasional lesions of this type have enlarged substantially during follow-up, suggesting either malignant change or erroneous initial clinical diagnosis.
Pathology
The lesion consists of an increased number of retinal pigment epithelial cells that are also larger than normal. The cells are densely packed with large spherical melanin granules. The overlying sensory retina is partially degenerated, and the retinal pigment epithelial cells extend into the sensory retina.
Differential diagnosis
Choroidal malignant melanoma
Unifocal hypertrophy of RPE
Management
No treatment is generally indicated for typical lesions of this type. However, enucleation is usually performed when malignant change is suspected on the basis of substantial or rapid tumor progression during follow-up.
Acquired Nonfamilial Retinal Hemangiomatous Lesion
The acquired nonfamilial retinal hemangiomatous lesion is a benign nodular retinal vascular tumor of the peripheral fundus that resembles a retinal capillary hemangioma. It typically occurs in older adults as a manifestation of peripheral chorioretinal degeneration with subretinal neovascularization, but similar lesions also occur in younger patients with certain underlying chorioretinal disorders.
P.286
Clinical Features
Laterality and focality
This lesion is almost exclusively unilateral and unifocal. It usually develops in the oral zone of the fundus adjacent to the ora serrata, most commonly in the inferotemporal quadrant.
Presenting signs and symptoms
The lesion is asymptomatic in most affected patients because of its peripheral location, but it can cause blurred vision if an exudative response extends to the macula or vitreous hemorrhage occurs.
Appearance of tumors
The typical lesion is a red to pink, globular oral zone mass associated with subretinal and intraretinal exudation (Fig. 7.57). Unlike the classic retinal capillary hemangioma, this lesion tends not to have any prominent dilated, tortuous retinal feeder and drainer blood vessels.
Ancillary diagnostic studies
Fluorescein angiography usually reveals slow filling of the retinal vascular network that ramifies on the surface of the tumor, but it also commonly suggests at least partial vascular supply from the underlying choroid. In the late frames, one usually observes profuse leakage of fluorescein into the overlying vitreous and surrounding subretinal fluid.
Natural history
The tumor characteristically changes minimally if at all in size during postdetection follow-up; however, the exudation associated with the vascular tumor can increase over time and result in exudative retinal detachment that threatens or involves the macula. In some cases, the lesion can also be the source of intermittent intravitreal bleeding.
Pathology
The few lesions of this type that have been examined histopathologically have consisted of hemangiomatous neovascular proliferations arising from the peripheral uvea via a defect in Bruch's membrane. They are composed of cavernous vascular channels lined by uveal-type endothelial cells, extravasated blood, and fibrous connective tissue.
 |
Figure 7.57. Typical acquired nonfamilial retinal hemangiomatous lesion in the inferior periphery with associated thick subretinal and intraretinal exudates. |
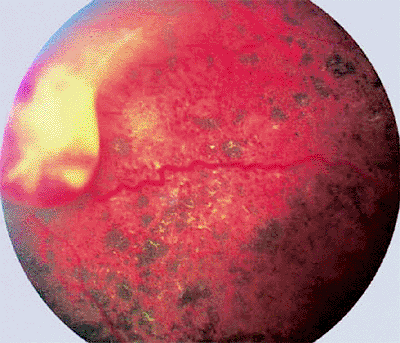 |
Figure 7.58. Acquired hemangiomatous lesion of peripheral retina in a young man with advanced retinitis pigmentosa. |
Association with chorioretinal disorders
Although most lesions of this type occur in eyes with peripheral chorioretinal degeneration of aging, some lesions that appear virtually identical have been associated with various forms of retinitis pigmentosa (Fig. 7.58), familial exudative vitreoretinopathy, and cicatricial retinopathy of prematurity.
Differential diagnosis
Retinal capillary hemangioma
Choroidal malignant melanoma with retinal invasion
Management
Most lesions of this type require no treatment. If progressive exudative retinal detachment or recurrent episodes of intravitreal bleeding develop, then transscleral cryotherapy may be advisable.
Bibliography
Alpek EK, Ahmed I, Hochberg FH, et al. Intraocular-central nervous system lymphoma: clinical features, diagnosis, and outcomes. Ophthalmology 1999;106:1805.
Arnold AC, Hepler RS, Yee RW, Maggiano J, Eng LF, Foos RY. Solitary retinal astrocytoma. Surv Ophthalmol 1985;30:173.
Baud O, Cormier-Daire V, Lyonnet S, et al. Dysmorphic phenotype and neurological impairment in 22 retinoblastoma patients with constitutional cytogenetic 13q deletion. Clin Genet 1999;55:478.
Bell D, Yang HK, O'Brien C. A case of bilateral cavernous hemangioma associated with intracerebral hemangioma. Arch Ophthalmol 1997;115:818.
Benz MS, Scott IU, Murray TG, et al. Complications of systemic chemotherapy as treatment of retinoblastoma. Arch Ophthalmol 2000;118:577.
Bhatnagar R, Vine AK. Diffuse infiltrating retinoblastoma. Ophthalmology 1991;98:1657.
Blach LE, McCormick B, Abramson DH. External beam radiation therapy and retinoblastoma: long-term results in the comparison of two techniques. Int J Radiat Oncol Biol Phys 1996;35:45.
P.287
Blodi CF, Russell SR, Pulido JS, Folk JC. Direct and feeder vessel photocoagulation of retinal angiomas with dye yellow laser. Ophthalmology 1990;97:791.
Blumenkranz MS, Ward T, Murphy S, et al. Applications and limitations of vitreoretinal biopsy techniques in intraocular large cell lymphoma. Retina 1992;12:S64.
Blumenthal EZ, Papamichael G, Merin S. Combined hamartoma of the retina and retinal pigment epithelium: a bilateral presentation. Retina 1998;18:557.
Bornfeld N, Schuler A, Bechrakis N, et al. Preliminary results of primary chemotherapy in retinoblastoma. Klin Padiatr 1997;209:216.
Bouzas EA, Parry DM, Eldridge R, Kaiser-Kupfer MI. Familial occurrence of combined pigment epithelial and retinal hamartomas associated with neurofibromatosis 2. Retina 1992;12:103.
Campochiaro PA, Conway BP. Hemangiomalike masses of the retina. Arch Ophthalmol 1988;106:1409.
Canning CR, McCartney ACE, Hungerford J. Medulloepithelioma (diktyoma). Br J Ophthalmol 1988;72:764.
Cardoso RD, Brockhurst RJ. Perforating diathermy coagulation for retinal angiomas. Arch Ophthalmol 1976;94:1702.
Chamot L, Zografos L, Klainguti G. Fundus changes associated with congenital hypertrophy of the retinal pigment epithelium. Am J Ophthalmol 1993;115:154.
Chan CC, Vortmeyer AO, Chew EY, et al. VHL gene deletion and enhanced VEGF gene expression detected in the stromal cells of retinal angioma. Arch Ophthalmol 1999;117:625.
Chan HSL, DeBoer G, Thiessen JJ, et al. Combining cyclosporin with chemotherapy controls intraocular retinoblastoma without requiring radiation. Clin Cancer Res 1996;2:1499.
Chang JH, Spraul CW, Lynn ML, et al. The two-stage mutation model in retinal hemangioblastoma. Ophthalmic Genetics 1998;19:123.
Cohen SY, Quental G, Guiberteau B, Coscas GJ. Retinal vascular changes in congenital hypertrophy of the retinal pigment epithelium. Ophthalmology 1993;100:471.
Cole EL, Zakov ZN, Meisler DM, et al. Cutaneous malignant melanoma metastatic to the vitreous. Arch Ophthalmol 1986;104:98.
Colvard DM, Robertson DM, Trautman JC. Cavernous hemangioma of the retina. Arch Ophthalmol 1978;96:2042.
Crino PB, Henske EP. New developments in the neurobiology of the tuberous sclerosis complex. Neurology 1999;53:1384.
Dithmar S, Holz FG, Volcker HE. Massive reaktive Gliose der Netzhaut. Klin Monatsbl Augenheilkd 1997;211:338.
el-Asrar A, al-Momen AK, Kangave D, et al. Correlation of fundus lesions and hematologic findings in leukemic retinopathy. Eur J Ophthalmol 1996;6:167.
el-Asrar A, al-Momen AK, Kangave D, Harakati MS. Prognostic importance of retinopathy in acute leukemia. Doc Ophthalmol 1995-96;91:273.
Eng C, Li FP, Abramson DH, et al. Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst 1993;85:1121.
Fishburne BC, Wilson DJ, Rosenbaum JT, Neuwelt EA. Intravitreal methotrexate as an adjunctive treatment of intraocular lymphoma. Arch Ophthalmol 1997;115:1152.
Friedrich CA. von Hippel Lindau syndrome: a pleomorphic condition. Cancer 1999;86:2478.
Garcia-Arumi J, Sararols LH, Cavero L, et al. Therapeutic options for capillary papillary hemangiomas. Ophthalmology 2000;107:48.
Gass JDM. Focal congenital anomalies of the retinal pigment epithelium. Eye 1989;3:1.
Glenn GM, Linehan WM, Hosoe S, et al. Screening for von Hippel Lindau disease by DNA polymorphism analysis. JAMA 1992;267:1226.
Gottlieb F, Fammartino JJ, Stratford TP, Brockhurst RJ. Retina angiomatous mass. A complication of retinal detachment surgery. Retina 1984;4:152.
Green WR. Vascular and circulatory conditions and diseases. In: Spencer WH. Ophthalmic pathology. An atlas and textbook. 3rd ed. Philadelphia: WB Saunders; 1985:1515.
Green WR. Peripheral retinal lesions, degenerations, and related conditions. In: Spencer WH. Ophthalmic pathology. An atlas and textbook. 3rd ed. Philadelphia: WB Saunders; 1985:837.
Griffiths PD, Martland TR. Tuberous sclerosis complex: the role of neuroradiology. Neuropediatrics 1997;28:244.
Grossniklaus HE, Thomas JW, Vigneswaren N, Jarrett WH. Retinal hemangioblastoma. A histologic, immunohistochemical, and ultrastructural evaluation. Ophthalmology 1992;99:140.
Guyer DR, Schachat AP, Vitale S, et al. Leukemic retinopathy. Relationship between fundus lesions and hematologic parameters at diagnosis. Ophthalmology 1989;96:860.
Hardwig P, Robertson DM. von Hippel Lindau disease: a familial, often lethal, multi-system phakomatosis. Ophthalmology 1984;91:263.
Haines JL, Short MP, Kwiatkowski DJ, et al. Localization of one gene for tuberous sclerosis within 9q32-9q34, and further evidence for heterogeneity. Am J Human Genetics 1991;49:764.
Haller JA, Knox DL. Vitrectomy for persistent vitreous hemorrhage from a cavernous hemangioma of the optic disk. Am J Ophthalmol 1993;116:106.
Hussain SE, Hussain N, Boniuk M, Font RL. Malignant nonteratoid medulloepithelioma of the ciliary body in an adult. Ophthalmology 1998;105:596.
Irvine F, O'Donnell N, Kemp E, Lee WR. Retinal vasoproliferative tumors. Surgical management and histological findings. Arch Ophthalmol 2000;118:563.
Jampel HD, Schachat AP, Conway B, et al. Retinal pigment epithelial hyperplasia assuming tumor-like proportions. Report of two cases. Retina 1986;6:105.
Kasner L, Traboulsi EI, Delacruz Z, Green WR. A histopathologic study of the pigmented fundus lesions in familial adenomatous polyposis. Retina 1992;12:35.
Kivela T, Tarkkanen A. Recurrent medulloepithelioma of the ciliary body. Immunohistochemical characteristics. Ophthalmology 1988;95:1565.
Kreusel KM, Bornfeld N, Lommatzsch A, et al. Ruthenium-106 brachytherapy for peripheral retinal capillary hemangioma. Ophthalmology 1998;105:1386.
Lafaut BA, Meire FM, Leys AM, et al. Vasoproliferative retinal tumors associated with peripheral chorioretinal scars in presumed congenital toxoplasmosis. Graefes Arch Clin Exp Ophthalmol 1999;237: 1033.
Landau K, Dossetor F, Hoyt WF, Muci-Mendoza R. Retinal hamartoma in neurofibromatosis 2. Arch Ophthalmol 1990;108:329.
Leonardy NJ, Rupani M, Dent G, Klintworth GK. Analysis of 135 autopsy eyes for ocular involvement in leukemia. Am J Ophthalmol 1990;109:436.
Leys AM, Van Eyck LM, Nuttin BJ, et al. Metastatic carcinoma to the retina: clinicopathologic findings in two cases. Arch Ophthalmol 1990;108:1448.
Lloyd WC, Eagle RC, Shields JA, et al. Congenital hypertrophy of the retinal pigment epithelium. Electron microscopic and morphometric observations. Ophthalmology 1990;97:1052.
Mack HG, Jakobiec FA. Isolated metastases to the retina or optic nerve. Int Ophthalmol Clin 1997;37:251.
Maher ER, Kaelin WG. von Hippel Lindau disease. Medicine 1997;76:381.
Maher ER, Moore AT. von Hippel Lindau disease. Br J Ophthalmol 1992;76:743.
McCabe CM, Mieler WF. Six-year follow-up of an idiopathic retinal vasoproliferative tumor. Arch Ophthalmol 1996;114:617.
P.288
McDonald HR, Schatz H, Johnson RN, et al. Vitrectomy in eyes with peripheral retinal angioma associated with traction macular detachment. Ophthalmology 1996;103:329.
Medlock RD, Shields JA, Shields CL, et al. Retinal hemangioma-like lesions in eyes with retinitis pigmentosa. Retina 1990;10:274.
Messmer EP, Heinrich T, Hoepping W, deSutter E, Havers W, Sauerweim W. Risk factors for metastases in patients with retinoblastoma. Ophthalmology 1991;98:136.
Messmer EP, Fritze J, Mohr C, et al. Long-term treatment effects in patients with bilateral retinoblastoma: ocular and mid-facial findings. Graefes Arch Clin Exp Ophthalmol 1991;229:309.
Meyers SM, Gutman FA, Kaye LD, Rothner AD. Retinal changes associated with neurofibromatosis 2. Trans Am Ophthalmol Soc 1995;93:245.
Moore AT, Maher ER, Rosen P, et al. Ophthalmological screening for von Hippel Lindau disease. Eye 1991;5:723.
Mohney BG, Robertson DM. Ancillary testing for metastasis in patients with newly diagnosed retinoblastoma. Am J Ophthalmol 1994;118:707.
Mullaney PB, Jacquemin C, Abboud E, Karcioglu ZA. Tuberous sclerosis in infancy. J Pediatr Ophthalmol Strabismus 1997;34:372.
Murphree AL, Villablanca JG, Deegan WF, et al. Chemotherapy plus local treatment in the management of intraocular retinoblastoma. Arch Ophthalmol 1996;114:1348.
Niemela M, Lemeta S, Sainio M, et al. Hemangioblastomas of the retina: impact of von Hippel Lindau disease. Invest Ophthalmol Vis Sci 2000;41:1909.
Nikaido H, Mishima H, Ono H, et al. Leukemic involvement of the optic nerve. Am J Ophthalmol 1988;105:294.
Nork M, Ghobrial MW, Peyman GA, Tso MOM. Massive retinal gliosis. A reactive proliferation of M ller cells. Arch Ophthalmol 1986;104:1383.
Nyboer JH, Robertson DM, Gomez MR. Retinal lesions in tuberous sclerosis. Arch Ophthalmol 1976;94:1277.
Ohkoski K, Tsiaras WG. Prognostic importance of ophthalmic manifestations in childhood leukemia. Br J Ophthalmol 1992;76:651.
O'Keefe M, Fulcher T, Kelly P, et al. Medulloepithelioma of the optic nerve head. Arch Ophthalmol 1997;115:1325.
Olsen TW, Frayer WC, Myers FL, et al. Idiopathic reactive hyperplasia of the retinal pigment epithelium. Arch Ophthalmol 1999;117:50.
Palmer JD, Gragoudas ES. Advances in treatment of retinal angiomas. Int Ophthalmol Clin 1997;37:159.
Paulino AC. Trilateral retinoblastoma: is the location of the intracranial tumor important? Cancer 1999;86:135.
Peterson K, Gordon KB, Heinemann MB, DeAngelis LM. The clinical spectrum of ocular lymphoma. Cancer 1993;72:843.
Peyman GA, Rednam KRV, Mottow-Lippa L, Flood T. Treatment of large von Hippel tumors by eye wall resection. Ophthalmology 1983;90:840.
Peterson K, Gordon KB, Heinemann MB, DeAngelis LM. The clinical spectrum of ocular lymphoma. Cancer 1993;72:843.
Pollack IF, Lunsford LD, Flickinger JC, Dameshek HL. Prognostic factors in the diagnosis and treatment of primary central nervous system lymphoma. Cancer 1989;63:939.
Reddy SC, Quah SH, Low HC, Jackson N. Prognostic significance of retinopathy at presentation in adult acute leukemia. Ann Hematol 1998;76:15.
Renardel de Lavalette VW, Cruysberg JRM, Deutman AF. Familial congenital grouped pigmentation of the retina. Am J Ophthalmol 1991;112:406.
Ridley ME, McDonald HR, Sternberg P, Blumenkranz MS, Zarbin MA, Schachat AP. Retinal manifestations of ocular lymphoma (reticulum cell sarcoma). Ophthalmology 1992;99:1153.
Ridley M, Green J, Johnson G. Retinal angiomatosis: the ocular manifestations of von Hippel Lindau disease. Can J Ophthalmol 1986;21:276.
Ruhswurm I, Zehetmayer M, Till P, et al. Kavern ses H mangiom der Papille: klinische und echographische Befunde. Klin Monatsbl Augenheilkd 1996;209:380.
Sahel JA, Frederick AR, Pesavento R, Albert DM. Idiopathic retinal gliosis mimicking a choroidal melanoma. Retina 1988;8:282.
Saleh RA, Gross S, Cassano W, Gee A. Metastatic retinoblastoma successfully treated with immunomagnetic purged autologous bone marrow transplantation. Cancer 1988;62:2301.
Schachat AP, Shields JA, Fine SL, et al. Combined hamartomas of the retina and retinal pigment epithelium. Ophthalmology 1984;91:1609.
Schachat AP, Markowitz JA, Guyer DR, Burke PJ, Karp JE, Graham ML. Ophthalmic manifestations of leukemia. Arch Ophthalmol 1989;107:697.
Shields CL, Santos MCM, Diniz W, et al. Thermotherapy for retinoblastoma. Arch Ophthalmol 1999;117:885.
Shields CL, Shields JA, De Potter P, et al. Plaque radiotherapy in the management of retinoblastoma: use as a primary and secondary treatment. Ophthalmology 1993;100:216.
Shields JA, Shields CL, Singh AD. Metastatic neoplasms in the optic disc. Arch Ophthalmol 2000;118:217.
Shields JA, Shields CL, Singh AD. Acquired tumors arising from congenital hypertrophy of the retinal pigment epithelium. Arch Ophthalmol 2000:118:637.
Shields JA, Eagle RC, Shields CL, De Potter P. Congenital neoplasms of the nonpigmented ciliary epithelium (medulloepithelioma). Ophthalmology 1996;103:1998.
Shields JA, Decker WL, Sanborn GE, Augsburger JJ, Goldberg RE. Presumed acquired retinal hemangiomas. Ophthalmology 1983;90:1292.
Singh AD, Santos MCM, Shields CL, et al. Observations on 17 patients with retinocytoma. Arch Ophthalmol 2000;118:199.
Smith BJ, O'Brien JM. The genetics of retinoblastoma and current diagnostic testing. J Pediatr Ophthalmol Strabismus 1996;33:120.
Traboulsi EI, Krush AJ, Gardner EJ, et al. Prevalence and importance of pigmented ocular fundus lesions in Gardner's syndrome. N Engl J Med 1987;316:661.
Tsai P, O'Brien JM. Combined hamartoma of the retina and retinal pigment epithelium as the presenting sign of neurofibromatosis-1. Ophthalmic Surg Lasers 2000;31:145.
Vadmal M, Kahn E, Finger P, Teichberg S. Nonteratoid medulloepithelioma of the retina with electron microscopic and immunohistochemical characterization. Pediatr Pathol Lab Med 1996; 16:663.
Watzke RC. Cryotherapy for retinal angiomatosis. A clinicopathologic report. Arch Ophthalmol 1974;92:399.
Webster AR, Maher ER, Moore AT. Clinical characterization of ocular angiomatosis in von Hippel Lindau disease and correlation with germline mutation. Arch Ophthalmol 1999;117:371.
Whitcup SM, deSmet MD, Rubin BI, et al. Intraocular lymphoma. Clinical and histopathologic diagnosis. Ophthalmology 1993;100: 1399.
Whitcup SM, Stark-Vancs V, Wittes RE, et al. Association of interleukin-10 in the vitreous and cerebrospinal fluid and primary central nervous system lymphoma. Arch Ophthalmol 1997;115: 1157.
Wittebol-Post D, Hes FJ, Lips CJM. The eye in von Hippel Lindau disease. Long-term follow-up of screening and treatment: recommendations. J Intern Med 1998;243:555.
Zimmer-Galler IE. Robertson DM. Long-term observation of retina lesions in tuberous sclerosis. Am J Ophthalmol 195;119:318.
EAN: 2147483647
Pages: 17
- The Second Wave ERP Market: An Australian Viewpoint
- The Effects of an Enterprise Resource Planning System (ERP) Implementation on Job Characteristics – A Study using the Hackman and Oldham Job Characteristics Model
- Data Mining for Business Process Reengineering
- Relevance and Micro-Relevance for the Professional as Determinants of IT-Diffusion and IT-Use in Healthcare
- Development of Interactive Web Sites to Enhance Police/Community Relations
- Chapter I e-Search: A Conceptual Framework of Online Consumer Behavior
- Chapter III Two Models of Online Patronage: Why Do Consumers Shop on the Internet?
- Chapter VI Web Site Quality and Usability in E-Commerce
- Chapter XIV Product Catalog and Shopping Cart Effective Design
- Chapter XVII Internet Markets and E-Loyalty