2. Genetics
Authors: Corwin, Elizabeth J.
Title: Handbook of Pathophysiology, 3rd Edition
Copyright 2008 Lippincott Williams & Wilkins
> Table of Contents > Unit I - Fundamental Mechanisms of Health and Disease > Chapter 2 - Genetics
Chapter 2
Genetics
Genetics is the study of genes. Genes are composed of sequences of deoxyribonucleic acid (DNA; see Chapter 1). Genes are packaged together as chromosomes and are passed from parent to offspring. It is our genes that determine who we are and how we function at the most basic cellular level. Transmission of genetic information is a balance between ensuring that genes are passed error-free between the generations and allowing enough diversity for the adaptation and survival of the species. Sometimes mistakes (mutations) are made that advance the species for example, mutations have resulted in several malaria-protective genes that offer evolutionary advantage but most times mutations cause significant disability or death.
Physiologic Concepts
The Genes
There are approximately 25,000 genes in the human genome, with each gene containing a few hundred to a few thousand base pairs of DNA. This is a much smaller number of genes than expected, given the complexity of the human genome. The DNA of a given gene includes coding portions, called exons, and non-coding portions. The coding portions of a gene carry the information needed to make a protein, often an enzyme. Enzymes and other proteins control the synthesis and function of each and every cell or tissue of the body. The function of the millions of molecules of non-coding DNA is unclear. Many genes grouped together make up the chromosomes.
P.37
Chromosomes
Chromosomes are made up of molecules of DNA, complexed with proteins called histones. Chromosomes together carry the genetic blueprint of an individual. All human somatic (body) cells contain 23 pairs of chromosomes, one pair from each parent, for a total of 46 chromosomes. Each human sex cell, an egg or a sperm, contains 23 unpaired chromosomes. Each chromosome is nearly identical (approximately 99.9%) across the human species in the genetic information it contains. The remaining variations are subtle but enough to make each one of us unique.
On February 28, 1953, James Watson and Francis Crick reported that the DNA of each chromosome is twisted into a double helix, and that it is by unraveling and copying this double helix that genes are activated and the secret of life is passed on.
Gene Activation
Although each somatic cell contains the same 23 pairs of chromosomes, only certain genes are activated in any given cell; therefore, only certain proteins or enzymes are produced by that cell. Which genes are activated in which cell is determined during embryologic development and throughout life by circulating growth factors, hormones, and chemical cues produced by a given cell and its neighboring cells. In addition, methylation of certain regions of a gene (adding a CH3 group) can turn off, or silence, that gene. Demethylating the region can activate that gene and lead to the production of the protein for which it codes. In general, cells that have similar genes turned on and off perform similar functions and group together as tissues.
It has recently become clear that many genes can code for more than one protein, a finding that helps explain how so few genes can result in so many different proteins. Recent research suggests that a process known as alternative splicing is responsible for this phenomenon. Alternative splicing refers to different combinations of exons on one gene that become active at different times, with each combination resulting in the production of one protein.
Cellular Reproduction
All cells reproduce during embryonic development, which allows for growth of the embryo and differentiation (specialization) of the cells making up tissues and organs. After birth and throughout adulthood, many cells continue to reproduce. Cells that reproduce throughout a lifetime include cells of the bone marrow, skin, and digestive tract. Liver and kidney cells reproduce when replacement of lost or destroyed cells is required. Special cells, called stem cells, are capable of reproducing indefinitely. Other cells, including nerve, skeletal muscle, and cardiac muscle cells, do not reproduce significantly after the first few months following
P.38
birth. Damage to these tissues generally cannot be repaired by growth of new cells (although nearby stem cells may differentiate into replacement cells).
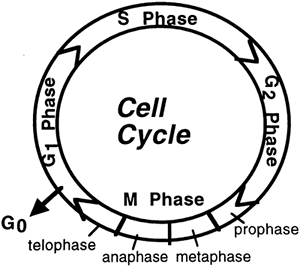 |
Figure 2-1. The cell cycle. |
The Cell Cycle
The cell cycle refers to a sequence of stages a cell goes through during its lifetime (Fig. 2-1). During embryogenesis, all cells go through all stages, as do adult cells that continue to reproduce. The rate at which a cell goes through its cell cycle depends on the given cell and the growth factors, hormones, and chemicals to which it is exposed. Cells that do not continue to reproduce after embryogenesis remain in a resting stage and do not cycle through the other stages. The cell cycle is divided into two parts: interphase and mitosis.
Interphase
When not actively dividing, a cell is said to be in interphase. There are three standard stages of interphase: G1, S, and G2. A fourth stage, Go, is a specialized resting stage. In these designations, the G stands for gap, referring to a time period the cell uses to check and recheck the preceding steps.
G1 is the stage during which a cell prepares for DNA replication by synthesizing new proteins and activating cytoskeletal components. During this stage, the cell monitors its environment to determine if the time is right for DNA replication. This stage is considered a checkpoint for the cell because if conditions are not right, the cell will not progress further through its cycle. A cell will be stimulated to progress through G1 when certain genes, including proto-oncogenes, are activated. S is the next stage, during which replication (copying) of the DNA occurs; DNA replication is described in Chapter 1. The third stage, G2, is the stage before cell division, during which the cell again undergoes protein synthesis, this
P.39
time in preparation for division. This stage is also a checkpoint because if the DNA has not been copied correctly, the cell has a second opportunity to stop its progression through the cycle before mitosis occurs. If an error has occurred, either it is repaired, and the cell re-enters the cell cycle, or the cell is stimulated to undergo apoptosis, i.e., programmed cell death. Genes that are activated at this stage to stop the progression of the cell through its cycle are known as tumor suppressor genes.
Go is a resting stage in which a cell in G1 that has not committed itself to DNA replication may pause. A cell may stay in Go indefinitely, but once a cell is stimulated to leave the Go stage, it will progress through the other stages, unless its progress is restricted at a subsequent checkpoint. The progression through interphase is a lengthy 10- to 22-hour process.
Mitosis
Mitosis (the M stage) is the stage of cell division. Mitosis is a shorter event than interphase; it lasts approximately 1 hour. During mitosis, the cell that has duplicated during interphase splits into two daughter cells that each contain the 23 pairs of chromosomes. Mitosis consists of the substages of prophase, metaphase, anaphase, and telophase.
Prophase
Prophase is the stage in which protein structures (centrioles) present in the cytoplasm of the cell begin to move toward opposite sides or poles of the cell. This stretches the nuclear membrane and causes it to break apart. The chromosomes are now in the cytoplasm rather than isolated in the nucleus.
Metaphase
Metaphase is the stage during which the chromosomes visibly become two sets of pairs lined up next to each other in the center of the cell. Microtubules extend from the centrioles to each chromosome pair.
Anaphase
Anaphase is the stage during which the microtubules begin to pull the chromosome pairs apart. One pair goes toward one centriole pole and one pair goes toward the other centriole pole.
Telophase
Telophase is the stage during which the cell splits down the middle and a new nuclear membrane develops around each of the two new cells, including the 23 pairs of chromosomes (46 total) present in each cell.
Control of the Cell Cycle
Cells that continually go through the cell cycle (i.e., cells of the gut and bone marrow) do so at an intrinsic rate that can be increased or decreased by internal and external cues. External cues that turn on the cell cycle may include neural or hormonal stimulation, or may come from cell products released in response to tissue injury and activation of the inflammatory
P.40
and immune systems. Brakes on the cell cycle may also include neural and hormonal stimulation and proteins synthesized by cells in response to activation of certain regulator genes, including the tumor suppressor genes. Uncontrollable cell growth and cancer (see Chapter 3) may occur with the destruction or inactivation of regulator genes or by excessive stimulation and activation of proto-oncogenes.
Another mechanism that serves to limit cell replication involves structures present on the chromosomes themselves, known as telomeres. A telomere is the end region of a chromosome that shortens with each replication. When the telomere shortens to a threshold length, after a certain number of cell cycles, it shuts off cellular replication. Shutting off cellular replication leads to replicative senescence, the characteristic that ensures normal somatic cells do not divide indefinitely. However, a few cells, including cancer cells and germ line cells, contain the enzyme telomerase; telomerase adds telomere sections back onto the chromosome. Telomerase stabilizes telomere length, functioning to immortalize these cells. Telomerase is discussed further in Chapter 3.
Meiosis
Meiosis is the process during which germ cells of the ovary (primary oocytes) or testicle (primary spermatocytes) give rise to mature eggs or sperm (Fig. 2-2). Meiosis involves DNA replication in the germ cell, followed by two cell divisions rather than one, which results in four daughter cells, each with 23 (unpaired) chromosomes. In males, all four daughter cells are viable and continue to differentiate into mature sperm. In females, only one viable daughter cell (egg) is formed; the other three cells become nonfunctional polar bodies. During fertilization, genetic information contained in the 23 chromosomes of the egg joins with genetic information contained in the 23 chromosomes of the sperm. This results in an embryo with 46 total chromosomes (two pairs of 23).
An interesting phenomenon occurs during DNA replication in the first meiotic stage. At this time, pieces of DNA may shift between the matched chromosome pairs, in a process called crossing-over. Crossing-over increases the genetic variability of offspring, and is one reason why siblings within a family may vary considerably in genotype and phenotype.
Genotype and Phenotype
Precise genetic information carried in the chromosomes of the offspring is termed the genotype. Physical representation of genetic information (tall or short, dark or light) is called the phenotype.
Single-Gene Inheritance
Some traits of the phenotype (e.g., eye color) are determined by a single gene. A gene that determines a specific trait is called an allele. For each
P.41
single-gene trait, there are two controlling alleles: one on the chromosome delivered from the mother and one on the chromosome delivered from the father.
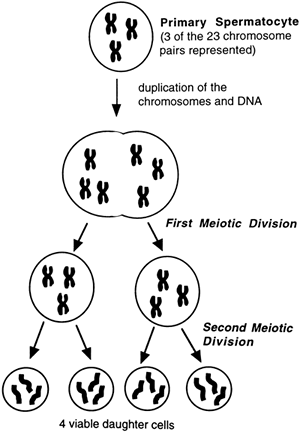 |
Figure 2-2. A schematic representation of meiosis in a male germ cell, leading to four daughter cells, called spermatids. Each spermatid will develop further to become mature sperm, as described in Chapter 20. |
Heterozygous and Homozygous Alleles
If an individual has two identical alleles (e.g., two alleles coding for brown eyes), that individual is said to be homozygous for the trait. If an individual has different alleles coding for a trait (e.g., one allele for brown eyes and one for blue), the individual is said to be heterozygous for the trait. One allele is usually dominant over the other, for instance, brown eyes over blue, but alleles are occasionally codominant (equally expressed). If a person is heterozygous for a single-gene trait, the phenotype will depend on which, if either, of the alleles is dominant. If the alleles are codominant, for example, those coding for the A and B red blood cell antigens, the individual will express both alleles (i.e., AB blood type).
P.42
Multifactorial Inheritance
Most phenotypic characteristics are influenced by several genes. Height, intelligence, and personality characteristics are among those traits termed multifactorial. They are inherited in a more complicated manner and usually involve many contributing genes present on the same or different chromosomes. The expression of these genes may be influenced by nongenetic (environmental) factors such as nutrition, family support, and exposure to various toxins or microorganisms. Ultimately, however, all human characteristics, including susceptibility to disease, are to some extent affected by our genes, even those characteristics clearly influenced by the environment.
Genetic Testing
Genetic testing, called cytogenetics, involves looking at the overall structure and number of the chromosomes. Genetic testing can be performed on any cell of the body, but in children and adults it is usually done by withdrawing white blood cells in a venous blood sample. For prenatal testing, fetal cells may be gathered during the processes of amniocentesis, at about 16 weeks of gestation, or during chorionic villi sampling, typically between 8 and 12 weeks of gestation. Even genetic testing of preimplantation embryos, obtained during in vitro fertilization procedures, is possible and allows clinicians to test for single gene disorders at the earliest stage of existence. Analysis of fetal chromosomes accounts for a large percentage of all tests.
For cytogenetic analysis, chemicals typically are added to the cultured cells to arrest the chromosomes during metaphase. The chromosomes are then spread out with members of a pair lined up together. The chromosomes are counted and the structures of the chromosomes under study are compared to control samples. The spread of 23 chromosome pairs is called the karyotype; the process is called karyotyping. The 23rd chromosome pair in males will show two chromosomes dissimilar in shape, the X and the Y. Females have two X chromosomes. An example of a normal male karyotype is shown in Figure 2-3.
Amniocentesis
Amniocentesis is performed by inserting a needle through the abdominal wall of a pregnant woman into the amniotic sac that surrounds the fetus. Amniotic fluid, into which fetal cells have been shed, is withdrawn. Chromosomes present in the fluid sample are then cultured and fixed, and their number and shape are analyzed for genetic integrity. This test is usually done at approximately 16 weeks' gestation and results are available in approximately 2 weeks. Performance of amniocentesis earlier in gestation has been attempted with mixed results.
Chorionic Villi Sampling
Chorionic villi sampling involves gathering cells of the chorion, the outer border of the fetal membranes. The cells are gathered by placing a needle
P.43
through the woman's lower abdomen or cervix between 8 and 12 weeks of pregnancy. The cells do not need to be cultured, so the chromosomal analysis is available in approximately 1 to 2 days. Chorionic villi sampling earlier than 8 to 12 weeks' gestation is possible, although after sporadic reports of limb or other abnormalities occurring after early testing, this particular procedure was discontinued.
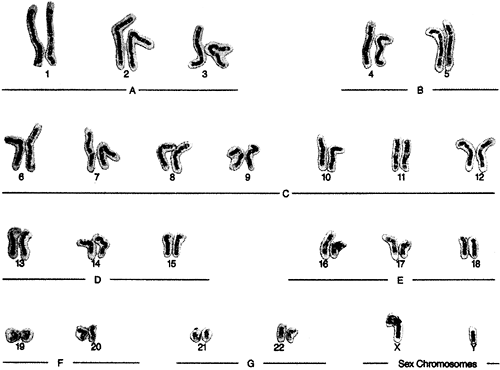 |
Figure 2-3. Karyotype of a normal human boy. (Courtesy of the Prenatal Diagnostic and Imaging Center, Sacramento, CA. Frederick W. Hansen, MD, Medical Director. In Porth, C. [2005]. Pathophysiology: Concepts of altered health states [7th ed.]. Philadelphia: Lippincott Williams & Wilkins. ) |
Genetic Engineering
Genetic engineering refers to experimental manipulation of the genome to produce certain characteristics. One technique of genetic engineering is gene splicing, which involves snipping a certain gene out of one cell and inserting it into another cell or into an attenuated (non-virulent) virus. The cell or virus may then be administered to an individual who suffers from a lack of that particular gene. This has been performed in clinical trials involving the gene for cystic fibrosis. The goal of gene splicing is that the genetically engineered gene will insert itself into the host's DNA, allowing the host to produce the missing protein or enzyme coded for by that gene.
Another procedure that takes advantage of the ability to manipulate genes is recombinant DNA technology. With this technology, a piece of DNA extracted from one organism can be inserted into another organism such as a single-celled bacterium. If the DNA incorporates into the
P.44
bacterium's own genome, it will start directing the bacterium to produce large amounts of a specific protein. In this way, valuable proteins can be produced for mass distribution. Examples of substances produced by recombinant DNA include growth hormone, insulin, clotting factors, and various vaccines.
The Human Genome Project
Several advanced techniques to identify the DNA code of specific genes have been developed in the last few decades. In 1990 a major national initiative, the Human Genome Project, was inaugurated in the United States, with joint funding from the National Institutes of Health and the Department of Energy. The goal of this initiative was to perform DNA mapping for each of the genes of the human genome by the year 2005. The project was completed two years early, in 2003, the 50th anniversary year of DNA's discovery by Watson and Crick.
Applications
The Human Genome Project has enormous significance for revealing the gene or genes responsible for thousands of genetic diseases and for synthesizing proteins or enzymes absent or deficient in each disease. Specific genes for many diseases, including cystic fibrosis, Huntington disease, and some inheritable types of breast and colon cancer, have already been identified.
Some predict that within 20 years, patients entering a medical facility will be subjected to a genomic scanning for pathogenic and epistatic genes. Epistasis refers to the tendency of some genes to interact and cross-over with genes other than their allelic partners. Epistasis appears to affect certain diseases positively or negatively and has the potential to result in variable intensities of disease in different ethnicities and individuals.
One area of genomics that has progressed more slowly than anticipated is that involving drug discovery based on specific gene targets unveiled by the Human Genome Project. Progress in this area has been slow for a number of reasons. First, the complexity of the human physiologic system has proven to be a bit daunting, perhaps accounting for the fact that most of the highly effective drugs on the market today, such as aspirin, act on multiple gene systems rather than on single gene disorders. The tendency of genes to experience epistatic interactions also has hindered the pace of drug production through gene targeting.
Ethical Implications
Results of the Human Genome Project will have enormous ethical implications for prenatal testing and selective abortion of defective embryos. Ethical implications are also involved in testing concerned adults who seek to know the likelihood of their developing a specific disease in the future. This is especially troubling if the identified disease is one for which
P.45
there is no treatment or cure, or if testing involves children. For adults and children with disease-causing mutations, future childbearing choices, the ability to purchase health insurance or life insurance, and the ability to find future employment are important considerations. The Human Genome Project has dedicated funding and time to explore the ethical factors involved in gene mapping. In the fall of 2005, a study was initiated by the National Human Genome Research Institute among healthy adult volunteers to sequence 100 to 300 genes that have been associated with various disease phenotypes. The aim is to provide information to individuals on potential genetic risk factors and to evaluate how this enormous amount of information is handled by the patients and their families.
Pathophysiologic Concepts
Mutation
A mutation is an error in the DNA sequence. Mutations can occur spontaneously, or after the exposure of a cell to radiation, certain chemicals, or various viral agents.
Most mutations will be identified and repaired by enzymes working in the cell. Other times, a mutation may lead to apoptosis. If a mutation is not identified or repaired, or if the cell does not undergo programmed death, that mutation will be passed on in all subsequent cell divisions. Mutations may result in a cell becoming cancerous. Mutations in the gametes (the egg or sperm) may lead to congenital defects in an offspring.
Congenital Defects
Congenital defects, also called birth defects, include genotypic and phenotypic errors occurring during embryogenesis and fetal development. Some congenital defects, such as cleft palate and limb abnormalities, may be apparent at birth, whereas other congenital defects, such as an abnormal or absent kidney and certain types of heart disease, may not be recognized immediately. Congenital defects may result from genetic mistakes made during meiosis of the sperm or egg, or from environmental insults experienced by the fetus during gestation. Examples of genetic mistakes include chromosomal breaks, unstable DNA, and mistakes in chromosome number. Environmental insults during gestation that are known to increase the likelihood of a congenital defect include maternal exposure to alcohol, certain drugs, and viruses. Environmental insults may lead to a genotypic or phenotypic error.
Chromosomal Breaks
During mitosis and meiosis, pieces of chromosomes may break off, be added inappropriately to other chromosomes, or be deleted entirely. If deletions or additions occur during meiosis in the egg or sperm, a congenital defect or death of the embryo may result. During fetal development and
P.46
throughout the life of an individual, mistakes may occur during mitosis in somatic cells. If deletions or additions of chromosomes occur during mitosis, the affected cell line will usually die out.
Hereditary Unstable DNA
Inheritance patterns of some genetic disorders are not easily explained. Recent evidence suggests that occasionally genes coding for a certain trait may not be passed down in a stable fashion from parents to offspring, but instead may have their effect magnified in succeeding generations of offspring. Other genes may be expressed only in certain members of a family, even though all members of the family may carry the gene. Whether an individual in the family expresses the trait coded for by these genes may depend on the individual's sex, the sex of the parent donating the unstable gene, or environmental conditions.
Fragile X Syndrome
A particularly striking finding concerning hereditary unstable DNA is that some diseases occur when a certain group of repeating codons (a set of three DNA bases grouped together) expands. For instance, the genetic disorder known as fragile X syndrome, the most common cause of inherited mental retardation, results from a mutation in the fragile X mental retardation 1 (FMR1) gene on the X chromosome. Features include elongated face, prominent jaw, large ears, macroorchidism, behavioral anomalies, and cognitive deficits. Fragile X syndrome results when the DNA codon CCG, normally repeated approximately 40 times in a gene near the top of the long arm of the X chromosome, begins to expand and is repeated excessively. Carriers of the disorder show 70 to 200 repeats on the chromosome, but are cognitively and behaviorally normal. However, offspring of the carriers can show the region expanding to greater than 200 repeats of the codon. The degree of mental retardation corresponds to the length of the repeated codon. The degree to which the syndrome is expressed in any one family member depends on whether the expanded codon is inherited from the mother or the father, and whether the offspring is male or female, with male offspring being more likely and more severely affected. The tendency for the pattern to repeat is caused by a fragility of the chromosome.
Errors in Chromosome Number
Any change from the normal human chromosome number of 46 chromosomes is called aneuploidy. An aneuploidy in which there are only 45 chromosomes is called a monosomy. An aneuploidy in which there are 47 chromosomes is called a trisomy. Having more than 47 chromosomes is possible but rare.
Monosomy
If any chromosome other than the X or Y is lost, the embryo will spontaneously abort. However, the loss of one of the sex chromosomes may
P.47
result in a viable offspring. Usually the Y chromosome is lost, resulting in 44 somatic chromosomes and one sex chromosome, for a total of 45 chromosomes (often expressed 45, X/O, to indicate no Y chromosome). The resulting disorder is called Turner's syndrome. Monosomy of any chromosome is a major cause of spontaneous abortion in the first trimester.
Trisomy
A trisomy occurs when somatic or sex chromosomes do not separate properly during meiosis. This is called non-disjunction. Most trisomies cause spontaneous abortion of the embryo, but rarely live births may result. Trisomies that may result in live births include trisomies of the sex chromosomes and trisomies of chromosomes 8, 13, 18, and 21. Trisomy 21 is called Down syndrome.
Teratogenesis
Teratogenesis is an error in fetal development that results in a structural or functional deficit (e.g., a deficit in brain function). Environmental stimuli that cause congenital defects are called teratogenic agents. Teratogenic agents can lead to genetic mutations or errors in phenotype. Common manifestations of teratogenic exposure include congenital heart disease, abnormal limb development, mental retardation, blindness, hearing loss, and abnormalities in growth. Some teratogenic agents, including x-rays and some viruses, are known to cause chromosomal breakage, additions, and deletions. Many drugs, including the anticoagulant coumadin (Panwarfin) and the anti-acne medication isotretinoin (Accutane), can also be teratogenic.
Alcohol
The most common teratogenic drug used in the United States is alcohol. Alcohol at any dose is capable of causing neurologic deficits and facial deformities ranging from mild to severe. Alcohol can adversely affect several cellular functions associated with fetal development, including DNA synthesis, protein synthesis, glucose uptake, and the development of neural signaling pathways. Human infants who experience a complex group of congenital effects as a result of exposure to alcohol may be diagnosed as suffering from fetal alcohol syndrome.
![]() ediatric Consideration
ediatric Consideration
Alcohol is the leading cause of birth defects in certain parts of the United States and is the top cause of mental retardation in this country. Each year, approximately 4,000 children are born in the United States with fetal alcohol syndrome and at least twice that number will have various forms of the disorder in the absence of the full syndrome. Fetal alcohol syndrome is 100% preventable.
P.48
The TORCH Group of Teratogens
Several different microorganisms are known to be teratogenic in humans. Many of these are described under the acronym TORCH, in which each letter stands for a particular microorganism that may infect the embryo or fetus. T stands for toxoplasmosis, R for rubella, C for cytomegalovirus, and H for the herpes simplex virus 2. The letter O stands for all other infections, especially syphilis, hepatitis B, mumps, gonorrhea, and varicella (chickenpox or shingles).
Effects of Teratogenic Agents
A newborn infected during gestation with any of the TORCH group of microorganisms may show microcephaly, hydrocephaly, mental retardation, or loss of hearing or sight. Congenital heart defects are common, especially with rubella. Radiation exposure may increase the risk that the child will later develop cancer. Certain drugs (e.g., thalidomide, sedatives, anti-seizure medications, and sleeping pills) may affect the growth of the embryo or fetus. Angiotensin-converting enzyme inhibitors, used to treat hypertension, may cause the embryo not to develop normal kidneys. Whether an embryo or fetus will be affected by any teratogenic agent depends on several factors, which include the timing and dose of exposure, and maternal and paternal health and nutritional status.
Timing of Exposure to a Teratogen
Because most organs and tissues are formed during the first trimester, teratogenic agents are most likely to cause structural defects at this time. This is especially true of rubella infection and exposure to drugs that interfere with development. However, the nervous system is always susceptible to a teratogen because it continues to develop even after birth. Infants exposed to an infectious agent in the third trimester or during the birth process are at increased risk of developing the disease. This is true for neonatal infection by hepatitis B virus or HIV. Primary maternal infection with the herpes simplex virus near the time of labor is associated with the occurrence of neonatal herpes and increased neonatal morbidity and mortality.
Dose of a Teratogen
The dose of exposure is important in determining the likelihood that a teratogenic agent will cause a congenital defect. Levels of radiation used in most diagnostic techniques or low concentrations of a drug may not produce any discernible effect on the fetus. Higher doses of radiation or a drug may adversely affect the fetus.
Maternal Health and Nutritional Status
Maternal health and nutritional status also play a role in determining teratogen effect. Infants born to women with diabetes or seizure disorders are at higher risk of fetal anomalies, the latter perhaps due to the
P.49
effects of both the seizures themselves on fetal growth and the medications used to treat the disorder. Maternal diets low in folic acid have been associated with development of neural tube defects such as spina bifida. Because most adults do not ingest adequate amounts of folic acid, folic acid supplementation (usually found in a One-A-Day or prenatal vitamin) is recommended for all women for at least 3 months before conception. Folic acid is required for full functioning of the DNA proofreading enzymes responsible for checking and rechecking DNA replication, which may explain its protective effect against certain congenital malformations.
Paternal Health and Chemical Exposures
Studies on the effect of diet, chemical exposure, or drug usage in fathers suggest that teratogenic effects may also be passed through damaged sperm. Some studies suggest paternal (and maternal) cigarette smoking may be associated with increased risk of childhood leukemia in offspring. Paternal occupational hazards, such as exposure to paint fumes, may increase the risk of spontaneous abortion.
Conditions of Disease or Injury
Single-Gene Disorders
Single-gene disorders are caused by a single gene mistake on the DNA strand. There are approximately 4,500 known single-gene disorders, some of which are identified in Table 2-1. Single gene disorders may be assessed by obtaining a comprehensive family history and preparing a three-generation pedigree, which is the most efficient way to assess hereditary influences on disease. A three-generation pedigree also offers the best chance of determining the likelihood that other members of the family may be at risk for certain genetic diseases.
Causes of Single-Gene Disorders
Single-gene disorders may result from a mistake in the copying of a single code letter. For example, the codon CCG can be transcribed incorrectly to CGG during DNA replication. Because each codon codes for a specific amino acid in a protein, these mistakes usually make the gene incapable of correctly directing the production of its protein.
How important it is to transcribe exactly each letter of each codon is apparent when one considers that, although there are approximately three billion code letters used in the human genome, a mistake in the copying of a single codon is what causes several of the disorders in Table 2-1. For example, sickle cell disease results when one A (adenine) in one gene is replaced by a T (thymine). The fatal neurologic disorder Huntington disease, the congenital bone disease osteogenesis imperfecta, and the
P.50
P.51
metabolic disorders phenylketonuria and Tay-Sachs disease also occur as a result of miscopying a single codon.
Table 2-1. Some Disorders of Single-Gene Inheritance and Their Significance | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
Codon meanings also will be destroyed and single-gene disorders may result if DNA bases are added or deleted inadvertently. Other single-gene disorders may result from a codon being repeated excessively, as described earlier for the X-linked disorder fragile X syndrome.
The Inheritance Pattern of Single-Gene Disorders
Single-gene disorders may be passed on as dominant or recessive genes. For a disease passed by a dominant gene to be expressed phenotypically, only one gene for the disease is required. For a disease passed by a recessive gene to be expressed phenotypically, either both the maternal and paternal chromosomes must carry the recessive gene or the individual must develop a spontaneous mutation during embryogenesis. Individuals who carry one defective recessive gene causing a particular disease are called carriers for the disease. Although they will not usually express the disease clinically, the gene may pass to their offspring. If their offspring receive a second defective gene from the other parent, they will be homozygous for the recessive gene and will express the disease as shown in Figure 2-4. In this example the capital D is normal, and the lowercase d is the defective gene.
Sex-Linked Single-Gene Disorders
Some single-gene disorders are considered sex-linked disorders because they are passed on the X or Y sex chromosomes. Most sex-linked disorders are passed on the X chromosome and are recessive traits. These disorders are usually seen in males because any woman carrying the defective gene on one X chromosome will most likely carry the healthy gene on her other X chromosome. A male has a 50% chance of inheriting the defective X chromosome if his mother is a carrier. Because his other sex chromosome is a Y, the recessive gene would be expressed as shown in Figure 2-5 (the X chromosome carrying the defective gene is shown in bold letters). Rarely, a female may inherit a defective X chromosome from her mother and father. She would then be homozygous for the defective gene and express the disorder.
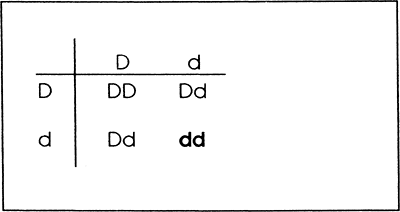 |
Figure 2-4. The inheritance of a single-gene recessive trait, identified as d. |
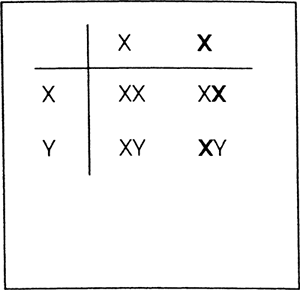 |
Figure 2-5. The inheritance of a sex-linked recessive gene on the X chromosome. Note that 50% of the daughters are carriers of the defect and 50% of the sons express the defect. |
P.52
Clinical Manifestations
Clinical manifestations depend on the specific altered or missing gene.
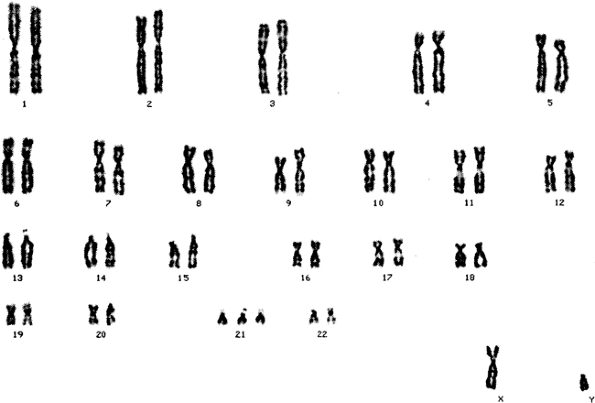 |
Figure 2-6. Trisomy 21 in the karyotype of a child with Down syndrome. All other chromosomes are normal. ( Rubin, E., & Farber, J. L. [2005]. Pathology [4th ed.]. Philadelphia: Lippincott Williams & Wilkins. ) |
Diagnostic Tools
Prenatal amniocentesis or chorionic villi sampling may identify a single-gene defect.
Karyotyping of cells from an adult or child may confirm a clinical diagnosis of a single-gene defect.
Treatment
Treatment for each disease may be supportive if no cure is available, as is the case for Huntington disease, phenylketonuria, fragile X syndrome, or sickle cell disease.
Treatment may involve replacing the missing protein or enzyme if possible. This has been tried for persons with hemophilia A.
Gene splicing may allow insertion of a correct copy of a defective gene into the host genome. Gene splicing has been tried in individuals suffering from cystic fibrosis and muscular dystrophy.
Multifactorial Disorders
Multifactorial disorders are polygenic in nature; they are caused by multiple genes, each having a small, additive effect. If the additive effects reach a threshold level, the disorder will be expressed. The severity of any combination of genetic errors in any one person cannot be predicted.
Some multifactorial disorders may be apparent at birth; others may develop during adulthood. Examples of multifactorial diseases apparent at birth include cleft palate, congenital heart disease, anencephaly, and club foot. Multifactorial disorders expressed in later childhood or adulthood include
P.53
hypertension, hyperlipidemia, diabetes mellitus, most autoimmune diseases, many cancers, and schizophrenia. Multifactorial disorders that develop during adulthood are usually strongly influenced by environmental factors.
Causes of Multifactorial Disorders
Multifactorial disorders result from the additive effects of many gene errors. Multifactorial disorders also may result from less-than-optimum expression of many different genes and not from any particular error. Environmental influences may increase or decrease the likelihood of a multifactorial disorder being expressed and to what degree it is expressed.
Predicting the Occurrence of a Multifactorial Disease
Whether an individual will develop a multifactorial disease cannot be accurately predicted. However, with complete mapping of the genome it may become possible to identify who is most likely to develop a disease. This would influence risk behavior and preventive screening measures.
For example, individuals with type 2 diabetes mellitus often have a strong family history of the disease, but not every member of the family will develop the disease. A strong predictor of developing type 2 diabetes is obesity, a clear example of the interdependency of genetics and environment. Similarly, various genes have been shown to contribute to the risk of developing cancer. However, whether an individual will develop cancer depends on a variety of personal behaviors, including exercise, smoking, and diet.
Ethical concerns abound for individuals at risk of developing certain multifactorial disorders. For example, certain high-paying jobs that expose workers to potential carcinogens may not be offered to an individual with a high risk of developing cancer.
Clinical Manifestations
Each disorder has unique clinical manifestations ranging from nonexistent to mild to severe.
Diagnostic Tools
In families at high risk for a multifactorial disease, genetic mapping may indicate which members of the family are likely to develop the disease and which are not.
Treatment
An individual at known risk for developing a multifactorial disorder based on genetic mapping or family history may modify his or her diet, toxin exposure, and exercise level to reduce additive environmental factors.
An individual at known risk for developing a multifactorial disorder based on genetic mapping or family history may consider not having children of his or her own.
P.54
Down Syndrome
Down syndrome is a genetic disorder caused by a trisomy of chromosome 21. Down syndrome is seen in 1 in 800 live births, making it the most common chromosomal disorder seen in live births. In 95% of cases, Down syndrome is caused by non-disjunction of maternal chromosome number 21 during meiosis. The incidence of Down syndrome related to non-disjunction increases with maternal age. Down syndrome occurs in 1 in 1,350 infants born to mothers younger than 24 years of age, but in 1 in 65 infants born to mothers 41 to 45 years old. Less than 5% of Down cases can be traced to an extra paternal chromosome. A third, uncommon cause of Down syndrome is a translocation of all or part of one of the normally duplicated chromosome 21 onto a different chromosome most commonly chromosome 13, 14, 15, 18, or 22, but other chromosomes may also be targeted. Children with Down syndrome have variable levels of mental retardation and can often be positively influenced by early child intervention programs. A karyotype of a male with Down syndrome is shown in Figure 2-6.
Clinical Manifestations
Variable levels of mental retardation.
Upward slanting of the eyes, short hands that have only one crease on the palm (a simian crease), and low-set ears.
Short stature.
Protruding tongue.
P.55
Diagnostic Tools
Prenatal genetic testing (amniocentesis or chorionic villi sampling) can diagnose fetuses with Down syndrome.
Maternal blood tests are available that can screen for fetuses at increased risk of having Down syndrome. In one test, called the quad test, four circulating maternal substances are measured during the second trimester of pregnancy. When considered together, they can predict up to 75% of Down syndrome cases in women under age 35 and up to 85% to 90% of Down syndrome cases in women age 35 years and older. These substances are:
Unconjugated estriol (uE3). uE3 is produced by the placenta. It is reduced by about 25% in maternal sera from pregnancies affected by Down syndrome compared to unaffected pregnancies.
Alpha-fetoprotein (AFP). AFP is the major serum protein of the fetus. It migrates from the fetal to the maternal circulation. Levels of AFP are reduced in the maternal serum of women carrying a Down syndrome fetus. AFP levels are also used to detect fetal neural tube defects and anencephaly; maternal AFP levels are increased with these defects.
Human chorionic gonadotropin (hCG). hCG is produced during pregnancy, first by the trophoblast and then by the placenta. Levels in maternal serum are higher in pregnancies with Down syndrome fetuses than in unaffected pregnancies.
Inhibin A. Inhibin A is a glycoprotein produced during pregnancy mainly by the placenta. Among women carrying a Down syndrome fetus, inhibin A is increased.
Ultrasound screening in the prenatal period may demonstrate physical suggestions of Down syndrome, especially involving abnormalities in nuchal thickness.
Genetic karyotyping after birth can confirm a clinical diagnosis of Down syndrome.
Complications
Congenital heart or other organ defects frequently occur in association with Down syndrome.
Risk of childhood leukemia is increased in children with Down syndrome. This is related to the observation that some forms of leukemia may be related to defects on chromosome 21. Development of Alzheimer disease in the fourth or fifth decade of life is common in individuals with Down syndrome. This is related to the observation that Alzheimer disease may occur partially as a result of a defect on chromosome 21.
Approximately 20% of fetuses with Down syndrome are spontaneously aborted between 10 and 16 weeks' gestation. Many others do not implant or are miscarried before 6 to 8 weeks' gestation.
P.56
Treatment
Surgery may be required if another congenital defect is present.
Early intervention programs may limit the degree of mental retardation.
Turner Syndrome
Turner syndrome is a monosomy of the sex chromosomes. Infants born with Turner syndrome have 45 chromosomes: 22 pairs of somatic chromosomes and 1 sex chromosome, usually the X (45, X/O). This disorder is common in spontaneously aborted fetuses, and is present in approximately 1 in 2,500 live births. Females with Turner syndrome lack ovaries.
Clinical Manifestations
Clinical manifestations may be nonexistent, mild, or moderate and include:
Short stature and webbing of the neck.
Lack of secondary sex characteristics and amenorrhea (no menstrual cycles) with associated sterility.
Diagnostic Tools
Prenatal genetic testing can identify fetuses afflicted with Turner syndrome.
Genetic karyotyping after birth can confirm the clinical diagnosis.
Complications
Congenital heart defects may accompany the sex chromosome monosomy.
Individuals with Turner syndrome are at increased risk of childhood bone fractures and adult osteoporosis due to lack of estrogen.
Some individuals may demonstrate signs of learning disability.
Treatment
Surgery may be required if a congenital heart defect is present.
Estrogen replacement for a female may increase growth and allow development of secondary sex characteristics. Growth hormone replacement may also stimulate skeletal growth.
Counseling to assist with the issue of infertility may be desired. There are incidents of successful pregnancies after in vitro fertilization.
Klinefelter Syndrome
Klinefelter syndrome is a polysomic disorder characterized by one or more extra X chromosomes in a genotypic male (47, X/X/Y; 47, X/X/X/Y). This
P.57
syndrome occurs in approximately 1 in 600 live births. Klinefelter syndrome may result from non-disjunction of the male or female X chromosome during the first meiotic division, at approximately equal rates in males and females.
Clinical Manifestations
Although the infant may appear normal at birth, he may show a decrease in male secondary sex characteristics during puberty.
Gynecomastia (breast enlargement) and other female patterns of fat deposit.
Infertility and sexual dysfunction.
Tall stature in adult life because decreased levels of testosterone do not contribute to epiphyseal bone plate closure.
Individuals may demonstrate reduced mental functioning, especially with increasing number of X chromosomes.
Treatment
Testosterone replacement.
Counseling may be required.
Selected Bibliography
Betz, U.A.K., Farquhar, R., & Ziegelbauer, K. (2005). Genomics: success or failure to deliver drug targets? Current Opinion in Chemical Biology 9, 387 391.
Collins, F.S., Green, E.D., Guttmacher, A.E., & Guyer, M.S. (2003). A vision for the future of genomics research. A blueprint for the genomic era. Nature 422, 1 13.
Corwin, E.J. (2004). The concept of epigenetics and its role in the development of cardiovascular disease. Biological Research for Nursing 6: 11 16.
Gomez-Lazaro, M., Fernandez-Gomez, F.J., & Jordan, J. (2005). Twenty-five years understanding the mechanism of genome protection. Journal of Physiology and Biochemistry 60, 287 307.
Hahn, W.C. (2005). Telomere and telomerase dynamics in human cells. Current Molecular Medicine 5, 227 231.
Kimberlin, D.W., & Whitley, R.J. (2005). Neonatal herpes: what have we learned. Seminar in Pediatric Infectious Disease 16, 7 16.
Koren, G., Pastuszak, A., & Ito, S. (1998). Drugs in pregnancy. New England Journal of Medicine 338, 1128 1137.
Kuehn, B.M. (2005). Genetic information: How much can patients handle? Journal of the American Medical Association 294, 295 296.
Kwiatkowski, D.P. (2005). How malaria has affected the human genome and what human genetics can teach us about malaria. American Journal of Human Genetics 77, 171 190.
Lambert-Messerlian G.M., & Canick J.A. (2004). Clinical application of inhibin a measurement: prenatal serum screening for Down syndrome. Semininar in Reprod Medicine 22, 235 242.
Nagel, R.L. (2005). Epistasis and the genetics of human diseases. C. R. Biologies 328, 606 615.
Nicklas, R.B. (1997). How cells get the right chromosomes. Science 275, 632 637.
Ooi, C., & Dayan, L. (2004). STIs in pregnancy. An update for GPs. Austrian Family Physician 33, 723 726.
Pastva, M., Corwin, E.J., & Morin, K. (2004). Down's syndrome with an unusual etiology. Journal of the Academy of Nurse Practitioners 16, 244 250.
Pennisi, E. (2005). Why do humans have so few genes? Science 309, 80.
Shahine, L.K., & Caughey, A.B. (2005). Preimplantation genetic diagnosis: the earliest form of prenatal diagnosis. Gynecologic and Obstetric Investigation 60, 39 46.
P.58
Shibley, I.A. Jr. & Pennington, S.N. (1997). Metabolic and mitotic changes associated with the fetal alcohol syndrome. Alcohol and Alcoholism 32, 423 434.
Stranc, L.C., Evans, J.A., & Hamerton, J.L. (1997). Chorionic villus sampling and amniocentesis. Lancet 249, 711 714.
von Zglinicki, T., & Martin-Ruiz, C.M.(2005). Telomeres as biomarkers for ageing and age-related diseases. Current Molecular Medicine 5, 197 203.
Wattendorf, D.J., & Hadley, D.W. (2005). Family history: the three generation pedigree. American Family Physician 72, 441 448.
Wattendorf, D.J., & Muenke, M. (2005). Diagnosis and management of fragile X syndrome. American Family Physician 72, 111 113.
EAN: 2147483647
Pages: 26