6.3 Genetic Engineering of Proteins for Device Applications
|
6.3 Genetic Engineering of Proteins for Device Applications
Genetic engineering is the systematic manipulation of the genetic code (i.e., DNA) of an organism to modify the traits of that organism. Material scientists and molecular electronic engineers view genetic engineering primarily as a tool for changing the properties of biological molecules, like proteins, for potential device applications. For example, although the quantum efficiency of the O → P photo conversion in wild-type bacteriorhodopsin is approximately 10−6, the ideal quantum efficiency for the volumetric memory is on the order of 10−3. Like chemical modification and organic cation replacement, as discussed above, mutagenesis is a powerful technique for modifying the properties of proteins. Genetic engineering has long been a standard technique in the fields of biochemistry, pharmaceuticals, and agriculture, but it has only recently become a standard method in bioelectronics. Although a comprehensive review of the techniques and theory of genetic engineering is beyond the scope of this work, a brief discussion is provided below. Our goal is to provide the reader with an appreciation for the basic methods and procedures as well as the inherent capabilities of these techniques.
Deoxyribonucleic acid (DNA) is the molecule that carries the genetic code for all organisms. DNA is a double-stranded helix biopolymer made up of four nucleotides: adenine (A), guanine (G), thymine (T), and cytosine (C). A region of DNA that encodes for a protein is called a gene. A gene can be isolated and transferred to a circular piece of DNA, called a plasmid, which contains only that gene and the genetic machinery required to express that gene. The average protein is 400 amino acids long, and the average gene is 1200 nucleotides long (Watson, Gilman, and Witkowski 1992). This relationship is due to the fact that three consecutive nucleotides make a codon, and each codon is ultimately translated to a single amino acid. More than one codon exists for most amino acids. For example, GGG codes for a glycine amino acid, but GGT, GGC, and GGA are synonymous. The amino acids are then constructed in the order of the codons on the DNA. Proteins are biopolymers of 20 different amino acid building blocks. A mutant protein has had the amino acid sequence modified from the naturally occurring (wild type) sequence. Mutations can take the form of site-specific or random replacements, additions of new amino acids (insertions), or deletions of amino acids within the primary structure. (For a review of mutagenesis, see Botstein and Shortle 1985; Smith 1985; Reidhaar-Olson and Sauer 1988.) Mutagenesis is routinely used to study structurefunction relationships existing in different proteins by biochemists and biophysicists.
Two common strategies used in the construction of site-specific mutations employ DNA "cassettes" or mismatched primers and the polymerase chain reaction (PCR). Restriction enzymes will cut DNA only at sites within a specific sequence. To perform cassette mutagenesis, two restriction sites unique to the plasmid must flank the location of the desired mutant, and the distance between the two restriction sites must be not more than 80 nucleotides. The sites must be unique in the plasmid because the DNA should be cut into no more than two pieces—a large fragment and a small fragment (figure 6.9). The synthetic fragments are limited to a length of about 80 nucleotides because this is the practical length limit of oligomeric synthesis. Once the small fragment is removed, a new synthetic oligonucleotide with the desired mutant is attached into place with an enzyme (ligase). Interestingly, Khorana and coworkers reported one of the first examples of cassette mutagenesis done on the bacteriorhodopsin gene (Lo et al. 1984).
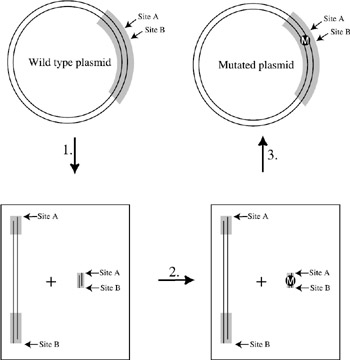
Figure 6.9: General scheme for cassette mutagenesis. The double circles represent a double-stranded plasmid, and the gray region indicates a gene. Restriction sites unique to the plasmid flank the region to be mutated. The distance from site A to site B should not be more than 80 nucleotides. In step 1, enzymes A and B are added to digest the plasmid at Sites A and B only, producing two linear pieces of DNA. The large fragment is then purified by gel electrophoresis, and added to a synthetic piece of DNA which contains the desired mutation (denoted by a M in a circle) (step 2). In the final step (step 3), the small synthetic fragment (containing the desired mutation) is ligated onto the large fragment. One end of the fragment then ligates with the other end to produce a circular mutant plasmid. The plasmid can then be expressed in bacteria to produce protein.
Cassette mutagenesis is not always possible, because unique restriction sites usually do not flank a desired mutation location. If many mutations are going to be performed on a gene (protein), a synthetic gene can be constructed with a unique restriction site placed approximately every 70 nucleotides throughout the gene. Incorporating silent (also called synonymous) mutations—that is, mutations that change the DNA sequence (to introduce a restriction site, for example) but leave the translated amino acid sequence unchanged—is facilitated with redundant codons (Feretti et al. 1986). Because producing synthetic genes is both labor intensive and expensive, PCR-based mutagenesis methodologies are more commonly used in the laboratory.
PCR-based mutational strategies using mismatched primers are generally applicable to any sequence (figure 6.10). Thermocyclers and polymerases required for PCR have gotten more accurate, robust, and inexpensive, so almost every protein biochemistry laboratory now uses PCR regularly. Many different techniques (and many commercially available kits) take advantage of the flexibility of this method. This alternative strategy is based on the fact that double-stranded DNA can be denatured (i.e., made single stranded) and renatured as a function of temperature. A 20–30 oligonucleotide primer is designed to complement the template DNA, except for the introduced mutation. A reaction mixture contains a relatively small amount (tens of nanograms) of template DNA and a larger amount (120 ng) of primer and nucleotides. The mixture is first heated to 95 C to melt the template DNA (making it single stranded). Upon cooling to 62 C, the primer anneals to the template DNA. A polymerase enzyme then elongates the primer with the complement to the template DNA at 72 C. Now two strands of DNA exist, the original (template DNA) and the new mutant extended primer. This process is repeated 20–30 more times, exponentially amplifying the mutant DNA (approximately by 2number of cycles). After 30 cycles, the reaction mixture will contain more than 268,400,000 times more mutant DNA molecules than template. The original template DNA is selectively digested (i.e., discarded) with enzymes, and the mutant DNA is transformed into a bacterium like Escherichia coli and expressed to obtain the mutant protein.
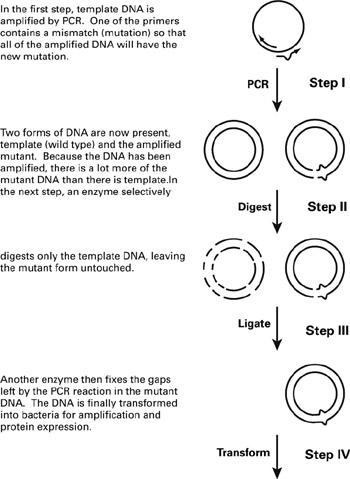
Figure 6.10: General schematic for mismatched primer mutagenesis. Although figure 6.9 is based on the ExSiteTM Mutagenesis kit (Stratagene, LaJolla, CA), the overall strategy used by this kit is similar to many PCRbased methods. In the first step, primers are designed to copy the wild type, template DNA. One of the primers is the complement to the wild type DNA, while the second primer contains the mutation. The example shown here is for a point mutation, but in fact, insertions and deletions can also be done with this method. In the first step, the template DNA is amplified with the new mutation incorporated. An enzyme that recognizes the template DNA cuts it into multiple fragments. In step III, another enzyme, ligase, circularizes the new, mutant DNA allowing it to transform into bacteria efficiently in step IV. Once in bacteria, the gene can be amplified or expressed (resulting in the production of mutant protein).
Evolution has optimized each protein for a specialized task in the cell. Some of these properties may be less than ideal for commercial applications. For example, proteins may not be stable at elevated temperatures. They may not be active in organic solvents, or their products may inhibit activity. As mentioned above, rationally modifying these properties in even the most studied and well-understood proteins would be nearly impossible to do—in part, because of the complexity of proteins. With 20 naturally occurring amino acids, there are 20N possible combinations of N amino acids in a protein—20248 potential combinations for a protein the length of bacteriorhodopsin. Directed evolution, sometimes referred to as in vitro evolution, mimics the methods used in nature during evolution (for general reviews of directed evolution see Arnold 1998; Arnold and Volkov 1999; Schmidt-Dannert and Arnold 1999). Conceptually, directed evolution is a combinatorial approach to genetic engineering (Zhao and Arnold 1997). The gene of interest is first modified by random mutagenesis, saturation mutagenesis, or combined with other homologous genes (homologous recombination). Then, from a sampling of hundreds of these DNAs, the proteins that provide the best properties are screened or selected for another round of mutagenesis or homologous recombination. These iterations continue until the protein has the desired function. Notable successes of directed evolution have modified an enzyme's substrate (Yano, Oue, and Kagamiyama 1998), introduced thermal stability (Miyazaki et al. 2000), and made an enzyme functional in organic solvent (Moore and Arnold 1996; Spiller et al. 1999).
The first step in directed evolution is to generate a sampling of random point mutations. Because some mutations may improve the desired function while others may be deleterious or silent, the optimum mutation rate is one mutation per gene per round of evolution (Miyazaki and Arnold 1999). The most common method for mutagenesis is to use error-prone PCR to randomly mutate sites within a gene (Zhao et al. 1999). If specific amino acids are known to be important, saturation mutagenesis works with degenerate primers to modify that site to every possible combination (Hayashi et al. 1994). Another way to modify a gene is by homologous recombination, the mixing of two or more similar genes (Giver and Arnold 1998). The genes may be related to one another by homology. Homologous cephalosporinases from four different species were recombined to make a protein 270–540fold more active than the original enzyme (Crameri et al. 1998). The genes may also be different mutants derived from a common parent. By randomly combining these genes and then screening or selecting the best products, better proteins are produced. Stemmer first introduced homologous recombination by using a DNAse to cut DNA into fragments and then randomly recombining them (Stemmer 1994, 1994b). A simpler and more efficient method has been developed. The staggered extension process (StEP) uses short bursts of extension times followed by denaturation during PCR of several genes (Shao et al. 1998; Zhao et al. 1998). A DNA primer is extended briefly on one template, denatured, bound to another template, elongated again, denatured, and so on. The result is a random recombination of the original templates (Michnick and Arnold 1999; Ness et al. 1999).
Once the modified gene is produced, the resulting proteins have to be tested by a screen or selection. Selection can be used when the protein of interest confers a growth advantage to the host organism (for a review of using selection in directed evolution see Kast and Hilvert 1997). Screens are used in all other cases (for a review of implementing screens for directed evolution, see Zhao and Arnold 1997). Choosing the most successful product from a large library of mutations is usually the most difficult step in directed evolution. New technologies like inexpensive microfabricated fluorescence-activated cell sorters (Fu et al. 1999) will make screening promising mutations cheaper and easier in the future.
Site-directed mutagenesis has been used to create rationally designed bacteriorhodopsin mutants with enhanced materials properties (Miercke et al. 1991; Hampp et al. 1992; Zeisel and Hampp 1992; Gergely et al 1993; Misra et al. 1997). For example, some mutants have enhanced the holographic properties of the protein by producing an M state with an extended lifetime (Gergely et al. 1993; Miercke et al. 1991; Hampp et al. 1992; Zeisel and Hampp 1992), while others improve the branched-photocycle memory by enhancing the yield of the O state (Misra et al. 1997). The challenge for material scientists is to predict a priori what amino acid sequence will create or enhance a specific protein property. At present, the vast majority of genetic engineering for materials applications is a trial-and-error process, due to the complexity of protein structure and function and the lack of satisfactory molecular modeling tools. However, directed evolution promises to provide new methods to solve previously limiting problems associated with available proteins. It is hoped that continued theoretical work will yield computer programs with the predictive capabilities comparable to the SPICE packages that are the cornerstone of electrical engineering. In this regard, bioelectronics is many years if not decades behind computer engineering.
|