9. The Endocrine System
Authors: Corwin, Elizabeth J.
Title: Handbook of Pathophysiology, 3rd Edition
Copyright 2008 Lippincott Williams & Wilkins
> Table of Contents > Unit III - Integrated Control and Dysfunction > Chapter 9 - The Endocrine System
function show_scrollbar() {}
Chapter 9
The Endocrine System
The endocrine system, along with the nervous system, allows for communication between distant sites in the body. There are three components to the endocrine system: endocrine glands that secrete chemical messengers into the bloodstream; the chemical messengers themselves, called hormones; and the target cells or organs that respond to the hormones.
Physiologic Concepts
Endocrine Glands
Endocrine glands are organs that synthesize, store, and secrete hormones into the bloodstream. There are many endocrine glands in the body, including the pancreas, thyroid, parathyroid, and some cells of the gut and kidney. The endocrine glands reviewed in this chapter are the hypothalamus, the anterior and posterior pituitary glands, and the glands that function as target organs for the pituitary hormones.
The Hypothalamus
The hypothalamus is a small area of the brain located in the section of the forebrain called the diencephalon. The hypothalamus is a neural and an endocrine organ. It is concerned with maintaining homeostasis that is, keeping the body's internal environment constant. The hypothalamus is also essential in controlling behavior and allowing appropriate responses to multiple incoming stimuli. It continually receives information from the
P.244
central and peripheral nervous systems concerning temperature, pain, pleasure, feeding, hunger, body mass, and metabolic status. It also receives input from other hormones of the body and receives neural extensions from other areas of the brain.
 |
Figure 9-1. The hypothalamic-pituitary system. The hypothalamus is connected through the blood to the anterior pituitary while the posterior pituitary is a neural outgrowth. |
The hypothalamus, in turn, responds to all the incoming stimuli by sending neural projections throughout the brain and by synthesizing and secreting its own hormones. Nerve cell bodies in the ventral hypothalamus synthesize several hormones and send them in axon projections to be released into the blood and delivered to the anterior pituitary gland. Other nerve cell bodies in the hypothalamus synthesize hormones that are sent down via axon projections to the posterior pituitary, where they are stored until they are eventually released into the bloodstream. These two routes by which the hypothalamus controls hormone release by the anterior and posterior pituitary are shown in Figure 9-1.
The Anterior Pituitary
The anterior pituitary, also called the adenohypophysis, is composed of non-neural tissue. It is anatomically separate from the hypothalamus, but functionally connected to it through its blood supply. The anterior pituitary receives its blood through venous drainage from the hypothalamus. When blood flowing in a vein breaks into another capillary network
P.245
instead of flowing back to the vena cava, the system is called a portal venous system. Thus, the hypothalamus and the anterior pituitary are connected by the hypothalamic-anterior pituitary portal blood flow system. Because this blood has already been used by the hypothalamus, it is poorly oxygenated but rich in hormonal messages put out by the hypothalamus into the median eminence (see later). The anterior pituitary is, therefore, a major target organ for hypothalamic hormones and responds to these hormones with release of its own hormones.
The Posterior Pituitary
The posterior pituitary, also called the neurohypophysis, is true neural tissue derived embryologically from the hypothalamus. There are three parts to the posterior pituitary: the median eminence (sometimes considered hypothalamic tissue), into which the hypothalamus secretes the anterior pituitary-releasing hormones; the infundibular stem connecting the hypothalamus with the posterior pituitary; and the infundibular process, which is the terminal end of the posterior pituitary.
Nerve cell bodies in the supraoptic and paraventricular nuclei of the hypothalamus synthesize two hormones: antidiuretic hormone, also called vasopressin, and oxytocin. The hypothalamus sends these hormones in axon projections through the infundibular stem to the infundibular process. They are stored there until the hypothalamus stimulates them to be released into the general circulation. Thus, the hormones released by the posterior pituitary are hypothalamic in origin and depend on the hypothalamus for their release.
Target Glands
The third group of endocrine glands discussed in this chapter consists of those outside the brain that respond to the anterior and posterior pituitary hormones with the release of their own hormones. These glands are the target organs of the pituitary hormones and include the thyroid gland, the adrenal gland, and the testes and ovaries. The pancreas, which secretes insulin, is also an endocrine gland and is discussed in Chapter 16.
Hormones
A hormone is a chemical messenger released by an endocrine gland into the circulation. Once released, a hormone travels in the bloodstream and affects only cells in the body that have receptors (binding sites) specific to it. Cells that respond to a particular hormone are called target cells for that hormone. Typically, a hormone is released in bursts from an endocrine gland in a pattern that often follows an inherent daily (diurnal) rhythm. The burst of hormone release can be increased or decreased above or below baseline level by various inputs to the gland. Inputs that affect hormone release involve: (1) stimulation by another hormone or neurotransmitter, or (2) stimulation caused by a decrease or increase in a certain ion or nutrient. Examples of
P.246
hormones that cause an increase or decrease in another hormone's release include all the hypothalamic hormones affecting the anterior pituitary. Examples of neurotransmitters affecting a hormone's release include the release of insulin in response to epinephrine and norepinephrine stimulation. Ions that influence the release of a hormone include calcium ion's effect on parathyroid hormone, and sodium ion's effect on aldosterone. Nutrients that affect the release of hormones include the amino acids that stimulate the release of insulin and growth hormone. Frequently, one endocrine gland is stimulated simultaneously by several different inputs.
There are three broad categories of hormones: peptide, steroid, and amino acid. Most hormones, including all the hypothalamic and pituitary hormones, are peptide hormones. The steroid hormones are made from cholesterol and are soluble across the cell membrane. The amino acid hormones are made from the amino acid tyrosine.
Peptide Hormones
Peptide hormones range in size from a few amino acids to relatively large protein complexes. Peptide hormones circulate in the plasma to their target organs and exert their effects by binding to specific receptors present on the outside of target cell membranes. By binding to its receptor, a protein hormone changes the cell's permeability to water, electrolytes, or organic molecules such as glucose, or causes the activation of intracellular messengers, which then causes enzyme activation or protein synthesis. Examples of intracellular messengers include the G proteins, which many protein hormones first activate during receptor binding, and the second messengers such as cyclic adenosine monophosphate (cyclic AMP) and calcium, which are subsequently activated by the G proteins. See Box 9-1 for a list of the peptide hormones.
Steroid Hormones
Steroid hormones are cholesterol-based, lipid-soluble molecules produced by the adrenal cortex and the sex organs. Because steroid hormones are lipid-soluble, they can cross the cell membrane and bind to receptors or carriers inside the cell. Once inside a cell, the steroid hormone travels to the cell nucleus, where it influences the cell by affecting DNA replication, transcription of DNA into RNA, or translation of RNA into proteins. Steroid hormones are discussed in this chapter and in Chapter 20. See Box 9-2 for a list of steroid hormones.
Amine Hormones
The amine hormones are derivatives of the amino acid tyrosine and include thyroid hormone and the catecholamines (epinephrine, norepinephrine, and dopamine). Epinephrine, norepinephrine, and dopamine also act as neurotransmitters in the central and peripheral nervous systems. The catecholamine hormones travel in the blood to their target cell and bind to the
P.247
plasma membrane at specific receptor sites. Binding of catecholamine activates the cyclic AMP second messenger system and alters enzyme activity or membrane permeability. Thyroid hormone travels in the blood mostly bound to carrier proteins with a smaller amount circulating free. Once at the target cell, free thyroid hormone crosses the cell membrane and binds to the nuclear DNA, directly affecting DNA transcription. Therefore, free
P.248
hormone, although lesser in quantity, is the active hormone. Box 9-3 lists the amine hormones.
Box 9-1. Peptide Hormones
Hypothalamic-Releasing and -Inhibiting Hormones and Factors
Thyrotropin-releasing hormone (TRH)
Corticotropin-releasing hormone (CRH)
Growth hormone releasing factor (GRF)
Somatostatin (growth hormone inhibiting hormone)
Gonadotropin-releasing hormone (GnRH)
Prolactin-inhibiting factor (PIF)
Prolactin-releasing hormone (PRH)
Substance P
Anterior Pituitary Protein Hormones
Thyroid-stimulating hormone (TSH)
Adrenocorticotropic hormone (ACTH)
Growth hormone (GH)
Follicle-stimulating hormone (FSH)
Luteinizing hormone (LH)
Prolactin
Melanocyte-stimulating hormone
Posterior Pituitary Hormones
Antidiuretic hormone (ADH)
Oxytocin
Hormones of Digestion and Metabolism (Chapters 15 and 16)
Insulin
Glucagon
Calcitonin
Parathyroid hormone
Cholecystokinin
Gastrin
Secretin
Hormone of Blood Pressure and Electrolyte Balance (Chapters 13 and 19)
Angiotensin II
Hormone for Red Blood Cell Development (Chapters 12 and 19)
Erythropoietin
Hormone to Modulate Stress and Pain (Chapters 6, 8 and 9)
Endorphin
Box 9-2. Steroid Hormones
Gonadal hormones
Estrogens
Progesterone
Androgens (primarily testosterone)
Hormones of the Adrenal Cortex
Aldosterone
Glucocorticoids (primarily cortisol)
Androgens (primarily testosterone)
Estrogens
Feedback
In the endocrine system, feedback refers to the response of a target tissue after stimulation by a specific hormone that then influences the continued release of that hormone. Each hormone is stimulated to be released by a specific signal. Once released, a hormone affects its target organ, causing a response that usually reduces further hormone release. This type of feedback, shown in Figure 9-2, is called negative feedback, and allows tight control over hormone levels. Positive feedback is uncommon and occurs when the response by a target tissue to hormonal stimulation increases the further release of that hormone.
Factors Controlling Hormone Secretion
Factors Controlling Anterior Pituitary Hormone Secretion
The stimuli that control the secretion of the pituitary hormones (except melanocyte-stimulating hormone) are the hormones secreted by the hypothalamus that travel in the portal blood to the anterior pituitary. These hormones are hypothalamic-releasing or hypothalamic-inhibiting hormones, depending on whether they increase or decrease the release of
P.249
the pituitary hormone they control. When a hypothalamic-releasing hormone is secreted, its corresponding anterior pituitary hormone is released. When a hypothalamic-inhibiting hormone is secreted, it inhibits synthesis and release of the anterior pituitary hormone over which it has control. Once secreted, the pituitary hormones act to stimulate another target organ or cell to perform a function or release a hormone of its own.
Box 9-3. Amine Hormones
Thyroid hormones
Epinephrine
Norepinephrine
Melatonin (from the anterior pituitary)
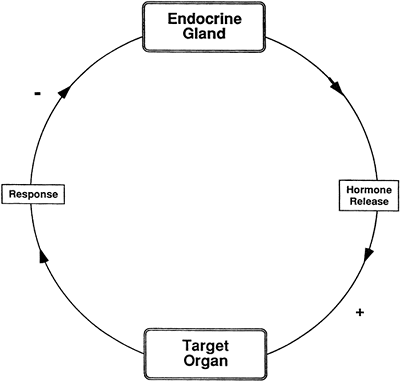 |
Figure 9-2. A typical negative feedback cycle in which an endocrine gland releases a hormone, which then stimulates (+) its target organ to respond in such a way that the further secretion of the hormone by the endocrine gland is reduced (-). |
The pituitary hormone and the subsequent response to it by its target organ may feed back on the hypothalamus to decrease further release of the hypothalamic hormone. The target organ response may also inhibit further release of the pituitary hormone.
Factors Controlling Hypothalamic Hormone Secretion
For the hypothalamic-pituitary hormonal system, the hypothalamus ultimately determines whether a hormone will be secreted. The hypothalamic-releasing or -inhibiting hormones are secreted at a baseline level that can be increased or decreased as a result of the integration of many neural inputs to the hypothalamus. The inputs are related to stress, pain, body weight, temperature, emotions, and various hormones released by target organs. All these influences can be excitatory or inhibitory for each releasing or inhibiting hormone.
P.250
Target Organ Hormones
Thyroid Hormone
Thyroid hormone (TH) is an amine hormone synthesized and released from the thyroid gland. It is made when one or two iodine molecules are joined to a large glycoprotein called thyroglobulin, which is synthesized in the thyroid gland and contains the amino acid tyrosine. These iodine-containing complexes are called iodotyrosines. Two iodotyrosines then combine to form two types of circulating TH, called T3 and T4. T3 and T4 differ in the total number of iodine molecules they contain (three for T3 and four for T4). Most (90%) of the TH released into the bloodstream is T4, but T3 is physiologically more potent. In passage through the liver and kidney, most T4 is converted to T3. T3 and T4 are carried to their target cells in the blood bound to a plasma protein, but enter the cell as free hormone. T3 and T4 collectively are referred to as TH.
Effects of Thyroid Hormone
Target cells for TH include almost all cells of the body. The primary effect of TH is to stimulate the metabolic rate of all target cells by increasing the metabolism of protein, fat, and carbohydrate. TH also appears to stimulate the rate of the sodium-potassium pump in its target cells. Both functions serve to increase utilization of energy by the cells, thereby increasing basal metabolic rate (BMR), burning calories, and increasing heat production by each cell.
Thyroid hormone also increases the sensitivity of target cells to catecholamines, thus increasing heart rate and causing heightened emotional responsiveness. TH increases the rate of depolarization of skeletal muscle, which increases the speed of skeletal muscle contractions, often leading to a fine tremor. TH is essential for normal growth and development of all cells of the body and is required for the function of growth hormone.
Factors Controlling Thyroid Hormone Secretion
The stimulus for the secretion of TH is thyroid-stimulating hormone (TSH), released into the bloodstream from the anterior pituitary. The stimulus for the release of TSH is thyroid-releasing hormone (TRH), secreted from the hypothalamus into the portal bloodstream. Thyroid hormone appears to act in a negative feedback manner on the hypothalamus, to decrease the further release of TRH, and on the pituitary, to decrease the release of TSH. TSH may also act on the hypothalamus to decrease further release of TRH.
Factors Controlling Thyroid-Releasing Hormone Secretion
The stimuli responsible for increasing TRH secretion include exposure of the body to cold temperature, physical and perhaps psychological stress, and low levels of TH. When the secretion of TRH is stimulated by cold temperature, the result is an increase in TH, which increases BMR, thereby
P.251
increasing body heat and reducing the demand for a further increase in TRH (Fig. 9-3). This is an example of negative feedback.
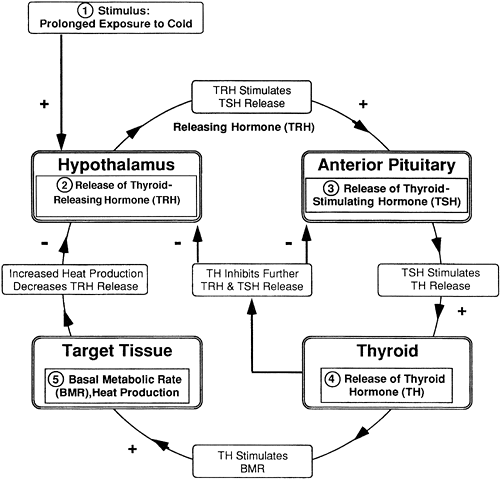 |
Figure 9-3. Feedback: thyroid hormone. |
Glucocorticoids
Glucocorticoids are steroid hormones released from the cortex (outer layer) of the adrenal gland that affect many aspects of metabolism, especially glucose metabolism. In humans, the main glucocorticoid is cortisol. The glucocorticoids also affect many other systems of the body, including the cardiovascular and immune systems. Glucocorticoids are released in a diurnal (daily) manner, peaking in the early morning hours.
Effects of the Glucocorticoids
The glucocorticoids increase the level of blood glucose by stimulating gluconeogenesis (conversion in the liver of fats and proteins into glucose). Glucocorticoids also increase blood glucose levels by stimulating muscle, adipose (fat), and lymphatic tissues to use free fatty acids for energy instead of glucose. Likewise, the glucocorticoids stimulate protein breakdown and inhibit protein synthesis in all body cells. They also stimulate hunger, promote fat buildup in the trunk and face, and inhibit growth by suppressing growth hormone and antagonizing the effects of growth hormone on protein synthesis. The glucocorticoids increase the effect of growth hormone on adipose tissue and increase the effect of thyroid
P.252
hormone on its target tissues. They also increase the effects of the catecholamines, causing increased heart rate and blood pressure. Many of these glucocorticoid effects are essential in times of trauma and stress. They allow one to survive blood loss, periods of hunger or starvation, or prolonged exposure to environmental extremes.
A non-metabolic effect that occurs with high circulating levels of cortisol is the inhibition of immune and inflammatory functions. The glucocorticoids exert this effect by blocking almost every component of the immune and inflammatory responses: they depress cytotoxic T-cell function and suppress the production, release, and activation of many chemical mediators of inflammation, including interleukins, prostaglandins, and histamine. Levels of cortisol high enough to inhibit immune and inflammatory function may be reached with pharmacologic administration of cortisol for immunosuppression, with tumors of the adrenal gland, or with long-term stress. In addition, cortisol and metabolites of cortisol such as cortisone appear to have a strong effect on emotional stability and mood.
Factors Controlling Glucocorticoid Release
Glucocorticoids are released from the adrenal gland in response to circulating adrenocorticotropic hormone (ACTH) from the anterior pituitary. ACTH is released in response to corticotropin-releasing hormone (CRH) carried in the portal blood from the hypothalamus. CRH also stimulates the release of endorphins by the anterior pituitary and perhaps elsewhere. When released, glucocorticoids feed back on the hypothalamus and on the anterior pituitary to decrease the further release of CRH and ACTH, respectively.
Factors Controlling Corticotropin-Releasing Hormone
Corticotropin-releasing hormone is secreted from the hypothalamus in a diurnal pattern that sets the subsequent release pattern of ACTH and cortisol. Stimuli for an increase in CRH include stress, hypoglycemia (low blood glucose), and decreased circulating levels of glucocorticoids. The feedback cycle of CRH release in response to hypoglycemia is shown in Figure 9-4.
Other Effects of Adrenocorticotropic Hormone
Adrenal androgens are released in response to ACTH stimulation of the adrenal gland. Adrenal androgens are the primary source of androgens in women and children. High levels of ACTH can result in masculinization of women and children. ACTH is similar in structure to another anterior pituitary hormone, melanin-stimulating hormone (MSH), which causes the cells of the skin to produce the tanning substance melanin. High levels of ACTH can have cross-over effects on the skin and cause bronzing. A limited amount of ACTH appears essential for the synthesis of another adrenal cortical hormone, aldosterone. Without aldosterone, salt wasting and death occur.
 |
Figure 9-4. Feedback: glucocorticoids. |
P.253
Growth Hormone
Growth hormone (GH), also called somatotropin, is a protein hormone released in a diurnal pattern over 24 hours. Approximately 70% of daily secretion occurs in a burst 1 to 4 hours after the onset of sleep. Accelerated GH release occurs during puberty and pregnancy.
Effects of Growth Hormone
Growth hormone increases protein synthesis in all cells of the body, especially muscle cells. GH stimulates the growth of cartilage and activity of osteoblasts, the bone-producing cells of the body. GH is essential for longitudinal bone growth and for the continual remodeling of bone that occurs throughout life. Effects of GH on bone and cartilage occur through intermediary peptides, called somatomedins or insulin-like growth factors (IGFs), released from the liver in response to growth hormone. GH directly stimulates the growth of almost all other organs of the body, including the heart muscle, skin, and endocrine glands.
Growth hormone causes breakdown of fats and subsequent use of fatty acids for energy. Because fats are being used as an energy source, GH results in increased circulating blood glucose. GH also induces insensitivity to insulin. With decreased sensitivity to insulin, most cells will not transport glucose intracellularly, further increasing plasma glucose levels.
P.254
Factors Controlling Growth Hormone Release
Growth hormone is released from the anterior pituitary in response to a balance between two hypothalamic hormones: growth hormone-releasing hormone (GHRH) and growth hormone inhibiting hormone, also called somatostatin. GH acts in a negative feedback manner on the hypothalamus to decrease further release of GHRH.
Factors Controlling Growth Hormone Releasing Hormone
Increased GHRH occurs in response to increased levels of circulating amino acids, hypoglycemia, fasting or starvation, physical and emotional stress, and decreased GH. Exercise stimulates the release of GHRH, directly or through the effects of hypoglycemia and physical stress. The reproductive hormones (estrogen and testosterone) appear to increase secretion of GH, either by acting directly on the pituitary or through stimulation of GHRH. The feedback pattern of GHRH secretion in response to increased plasma amino acids is shown in Figure 9-5.
Factors Controlling Somatostatin Release
The hypothalamus releases an inhibitory hormone for GH, called somatostatin. Somatostatin is released in response to high blood glucose, free
P.255
fatty acids, obesity, and cortisol. Emotional influences including stress stimulate somatostatin, most likely through increased cortisol, thereby reducing growth.
 |
Figure 9-5. Feedback: growth hormone. |
![]() ediatric Consideration
ediatric Consideration
Children release more of their GH in the nocturnal burst than do adults. In children, GH is essential for the longitudinal bone growth that occurs throughout gestation, infancy, childhood, and puberty. Infants and children suffering emotional or physical neglect may develop failure to thrive syndrome, characterized by a decrease in longitudinal growth and weight gain. It has been suggested that a stress-induced increase in the release of somatostatin as a result of neglect may play a role in failure to thrive syndrome.
Gonadotropins
The gonadotropins include two anterior pituitary hormones: follicle-stimulating hormone (FSH) and luteinizing hormone (LH). Target tissues of FSH and LH are the ovary in women and the testis in men (Chapter 20).
Effects of the Gonadotropins
In response to FSH and LH in women, the ovary secretes the steroid hormones estrogen and progesterone. Estrogen feeds back on the hypothalamus and anterior pituitary in a complicated manner, with a negative effect on increasing the release of FSH and a positive effect on the release of LH, ultimately resulting in ovulation (the rupture of an ovarian follicle). With ovulation, the egg, also called the ovum, is released (Chapter 20) and becomes available for fertilization by a sperm. Progesterone appears to feed back on the anterior pituitary to limit secretion of FSH and LH.
In men, FSH stimulates cells of the testis to initiate and support spermatogenesis (production of sperm). The cells of the testis predominantly affected by FSH in the male are Sertoli cells. Sertoli cells make up the inner lining of the seminiferous tubules, the site of spermatogenesis, and are important in providing nutrients to the developing sperm (Chapter 20). A hormone produced by Sertoli cells, inhibin, influences the production of testosterone by acting directly on the pituitary gland to decrease the release of FSH.
LH also is released from the anterior pituitary in men. LH causes interstitial cells of the testes to produce and secrete testosterone. Estrogen and testosterone are also synthesized by the adrenal gland, in men and women, in response to stimulation by ACTH.
Factors Controlling Gonadotropin Release
The gonadotropins are released from the pituitary in response to gonadotropin-releasing hormone (GnRH) from the hypothalamus. It appears that one hypothalamic hormone controls the release of both of
P.256
the pituitary gonadotropins. GnRH is sometimes referred to as luteinizing hormone releasing hormone (LHRH). An increase in GnRH synthesis and release causes the onset of puberty.
Factors Controlling Gonadotropin-Releasing Hormone
Before puberty, circulating GnRH is very low. With maturation of the hypothalamus and perhaps attainment of a certain body mass, GnRH increases and initiates puberty. After sexual maturation has been established, the circulating level of GnRH is controlled in a negative feedback manner by estrogen and testosterone. Stress, starvation, and fear may affect the release of GnRH at any time, influencing the release of estrogen and progesterone in females and testosterone in males, and altering reproductive function.
Estrogens
Estrogens are steroid hormones that affect their target tissues by altering the rate of DNA replication, DNA transcription, or RNA translation. Although the effects of estrogens are most apparent in females, males also produce and are affected by estrogens. There are three main types of estrogens in humans: estrone, estradiol, and estriol.
Effects of estrogens include the following:
Development in utero of female internal and external sex organs.
Female distribution of body fat.
Pigmentation of the nipples.
Stimulation of breast development during pregnancy.
Stimulation of growth of the endometrial lining of the uterus each month to prepare for implantation of the embryo.
Maintenance of pregnancy.
Stimulation of lactation.
Stimulation of bone formation throughout life in males and females.
Limiting bone resorption (breakdown) by direct action on bone or by limiting bone response to parathyroid hormone in males and females.
Affecting liver protein production of lipoproteins (stimulates HDL, decreases LDL), coagulation factors, and carrier molecules for steroid hormones and thyroxine in males and females.
Acting to reduce the risk of coronary artery disease, most likely as a result of increasing HDL, in males and females.
Stimulating the kidneys to retain sodium in males and females.
Influencing brain neural signaling in males and females, affecting behavior and mood.
Estrogen excess in men can cause gynecomastia (breast enlargement).
![]() ediatric Consideration
ediatric Consideration
Estrogen acts during puberty to cause the development of female secondary sex characteristics, including the development of breasts and growth of axillary and pubic hair. Estrogen also acts along with growth hormone and the androgens to cause skeletal growth during puberty, and causes the closure of the epiphyseal bone plates to halt growth in males and females at the end of puberty. Estrogen can pathologically affect children, causing precocious (early) onset of menstruation in girls and breast development in girls and boys.
![]() eriatric Consideration
eriatric Consideration
Menopause is considered to have occurred when a woman has not experienced a menstrual period for a year. Menopause happens when aging ovaries no longer respond to the signals of the gonadotropins to synthesize and secrete estrogen. As estrogen levels decrease, LH, FSH, and GnRH levels increase because any negative feedback by estrogen has been removed. Although menopause is a normal developmental stage, lack of estrogen in postmenopausal women causes decreased bone density, increased risk of cardiovascular disease, drying of skin and vaginal membranes, and hot flashes or flushes. Most women in developed countries experience menopause in their late 40s or early 50s. Hormonal and cytotoxic treatments used for breast cancer may induce early menopause in some women.
Several randomized clinical trials carefully investigated the safety of hormone replacement therapy (HRT) in postmenopausal women, including estrogen given both alone and in combination with progesterone. Results indicated that combination hormone therapy was associated with an increased risk of breast cancer, cardiovascular disease, and stroke among postmenopausal women. The effect of estrogen alone is still being investigated, although estrogen alone is contraindicated for women with an intact uterus due to an increased risk of endometrial cancer. As a result of these studies, which have been termed the Women's Health Initiative, clinical experts recommend that HRT either not be prescribed at all or be prescribed for as short a period of time as necessary to control menopausal symptoms.
P.257
Progesterone
Progesterone, like estrogen, is a steroid hormone. In women, progesterone is synthesized by thecal cells of the developing follicle, and later the corpus luteum, in response to stimulation by LH and, to a lesser extent, FSH.
Effects of progesterone include the following:
Progesterone is released from an ovarian follicle after the follicle has ruptured during ovulation. It causes the endometrial lining of the uterus to become secretory in anticipation of fertilization of the ovum and embryo implantation, with the result that blood vessels in the endometrium begin to branch and glands begin secreting a thin glycogen-rich fluid. The ruptured follicle becomes the corpus luteum, which continues progesterone secretion.
If the ovum is fertilized and the embryo implants in the uterus, the corpus luteum and later the placenta maintain the pregnancy by secreting progesterone. If progesterone decreases, the pregnancy terminates.
If pregnancy does not occur, the corpus luteum degenerates over the next 14 days, progesterone levels decline, and menstruation (sloughing off of the uterine lining) occurs.
Progesterone works with estrogen and prolactin to stimulate breast development during puberty and pregnancy.
Progesterone relaxes smooth muscles, including the uterus and the vascular smooth muscle of the arterioles.
Progesterone appears to be protective against some cancers.
P.258
Testosterone
Testosterone, also a steroid hormone, is the most abundant of the powerful androgen hormones. Testosterone synthesis occurs in specialized cells of the testes called Leydig cells, and in the adrenal gland in women.
Effects of testosterone include the following:
Development in utero of male internal and external sex organs.
Maintenance of sperm production throughout a man's lifetime.
Stimulation and maintenance of male distribution of muscle.
Stimulation of bone formation throughout life in males and females.
Stimulation of red blood cell formation in males and females.
Stimulation of anabolism (buildup) of proteins in males and females.
Involvement in brain neural signaling, affecting behavior and mood, in males and females.
Testosterone excess in women can cause clitoral enlargement, voice deepening, and beard development.
![]() ediatric Consideration
ediatric Consideration
Testosterone acts during puberty to cause the development of male secondary sex characteristics, including growth of the penis and scrotum and the development of male axillary and pubic hair patterns. Testosterone is also important for skeletal growth during puberty, especially in males. Excess testosterone in girls can cause voice deepening, acne, male pattern muscle development and clitoral enlargement.
![]() eriatric Consideration
eriatric Consideration
The testes continue to respond to the gonadotropins as a man ages, albeit at a reduced level. Testosterone synthesis and release by the testes continues, as does sperm production, throughout a man's lifetime, albeit at some declining rate. Testosterone levels adequate to maintain sperm production and muscle mass continue into a man's seventh decade, at least.
Prolactin
Prolactin is a protein hormone released from the anterior pituitary.
P.259
Effects of Prolactin
When a girl reaches puberty, prolactin acts in concert with estrogen, progesterone, and GH to promote breast tissue development. Each of these hormones increases dramatically during pregnancy, resulting in further stimulation of breast development. After the birth of an infant, prolactin acts on the breast to stimulate lactation (milk production), allowing the infant to breastfeed. In postpartum women, the posterior hypothalamic hormone oxytocin works in concert with prolactin and is required for successful breastfeeding.
In non-pregnant women, high prolactin levels inhibit the release of two other anterior pituitary hormones: FSH and LH. Because FSH and LH are essential for ovulation and pregnancy, high prolactin in women who breastfeed full-time may offer some protection against another pregnancy occurring.
The role of prolactin in men has not been identified, although recent evidence suggests that, in men and women, prolactin may affect the immune system, possibly by modulating the release of certain cytokines.
Factors Controlling Prolactin Release
The secretion of prolactin from the anterior pituitary is controlled by release of a prolactin-inhibitory hormone (PIH) from the hypothalamus, recently identified as the catecholamine dopamine. A decrease in the release of dopamine stimulates prolactin release. There may also be a prolactin-stimulating hormone released from the hypothalamus, although it is yet to be identified.
Stimulation for increased prolactin during pregnancy appears to be an estrogen-dependent decrease in the hypothalamic release of PIH. The suckling of the mother's nipple during breastfeeding by the infant stimulates prolactin release after pregnancy stimulation of the nipple by suckling appears to cause increased prolactin by decreasing the hypothalamic release of PIH.
The major hypothalamic and anterior pituitary hormones and their target organ effects are listed in Figure 9-6.
 |
Figure 9-6. The major hypothalamic and anterior pituitary hormones. Note that decreased PIH increases prolactin release. |
P.260
Antidiuretic Hormone
Antidiuretic hormone (ADH) is a protein hormone made in the supraoptic nuclei of the hypothalamus and stored in and released from the posterior pituitary. It is also called vasopressin, which means vascular tensor.
Effects of Antidiuretic Hormone
ADH causes the cells of the renal collecting ducts to become more water permeable. This increases the reabsorption of water into the blood, decreasing urine diuresis (flow). This is the antidiuretic effect of ADH (Chapter 18). At very high levels, ADH causes vascular smooth muscle contraction, increasing total peripheral resistance and blood pressure (Chapter 13). This is the major vasoactive effect.
Factors Controlling Antidiuretic Hormone Release
The major stimulus for ADH release is increased plasma osmolality (increased solute concentration). Increased plasma osmolality is sensed by osmoreceptors in the hypothalamus. Normal plasma osmolality is approximately 280 mOsm/kg. ADH-induced antidiuresis returns a high plasma osmolality toward normal by diluting the plasma (increasing its water concentration), as shown in Figure 9-7.
Other stimuli for ADH release include decreased blood pressure (sensed by the carotid and aortic baroreceptors), stress, pain, and exercise.
P.261
ADH secretion is inhibited by decreased plasma osmolality, increased blood pressure, and alcohol.
 |
Figure 9-7. Feedback: antidiuretic hormone. |
Oxytocin
Oxytocin is a protein hormone made in the paraventricular nuclei of the hypothalamus and stored in and released from the posterior pituitary.
Effects of Oxytocin
Oxytocin stimulates contraction of the smooth muscle lining of the milk ducts of the breast, causing increased intra-mammary pressure and subsequent letdown of stored milk into the nipples.
Oxytocin also stimulates contraction of the uterine smooth muscle. Its exact role in initiating labor in a pregnant woman is unclear. However, it causes increased intensity of uterine contractions as labor progresses and delivery approaches. The drug Pitocin is a derivative of oxytocin and is used clinically to initiate and speed labor.
Factors Controlling Oxytocin Release
The primary stimulus for the release of oxytocin is suckling on the nipple of the breast in women. As shown in Figure 9-8, suckling leads to milk letdown, which allows the infant to feed. As the drive for suckling is reduced, the stimulus for oxytocin release is decreased and milk letdown slows; this is a clear example of negative feedback. Stress or fear may inhibit synthesis of oxytocin.
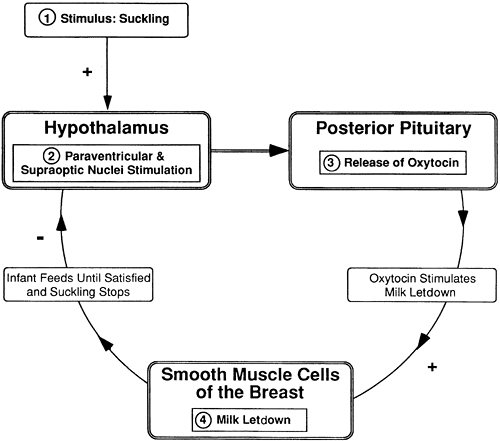 |
Figure 9-8. Feedback: oxytocin. |
P.262
Pathophysiologic Concepts
Hypopituitarism
Hypopituitarism refers to low secretion of any anterior pituitary hormone. Panhypopituitarism refers to low secretion of all anterior pituitary hormones. Clinical manifestations include reproductive malfunction, growth failure, decreased bone density, and morbid obesity. It can be misinterpreted as child abuse or neglect. Hypopituitarism can result from malfunction of the pituitary gland or the hypothalamus. Causes include:
Infection or inflammation.
Autoimmune disease.
A tumor (adenoma). Typically, a tumor of one type of hormone-producing cell expands to the point that it begins to interfere with the function of other hormone-producing cells, leading to reduced secretion of one or more other hormones.
Feedback from a malfunctioning target organ; for example, reduced secretion of TSH from the pituitary would occur if a diseased thyroid gland were secreting excessively high levels of TH.
Hypoxic necrosis (death caused by lack of oxygen) of the pituitary or hypothalamus resulting from decreased blood flow or decreased oxygenation. Hypoxia can destroy any or all of the hormone-producing cells. An example of this is Sheehan's syndrome, which develops following maternal hemorrhage during or after birth.
Hyperpituitarism
Hyperpituitarism is the excess secretion of an anterior pituitary hormone. Hyperpituitarism typically involves just one of the pituitary hormones; the other pituitary hormones are often secreted in reduced levels. Hyperpituitarism can result from malfunction of the pituitary gland or the hypothalamus. Causes include:
A primary adenoma of one type of hormone-producing cell, usually GH, ACTH, or the prolactin-producing cells.
A lack of feedback from a target gland; for example, increased TSH may occur in response to decreased or absent secretion of TH by the thyroid gland.
Conditions of Disease or Injury
Hypothyroidism
Hypothyroidism results from decreased levels of circulating thyroid hormone. Hypothyroidism is characterized by myxedema, the non-pitting, boggy edema that develops around the eyes, feet, and hands and
P.263
infiltrates other tissues as well. Hypothyroidism may result from malfunction of the thyroid gland, the pituitary, or the hypothalamus. If it results from thyroid gland malfunction, low TH levels are accompanied by high TSH and high TRH because of the lack of negative feedback on the pituitary and hypothalamus by TH. If hypothyroidism results from pituitary malfunction, low levels of TH are caused by low TSH. TRH from the hypothalamus is high because there is no negative feedback on its release by TSH or TH. Hypothyroidism caused by hypothalamic malfunction results in low TH, low TSH, and low TRH. Medically-induced hypothyroidism may follow previous thyroid therapy or surgery, radioiodine therapy, or drugs such as cytokines, amiodarone, and lithium. See page C5 for illustrations relating to hypothyroidism.
Diseases of Hypothyroidism
Hashimoto's disease, also called autoimmune thyroiditis, results from autoantibody destruction of thyroid gland tissue. This results in decreased TH, with increased TSH and TRH levels caused by minimal negative feedback. The cause of autoimmune thyroiditis is unknown, but there appears to be a genetic tendency to develop the disease.
Endemic goiter is hypothyroidism caused by a dietary deficiency of iodide. A goiter is an enlargement of the thyroid gland. Goiter occurs with a deficiency of iodide because the thyroid cells become overactive and hypertrophic (larger) in an attempt to sequester all possible iodide from the bloodstream. Low TH levels are accompanied by high TSH and TRH because negative feedback is minimal.
Thyroid carcinoma may cause hypothyroidism or hyperthyroidism. Treatment of this rare cancer may include thyroidectomy, TSH suppression drugs, or radioactive iodine therapy to destroy thyroid tissue. All of these treatments may result in hypothyroidism. Exposure to radiation, especially during childhood, is a cause of thyroid cancer. Iodine deficiency may also increase the risk of developing thyroid cancer because it stimulates thyroid cell proliferation and hyperplasia.
Clinical Manifestations
Sluggishness, slow thinking, and clumsy, slow movements.
Decreased heart rate, enlarged heart (myxedemic heart), and decreased cardiac output.
Bogginess and edema of the skin, especially under the eyes and in the ankles.
Intolerance to cold temperatures.
Decreased metabolic rate, decreased caloric requirements, decreased appetite and nutrient absorption across the gut.
Constipation.
Change in reproductive function.
Dry, flaky skin and brittle, thin body and head hair.
P.264
Diagnostic Tools
A good history and physical examination will help diagnose hypothyroidism.
Blood tests measuring levels of TH (both T3 and T4), TSH, and TRH will allow diagnosis of the condition and localization of the problem at the level of the central nervous system or the thyroid gland.
Complications
Myxedema coma is a life-threatening situation characterized by exacerbation (worsening) of all symptoms of hypothyroidism, including hypothermia without shivering, hypotension, hypoglycemia, hypoventilation, and a decrease in consciousness resulting in coma.
Death can occur without TH replacement and stabilization of symptoms.
There are also risks associated with the treatment of thyroid deficiency. These risks include hormone over-replacement, anxiety, muscle wasting, osteoporosis and atrial fibrillation.
Treatment
Treatment always includes replacement of thyroid hormone with synthetic thyroxine.
For endemic goiter, iodide replacement may relieve symptoms.
If the cause of hypothyroidism is related to a central nervous system tumor, it may be treated with chemotherapy, radiation, or surgery.
![]() ediatric Consideration
ediatric Consideration
Infants born without a thyroid gland or with defects in TH synthesis will develop congenital hypothyroidism, a disease sometimes referred to as cretinism. Congenital hypothyroidism is characterized by low TH, with high TSH and TRF. TH is permissive (necessary) for functioning of all cells of the body, including cells of the central nervous system (CNS). Development of the CNS occurs in utero and for approximately 1 year after birth. Because an infant with congenital hypothyroidism was exposed to maternal TH in utero, it will be born neurologically intact. If the condition is unrecognized after birth and TH is not replaced pharmacologically, further development of infant CNS will be compromised and severe mental retardation will result. Growth will be stunted and skeletal deformity will develop. Many states require measurement of TH levels at birth. With thyroxine replacement, CNS damage can be avoided.
Hypothyroidism at birth may also occur if maternal antithyroid antibodies attack the fetal thyroid during pregnancy. Likewise, if a pregnant woman is severely deprived of iodide, her infant may also have hypothyroidism after birth. Long-term neurologic prognosis for either of these conditions depends on the extent of thyroid deficit.
![]() eriatric Consideration
eriatric Consideration
Myxedema coma is usually seen in elderly persons who are not being adequately treated for hypothyroidism. It is more common in older women with autoimmune thyroiditis. It may also occur after an acute illness in this population. Prolonged exposure of an elderly individual to cold weather may precipitate the disorder.
P.265
Hyperthyroidism
Hyperthyroidism is excessive levels of circulating TH. This disorder can result from dysfunction of the thyroid gland, pituitary, or hypothalamus. Increased TH caused by malfunction of the thyroid gland is accompanied by decreased TSH and TRF, as a result of the negative feedback on their release by TH. Hyperthyroidism caused by malfunction of the pituitary results in high TH and high TSH. TRF is low because of negative feedback from TH and TSH. Hyperthyroidism caused by malfunction of the hypothalamus shows high TH accompanied by excess TSH and TRH. See page C5 for illustrations related to hyperthyroidism.
Diseases of Hyperthyroidism
Graves' disease, the most common cause of hyperthyroidism, is an autoimmune disorder usually characterized by production of autoantibodies that mimic the action of TSH on the thyroid gland. These IgG autoantibodies, termed thyroid-stimulating immunoglobulins, turn on the production of TH, but are not inhibited by rising levels. TSH and TRH levels are low because they are inhibited by high TH. The cause of Graves' disease is unknown; however, there appears to be a genetic predisposition to autoimmune disease. Women in their 20s and 30s are most often diagnosed, although the disease may start during the teen years.
Nodular goiter is an increase in the size of the thyroid gland caused by increased demand for thyroid hormone. Increased demand for thyroid hormone occurs during periods of growth or excess metabolic demand such as puberty or pregnancy. In these cases, increased TH is caused by metabolically driven activation of the hypothalamus, and therefore is accompanied by increased TRH and TSH. When the demand for thyroid hormone is lessened, the thyroid gland usually returns to its previous size. Occasionally, irreversible changes may have occurred and the gland does not regress. The enlarged thyroid may continue to produce excess TH. If the individual remains hyperthyroid, the condition is referred to as a toxic nodular goiter. Pituitary adenomas of TSH-producing cells or hypothalamic diseases rarely occur.
Clinical Manifestations
Increased heart rate.
Increased muscle tone, tremors, irritability, increased sensitivity to catecholamines.
Increased basal metabolic rate and heat production, intolerance to heat, excess sweating.
Weight loss, increased hunger.
A staring appearance.
Exophthalmos (bulging of the eyes) may develop.
Increased number of bowel movements.
Goiter (usually), which is an increase in the size of the thyroid gland.
Changes in skin and hair condition may occur.
Reproductive irregularities.
P.266
Diagnostic Tools
A good history and physical examination will help diagnose hyperthyroidism.
Blood tests measuring levels of TH (both T3 and T4), TSH, and TRH will allow diagnosis of the condition and localization of the problem at the level of the CNS or the thyroid gland.
Decreased serum lipids may accompany hyperthyroidism.
Decreased sensitivity to insulin, which may result in hyperglycemia.
Complications
Arrhythmias are common in patients with hyperthyroidism and may be the presenting symptom of the disorder. Any person complaining of arrhythmia should be evaluated for thyroid disorder.
A life-threatening complication of hyperthyroidism is thyrotoxic crisis (thyroid storm), which may develop spontaneously in patients with hyperthyroidism undergoing therapy or during surgery on the thyroid gland, or may occur in undiagnosed patients with hyperthyroidism. The result is a large burst of TH release that causes tachycardia, agitation, tremors, hyperthermia (up to 106 F), and, if untreated, death.
Treatment
Treatment depends on the site and cause of hyperthyroidism.
If the problem is at the level of the thyroid gland, treatment usually involves anti-thyroid drugs that block TH production or beta-blocking drugs to decrease sympathetic hyper-responsiveness. Drugs that destroy thyroid tissue also may be used. For instance, radioactive iodine (I131) administered in oral form is actively taken up by hyperactive thyroid cells. Once incorporated, I131 destroys the cells. This is a permanent treatment for hyperthyroidism and frequently results in the individual becoming hypothyroid and requiring lifelong TH replacement.
Partial or total thyroidectomy may be a treatment choice. Total thyroidectomy results in hypothyroidism, as may partial thyroidectomy.
Percutaneous ethanol injection of the thyroid is used for patients with benign thyroid nodule and those with increased surgical risk due to cardiac or pulmonary diseases, advanced age, multimorbidity, or dialysis.
P.267
Adrenal Insufficiency
Adrenal insufficiency is a decrease in the circulating level of the glucocorticoids. The mineralocorticoid aldosterone may also be reduced. Adrenal insufficiency may be caused by dysfunction of the adrenal gland, called primary adrenal insufficiency, or by dysfunction of the pituitary or hypothalamus. Both of these latter conditions cause secondary adrenal insufficiency.
Primary adrenal insufficiency is characterized by low levels of glucocorticoids, especially cortisol, accompanied by high ACTH and high CRH because there is no negative feedback on their release. Adrenal androgens and aldosterone levels may be normal, increased, or decreased depending on the cause of the glucocorticoid deficiency.
If the entire adrenal gland is destroyed or malfunctioning, adrenal androgens and aldosterone will be low. If only the glucocorticoid-secreting cells are malfunctioning, the high ACTH levels that accompany primary adrenal insufficiency will cause high levels of circulating adrenal androgens. Aldosterone secretion is primarily determined by the renin-angiotensin system, but may be slightly increased by elevated ACTH.
If the cause of adrenal insufficiency is secondary to a pituitary dysfunction, low glucocorticoids will be accompanied by low ACTH and high CRH. In this case, adrenal androgens will also be low. If there is zero ACTH, aldosterone levels will be reduced. If adrenal insufficiency is caused by a hypothalamus malfunction, the glucocorticoids, ACTH, and CRH will be low.
Diseases of Adrenal Insufficiency
Primary adrenal insufficiency, called Addison's disease, occurs from destruction of the adrenal cortex. The disease is usually autoimmune, and results from IgG antibodies directed against all or part of the adrenal gland. Addison's disease also may result from infections such as tuberculosis. Tuberculosis of the adrenal gland is a common cause of adrenal insufficiency in developing countries and does not typically resolve with treatment of the infection. Destructive adrenal gland tumors also may lead to adrenal insufficiency.
Addison's disease is characterized by low glucocorticoid levels accompanied by high ACTH and high CRH. Total loss of the adrenal gland results in the loss of adrenal androgens and aldosterone as well. Aldosterone deficiency leads to increased loss of sodium in the urine, resulting in hyponatremia (decreased sodium in the blood), dehydration, and hypotension (because water loss in the urine frequently accompanies the loss of sodium). Decreased potassium excretion in the urine will lead to hyperkalemia (increased potassium concentration in the blood).
Secondary adrenal insufficiency can occur as a result of hypopituitarism or hypothalamic dysfunction. With secondary adrenal insufficiency, ACTH is not released, so the adrenals do not secrete glucocorticoids or androgens. Aldosterone synthesis may also be affected.
P.268
Secondary adrenal insufficiency can occur if cortisol is used therapeutically for anti-inflammatory purposes. When one takes pharmacologic levels of corticosteroids, the pituitary secretion of ACTH is inhibited in a negative feedback manner. If the prescribed medication is abruptly discontinued, the pituitary remains in a refractory period and does not secrete ACTH for an extended period of time. Even a few weeks of oral glucocorticoid therapy can result in the suppression of ACTH, and hence secondary adrenal insufficiency, for several months. Judicious treatment of inflammatory illnesses with glucocorticoids for fewer than approximately 10 days will not result in pituitary suppression.
Clinical Manifestations
Depression, because cortisol levels influence mood and emotions.
Fatigue, related to hypoglycemia, and decreased gluconeogenesis.
Anorexia, vomiting, diarrhea, and nausea.
Hyperpigmentation of the skin if ACTH levels are high (primary adrenal insufficiency) as a result of ACTH having melanin-stimulating hormone like effects on the skin.
Sparse body hair in women, if the adrenal cells producing androgens are destroyed or if ACTH levels are very low.
Inability to respond to stressful situations, perhaps leading to severe hypotension and shock.
Diagnostic Tools
A good history and physical examination will help diagnose glucocorticoid deficiency.
Blood tests measuring levels of CRH, ACTH, and different glucocorticoids will allow diagnosis of the condition and localization of the problem at the level of the CNS or adrenal gland.
Hyponatremia, hyperkalemia, and hypotension may be present if the adrenal cells that produce aldosterone are destroyed or if ACTH levels are undetectable.
Complications
Adrenal crisis may occur after physical or mental stress in an affected individual. This can be life-threatening and is characterized by volume depletion, hypotension, and vascular collapse.
Treatment
Glucocorticoid replacement such as the use of hydrocortisone or cortisone acetate is required.
Health providers should monitor the history of glucocorticoid dose adjustments; potential adverse events including any crisis since last visit; the individual's ability to cope with daily stressors; the individual's body weight; and signs that suggest over-replacement or under-replacement.
Monitoring blood pressure, peripheral edema, serum sodium, serum potassium, and plasma renin activity provides clues to treatment efficacy.
Aldosterone replacement (only in primary adrenal insufficiency) may be necessary.
Glucocorticoid administration may need to be increased during periods of stress, including infection, trauma, and surgery. Morbidity and mortality are high without treatment.
If the cause of adrenal insufficiency is related to a pituitary tumor, it may be treated with chemotherapy, radiation, or surgery.
P.269
Glucocorticoid Excess
Glucocorticoid excess refers to any condition in which there are very high levels of circulating glucocorticoids. The cause of glucocorticoid excess may reside at the level of the adrenal gland or at the pituitary/hypothalamic level. If the cause of glucocorticoid excess is primary adrenal gland hypersecretion, there is usually an adrenal tumor present. In this situation, low ACTH and low CRH levels will be present as a result of negative feedback from high glucocorticoids. Adrenal androgen levels will be low because ACTH is low. Bronzing of the skin will not occur.
If glucocorticoid excess results from an adenoma of the pituitary cells producing ACTH, elevated ACTH also will cause excess adrenal androgen production. Bronzing of the skin will occur because of crossover effects between ACTH and melanin-stimulating hormone. CRH levels will be low as a result of negative feedback from ACTH and the glucocorticoids.
Excess ACTH also may occur as a result of the production of ACTH by a tumor outside of the pituitary or hypothalamus. This is referred to as an ectopic (abnormal) source of ACTH. Many tumors demonstrate ectopic production of ACTH, especially lung tumors. Excess adrenal androgens and skin bronzing will accompany ACTH-secreting tumors.
High levels of glucocorticoids also may result from chronic administration of high-dose corticosteroids, especially cortisol, for treatment of inflammatory conditions. Disease states in which long-term administration of corticosteroids occurs include asthma and several different autoimmune diseases.
Diseases of Excess Glucocorticoids
Cushing's syndrome refers to any condition of high glucocorticoids and includes glucocorticoid excess caused by therapeutic administration of corticosteroids.
Cushing's disease refers to high glucocorticoids caused specifically by malfunction of the anterior pituitary resulting in excess ACTH.
Clinical Manifestations
Altered fat metabolism leading to fat pads on the back (subclavian buffalo hump), moon face, protruding abdomen with thin extremities,
P.270
and stretch marks on breasts, thighs, and abdominal surface. Muscle weakness from protein breakdown.Hypertension as a result of increased catecholamine responsiveness.
Weight gain resulting from strong appetite stimulation. Because of effects on hepatic gluconeogenesis, a reversible form of diabetes mellitus may result.
Inhibition of immune and inflammatory reactions, leading to poor wound healing.
Extreme emotional swings (lability), sometimes causing psychosis and occasionally resulting in suicide.
Masculinization of women and children as a result of adrenal androgen stimulation if ACTH levels are high.
Bronzing of the skin if ACTH levels are high.
Diagnostic Tools
A good history and physical examination will help diagnose glucocorticoid excess.
Blood tests measuring levels of CRH, ACTH, and different glucocorticoids will allow diagnosis of the condition and localization of the problem at the level of the CNS or adrenal gland.
Loss of normal diurnal (morning) pattern of cortisol release.
Hyperglycemia, hypernatremia and hypokalemia may be present because of aldosterone-like properties of the glucocorticoids. This can contribute to hypertension and cardiac and neural irregularities.
A dexamethasone challenge test is commonly used in clinical practice to evaluate states of glucocorticoid excess. In healthy individuals, a low dose of dexamethasone will suppress ACTH secretion; in those with Cushing's syndrome, suppression does not occur.
Complications
There are many complications of excess glucocorticoid levels. Morbidity and mortality are high without treatment and approximately 50% of individuals die within 5 years. Causes of death include suicide, overwhelming infections, and coronary artery disease from severe hypertension.
Insulin resistance and hypertension may develop in those with glucocorticoid excess. These may be due to abnormal changes in hepatic fatty acid metabolism.
Treatment
Correction of high glucocorticoid levels depends on the cause of the problem.
Surgery for tumors of the adrenal, pituitary, or other tissue (e.g., the lung) is frequently performed. Radiation therapy is done if a tumor is present.
Drugs that block steroid synthesis may be used if the tumor is inoperable.
Discontinue corticosteroid therapy, by weaning down, if syndrome is caused by medication.
P.271
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia is the total or relative unresponsiveness of the adrenal glucocorticoid-producing cells to ACTH during gestation, resulting in very low levels of glucocorticoids and masculinization of the genitalia in a female fetus. Female masculinization occurs because the androgen-producing cells of the adrenal gland respond to the continuously high levels of ACTH resulting from little or no negative feedback on ACTH release. Congenital adrenal hyperplasia occurs as a result of an autosomal-recessive genetic alteration whereby there is a deficiency in one or more of the five enzymes needed to produce cortisol. Mineralocorticoid production (aldosterone) may be affected. The most common enzyme deficiencies are of 21-hydroxylase or 11 -hydroxylase. Enzyme deficiencies may be partial or total. Carriers of the disorder do not appear to be affected.
Clinical Manifestations
Masculinization of the female infant is apparent at birth and may include ambiguous genitalia with an enlarged clitoris, fused labia, and malformation of the urogenital area. The degree of abnormality is variable. Male fetuses are usually normal at birth or have slightly enlarged genitalia.
If aldosterone production is blocked, salt wasting, dehydration, vomiting, hyperkalemia, and hypotension develop. In the rare case of enzyme 11-deoxycorticosterone deficiency, mineralocorticoid levels increase, resulting in salt retention, hypokalemia, and hypertension.
Diagnostic Tools
Physical examination at or soon after birth will help diagnose the condition in females.
Blood tests will demonstrate enzyme deficiency in either sex.
Complications
Because cortisol is essential to surviving even relatively minor stresses, illnesses or surgeries in the newborn period may be fatal if the diagnosis of congenital adrenal hyperplasia has not been made.
Psychological distress to the child and family may result from delayed diagnosis.
Treatment
Cortisol, and possibly aldosterone, replacement therapy will be required throughout life. Therapy must be monitored and adjusted appropriately for growth and in times of excess physical stress.
Masculinized females may require reconstructive surgery. With successful treatment, sexual functioning and fertility will be unaffected.
P.272
Growth Hormone Deficiency
Growth hormone deficiency is a decrease in circulating levels of GH. Most cells of the body will be affected. GH deficiency is usually clinically recognized only in children. Usually, growth hormone deficiency is caused by a pituitary adenoma of another anterior pituitary hormone producing cell type. It can also be a result of hypoxic necrosis (death caused by lack of oxygen) and inflammation of the pituitary. The cause of GH deficiency may also be at the hypothalamic level, resulting from malnutrition, sleep deprivation, or stimulation of somatostatin released during periods of prolonged physical or emotional stress. For example, some studies suggest that growth potential may be reduced in adolescent female athletes as a result of intense physical exercise and reduced nutritional intake caused by dieting. Low estrogen levels are frequently seen in female athletes, which may also affect growth. GH deficiency also may result from genetic abnormality, from defects of the brain present congenitally or following infection or trauma, or from cranial irradiation used in treatment for a brain tumor or for leukemia prophylaxis.
Diseases of Growth Hormone Deficiency
Dwarfism.
A reduction of growth potential may occur in children.
Alteration in metabolic functioning, including insulin resistance and abnormal lipid profile, may occur in children and adults.
Clinical Manifestations
In children, GH deficiency results in proportional short stature (below the third percentile for their age). Affected children have decreased muscle mass and increased subcutaneous fat stores. They are typically bright mentally.
Short stature different from predicted based on familial patterns may be observed if a reduction in growth potential occurs.
Delayed onset of puberty may accompany GH deficiency, especially if abnormalities in the gonadotropins occur concomitantly.
Adult-onset GH deficiency may result in nonspecific changes in functioning, including alterations in physical and mental well-being, cardiac function and metabolic parameters.
Adults with GH deficiency may experience lower levels of energy and libido.
Diagnostic Tools
A good history and physical examination will help diagnose growth hormone deficiency.
Blood tests measuring decreased levels of GH will support diagnosis of the condition.
Neuroimaging tests to identify pituitary tumors can improve diagnosis.
Lack of responsiveness to GH provocation will assist in confirming GH deficiency.
P.273
Treatment
Treatment of GH deficiency in children involves subcutaneous injections of recombinant GH several times per week during the pubertal years or earlier. Success is greater in children treated early.
GH deficiency in adults may also be treated with GH injections.
![]() ediatric Consideration
ediatric Consideration
Most children with short stature do not have an endocrine or genetic abnormality, but rather have a normal genetic predisposition to be short (i.e., short parents). However, any child who presents with short stature should be carefully assessed to rule out underlying illnesses, including renal, cardiac, or gastrointestinal disease, and certain genetic abnormalities such as Turner's syndrome. Medications should be assessed, since, for example, oral steroids used to treat chronic asthma and medications used to treat attention-deficit disorder have been reported to reduce growth. The term constitutional short stature is used to describe children who are shorter than others of their age, and those growing at a reduced pace without a known cause. Under certain circumstances, treatment with exogenous GH may be used to increase final growth in this population as well. In general, GH treatment appears to be safe for most children without other contraindications. Patients who have growth hormone insensitivity syndrome (e.g., receptor defects) do not respond to exogenous growth hormone treatments.
Growth Hormone Excess
Growth hormone excess is the increase in circulating levels of GH. Increased levels of GH result in increased somatomedin levels and increased growth of bone, cartilage, and other tissues. Direct effects of GH on the breakdown of carbohydrates and on protein synthesis also occur. Growth hormone excess is usually caused by a GH-secreting tumor of the anterior pituitary.
Diseases of GH Excess
Gigantism, a disease of excess longitudinal growth of the bones of the skeleton, is seen as a result of GH excess before puberty.
Acromegaly, a disease of connective tissue proliferation, is seen in adults with GH excess. Because long bone growth has stopped in adults, GH excess cannot cause growth of the skeleton. It is associated with growth of the cartilage of the hands, feet, nose, jaw, chin, and facial bones. Connective tissue proliferation of internal organs, including the heart, also occurs.
P.274
Clinical Manifestations
Tall stature with gigantism.
Thickening of the fingers, jaw, forehead, hands, and feet with acromegaly.
Because GH excess is usually caused by an aggressively growing adenoma, other hormone-secreting cells of the anterior pituitary are frequently destroyed. Thus, symptoms of GH excess often include those associated with deficiencies of other hormones. For example, if the growing tumor displaces the gonadotropin-secreting cells of the anterior pituitary, decreased reproductive function may occur. If the tumor affects any other hormone-producing cells, manifestations particular to that hormone will prevail. Increased intracranial pressure can also occur with a growing tumor. Symptoms include headache, vomiting, and papilledema (swelling where the optic nerve enters the eye chamber).
Diagnostic Tools
A good history and physical examination will help diagnose growth hormone excess.
Blood tests measuring increased levels of GH will support diagnosis of gigantism or acromegaly.
Increased blood glucose levels may be present with either condition.
The secretory pattern of GH release is no longer predictable and is unrelated to sleep with either condition.
Complications
Complications of acromegaly include cardiac hypertrophy and hypertension. Diabetes mellitus can occur from the tendency of GH to increase blood glucose and decrease cellular insulin sensitivity.
Treatment
Treatment of GH excess is usually by surgical excision of the GH-secreting tumor. Radiation therapy may also be applied.
Bromocriptine, a dopamine antagonist, may be effective in decreasing GH levels.
![]() ediatric Consideration
ediatric Consideration
Elevated growth in children may occur as a result of a normal genetic predisposition (i.e., tall parents) or may accompany certain genetic abnormalities such as Marfan's syndrome and Klinefelter's syndrome. The term constitutional tall stature is used to describe children who are taller than others their age, and those growing at an accelerated velocity. Occasionally, treatment may involve the judicial administration of sex hormones (birth control pills in girls) to retard excess growth.
P.275
Gonadotropin Deficiency
Gonadotropin deficiency is a decrease in circulating levels of FSH and LH. Gonadotropin deficiency is usually caused by pressure exerted on the gonodotropin-producing cells by a pituitary tumor of another hormone-producing cell type. Oversecretion of the target gland hormones estrogen, progesterone, or testosterone can also act in a negative feedback manner to cause gonadotropin deficiency. Prolactin is known to inhibit pituitary secretion of the gonadotropins, and prolactin-secreting tumors can cause gonadotropin deficiency. Finally, the hypothalamus may decrease its secretion of gonadotropin-releasing hormones under periods of physical stress, obesity, starvation, or emotional trauma.
Clinical Manifestations
Amenorrhea (lack of menstrual periods), vaginal, uterine, and breast atrophy in women.
Testicular atrophy and reduction in beard growth in men.
Patients with hypogonadotropic hypogonadism manifest decreased testosterone levels and interruption of spermatogenesis.
Diagnostic Tools
Blood tests measuring the levels of estrogen, testosterone, and the gonadotropins will allow diagnosis of the condition and localization of the problem at the level of the CNS or the ovary or testicle.
Treatment
Surgery if a tumor is present.
Gonadotropin, estrogen, or testosterone replacement may be considered.
Stress reduction, weight gain, or weight loss.
Hypoprolactemia
Hypoprolactemia is a decrease in circulating levels of prolactin. Hypoprolactemia may occur as a result of hypothalamic dysfunction leading to increased release of prolactin-inhibiting hormone. It may also occur because of dysfunction of the prolactin-secreting cells of the pituitary. Dysfunction of pituitary cells may be caused by increased pressure from a pituitary tumor of another cell type. More commonly, hypoprolactemia is diagnosed after an episode of pituitary ischemia and necrosis.
Diseases of Hypoprolactemia
Sheehan's syndrome is a condition of hypopituitarism resulting from an intrapartum or postpartum hemorrhage (during or after delivery of an infant). With a significant loss of blood volume during the birth process, blood flow to the anterior pituitary may be reduced. Complicating the problem further is
P.276
that during pregnancy the anterior pituitary grows and becomes very active metabolically. This is especially true for cells that produce prolactin, TSH, and GH. The result is a very high oxygen demand. In addition, anterior pituitary blood flow is venous blood coming from the hypothalamus through the hypothalamic-pituitary portal system, and is therefore relatively deoxygenated. Thus, the anterior pituitary is particularly susceptible to ischemic damage with a birth hemorrhage. Sheehan's syndrome may manifest after delivery of the infant when the woman experiences an inability to breastfeed. Other pituitary hormones may also be deficient.
Clinical Manifestations
Inability to breastfeed in women.
In Sheehan's syndrome, other symptoms will depend on which hormone-producing cells were affected by the ischemia.
Diagnostic Tools
Blood tests measuring decreased levels of prolactin will allow diagnosis of the condition.
Treatment
Treatment is related to needs of the individual and may involve hormone replacement therapy.
Hyperprolactemia
Hyperprolactemia is an increase in circulating levels of prolactin. Hyperprolactemia may be caused by a decrease in secretion of prolactin-inhibiting hormone by the hypothalamus, or as a result of a prolactin-secreting tumor of the pituitary (prolactinoma). Certain phenothiazine drugs, used to treat psychosis, sometimes cause hyperprolactemia, probably by affecting the hypothalamus. It may also occur with pregnancy and during hypothyroidism.
Clinical Manifestations
Infertility, hypogonadism, anovulation, and amenorrhea in women as a result of prolactin-mediated decreases in LH or FSH secretion by the pituitary. This may result in osteopenia.
Galactorrhea (lactation not associated with childbirth or nursing) may develop.
No clinical signs are apparent in men.
Diagnostic Tools
Blood tests measuring the increased level of prolactin will allow diagnosis of the condition.
Imaging of the sella turcica may provide evidence of tumor.
P.277
Treatment
A prolactin-secreting tumor may be surgically resected.
If the condition is drug related and the patient is concerned about her reproductive status, further use of the drug should be evaluated.
Dopamine agonists (cabergoline and bromocriptine) to inhibit prolactin secretion may be prescribed.
Syndrome of Inappropriate Antidiuretic Hormone (ADH)
Syndrome of inappropriate ADH (SIADH) is characterized by increased release of ADH from the posterior pituitary in the absence of normal stimuli for ADH release. Increased ADH release usually occurs in response to increased plasma osmolality (a decrease in plasma water concentration) or, to a lesser extent, decreased blood pressure. With SIADH, ADH is high in the face of low plasma osmolality. Plasma osmolality continues to decrease because of ADH stimulating water reabsorption by the kidneys. Release of ADH continues without feedback control, in spite of low osmolality and increased blood volume.
SIADH is most commonly induced by drugs. Other causes of SIADH include disease, injury, or tumors of the CNS, pain, stress, and temperature extremes. Surgery may result in a transient occurrence of SIADH. Tumors outside the CNS, especially bronchogenic carcinomas, frequently produce ADH ectopically.
Clinical Manifestations
Water retention and weight gain.
Decreased urinary output.
Nausea and vomiting worsening with the degree of water intoxication.
Diagnostic Tools
Blood tests measuring increased levels of ADH with decreased plasma osmolality and hyponatremia (decreased sodium concentration, mild: serum sodium decreased to 130 mEq/L; severe: serum sodium below 126 mEq/L) will allow diagnosis of the condition.
Complications
Neurologic symptoms may range from headache and confusion to muscle twitching, seizures, coma, and death as a result of hyponatremia and water intoxication.
Treatment
For mild cases, fluid restriction is adequate to control symptoms until the syndrome spontaneously regresses.
If the condition is more severe, diuretics and drugs that block ADH action on the collecting tubules will be administered. A hypertonic solution of sodium chloride may occasionally be used to increase plasma sodium concentration.
If ADH is arising from ectopic tumor production, treatment will be aimed at eliminating the tumor.
P.278
Diabetes Insipidus
Diabetes insipidus is a disease of decreased ADH production, secretion, or function. The term diabetes insipidus refers to the quantity and quality of the urine: the disease is associated with copious amounts of dull, or tasteless, urine. Without ADH, the renal-collecting tubules cannot reabsorb water and cannot concentrate the urine. Diabetes insipidus may result from a partial or total lack of ADH production by the hypothalamus, or decreased release of ADH from the posterior pituitary. These deficits may result from a tumor or head injury. Diabetes insipidus may also result from the kidney not responding to circulating ADH because of a receptor or second messenger deficit. This type of diabetes insipidus is called nephrogenic, that is, originating in the kidney. Causes of nephrogenic diabetes insipidus include a genetic, X-linked recessive trait, kidney disease, hypokalemia, and hypercalcemia.
Clinical Manifestations
Large volumes of dilute urine.
Polydipsia (excessive thirst).
Diagnostic Tools
Blood tests measuring decreased levels of ADH with increased plasma osmolality and hypernatremia will allow diagnosis of the condition.
Complications
Severe dehydration may occur if large volumes of drinking water are unavailable.
Treatment
Drugs are available that mimic the action of ADH. The most commonly used drug of this category, desmopressin, was previously offered only as a nasal spray for home use but is now available in pill form.
For nephrogenic diabetes insipidus, thiazide diuretics are administered. These seem to work by decreasing glomerular filtration rate, allowing increased amounts of fluid to be reabsorbed at the proximal, rather than collecting, tubule.
P.279
Selected Bibliography
Arlt, W., & Allolio, B. (2003). Adrenal insufficiency. Lancet 361, 1881 1893.
Arlt, W., Walker, E.A., Draper, N., Ivison, H.E., Ride, J.P., Hammer, F., et al. (2004). Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet 363, 2128 2135.
Cameron, D.R., & Braunstein, G.D. (2004) Androgen replacement therapy in women. Fertility and Sterility 82, 273 289.
Dattani, M., & Preece, M. (2004). Growth hormone deficiency and related disorders: insights into causation, diagnosis, and treatment. Lancet 363, 1977 1987.
Dubey, P., Raymond, G.V., Moser, A.B., Kharkar, S., Bezman, L., & Moser, H.W. (2005). Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. Journal of Pediatrics 146, 528 532.
Ehrmann, D.A. (2005). Polycystic ovary syndrome. (2005). New England Journal of Medicine 352, 1223 1236.
Hersh, A.L., Stefanick, M.L., & Stafford, R.S. (2004). National use of postmenopausal hormone therapy. Annual trends and response to recent evidence. JAMA 291, 47 53.
Holmberg, L., & Anderson, H. (2004). HABITS (Hormonal replacement after breast cancer - Is it safe?), a randomized comparison: Trial stopped. Lancet 363, 453 455.
Kovacs, K. (2003). Sheehan syndrome. Lancet 361, 520 522.
Laurberg, P., Andersen, S., Pedersen, I.B., & Carle, A. (2005). Hypothyroidism in the elderly: pathophysiology, diagnosis and treatment. Drugs and Aging 22, 23 38.
Marcus, B.J., & Collins, K.A. (2004). Childhood panhypopituitarism presenting as child abuse: A case report and review of literature. American Journal of Forensic Medicine and Pathology 25, 265 269.
Miehle, K., & Paschke, R. (2003). Therapy of hyperthyroidism. Experimental and Clinical Endocrinology and Diabetes 111, 305 318.
Minhtri, K., & Nguyen, I.K. (2005). An analysis of current quantitative approaches to the treatment of severe symptomatic SIADH with intravenous saline therapy. Clinical Experimental Nephrology 9, 1 4.
Roberts, C.G.P., & Ladenson, P.W. (2004). Hypothyroidism. Lancet 363, 793 803.
Samuels, M.H. (2004). Sheehan's syndrome. Endocrinologist 14, 25 30.
Semenkovich, C.F. (2004). Fatty acid metabolism and vascular disease. Trends in Cardiovascular Medicine 14, 72 76.
EAN: 2147483647
Pages: 26