7. Neuroendocrine - Immune Interaction
Authors: Corwin, Elizabeth J.
Title: Handbook of Pathophysiology, 3rd Edition
Copyright 2008 Lippincott Williams & Wilkins
> Table of Contents > Unit II - Effective and Ineffective Health Protection > Chapter 7 - Neuroendocrine-Immune Interaction
Chapter 7
Neuroendocrine-Immune Interaction
Joseph Cannon
A rapid decline in health or even the death of an elderly person after the loss of a spouse is a familiar situation. Many younger individuals also have noticed that their own resistance to infectious illness seems particularly labile during stressful periods. Recognizing the harmful effects of stress, caregivers intuitively strive to provide a supportive, stress-free environment for patients. The mechanistic explanations for these real-life empirical observations can be found in the anatomic and functional connections between the central nervous system, the endocrine system, and the immune system. This field of research is sometimes called psychoneuroimmunology.
The exact biologic mechanisms that tie the central nervous, endocrine, and immune systems together continue to be clarified. One common stimulus that appears to integrate these systems is stress; the more intransigent aspect of this research may be understanding stress itself. The difficulty in reaching an understanding of stress is related to its subjective and inconsistent nature. For example, common experience has shown that conditions perceived as oppressive by some have no effect on others. Likewise, in the laboratory, exposing mice to inescapable shock causes suppression of certain immune parameters. However, if the mice have a means to escape the shock, they do not become immunosuppressed, even if the duration of exposure to the shock and the shock frequency is kept constant. Thus, stress is subjective. Providing the means for a patient to avoid or to escape stress may represent the potential for therapy and for improved care giving.
P.174
Physiologic Concepts
Neuroendocrine Control of the Immune System
Controlled experiments have shown that conditioned (Pavlovian) immune responses could be induced in animals by pairing the administration of an immunosuppressive drug (cyclophosphamide) with a neutral conditioning stimulus (saccharine in the drinking water). After sufficient paired conditioning, rats given the saccharine alone developed significantly reduced antibody titers when exposed to antigen, compared with unconditioned animals. This indicated a psychological influence on immune function unrelated to an infectious or inflammatory condition, thereby suggesting a neural or an endocrine effect.
An effect on the immune system by neural or endocrine activation also has been shown in controlled human studies. Psychological stress has been associated with suppression of certain immune parameters. For example, the ability of lymphocytes to respond to mitogens (substances that initiate cell division) was depressed in blood samples taken from students just before oral fellowship exams. Lymphocyte responsiveness returned to normal several weeks after the examinations.
Neuronal Connections to the Immune System
If immune function is controlled by the brain, then interrupting (lesioning; i.e., cutting or damaging) neuronal pathways should disrupt normal immune function. Experiments have proved this. For example, lesioning the preoptic anterior hypothalamus (the structure that controls body temperature, eating, sleeping, and other vegetative functions) reduces the number of leukocytes in the spleen and thymus, reduces natural killer-cell function, and suppresses antibody production. Lesions to the limbic system (which is involved in emotion and motivation) have the opposite effect on the spleen and thymus. Interestingly, lesions to the cerebral cortex exhibit lateralized influences: lesions on one side of the cortex increase T-cell number and function in the spleen and thymus, and lesions on the other side have the opposite effect.
Electron microscopy studies have confirmed that the secondary lymphoid tissues (thymus, spleen, lymph nodes) are innervated by the autonomic nervous system. In addition, all the elements necessary for synaptic signal transmission have been identified at the interface between nerve terminals and leukocytes in these tissues. Several neurotransmitters have been identified in the neurons innervating lymphoid tissue. Corresponding receptors for these neurotransmitters have been found on leukocytes, and leukocyte function is altered in vitro in response to these substances. As with any synapse, a mechanism needs to be in place to break down the neurotransmitters and thus terminate the individual signal; leukocytes possess appropriate enzymes to accomplish this. In addition, neurons have cytokine receptors (chemicals released by leukocytes) that make them susceptible to
P.175
feedback control by leukocytes. And finally, leukocytes themselves make neuropeptides that can presynaptically influence neuronal function and signaling, further emphasizing the feedback loop. Examples of neurotransmitters that bind to immune tissue and their effects include:
Substance P: Increases T-cell proliferation, B-cell antibody synthesis, the production of cytokines (including interleukin-1 and tumor necrosis factor) and reactive oxygen species by monocytes and macrophages, and neutrophil chemotaxis and phagocytosis. Substance P also increases histamine release from mast cells.
Somatostatin: Inhibits lymphocyte proliferation and antibody production.
Norepinephrine: Enhances lymphocyte proliferation (by means of alpha-adrenergic receptors) when used in low concentrations; diminishes proliferation (by means of beta-adrenergic receptors) when used in high concentrations.
Circulating epinephrine: Mobilizes preformed neutrophils and lymphocytes from storage depots (existing in the bone marrow and loosely adhering to the walls of large veins). In addition, low- or high-dose epinephrine alternately stimulates or inhibits monocyte function.
Acetylcholine: Inhibits macrophage function.
Cytokine Connections
Another critical linkage between the neuroendocrine and the immune systems involves cytokines, the chemical mediators of inflammation described in Chapter 5. Neuroendocrine signals can influence cytokine synthesis; conversely, cytokines can alter neuronal function and endocrine secretion. Since cytokines control the proliferative, cytotoxic, and synthetic activities of all the different leukocyte populations, neuroendocrine modification of cytokine synthesis or cytokine receptor expression can influence immune function more profoundly than can direct neuroendocrine action on individual leukocytes. Cytokines, in turn, modify neuroendocrine function (such as the hypothalamic-pituitary-adrenal axis) during times of infection or stress.
Hypothalamic-Pituitary-Adrenal Axis
The effector mechanisms that fight infection (proteolytic enzymes, reactive oxygen species, membrane-disrupting factors) are very destructive, and if these mechanisms are not controlled, they cause damage to a host's own cells. Cortisol, a hormone released from the adrenal gland, has pervasive immunomodulatory influences that provide this essential control at many levels. Cortisol is released both with stress and as part of the normal feedback control of immunologic and inflammatory processes. High physiologic and pharmacologic concentrations of cortisol keep the immune system under control by:
Directly inhibiting macrophage phagocytosis, reactive oxygen species generation, and proteolytic enzyme release;
Indirectly inhibiting reactive oxygen species production, proteolytic enzyme release and other cytotoxic mechanisms by suppressing production of the proinflammatory cytokines interleukin-1 and tumor necrosis factor;
Inhibiting antibody synthesis and lymphocyte proliferation;
Down-regulating expression of substance P receptors;
Stimulating expression of enzymes that break down substance P and other neuropeptides;
Stimulating beta-adrenergic receptor expression.
P.176
Because of its ability to control so many aspects of inflammation, synthetic preparations of cortisol often are given clinically to reduce inflammation.
The connection between infection and stress and the release of cortisol involves a feedback loop between the hypothalamus, the pituitary gland, and the adrenal gland. This cycle is shown in Figure 7-1. Infectious microorganisms or antigens stimulate secretion of interleukin-1 and other cytokines from macrophages. These cytokines travel to the hypothalamus and stimulate the release of corticotropin-releasing hormone (CRH). From here, the normal endocrine circuit begins with successive release of adrenocorticotropic hormone (ACTH) from the pituitary and cortisol secretion by the adrenal cortex. Cortisol then feeds back on the macrophages and inhibits further cytokine release, completing the loop. As such, cortisol is an integral part of a normally functioning immune system. If this cytokine-cortisol loop is disrupted by adrenal cortical insufficiency or the actions of drugs, then exaggerated and damaging inflammatory responses
P.177
can occur. This endocrine loop is vitally important but relatively slow (taking hours to develop). In contrast, a neural control loop also exists that acts within seconds or minutes.
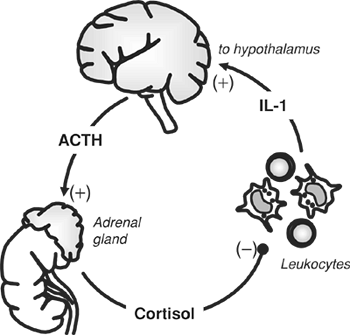 |
Figure 7-1. Hormonal feedback loop involving the hypothalamic-pituitary-adrenal axis. |
Vagal Inflammatory Reflex
The vagus is a cranial nerve containing a combination of afferent sensory fibers and efferent parasympathetic fibers. Its name comes from the Latin word for wandering, which is appropriate because its branches reach out to the heart, lungs, gastrointestinal tract, liver, spleen, and kidneys. With the exception of the heart, these organs constitute major elements of the reticuloendothelial system; i.e., they contain particularly high concentrations of phagocytic, macrophage-like cells. The sensory fibers of the vagus can detect interleukin-1 and tumor necrosis factor released locally by the reticuloendothelial cells at the site of infection. This neural information is transmitted to the brainstem, where it stimulates the centers controlling the parasympathetic nervous system (Fig. 7-2). The efferent parasympathetic postganglionic fibers (including the vagus) release acetylcholine (as described in Chapter 8), which binds to nicotinic receptors on macrophages distributed throughout the reticuloendothelial system. Binding of acetylcholine to the
P.178
macrophages inhibits the secretion of proinflammatory cytokines, such as interleukin-1 and tumor necrosis factor, but has no influence on anti-inflammatory cytokines, such as interleukin-10. Studies with animals have shown that exaggerated and damaging inflammatory responses can occur if the vagus nerve is severed.
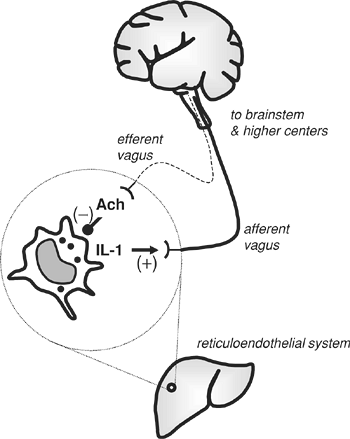 |
Figure 7-2. Neuronal feedback loop involving the vagus nerve. |
Hypothalamic-Pituitary-Gonadal Axis
Women have a more responsive immune system than men. Thus, women are more resistant to infections, but they are also more susceptible to autoimmune diseases. Androgens, estrogen, and progesterone all have profound influences on immune function, but only estrogen receptors have actually been found on lymphocyte subpopulations. There is evidence that receptors for the other steroid hormones are located on the thymic epithelium and the bone marrow stromal cells that nurture leukocyte development from stem cell precursors. In addition, progesterone may influence mature leukocytes through glucocorticoid (cortisol) receptors that are expressed on all leukocytes (Fig. 7-3).
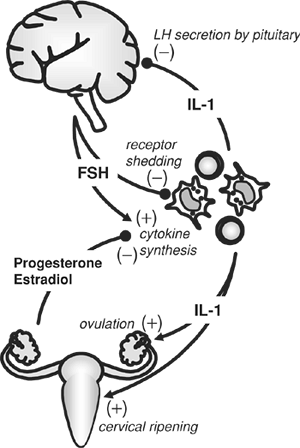 |
Figure 7-3. Reproductive-immune communication. |
P.179
Estrogen and progesterone have complicated, concentration-dependent influences on cytokine production. Low concentrations of estrogen stimulate interleukin-1 production (compared with no estrogen at all). But the increase of concentrations through the normal range experienced during a menstrual cycle leads to dose-related inhibition of interleukin-1 secretion. Interleukin-1 synthesis is stimulated in a dose-related manner through the normal menstrual range of progesterone concentrations. However, high concentrations of progesterone, such as those reached during pregnancy, are inhibitory. This inhibitory influence of progesterone on the immune system is considered vital to the development of the immune tolerance that prevents the foreign tissue of the fetus from being rejected.
Although steroid hormones have received the most attention, the pituitary hormones involved in reproduction also have significant influences on immune function. Prolactin appears to have a supportive role in lymphocyte proliferation and can counteract cortisol-mediated inhibition. Stress can decrease circulating prolactin concentrations. Follicle-stimulating hormone also may stimulate interleukin-1 production.
Pathophysiologic Concepts
Hyporesponsive and Hyperresponsive Immune Reactivity
A hyporesponsive immune system can lead to pathologic conditions because the host is unprotected from infectious microorganisms or tumors. A hyperresponsive immune system can be pathologic owing to nonspecific damage to host tissues caused by the overproduction of proteolytic enzymes and reactive oxygen species, or to specific damage to host cells triggered by autoantibodies.
Stress-Related Immunosuppression
Physiologically and emotionally stressful situations stimulate autonomic, limbic, and, possibly, cerebral cortical inputs to the hypothalamus that lead to the release of CRH and, subsequently, of cortisol. During prolonged or particularly severe stress, chronic elevations of cortisol are suspected of mediating immune dysfunction or, in some cases, immunosuppression. However, this is not at all a straightforward relationship. For example, plasma cortisol concentrations increase during exercise, but exercise also decreases the sensitivity of leukocytes to cortisol. Thus, leukocyte inhibition is not an automatic consequence of increases in cortisol concentration. Stress-related immunosuppression is more likely caused by a combination of factors (including, for example, decreased prolactin concentrations) than strictly by increased cortisol concentrations.
P.180
Age-Related Immunosuppression
Aging is associated with increased risk of cancer and reduced resistance to infectious disease. The critical lymphoid organ for T-cell development the thymus involutes (shrinks) with advancing age. This involution is characterized by reduction of the overall size of the thymus and, more importantly, by a proportionally greater loss of functional cells, with the mass replaced by inert fatty tissue. T-cell proliferation, T-killer-cell function, and T-cell enhancement of antibody synthesis all decline as well. These changes parallel age-associated declines in growth hormone. In animals, thymus involution can be reversed by growth hormone treatment (possibly in concert with other pituitary factors). These results raise hopes that hormone replacement therapy for senescent human immune systems can be devised that will improve immune surveillance against tumors and bolster resistance to infectious microorganisms.
Immune Activation and Reproductive Dysfunction
Increased concentrations of interleukin-1 in the brain can inhibit the luteinizing hormone (LH) surge, and therefore prevent ovulation. This may be an important protective mechanism that prevents conception during periods of infection or illness. On the other hand, recurrent spontaneous abortions have been associated with abnormally high uterine concentrations of cytotoxicity-promoting cytokines such as tumor necrosis factor and interferon-gamma. These cytokines are thought to help drive an inappropriately robust immunologic response that leads to rejection of the fetus as a foreign intruder.
Conditions of Disease
Autoimmune and Hyperimmune Disease
Neuroendocrine influences on the immune system are most drastically represented by diseases that have a strong gender bias or that wax and wane with stressful events. All of the disease conditions identified below are discussed in detail elsewhere in this book. A brief enumeration of them is as follows:
Asthma (Chapter 14) is a condition of airway hyperresponsiveness characterized by bronchoconstriction (wheezing) caused by mast cell release of histamine into the pulmonary tissues and by a subsequent inflammatory reaction. Although many factors are probably involved in an asthmatic response, inappropriate or exaggerated neuronal release of substance P, which stimulates mast cell degranulation, has been implicated, possibly in conjunction with insufficient production or release of another peptide, vasoactive intestinal peptide (VIP), which inhibits histamine release.
Crohn's disease and ulcerative colitis are inflammatory bowel diseases (Chapter 15). Intestinal biopsies from patients indicate a possible neurogenic component in the inflammatory process such that abnormally high substance P and substance-P receptor concentrations exist and VIP concentrations seem lower than normal. Exacerbations are stress-related.
Systemic lupus erythematosis (SLE) is an autoimmune inflammatory disease of connective tissue that can include skin rashes, joint pain, and fatigue (Chapter 5). The disease is more common in women than in men by a factor of 10:1. Androgens alleviate symptoms of SLE, and estrogen exacerbates the condition. Symptoms worsen during the luteal phase of the menstrual cycle but are not influenced to any great degree by pregnancy.
Rheumatoid arthritis (RA) is an autoimmune disorder that results in the destruction of the synovial lining of the joints (Chapter 10). RA is more common in women than in men by a factor of 3:1, but it is relieved during the luteal phase of the menstrual cycle and during pregnancy the high concentrations of progesterone at these times are thought to inhibit several of the inflammatory processes involved. Many cell types and inflammatory mediators have been implicated in this disease, but some possible neural-cytokine interactions deserve special note. Interleukin-1 and tumor necrosis factor concentrations are high in the synovial fluid of patients who have RA. These cytokines stimulate release of substance P, which in turn stimulates several proinflammatory processes, including increased production of interleukin-1 and tumor necrosis factor. Thus, a destructive positive feedback cycle is established.
P.181
Clinical Manifestations
Clinical manifestations of the above disease states are described in the chapters identified.
Diagnostic Tools
Mechanisms for diagnosis of the above disease states are described in the chapters identified.
Treatment
A better understanding of the relation between neuroendocrine factors and immune or inflammatory effectors should lead to improved therapies for these and other pathologic conditions.
Steroid hormone activity can be modulated by receptor antagonists and other drugs.
Neuropeptide release can be influenced pharmacologically as well as behaviorally by avoidance of exposure to stressors or, when exposure is unavoidable, by adoption of relaxation techniques and coping strategies. For example, people are able to use biofeedback to lower their heart
P.182
rate (through increased efferent vagal signals to the heart). Perhaps bio-feedback can also be used to increase vagal anti-inflammatory signaling to the reticuloendothelial system.Antibodies and soluble receptors against tumor necrosis factor and receptor antagonists against interleukin-1 have been used clinically to reduce the rate of tissue destruction and relieve signs and symptoms of rheumatoid arthritis, but these treatments can also increase risks for serious infection. It can be difficult to determine an optimal anti-cytokine dose that diminishes inappropriate destruction without seriously compromising host defense. Careful screening of patients for concurrent infection or use of immunosuppressive drugs including glucocorticoids seems necessary.
Selected Bibliography
Ader, R., Felton, D.L., & Cohen, N. (Eds.). (1991). Psychoneuroimmunology (2nd ed.). San Diego: Academic Press.
Baker, J.R., Jr., & Zylke, J.W. (Eds.). (1997). Primer on allergic and immunologic diseases. Special issue of the Journal of the American Medical Association 278, 1799 2034.
Berczi, I., Chow, D.A., Baral, E., & Nagy, E. (1998). Neuroimmunoregulation and cancer (review). International Journal of Oncology 13, 1049 1060.
Cannon, J.G., & St. Pierre, B.A. (1997). Gender differences in host defense mechanisms. Journal of Psychiatric Research 31, 99 113.
Dantzer, R. (2005). Somatization: A psychoneuroimmune perspective. Psychoneuroendocrinology 30, 947 952.
Dimitrov, S., Lange, T., Fehm, H.L., & Born, J. (2004). A regulatory role of prolactin, growth hormone, and corticosteroids for human T-cell production of cytokines. Brain, Behavior, and Immunity 18, 368 374.
Straub, R.H., & Besedovsky, H.O. (2003). Integrated evolutionary, immunological, and neuroendocrine framework for the pathogenesis of chronic disabling inflammatory diseases. FASEB Journal 17, 2176 2183.
Tracey, K.J. (2002). The inflammatory reflex. Nature 420, 853 859.
Tsigos, C., & Chrousos, G.P. (2002). Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. Journal of Psychosomatic Research 53, 865 871.
EAN: 2147483647
Pages: 26