8 - Intraocular Inflammation
Editors: Tasman, William; Jaeger, Edward A.
Title: Wills Eye Hospital Atlas of Clinical Ophthalmology , The, 2nd Edition
Copyright 2001 Lippincott Williams & Wilkins
> Table of Contents > Chapter 8 - Intraocular Inflammation
function show_scrollbar() {}
Chapter 8
Intraocular Inflammation
Jonathan B. Belmont
David H. Fischer
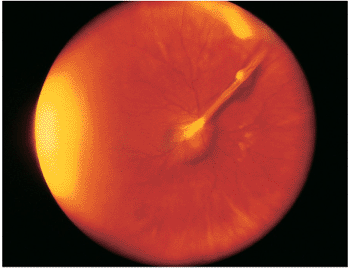 |
| Wide-angle postvitrectomy photograph of a young patient with ocular toxocariasis. The characteristic falciform fold still is evident and the three yellowish areas along the fold and peripheral to it represent areas of residual scar from the release of traction attachments during vitrectomy. There is a serous detachment around the disc. |
P.290
Uveitis is a general term denoting intraocular inflammation that either primarily or secondarily involves one (or more) components of the uveal tract. Although components of the uveal tract are comprised of the iris, ciliary body, and choroid, all layers of the eye may be involved by inflammation. Because the uveal tract is supplied by the systemic circulation, systemic diseases and local diseases that affect blood vessels can involve the uvea as well as contiguous ocular tissues and structures.
Classification of uveitis relates to the area or anatomic structure(s) involved, as well as to time course (i.e., acute, chronic, acute recurrent, and chronic recurrent), laterality (i.e., unilateral or bilateral), character of inflammation (i.e., nongranulomatous or granulomatous), and associated findings (e.g., glaucoma, cystoid macular edema, etc.). Uveitis may also be classified as either endogenous or exogenous in nature. Frequently, all of the above parameters are combined to provide a detailed and precise description of the ocular inflammation present in any given patient, a process called naming. Naming of the inflammatory process is the first step in diagnosis and management in that it allows narrowing of the differential diagnosis by providing a specific and discrete set of clinical findings to match against the known clinical findings of various uveitic entities and syndromes. For example, a patient presenting with acute, unilateral, nongranulomatous iridocyclitis would be matched with those clinical entities known to present with a compatible clinical picture, narrowing the differential diagnosis, helping to direct a diagnostic workup, and assisting in formulation of initial therapy.
From the standpoint of uveitis classification, it is also frequently helpful to think of eye inflammation by compartmentalizing the eye. The anterior segment has a specific subset of diseases that are common to that area; acute iritis and iridocyclitis are the most common forms of anterior uveitis. Most forms of anterior uveitis are characterized by episodes of pain, redness, and photophobia, and a tendency for synechiae formation. Iritis refers to inflammation of the iris. If the ciliary body is also involved, the condition is called iridocyclitis. Inflammation of the intermediate area or midzone of the eye is classified as intermediate uveitis; the pars planitis syndrome is an example. Posterior uveitis affects the deeper tissues, such as the retina and choroid; ocular toxoplasmosis and cytomegalovirus (CMV) retinitis are examples of posterior uveitis. Posterior uveitis syndromes characteristically occur without episodes of pain, redness, or photophobia, and floaters with or without blurring of vision frequently herald their onset. Choroiditis refers to inflammation primarily in the choroid. Because the choroid is contiguous with (and may involve) the retina, the condition may also be called chorioretinitis or, if the retina is the primary locus of inflammation, retinochoroiditis. Diffuse uveitis refers to intraocular inflammation that affects the front and back of the eye; conditions such as sarcoidosis can manifest in this manner. The clinical features of some diseases may fit more than one of these compartmental classifications.
The terms nongranulomatous and granulomatous are pathologic terms that denote Boyd's cellular response to an inciting agent. Lymphocytes, polymorphonuclear leukocytes, and plasma cells are found in nongranulomatous uveitis, and epithelioid and giant cells characterize granulomatous disease. A typical clinical picture is associated with each form. Nongranulomatous uveitis is usually anterior, has an acute onset, exhibits fine keratic precipitates (KPs), and often resolves with aggressive corticosteroid therapy or, sometimes, even spontaneously. The onset of granulomatous uveitis is often insidious, and the disorder frequently has a chronic course and is characterized by large mutton-fat KPs, Koeppe and Busacca nodules, and retinal vascular sheathing (Figs. 8.1, 8.2, 8.3). There is considerable overlap
P.291
in the clinical presentation of these types, but it is still valuable for the clinician to think in these terms. This method of classification suggests certain underlying causes, enabling a more focused diagnostic evaluation.
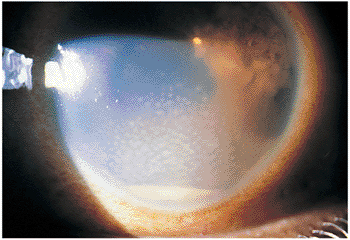 |
Figure 8.1. Anterior granulomatous uveitis in the left eye of a 26-year-old African-American man is indicated by a dependent accumulation of white cells (i.e., hypopyon), mutton-fat keratic precipitates, and a superotemporal Busacca nodule. |
The causes of uveitis include infectious agents (e.g., bacteria, protozoa, viruses, and spirochetes) and immunologic conditions in which the immune response initiates and potentiates acute and/or chronic inflammation. Underlying conditions may be related to the intraocular inflammation. Systemic diseases such as sarcoidosis or rheumatoid arthritis and infectious conditions such as syphilis or CMV infection in patients with acquired immunodeficiency syndrome (AIDS) may manifest primarily in the eye. Clinical and laboratory workups may help to elucidate the underlying problem.
In many cases, prompt and accurate diagnosis and appropriate therapeutic intervention can control or resolve inflammation and prevent the inflammatory sequelae of intraocular scarring, which may otherwise lead to complications or visual loss. Primary immune-related processes are usually treated with antiinflammatory agents. Corticosteroids are most commonly used for eye disease, and can be administered topically, in the form of periocular injections, or systemically through oral or intravenous routes. For some conditions, antiinfectious and antiinflammatory agents may be combined. The rationale is to treat the infectious and inflammatory components together to eradicate the disease promptly and reduce intraocular scarring. A biopsy of appropriate eye tissues may yield useful information in selected cases. Although uncommon, anterior chamber taps, vitreous taps, or partial-thickness eye wall biopsies may help to identify potentially irreversible, vision-threatening forms of disease.
 |
Figure 8.2. The right eye of the patient in Figure 8.1, showing typical granulomatous keratic precipitates on the posterior corneal surface inferiorly and a large iris granuloma (i.e., Busacca nodule) superiorly. Although cellular haze prevents sharp focus, synechiae formation around the pupil from Koeppe nodules completes the classic picture of granulomatous inflammation. |
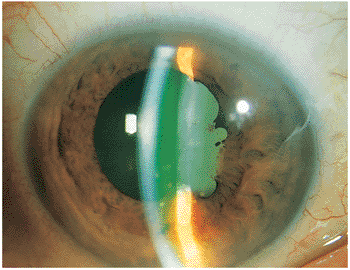 |
Figure 8.3. Slit lamp photograph demonstrating the characteristic anterior segment findings of granulomatous uveitis including keratic precipitates, posterior synechiae, and Koeppe nodules along the pupillary border. The presence of flare and cells is demonstrated by fluorescein in the anterior chamber. |
Anterior Uveitis
Acute Nongranulomatous Anterior Uveitis
Acute nongranulomatous anterior uveitis, commonly referred to as iritis or iridocyclitis, affects the front part of the eye. This form is the most common of all uveitis syndromes. Acute nongranulomatous anterior uveitis often is associated with the histocompatibility antigen HLA-B27 and linked with a predisposition for developing ankylosing spondylitis. Ninety percent of the patients who have the two diseases concomitantly are also positive for the HLA-B27 marker. Other diseases associated with this form of acute ocular inflammation include ulcerative colitis, Crohn's disease, Reiter's syndrome, psoriatic arthritis, and gastroenteropathies secondary to Yersinia and Klebsiella pneumoniae infections. These disease associations should be looked for in any individual presenting with acute nongranulomatous anterior uveitis. The HLA-B27 genotype is often the link that binds them together.
P.292
Clinical Features
The hallmark of acute nongranulomatous anterior uveitis is acute inflammation characterized by intense redness, pain, photophobia caused by ciliary spasm, and diminished vision, which usually prompts the individual to visit a physician. Symptoms generally evolve over hours to days. Findings include hyperemia of the conjunctival vessels, especially the ciliary zone; multiple fine clear keratic precipitates; intense cell and flare reaction in the anterior chamber; and in severe cases a dependent accumulation of cells, called hypopyon (see Fig. 8.1). If untreated, posterior synechiae form between the pupil margin and the lens, fixing the pupil in a moderately miotic position (Fig. 8.4). If the posterior synechiae encompass 360 degrees of the pupil, aqueous flow into the anterior chamber is blocked, resulting in the iris bowing forward, a condition called iris bomb (Fig. 8.5). The resulting closed anterior chamber angle causes an acute rise in intraocular pressure. In most cases of acute anterior uveitis, the intraocular pressure is diminished because of ciliary body dysfunction. Occasionally, intense inflammation can cause a rise in intraocular pressure despite an open angle because of acute trabeculitis. Acute inflammation of the iris vessels may cause their dilation and confuse the diagnosis with rubeosis iridis (Fig. 8.6). Prompt antiinflammatory therapy restores normal architecture.
The inflammatory cells and debris in the anterior segment can make fundus examination difficult. The posterior segment of the eye is generally normal, although cystoid macular edema may occur, especially in patients with severe anterior segment inflammation. If the inflammation is only partially treated or becomes subacute, spillover of inflammatory cells and debris into the vitreous may occur, and may contribute to blurring of vision. Most patients seek attention promptly because of the pain or an acute decrease in vision.
Management
The management of acute nongranulomatous anterior uveitis involves the intensive use of cycloplegic and steroidal agents, and the secret to successful therapy is prompt aggressive treatment. Cycloplegics such as 1% Cyclogyl administered four times daily or, in severe cases, 1% Atropine administered four times daily help to dilate the pupil, relieve ciliary spasm, and reduce pain. However, a side effect of this therapy is blurred near-range vision, which may incapacitate younger individuals. Fortunately, these effects are dose dependent. Topical steroids are the primary antiinflammatory agents, with an initial treatment rate of one drop every hour while awake. If no improvement occurs within 24 to 48 hours, high-dose topical steroid therapy can be supplemented with subconjunctival injections of intermediate-acting steroids or occasionally supplemented with a short course of high-dose oral steroid therapy (60 to 100 mg/day in divided doses). If an infectious process is identified, appropriate antibiotic or antiviral therapy should be started.
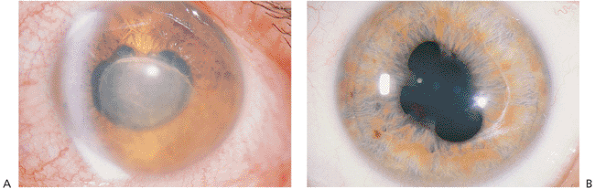 |
Figure 8.4. A: The rapid onset of pain, photophobia, redness, tearing, and diminished vision are classic features of acute iridocyclitis. The photograph shows the fibrin membrane generated by transudation from acutely inflamed tissues. Synechiae involve most of the lens but are partially broken superiorly. B: Inactive iridocyclitis. The eye is white and noninflamed, but telltale synechiae formation and pigment are seen on the lens capsule, producing a scalloped or irregular pupil that is characteristic of a prior acute uveitis process. |
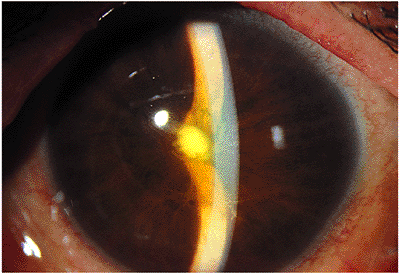 |
Figure 8.5. Iris bomb . |
The prognosis for the cessation of inflammation is uniformly good, but in severe or recalcitrant cases it may take weeks to months for the inflammatory cycle to resolve completely. Recurrences of inflammation, usually developing in
P.293
a stereotypical manner, are not uncommon, and the patient should be counseled to seek prompt attention if this occurs.
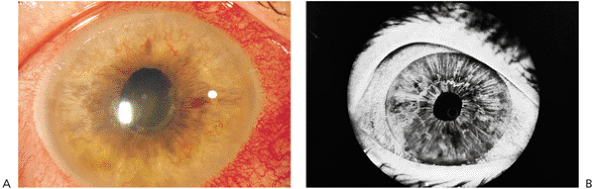 |
Figure 8.6. A: Acute iridocyclitis. Dilated iris vessels can often change the coloration of the iris. The conjunctiva, especially along the limbus, is intensely red. Iris details are slightly hazy because of anterior segment inflammation. Synechiae are seen with the fibrin membrane along the inferior aspects of the pupil. B: An iris fluorescein angiogram of the patient in (A) demonstrates the dilated vessels but normal anatomy of the iris. This is a common sequela of subacute iridocyclitis. |
The disease process is usually seen in teenagers and young or middle-aged adults, and it has a tendency to become less intense in the later years. Sometimes, only one or two bouts occur during a lifetime, although a higher recurrence rate is more common.
Chronic Iridocyclitis of Juvenile Rheumatoid Arthritis
Chronic iridocyclitis associated with juvenile rheumatoid arthritis is an indolent, smoldering disease that usually begins between ages 3 and 15, occurring more often in girls than boys.
Clinical Features
Affected individuals are frequently unaware of the low-grade inflammation, because minimal redness or pain occurs. A parent often notices an irregular pupil, or a school screening finds diminished vision, bringing the individual under medical supervision. The clinical findings are the direct antithesis of acute anterior nongranulomatous uveitis. Evaluation discloses a low-grade inflammatory process with small, white KPs, sometimes with a low-grade granulomatous response (Fig. 8.7). Examination often reveals synechiae, and the pupil can be small or irregular, with scalloped edges seen on dilation. Band keratopathy is frequently noted in patients with longstanding chronic inflammation.
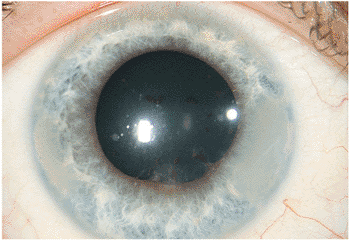 |
Figure 8.7. Chronic iridocyclitis secondary to juvenile rheumatoid arthritis. Patchy, irregular pupillary dilation with pigment along the lens capsule suggests breakage of old synechiae. The media are relatively clear, although band keratopathy is evident at the 3- and 9-o'clock positions on the peripheral cornea. The findings of a white eye and band keratopathy are characteristic of chronic iridocyclitis secondary to juvenile rheumatoid arthritis. |
This form of anterior uveitis is chronic and indolent in nature, often with minimal symptoms manifested over months to years. Cataract formation is not an unusual finding, and secondary glaucoma of an indolent nature is a feared complication (Fig. 8.8). The hallmark of this low-grade intraocular inflammation and systemic disease is the
P.294
pauciarticular form of rheumatoid arthritis seen in juveniles. Although the joint involvement usually precedes eye inflammation, it may occur years after the uveitis is diagnosed. Generally, the wrists, ankles, or knees are involved by mild inflammation. Two to five joints may be affected.
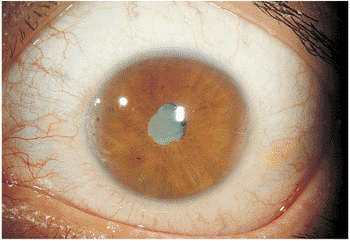 |
Figure 8.8. Secondary cataract as a result of chronic iridocyclitis in juvenile rheumatoid arthritis. The white, noninflamed eye has some subtle limbal calcium deposits at the 3- and 9-o'clock positions. Synechiae have formed, except for a pupillary area at the 12-o'clock position. The advanced cataract is characteristic of the late-stage sequelae of chronic inflammation related to juvenile rheumatoid arthritis. |
The diagnosis is based on the characteristic ocular and systemic clinical features and on serology testing. These individuals are generally rheumatoid factor negative and antinuclear antibody positive. Conditions that must be excluded include sarcoidosis and spirochetal diseases.
Management
The management of this form of chronic iridocyclitis involves the use of cycloplegics to prevent synechiae formation. Because this is a long-term disease process, the extended use of a cycloplegic, such as Atropine, may be indicated to keep the pupil from binding and producing secondary complications such as iris bomb . Typically, one to four drops daily of low-dose topical steroids are needed to reduce the potential for side effects of the inflammation. Long-term steroid use, however, is associated with the risks of cataract formation and glaucoma. Over many years of treatment, these are the two most feared complications. The former may be successfully treated with intraocular surgery. If topical antiglaucoma medications are ineffective, surgery may also correct the latter condition. In end-stage disease, often occurring in the patient's later years, ciliary dysfunction and hypotony are the more sinister problems. This expected course has prompted some ophthalmologists to treat the initial and long-term disease aggressively with oral nonsteroidal antiinflammatory agents or even with cytotoxic or alkylating medications to attempt to resolve all traces of inflammation. The rationale of aggressive treatment is that intraocular integrity may be better maintained over the many years the disease is present, but the added risks of aggressive treatment make this approach controversial.
Intermediate Uveitis
Pars Planitis
Pars planitis syndrome is a bilateral ocular inflammatory disease that usually affects otherwise healthy young people of grade-school through college age. The condition goes by many names, including intermediate uveitis, peripheral uveitis, and chronic cyclitis. The cause of pars planitis is usually idiopathic, although a small percentage of individuals have or may later develop associated demyelinating disease. Lyme disease has also been implicated in some patients.
Clinical Features
The main patient complaint concerns floaters, which are caused by a cellular reaction in the vitreous. There is usually minimal pain, redness, and photophobia. Slit lamp examination often reveals minimal aqueous cell and flare although moderate iritis may be present, especially during a flare-up of inflammation. Posterior synechiae are uncommon, but may develop. Inflammatory cells are typically noted in the vitreous, posterior to the lens. The chronic, low-grade inflammation results in a buildup of cellular material at the vitreous base and over the pars plana (Fig. 8.9). This initially results in a whitish and congealed-appearing accumulation of cells in the inferior pars plana, a finding called snowbanking that is the distinctive feature of pars planitis (Fig. 8.10). Pars plana snowbanking is best visualized with indirect ophthalmoscopy and scleral depression or with a Goldman three-mirror lens. Retinal periphlebitis and edema of the disc and macula frequently exist in patients with active pars planitis.
Pars planitis generally pursues an indolent course, often punctuated with spontaneous exacerbations and remissions. For many individuals, the effects of the inflammation are annoying but do not significantly impair vision, although central vision may be severely compromised if macular edema occurs.
Management
Treatment usually consists of cautious observation in patients who do not have significant cystoid macular edema, severe or disabling floaters, or significant anterior segment inflammation. In those patients who do require treatment, use of intensive high-dose topical steroid therapy is often effective in gradually achieving control of the inflammation, and may be very useful in unmasking steroid responders for whom treatment with periocular injection with depot corticosteroid agents is contraindicated. Subtenons steroid injections and/or oral steroid therapy are often helpful in resolving or controlling inflammation that has not responded satisfactorily to high-dose topical steroid therapy. Treatment with stronger agents such as cyclosporine or alkylating agents is rarely indicated.
Complications include cataracts and a slightly higher incidence of retinal detachment, which can be treated surgically. The long-term prognosis is good, especially if the inflammation is mild. The disease may resolve, sometimes after many years' duration.
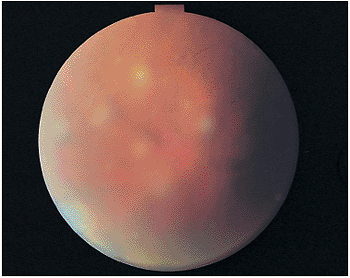 |
Figure 8.9. Pars planitis puffballs are usually found inferiorly and associated with peripheral periphlebitis. These fluffy exudates in the vitreous are commonly seen in patients with the pars planitis syndrome. |
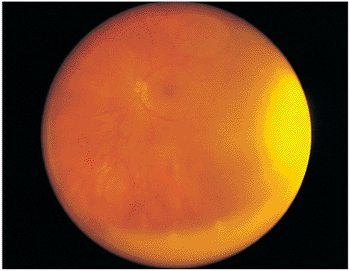 |
Figure 8.10. Pars planitis syndrome causes inferior snowbanking and precipitates along the inferior edge of the vitreous base, as seen in the wide-angle photograph with the transillumination light reflex at the periphery. Indirect ophthalmoscopy and scleral depression commonly are needed to evaluate this area. |
P.295
Fuchs' Heterochromic Cyclitis
Fuchs' heterochromic cyclitis is a form of anterior and intermediate uveitis. The condition is usually unilateral and chronic in nature, and eventually results in iris heterochromia (a change in the color of the iris), as well as posterior subcapsular cataract in many patients. The cause of this disorder is unclear; it may be an immune-related process or a manifestation of a low-grade degenerative process.
Clinical Features
The classic presentation usually involves minimal symptoms and the findings of a low-grade aqueous cell and flare reaction; stellate, white, granulomatous KPs involving the entire endothelial surface; and over many years, a characteristic change in the surface contours and the coloration of the iris (Fig. 8.11). The age of onset is usually in the twenties to forties, with women slightly more affected than men. Occasionally, floaters may be the presenting symptom. Loss of yellowish pigment within the iris stroma and subtle blunting of surface details of the iris stroma are seen early. With time, frank iris heterochromia develops, with generalized thinning and atrophy of the iris stroma. In blue-eyed individuals, this often results in the involved iris appearing darker than the noninvolved iris, since atrophy of the iris stroma allows the darkly pigmented underlying iris pigment epithelium to show through. In brown-eyed individuals, the involved iris often appears lighter in coloration than the iris in the noninvolved eye. The heterochromic iris is appreciated better in daylight. Although posterior synechiae are rare, glaucoma and cataract formation are long-term complications of the disease. Rare occurrences of occult-retained metallic intraocular foreign body may cause heterochromia from siderosis and may mimic iris heterochromia associated with Fuchs' heterochromic iridocyclitis; x-ray and computed axial tomography scan findings are generally useful in ruling out this entity.
 |
Figure 8.11. Stellate keratic precipitates from Fuchs' heterochromic cyclitis. Although difficult to photograph, these small, white, stellate keratic precipitates, with pseudopods along the edges, extend over the entire endothelial surface of the cornea. In conjunction with heterochromia, this is a classic feature of Fuchs' uveitis syndrome. |
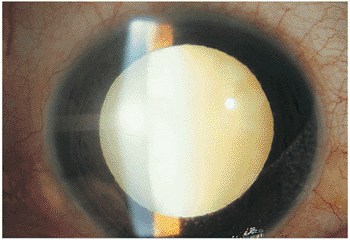 |
Figure 8.12. Fuchs' heterochromic cyclitis and cataract. This dense, mature cataract in a young individual is characteristic of one of the more common complications of Fuchs' heterochromic cyclitis. |
Management
Fuchs' heterochromic cyclitis responds variably to topical steroids and cycloplegic or systemic steroids. The complications of long-term use of these drugs may at times outweigh their potential benefits. Many affected individuals can be followed routinely without treatment and reevaluated at 6-month intervals, although at times the iritis may be sufficiently active to warrant judicious treatment with topical steroids. The primary complications include cataract and secondary glaucoma (Fig. 8.12). Cataracts respond well to most forms of intraocular surgery, including the standard extracapsular procedures with intraocular lens implantation. Glaucoma control may be somewhat more problematic, with surgical options indicated for later forms of the disease. The long-term prognosis is good.
P.296
Posterior Uveitis
Toxoplasmosis
Ocular toxoplasmosis, a condition caused by an obligate intracellular parasite, is one of the more common forms of posterior uveitis.
Clinical Features
The toxoplasmosis organism, Toxoplasma gondii, is a protozoa that has a predilection for the retina. The classic finding is retinochoroiditis. The organism reaches the eye through hematogenous spread. Eating uncooked meats that are infected with T. gondii and ingesting the organism from aerosolized fecal material of infected cats are the more common ways in which the organism enters the human body. Usually, the immune system can bring the infection under control, and the infected individual may notice only flu-like symptoms. However, the organisms may enter the eye and cause localized disease.
The primary lesion is a localized zone of inflammation in the retina, manifested as a whitish-yellow area of retinal swelling with indistinct borders (Fig. 8.13). Cellular exudation into the overlying vitreous results in a hazy ophthalmoscopic view. The lesion may occur anywhere in the retina. In immunocompetent individuals, the infectious process usually heals spontaneously, with characteristic chorioretinal scarring and pigment proliferation (Fig. 8.14). Sometimes, months to years later, the edge of the scar may become reactivated. This clinical picture is the most common (Figs. 8.15 and 8.16).
The clinical findings are usually characteristic, but serologic testing may be helpful. In a primary infection, test results for immunoglobulin M (IgM) antibodies are usually positive for the first few months, followed by elevated immunoglobulin G (IgG) levels. However, over many years, because of the low level of systemic antibodies, the IgG level may be quite low, and a high index of suspicion based on the clinical findings may be more important than results of the serologic studies.
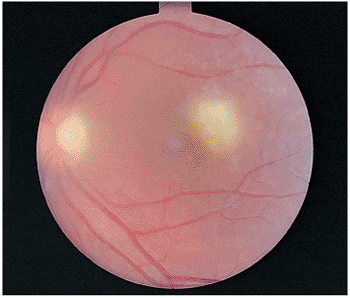 |
Figure 8.13. Acute toxoplasmosis retinochoroiditis in the macula of a 15-year-old Caucasian girl. Old toxoplasmosis scars were observed in the other eye. (Courtesy of Peter V. Palena, M.D., Philadelphia, PA.) |
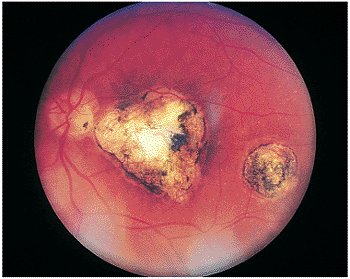 |
Figure 8.14. Full-thickness chorioretinal scars in a patient with recurrent ocular toxoplasmosis. The patient's vision has been reduced to the level of legal blindness. A peripheral scar is seen temporally in the macular area, but there is no active inflammation or infection. |
In immunocompetent individuals, the body's own defense mechanisms are often capable of bringing the intraocular infection under control. However, the scars resulting from the infection may damage the eye. If the infection affects the macular area, irreversible visual loss may occur. If infection occurs near the optic nerve, secondary neuritis may develop and cause optic atrophy and sectoral visual field loss (Fig. 8.17). Lesions along blood vessels can obstruct arteries or veins, resulting in hemorrhage, macular, edema, and visual field loss (see opening figure). Peripheral lesions may heal without any apparent symptoms.
Management
Treatment is aimed at prompt resolution of the infectious process in vision-threatening areas. Classically, sulfadiazine and the folic acid antagonist pyrimethamine have been used. Folinic acid is administered to reduce the bone marrow toxicity of pyrimethamine. Antibiotics such as clindamycin, trimethoprim-sulfamethoxazole, and tetracycline, which affect the organism's pathways
P.297
of protein transcription, have been used. Corticosteroids have also been advocated for severe inflammation, although caution is indicated because high doses of these drugs, especially in the form of periocular injections, may be contraindicated because of their immunosuppressive action, which may potentiate the infectious process (Fig. 8.18).
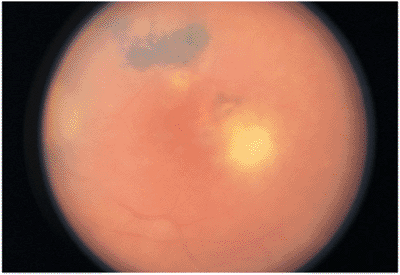 |
Figure 8.15. Acute, recurrent toxoplasmosis at the edge of a macular scar. The cellular reaction in the overlying vitreous results in hazy ophthalmoscopic views and gives the classic appearance of a headlight in fog. (Courtesy of Peter V. Palena, M.D.) |
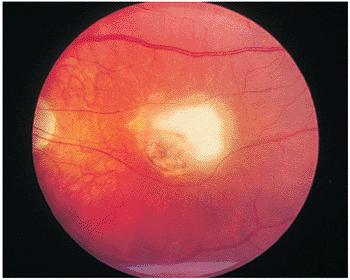 |
Figure 8.16. In this case of ocular toxoplasmosis with recurrent, active retinochoroiditis along the edge of an old macular scar in an early resolution stage, the overlying cellular exudation has cleared. |
The drugs used in the treatment of ocular toxoplasmosis may have significant side effects, and patients undergoing treatment must be closely monitored. Because the folic acid antagonists may cause bone marrow suppression, a platelet and complete blood count should be obtained at least weekly. With sulfadiazine, the rare but devastating complication of Stevens-Johnson syndrome must be watched for. Clindamycin may cause necrotizing enterocolitis, and tetracycline should not be prescribed for pregnant patients. Corticosteroids given topically or systemically are not without hazards. These treatment regimens, however, are often effective in reducing intraocular scarring and preserving visual function. For small, peripheral lesions or those that do not threaten vision, observation only may be a viable option.
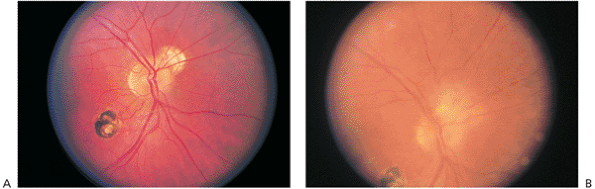 |
Figure 8.17. A: Juxtapapillary toxoplasmosis scar at the superonasal aspect of the right optic disc. B: Three years later, a lesion developed in the same area and involved the optic disc. (Courtesy of Peter V. Palena, M.D., Philadelphia, PA.) |
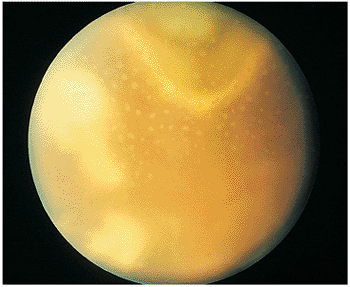 |
Figure 8.18. Chronic or recurrent ocular toxoplasmosis with endophthalmitic findings. The large geographic zones of retinochoroiditis shown superiorly are characteristic of chronic, low-grade endophthalmitic toxoplasmic choroiditis, which often occurs secondary to chronic steroid use. The small, white dots are granulomatous deposits of cells located along the back surface of the posterior hyaleid, analogous to the granulomatous keratic precipitates seen on the corneal endothelium. |
Toxocariasis
Ocular toxocariasis is caused by the second stage larvae of Toxocara canis and Toxocara cati. Toxocara canis is a common intestinal parasite in dogs, and Toxocara cati is found in cats. Young individuals, from toddlers to teenagers, most frequently acquire the disease. Classically, the organism enters the body through the ingestion of dirt contaminated with Toxocara ova. The animal vector is usually an infected puppy.
Clinical Features
Unilateral blurring of vision is often the presenting symptom in ocular toxocariasis infection in children older than 2 years of age. In preverbal patients, leukokoria may be the only presenting symptom, and retinoblastoma must be excluded. A characteristic whitish dome-shaped granuloma is noted ophthalmoscopically (Fig. 8.19). Granuloma formation within the choroid results from hematogenous spread of Toxocara to the eye; anterior
P.298
uveitis and vitritis may be present or absent, but are generally severe in the endophthalmitis form of ocular toxocariasis. The granuloma may be peripheral or central (i.e., near or involving the macula; Fig. 8.20) or may involve the optic nerve head. Rarely, the organism may travel beneath the retina, creating snail tracks. Most of the intraocular inflammation in ocular toxocariasis occurs after the death of the Toxocara larva and occurs as the eye reacts immunologically to antigens released from the dead or dying organism. Chronic inflammation leads to localized granuloma formation and may be associated with formation of retinal folds, vitreous traction bands, or generalized diffuse whitish inflammatory debris throughout the vitreous cavity (Fig. 8.21). In eyes with central lesions, destruction of the macular region or the optic nerve may cause a loss of vision.
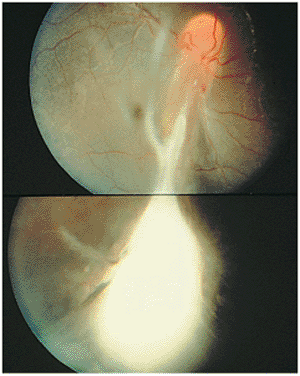 |
Figure 8.19. Traction retinal detachment with inferior peripheral granuloma secondary to ocular toxocariasis. A falciform fold has been created, dragging the optic nerve vessels inferiorly. The macular area is also dragged inferiorly, resulting in a secondary strabismus. (Courtesy of Jerry Shields, M.D., Philadelphia, PA.) |
Ocular toxocariasis is not associated with visceral larval migrans, a disease in which fever, hepatosplenomegaly, and systemic involvement occur. Ova and parasites are not present in stools, and the peripheral eosinophil count is characteristically normal. Intraocularly, eosinophilia may be found in the aqueous and vitreous if the eye fluids are sampled. The Toxocara antigen and antibody can also be detected in this manner. Unless secondary problems such as traction retinal detachment or opaque vitreous are encountered, intraocular biopsy for diagnostic and therapeutic reasons is usually unnecessary.
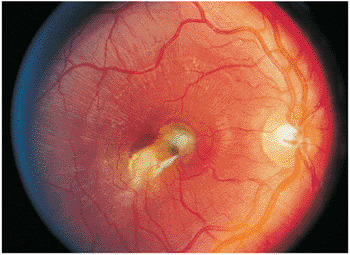 |
Figure 8.20. A full-thickness macular granuloma resulted from Toxocara canis infection and caused severe visual loss. All layers of the retina are involved by this condition. Secondary choroidal neovascularization may be a late sequela, as occurred in this case, producing the greenish appearance next to the old scar and mild degrees of subretinal blood. |
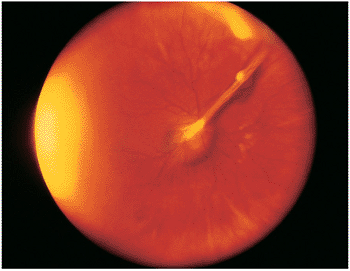 |
Figure 8.21. Wide-angle postvitrectomy photograph of a young patient with ocular toxocariasis. The characteristic falciform fold is still evident and the three yellowish areas along the fold and peripheral to it represent areas of residual scarring from the release of traction attachments during vitrectomy. There is a serous detachment around the disc. |
The outcome, depending on the intraocular location of the organism, can be quite good, and the disease is generally self-limited after the inflammatory process is quieted.
Management
Thiabendazole, the treatment of choice for visceral larval migrans, is not indicated for treatment of ocular toxocariasis because the organism has generally perished by the time that the eye changes are noted; secondary inflammation is responsible for the ocular symptoms and findings. Ocular inflammation may be self-limited in many cases, although active iritis and/or vitritis may require treatment with topical and/or periocular steroid injection. Vitrectomy may become a consideration in patients with chronic smoldering active inflammation, and removal of the antigen-laden vitreous in these patients often results in permanent quiescence of intraocular inflammation.
Candida Retinitis
Candida albicans retinitis is an uncommon disease seen in a specific clinical population that includes patients who are immunosuppressed as a result of disease or treatment of a disease that requires immunosuppressive therapy. Cancer patients receiving immunosuppressive agents, AIDS patients, patients with long-term indwelling catheters (e.g.,
P.299
hemodialysis, hyperalimentation), intravenous drug abusers, and debilitated patients are the most susceptible. The infection is usually indolent elsewhere in the body but spreads hematogenously to the eye.
Clinical Features
Multiple infectious foci initially develop within the choroid and secondarily involve the retina, appearing as multiple yellow-white spots. The infection subsequently spreads into the vitreous cavity. Intraocular inflammation evolves slowly, usually developing over a period of days to weeks, and the classic findings at diagnosis are small puffballs of localized fungal infection within the vitreous gel (Fig. 8.22). Symptoms may be minimal or nonexistent initially, although as the disease progresses, floaters become the most common complaint. If the organism affects the macular or parafoveal areas, diminished vision may also be a presenting symptom. Ocular candidal infection is often bilateral.
Management
The diagnosis is usually made by the characteristic clinical findings of puffballs in the vitreous, and is often confirmed by blood cultures or culture of a wound or catheter sample. If a diagnosis cannot be firmly established on the basis of blood, wound, or catheter cultures, diagnostic vitrectomy with staining and culture of the biopsy specimen usually demonstrates fungal organisms. Treatment involves administration of intravenous amphotericin B or oral fluconazole. Intravitreal injection of amphotericin B is often useful at the time of intraocular biopsy. The active infectious process generally responds promptly to antibiotic therapy. Depending on the intraocular location of the infection, scarring may be minimal, with complete return of vision. However, scars in the macular region may result in irreversible visual loss (Fig. 8.23). Because the affected individuals usually are quite ill from other causes, the ocular findings may not come to light until the systemic problems have been addressed. The ophthalmologist can be of great help in reducing visual problems by being alert to the characteristic pattern of the disease.
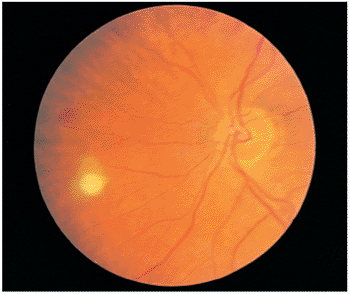 |
Figure 8.22. Candidal endophthalmitis. A candidal puffball is observed nasally in this immunocompromised patient on long-term intravenous therapy. The only complaint expressed by the patient was occasional floaters. |
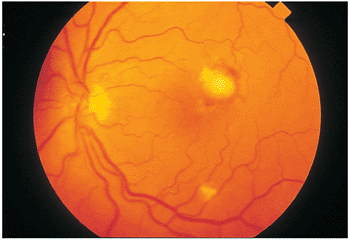 |
Figure 8.23. Candidal endophthalmitis with a macular lesion. Macular candidal infection causes gradual, progressive visual loss. Secondary scarring often prohibits good visual function. The small zone of blood at the superior edge suggested secondary choroidal neovascularization, which was confirmed angiographically. |
Cytomegalovirus Retinitis
CMV infection of the retina and surrounding ocular structures is a condition usually found in immunosuppressed individuals. The immune system may be compromised by the disease itself or by the use of immunosuppressive agents. CMV retinitis is the most common cause of visual morbidity in persons with AIDS. In renal transplant patients, the most common infectious complication is systemic CMV infection, which may involve the retina and cause a necrotizing retinitis.
Clinical Features
The onset of CMV retinitis is frequently insidious in nature. Early symptoms may include low-grade floaters and/or painless worsening of vision in one or both eyes. Anterior segment reaction is usually absent. Ophthalmoscopic examination reveals patchy, bone-white or grayish-white areas of full-thickness retinitis. Hemorrhage is occasionally noted both within and along the margins of areas of retinitis. Retinitis may begin peripherally or posteriorly (Fig. 8.24A) and characteristically progresses by contiguous spread into adjacent areas of retina. As the infection spreads contiguously it leaves atrophic necrotic retina and retinal pigment epithelium in its wake, with retinal pigment epithelial hyperplasia and atrophy as well as retinal gliosis. Generally, the edges of the areas of infection are the areas of active infection and, given the observed pattern of progressive contiguous spread and advancement, are often referred to as a brushfire border. Monitoring of the advancing edge of active infection is important in the management of CMV retinitis, since retinal function is effectively irretrievably lost in involved areas (Fig. 8.24B). Large areas of necrotic retina may result in retinal detachment. As in most cases of intraocular infection in immunocompromised patients, other opportunistic causes of infection should be excluded.
Management
Control of CMV retinitis depends on
P.300
prompt diagnosis. Indications for treatment are changing as new medications are discovered. Intravenous ganciclovir and foscarnet (Foscavir) remain the primary antiviral agents of choice. Oral ganciclovir has also been approved for use and in some patients can control advancing retinitis while avoiding both the inconvenience and the potential complications of intravenous drug delivery. Although they are virostatic, these agents are effective in controlling the progressive nature of the disease in many individuals. Remission usually occurs, but there may be exacerbations. The intraocular ganciclovir sustained-release implant (Vitrasert) can also be used to provide localized antiviral therapy for CMV retinitis, and intravitreal injections of ganciclovir are helpful if the implant is not available. Pars plana vitrectomy with silicone oil injection is often the initial surgical approach used for treating CMV-associated retinal detachments. Newer therapeutic agents are also under investigation.
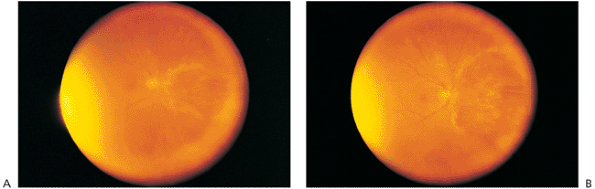 |
Figure 8.24. A: Cytomegalovirus (CMV) retinitis. Individuals who are immunocompromised because of disease or drug treatment are at risk of developing CMV retinitis. The wide-angle photograph demonstrates an area of retinitis nasal to the disc, with the active edge displaying a yellowish, patchy distribution. The central area of the lesion denotes destroyed retina, and the main ocular complaint was temporal visual loss, although central vision was 20/20. The transillumination light is evident at the periphery. B: CMV retinitis. The same individual, 2 months later, had progressive temporal visual loss and spreading of the active edge of the retinitis in a brushfire fashion. |
The visual prognosis depends on the time of diagnosis and the amount of retina that has been damaged or destroyed. If the immune status can be returned to normal, CMV usually disappears through normal immune responses. In renal transplantation cases, reducing immunocompromising drugs can often halt the progression of CMV retinitis.
Acute Multifocal Posterior Placoid Epitheliopathy
Acute multifocal posterior placoid pigment epitheliopathy (AMPPE) is a descriptive term given to an unusual and relatively uncommon posterior segment inflammatory process. AMPPE generally occurs in adults, and often has its onset after a relatively mild nonspecific viral illness.
Clinical Features
The presenting symptoms are spots, floaters, and blurred vision in one or both eyes. Fundus examination reveals multiple yellowish to cream-colored areas that are approximately 1 disc diameter or smaller, scattered throughout the posterior pole, and occasionally seen in the periphery (Fig. 8.25). There may be associated low-grade vasculitis with a cellular reaction and vitritis. Occasionally, iridocyclitis is also seen. Symptoms may be severe if the plaques involve the central foveal zone. If the lesions are extrafoveal, the symptoms may be minimal. The disease is often bilateral.
Management
The disease is usually self-limited and resolves within 6 to 8 weeks, leaving subtle pigmentation in the areas of the cream-colored plaques. Treatment other than observation is controversial; corticosteroids may have a place in therapy if there are vision-threatening areas of involvement.
P.301
Fluorescein angiography usually shows early blocked fluorescence and late hyperfluorescence and staining in the affected areas (Fig. 8.26). Rarely, cerebral vasculitis may be associated with the condition, and the clinician needs to be alert for this finding. The prognosis is good for the return of good functional vision, depending on the location of the initial lesions. In a small subgroup, the disease process may recur months to years later.
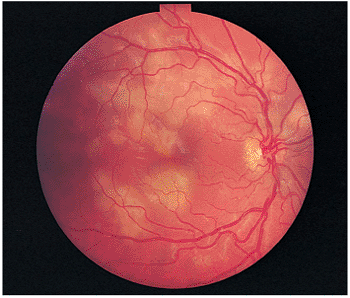 |
Figure 8.25. Acute, multifocal, posterior placoid epitheliopathy presents with cream-colored lesions at the level of the pigment epithelium and choriocapillaris, often after a viral illness. This individual's main complaint was fixed spots surrounding the central visual area. His acuity was 20/20 and remained so after the process healed. |
Acute Retinal Necrosis Syndrome
Acute retinal necrosis syndrome is a viral disease that affects immunocompetent individuals of any age. The condition is bilateral in one third of cases. The cause is unknown, although the varicella-zoster virus is considered the offending agent in most cases.
Clinical Features
Patients present with blurred vision and ocular pain in one or both eyes. The hallmark findings are small, opaque spots of grayish-white full-thickness retinitis in the retinal periphery and secondary vasculitis that is often obstructive (Fig. 8.27). Over a period of days, these spots increase in size and coalesce, resulting in a 360-degree involvement (Fig. 8.28) with subsequent extension into the posterior pole. In rare cases, the posterior pole can be affected earlier in the disease. Blindness can occur within weeks as the posterior pole becomes involved. Retinal detachment occurs in as many as one quarter of affected individuals (Fig. 8.29).
Management
Acyclovir, the agent of choice, is given intravenously for 7 to 10 days, and high-dose oral or parenteral steroid therapy is often employed. Concomitant aspirin therapy is recommended by many, and is felt to counter and minimize retinal ischemia due to associated retinal vasculitis. Prompt treatment with acyclovir is felt to minimize the chances for involvement of the fellow eye, and prolonged treatment with oral acyclovir for least several months is often recommended even after the acute phase of retinitis has responded to initial parenteral acyclovir therapy.
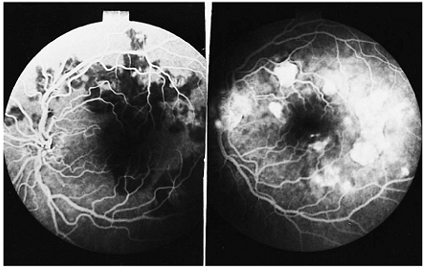 |
Figure 8.26. Fluorescein angiographic findings of acute, multifocal, posterior placoid epitheliopathy. The picture on the left shows the characteristic early-phase blockage of fluorescein caused by edema of the choriocapillaris, and the right photograph shows late staining of the inflamed areas. The person's visual acuity was 20/20. |
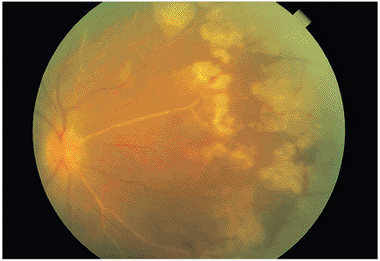 |
Figure 8.27. Acute retinal necrosis demonstrating the hallmark finding of grayish-white patches in the retinal periphery prior to becoming confluent. The arterioles are white due to immune reactants staining the arterial wall. |
The visual prognosis can be good if the disease is discovered early. A high index of suspicion is critical, because a delayed diagnosis can result in visual loss. Small, peripheral retinal detachments can be walled off by photocoagulation. In severe cases, proliferative vitreoretinopathy may develop because of the large amount of tissue loss and pigment proliferation. Pars plana vitrectomy with silicone oil injection may be the only way to reattach the retina in this subgroup of patients.
Bird-Shot Vitiliginous Chorioretinitis
Bird-shot choroidopathy (sometimes referred to as vitiliginous choroidopathy) is an unusual and rare syndrome. Although its cause is unknown, this syndrome is frequently associated with positive test results for the HLA-29 antigen, and it usually affects middle-aged women.
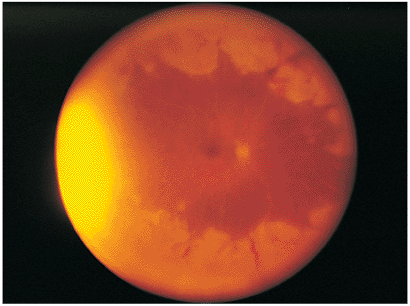 |
Figure 8.28. Acute retinal necrosis syndrome. This wide-angle photograph reveals the characteristic peripheral whitening of the retina from small spots of retinitis coalescing into broad areas. Vasculitis is also seen with whitening of the vessels and blood along the vessel borders. |
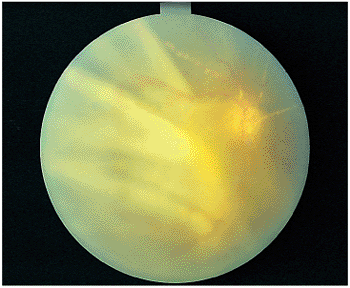 |
Figure 8.29. Acute retinal necrosis (ARN) syndrome and secondary retinal detachment. The classic findings of retinal detachment, seen in two thirds of individuals with ARN, are early fixed folds caused by a severe pigment epithelial response; white plaques in the arteries, thought to be immune reactants; and minimal compromise of the arterial circulation. |
P.302
Clinical Features
The classic diagnostic feature of bird-shot vitiliginous chorioretinitis is cream-colored spots, often as large as 0.5 to 1 disc diameter, that are scattered throughout the fundus (Fig. 8.30). With time, these spots may coalesce. Associated with this is a low-level vasculitis and cystoid macular edema, with disc leakage demonstrated on fluorescein angiography. Iridocyclitis and a vitreous cellular reaction are other common features. The condition often lasts months to years and may be progressive. The complications of chronic ocular inflammation such as cystoid macular edema (CME) and cataract formation may adversely affect vision.
The diagnosis depends on the clinical picture. The HLA-29 genotype has been associated with bird-shot vitiliginous chorioretinitis in as many as 80% of affected persons. Other causes of chorioretinitis, such as spirochetal disease, sarcoidosis, and tuberculosis, must be excluded.
Management
Treatment consists of antiinflammatory drugs, including corticosteroids and, in recalcitrant cases, cyclosporine or other immunosuppressive agents. Although acyclovir has been suggested as concomitant therapy, controversy persists about adding this drug to the regimen. The poor prognosis reflects the chronicity of the disorder and frequent involvement of the macula. A thorough understanding of the cause of the condition is needed to design effective therapy.
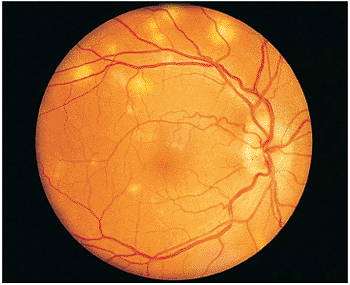 |
Figure 8.30. Bird-shot choroiditis. The classic finding is a creamy yellow, multifocal choroiditis. The central vision is excellent; the patient's main complaints were floaters and peripheral spots. |
Ocular Histoplasmosis Syndrome
The ocular histoplasmosis syndrome has been used to describe a primary choroidopathy thought to be caused by the fungus Histoplasma capsulatum. This organism is found most commonly in the Ohio River valley area but is also present in the Mid-Atlantic and Southeastern states. It is found in surface soil, particularly that which contains the droppings of certain birds. The organism is thought to be inhaled with the dust from a contaminated area.
Clinical Features
The classic findings are limited to the fundus and are manifested as the classic triad of small, whitish punched-out areas, peripapillary scarring, and choroidal neovascularization in the posterior segment (CNV; Fig. 8.31). The punched-out areas are less than 1 mm in diameter and can occur anywhere in the fundus. Vitreous inflammation is notably absent. The scarring adjacent to the disc is manifested by chorioretinal atrophy and a rim of pigment at the disc margin. Submacular choroidal neovascularization is manifested initially as a thickened, gray-green area deep to the retina. Fluorescein angiography is essential in delineating the location and extent of the choroidal neovascular membrane. Hemorrhage from the choroidal neovascular membrane and subsequent disciform scarring are vision-threatening complications (Fig. 8.32).
Patients are asymptomatic unless macular involvement develops, in which case central vision can be lost. There is no anterior segment reaction.
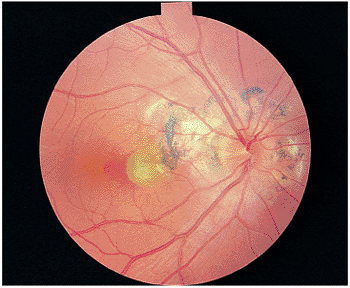 |
Figure 8.31. Presumed ocular histoplasmosis syndrome. The classic findings of juxtapapillary chorioretinal scars and macular scarring, with a greenish yellow area and blood along the edge, fulfill the clinical picture of choroidal neovascularization. Peripheral scars are often seen. |
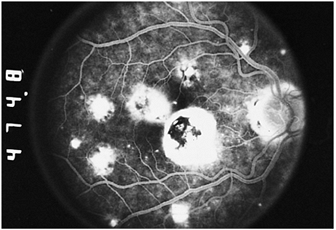 |
Figure 8.32. Fluorescein angiogram of an eye with presumed ocular histoplasmosis syndrome. Macular scars are present from old choroidal neovascularization, which has cicatrized but has destroyed central vision. Peripheral scars and juxtapapillary chorioretinal scars are also seen in this photograph. |
P.303
Management
Careful and ongoing assessment of macular function using an Amsler grid and careful periodic inspection of the macula are necessary for patients suspected of having choroidal neovascular involvement and for those previously treated for choroidal neovascularization. Fluorescein angiography plays a crucial role in the diagnosis and follow-up of these patients.
Choroidal neovascularization in the ocular histoplasmosis syndrome is treated with laser photocoagulation, similar to that used in age-related macular degeneration. Vitrectomy with extraction of the choroidal neovascular membrane is often successful in restoring macular function in patients with subfoveal choroidal neovascularization that cannot be treated with conventional laser therapy. Photodynamic therapy may also be a useful treatment option in patients with subfoveal choroidal neovascularization. Systemic antifungal treatment has not proven to be helpful.
Diffuse Uveitis
Sarcoidosis
Sarcoidosis is a multisystem systemic disease of unknown cause in which lymphoid tissue develops noncaseating, granulomatous inflammation. Usually, sarcoidosis is a disease that affects 20- to 40-year-old individuals, with persons of African descent and women more predisposed than other patient subgroups. The lungs are the most frequently involved organs, but the skin, liver, spleen, joints, and heart may also be involved. Ocular involvement occurs in 17% of cases of systemic involvement. Ocular sarcoidosis can take many forms, but it is often the presenting sign of the systemic disease.
Clinical Features
Acute iridocyclitis may be the earliest form of sarcoid uveitis. Patients complain of ocular pain, photophobia, and redness, and on slit lamp examination, flare and cells are seen in the anterior chamber. The uveitis may be granulomatous or nongranulomatous. Fine, gray KPs initially may be seen on the posterior surface of the cornea. The condition often becomes chronic, and mutton-fat KPs develop as the disease process continues. The granulomatous nature of sarcoidosis is responsible for some of the ocular features, including conjunctival granulomas, mutton-fat KPs, Busacca nodules within the iris stroma, and Koeppe nodules along the pupil margin (Fig. 8.33).
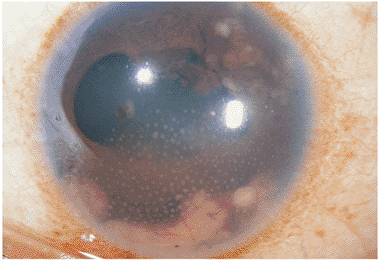 |
Figure 8.33. Anterior segment granulomatous involvement in a case of ocular sarcoidosis. White, mutton-fat, granulomatous keratic precipitates; nodules in the iris stroma (i.e., Busacca nodules); a low-grade cellular response; and adhesions of the pupil to the lens (i.e., synechiae formation secondary to Koeppe nodules) were found in this African-American patient with chronic sarcoid iridocyclitis. |
Posterior segment findings in ocular sarcoidosis may include vitritis, pars plana snowbanking, cyclitis, choroiditis, optic neuritis, cystoid macular edema, retinal arteritis, retinal phlebitis, and optic nerve granulomas (Figs. 8.34; 8.35A, B). All or none of these may be found. White, fluffy opacities may be seen in the inferior vitreous, and yellow-white exudates, called candle-wax drippings, and sheathing of the peripheral retinal veins may be seen (Fig. 8.36).
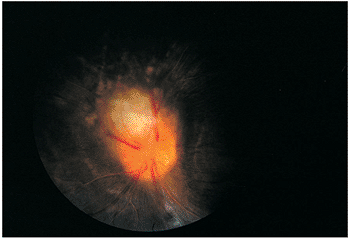 |
Figure 8.34. Optic nerve granuloma. Granulomatous disease can involve the optic nerve, producing a large blind spot and diminished visual function. A granuloma is seen off the superior border of the papilla. |
 |
Figure 8.35. A: Scattered peripheral multifocal lesions characteristic of ocular sarcoidosis. B: The macula of the same patient demonstrating a choroidal neovascular membrane. |
P.304
The characteristic granulomatous anterior, posterior, or diffuse uveitis should alert the clinician to search for systemic involvement. The workup should include a chest x-ray, serum angiotensin-converting enzyme assay, and if necessary, a gallium scan limited to the head, neck, and mediastinum. Biopsy of ocular tissues such as conjunctival granulomas (Fig. 8.37) or lacrimal gland granulomas may be helpful in confirming the diagnosis.
Management
The management of sarcoidosis is related to the level of eye inflammation and its structural damage. Topical, periocular, or systemic corticosteroids are the basic elements of treatment. Cycloplegic agents may also be helpful in reducing synechiae formation. The ophthalmologist should work with the internist to determine whether systemic therapy would benefit extraocular areas.
Oral steroid therapy may be needed initially in patients with severe inflammation, and chronic low-dose oral steroid therapy, such as 20 mg of prednisone daily, may be needed to control chronic inflammation or reduce exacerbations. If inflammation is not controlled, secondary scarring can occur. This can result in corneal scars, iris bomb from synechiae between the pupillary margin and the lens surface, cataract formation, chronic cystoid macular edema, optic nerve dysfunction, and vascular ischemic events caused by inflammatory obstructive phenomena.
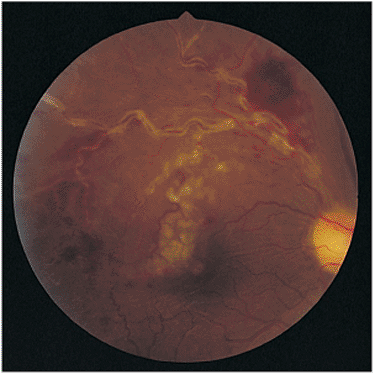 |
Figure 8.36. The classic findings of candle-wax drippings in ocular sarcoidosis are observed near the superior arcade. Because of the intense periphlebitis, capillary bleeding has occurred superiorly, and macular function is diminished because of edema. |
Sympathetic Ophthalmia
Sympathetic ophthalmia is a rare condition seen after penetrating trauma to one eye (called the inciting eye) that results in an inflammatory reaction in the other eye (called the sympathizing eye). It occurs after approximately one of 500 cases of penetrating eye injury and after one of 10,000 cases of intraocular surgery.
The most common sequence involves a traumatic rupture of one eye followed by inflammation in the second eye usually beginning 2 weeks to 1 year later, although cases have occurred within 5 days to as long as many years later (Figs. 8.38 and 8.39). The injury to the inciting eye may be as minimal as an intraocular penetration occurring during anterior chamber paracentesis. The cause is unclear but may include microbial hypersensitivity and immune reactivation
P.305
of intraocular antigens. Intraocular surgery is a form of controlled trauma that may result in sympathetic ophthalmia, although this is a rare event.
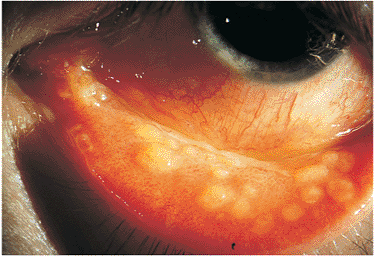 |
Figure 8.37. Conjunctival granulomas in ocular sarcoidosis. The classic yellowish follicular deposits are characteristic of ocular sarcoidosis and are easy to biopsy in an office setting. |
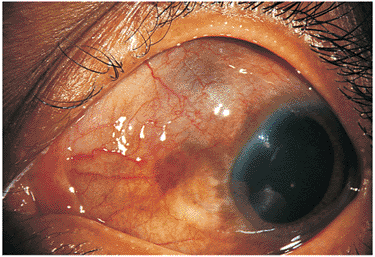 |
Figure 8.38. Sympathetic ophthalmia. This young person suffered trauma 3 weeks before evaluation. Examination of the traumatized eye revealed an occult scleral rupture superotemporally, low-grade inflammation, and irregular healing. The visit was prompted by acute uveitis in the sympathizing eye. |
Clinical Features
The patient presents with symptoms of iritis, including ocular pain, photophobia, redness, and decreased vision in both eyes. Sympathetic ophthalmia produces a diffuse granulomatous uveitis in both eyes. The result of this process may be mutton-fat KPs, granulomatous nodules on the iris, and focal areas of inflammation at the level of the retinal pigment epithelium, called Dalen-Fuchs nodules. The choroid is markedly swollen from increasing cellular infiltration, and choroidal exudation may cause serous retinal detachment. Over time, the granulomatous inflammation can cause chorioretinal scarring and poor vision if not treated aggressively (Fig. 8.40A, B).
Management
The clinician must remain alert to the possibility of sympathetic ophthalmia after ocular trauma. Prompt treatment of ruptures with primary closure and meticulous attention to cleanliness of the wound, with all uveal tissues cleaned from wound edges, may reduce the risk of sympathetic ophthalmia. Rarely, enucleation of the inciting eye may be indicated, especially if the condition is caught within 2 weeks or less from the inciting event or if the inciting eye is blind or painful. Evidence suggests this may reduce inflammation in the sympathizing eye. However, observation is indicated if the inciting eye has good vision, because it may have better visual function at the end of the disease.
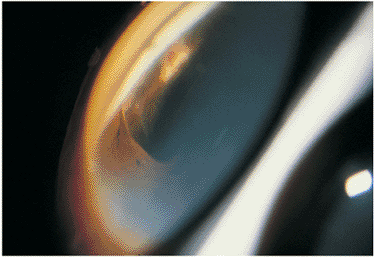 |
Figure 8.39. A superotemporal gonioscopic photograph of the eye in Figure 8.38 shows the internal scleral rupture that resulted in sympathetic ophthalmia. |
Treatment includes high-dose systemic corticosteroids along with periocular or topical medications and longer-acting immunosuppressive medications such as chlorambucil, methotrexate, or cyclosporine. Although treatment may require months to years, the final outcome can be good.
Vogt-Koyanagi-Harada Syndrome
Vogt-Koyanagi-Harada (VKH) syndrome is a rare and unusual form of diffuse granulomatous uveitis. This condition is usually more severe in more highly pigmented individuals. In the United States, the VKH syndrome is seen most commonly in individuals of Native American or Asian ancestry.
Clinical Features
Acute serous detachment of the retina is the hallmark of the VKH syndrome (Fig. 8.41) and may be accompanied by multiple areas of yellowish or yellowish-white edema of the retinal pigment epithelium, as well as by retinal vasculitis and by swelling of the optic disc. Associated anterior iritis may be severe and may be either granulomatous or nongranulomatous in appearance. Some patients will experience associated headache and stiffness of the neck or other neurologic symptoms such as tinnitus. With time, many patients will manifest associated dermatologic changes including poliosis (whitening of the lashes) and vitiligo (patchy depigmentation of the skin). Neither the vitiligo nor poliosis is generally present at the outset of the disease.
Management
Successful management of VKH requires a high index of suspicion for the disease based on the characteristic physical findings. A spinal tap may reveal leukocytosis, and other laboratory tests can exclude other granulomatous conditions. Affected individuals are often positive for the Dw-54 marker.
Treatment of this immunologic condition includes the use of systemic corticosteroids and sometimes includes immunosuppressive agents such as chlorambucil or methotrexate. Cyclosporine may also be used in the treatment of this condition. The final outcome depends on the control of inflammation and prevention of scar formation. Visual function may remain quite good, although the course may be long term and indolent (Fig. 8.42).
Beh et's Disease
Beh et's disease is a form of diffuse uveitis of unknown cause that is associated with significant dermatologic and vascular features. It is found in young adults, and although
P.306
rare in the United States, it is the most common form of uveitis in Japan. The ophthalmologist may be the first to recognize the condition because of the unusual presentation and clinical findings.
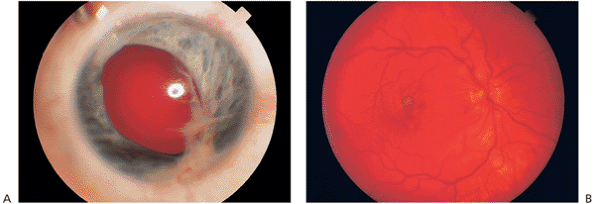 |
Figure 8.40. A: An injured eye in a 5-year-old that developed sympathetic ophthalmia. B: Dalen-Fuchs nodules in the macular region of the same patient that have gone on to choroidal neovascularization with leakage of fluid and bleeding. |
Clinical Features
The most common ocular findings are a posterior uveitis and occlusive vasculitis that may involve both arteries and veins (Fig. 8.43), although some patients may present initially with a hypopyon iridocyclitis without posterior segment findings. Clinically, the hypopyon may be seen in an eye that is otherwise relatively noninflamed. Beh et's disease can be severe, and irreversible damage can easily lead to blindness if the condition is not diagnosed or if it remains untreated.
The features of painful oral aphthous ulceration, genital ulceration, and uveitis fulfill the triad of physical findings characteristic of Beh et's disease. Other associated systemic findings may include phlebitis, polyarthritis, and cerebral vasculitis. Because of this systemic involvement, Beh et's disease may be life-threatening.
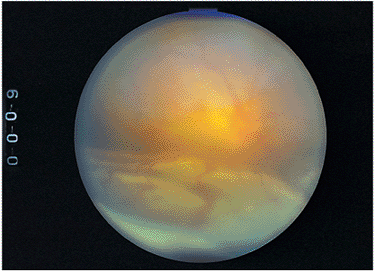 |
Figure 8.41. The hallmark of Vogt-Koyanagi-Harada syndrome is an exudative retinal detachment, shown here with hazy vitreous, dilation of the vessels and optic nerve, and the retina separated, with subretinal fluid puddling inferiorly because of dependent positioning. |
Management
Successful treatment requires prompt recognition and suppression of inflammation. Laboratory studies may be helpful, and patients commonly have the HLA-B5 marker. Therapeutic agents include systemic corticosteroids, cyclosporine, and immunosuppressive agents such as chlorambucil. The final outcome depends on the amount of damage caused by the initial vasculitis, which often results in arterial occlusion and retinal necrosis. The end result, however, may be satisfactory if the process is diagnosed and treated promptly. Localized treatment, such as laser therapy for the ischemic complications of vasculitis, is sometimes helpful, and the proper management of Beh et's disease should include consultation with a skilled internal medicine specialist.
Lyme Disease
Lyme disease is a spirochetal infection (Borrelia burgdorferi) that is transmitted through a deer tick vector. Lyme disease
P.307
may involve many organ systems, and although it rarely involves the eye, the ophthalmologist may be the first to diagnose the disease.
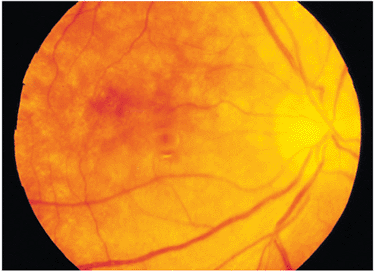 |
Figure 8.42. Resolution of an exudative retinal detachment in a patient with Vogt-Koyanagi-Harada syndrome is often associated with pigment dispersion, a condition called a sunset fundus. If the macular area has been maintained during treatment, the end result may be good vision. |
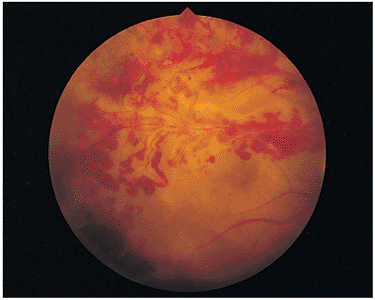 |
Figure 8.43. Acute retinal vasculitis in a patient with Beh et's syndrome. The central vision remained good, although an inferior visual field defect resulted from the vasculitis in the superior arcade. |
Clinical Features
The eye findings are protean and reflect low-grade inflammation, which may be granulomatous in nature. Ocular findings may include corneal disease in the form of a nummular keratitis, low-grade iridocyclitis, pars planitis syndrome (which may be one of the more common presentations), or low-grade optic neuritis and chorioretinitis (Fig. 8.44). Because of the wide array of ocular findings, some degree of suspicion is needed for the correct diagnosis. Serologic testing and DNA probes are improving Lyme disease diagnosis.
Management
The management of Lyme disease depends on the systemic and ocular findings. Systemic and ocular treatments include antibiotic use orally or intravenously. There is some controversy about the optimum duration of antibiotic treatment, which can range from weeks to months. Treatment of the ocular manifestations also usually involves the use of corticosteroids given topically, periocularly, or systemically to reduce the inflammatory complications of the disease. The extent of steroid use depends on the severity and vision-threatening nature of the ocular findings. The outcome can be good if the condition is caught early and scarring is minimal.
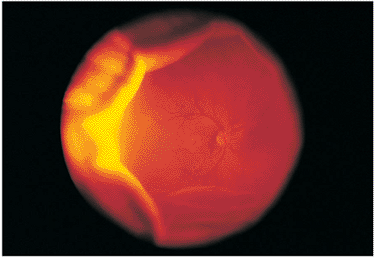 |
Figure 8.44. A wide-angle photograph of a peripheral choroidal detachment with puddled subretinal fluid and a secondary exudative retinal separation inferiorly in a patient with Lyme disease also shows a macular lesion. Prompt resolution occurred with corticosteroids and antibiotic treatment. |
Syphilis
Ocular syphilis may occur as both an acquired and a congenital form of disease, and can confuse the uveitis diagnosis. In acquired syphilis, eye involvement can occur in the secondary, latent, and tertiary phases of the disease. Untreated acquired syphilis and congenital syphilis are associated with multisystem abnormalities.
Clinical Features
The protean ocular findings of acquired syphilis may be acute or chronic in nature, and are often granulomatous in character. Any portion of the ocular system may be involved. Some characteristic findings of acute ocular infection are anterior uveitis, diffuse and focal chorioretinitis, retinal vasculitis, and optic neuritis (Fig. 8.45). Late findings include interstitial keratitis, chorioretinal scars, salt-and-pepper retinal pigment epithelial scarring, and optic atrophy. The Argyll-Robertson pupil, which fails to react to light but preserves accommodation, classically has been associated with syphilis. The systemic complications of tertiary syphilis include cardiovascular and meningovascular disease, generalized paresis, and tabes dorsalis.
Ocular findings associated with congenital syphilis may include anterior uveitis, chorioretinitis, interstitial keratitis, and optic atrophy. The interstitial keratitis develops in the first or second decade of life and is bilateral. There is cellular infiltration and vascularization of the corneal stroma. Eventually, the cornea becomes thin, with patchy opacification and residual ghost vessels in the stroma (Fig. 8.46). The
P.308
chorioretinitis results in pigment clumping interspersed with atrophic white areas, producing the classic salt-and-pepper fundus of ocular syphilis. The systemic features of congenital syphilis include Hutchinson's teeth (i.e., widely spaced, peg shaped), saddle nose, nerve deafness, arthropathy, and mental retardation.
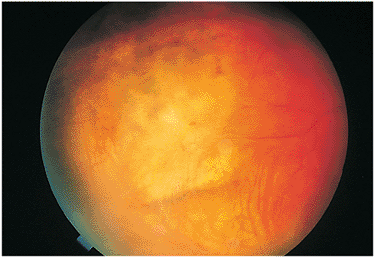 |
Figure 8.45. Resorbing retinitis secondary to ocular syphilis. This patchy retinitis with pigment along the superior border may be found in early, latent, or late ocular syphilis. The patient previously presented with an acute loss of vision secondary to severe vitritis. The patchy, focal retinitis was noticed after a course of intraocular antibiotics. |
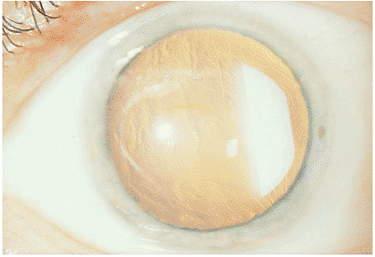 |
Figure 8.46. Interstitial keratitis in secondary syphilis. The interstitial keratitis, revealed in vessels by transillumination, extends into the corneal surface. |
The workup of patients suspected of having ocular syphilis begins with a careful history of sexual activity, including eliciting a history of chancre, which is a manifestation of the primary infection. Because history alone is frequently unproductive, serologic testing using the Venereal Disease Research Laboratory (VDRL) test and fluorescent treponemal antibody absorption (FTA-ABS) test is required. The VDRL test result is usually positive during the acute phase of the disease but can become negative over many years or if the condition is in remission. The FTA-ABS result, however, usually remains positive for life. Both tests should be ordered. Theoretically, a negative VDRL test result with a positive FTA-ABS result can be compatible with ocular syphilis. A VDRL assay of spinal fluid may also be helpful, although a negative result does not preclude central nervous system syphilis.
Management
Ocular syphilis is treated with oral, intramuscular, or intravenous antibiotics. A full systemic course is indicated for ocular and extraocular involvement. If macular scarring is minimized, good vision may be obtained. The VDRL test result falls slowly after treatment and is useful in following patients over time. Because the spirochetes are slow-growing organisms, recurrences are possible, and follow-up VDRL testing is indicated.
Tuberculosis
Tuberculosis is a chronic disease caused by Mycobacterium tuberculosis. Any organ system, including the eye, can be affected. Ocular tuberculosis is rare, although it is becoming more common with the advent of AIDS and the use of immunosuppressive agents in the treatment of other diseases.
Clinical Features
A posterior granulomatous choroiditis is the most common finding in ocular tuberculosis (Fig. 8.47). Retinal vasculitis in the form of sheathing of the veins and arteritis is common. Blurred vision is usually the presenting symptom and is generally related to vitritis. Chronic iridocyclitis may also occur, although it may be solely immunologic in nature, without active organisms. In overwhelming systemic infections, tuberculous endophthalmitis has been reported.
As with many of these rare conditions, a high index of suspicion is necessary for the initial diagnosis. Skin testing and a chest radiograph are indicated as part of the ophthalmologist's workup, but an internal medical consultation also is indicated before determining the diagnosis and treatment.
Management
Treatment is accomplished with antitubercular medications, which include isoniazid and rifampin. Because resistant strains commonly develop, single drug therapy should not be used. Topical corticosteroids and cycloplegics may be used to treat anterior uveitis. Posterior uveitis may be treated with the judicious use of systemic corticosteroids after consultation with the internist.
Acquired Immunodeficiency Syndrome
The human immunodeficiency virus (HIV) may affect the eye directly, but more frequently because of the underlying systemic immunodepression the eye becomes vulnerable to a number of opportunistic infections and neoplastic conditions, including CMV retinitis, Candida retinitis, herpes zoster infection, toxoplasmosis, Pneumocystis carinii infection, and Kaposi's sarcoma.
AIDS is a bloodborne and sexually transmitted disease. Groups at increased risk include homosexual or bisexual men, intravenous drug users, transfusion recipients, hemophiliacs, prostitutes, healthy sex partners of those with
P.309
AIDS, and infants born to mothers with AIDS. The ophthalmologist may be the first to diagnose AIDS in a seemingly healthy individual.
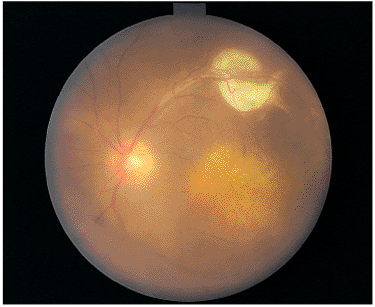 |
Figure 8.47. Granulomas secondary to ocular tuberculosis. The media are slightly hazy because of low-grade vitritis. Off the superior arcade is an old granuloma that has healed with glial scarring. In the macular region is an orange-brown macular granuloma, causing a drop in vision to 20/50. This patient had been treated 5 years previously for Pott's disease. |
Clinical Features
The cotton-wool spot (i.e., a retinal nerve fiber layer infarct) is the most common eye finding of ophthalmic examination (Fig. 8.48). These localized areas of ischemia are usually located in the posterior pole and may be related to circulating immune complexes. Cotton-wool spots may be found in as many as 100% of HIV-positive individuals over the course of the disease. In most cases, cotton-wool spots are asymptomatic. The diagnosis must exclude other causes of cotton-wool spots, such as diabetes, high blood pressure, collagen vascular disease, and retinal venous occlusion.
Management
Serologic testing is necessary to confirm the diagnosis of AIDS, and a physician skilled in the treatment of this patient population should be consulted. The cotton-wool patches usually are evanescent, and although there may be some mild associated microangiopathy, no treatment is indicated.
AIDS-Related Toxoplasmosis
Clinical Features
Toxoplasmosis may be seen in individuals with HIV infection. If the immunosuppression is moderate to severe, the toxoplasmic retinochoroiditis may be multifocal and endophthalmitic in nature with numerous foci of retinochoroiditis and with marked vitritis (Fig. 8.49). This is a different presentation than that seen in the immunocompetent patient.
Management
The management of ocular toxoplasmosis in patients with HIV-related conditions is somewhat more aggressive than in immunocompetent individuals. Sulfa compounds, clindamycin, and Daraprim have been advocated for the therapy of intraocular infections and are given in high doses to these patients.
The complications of the ocular disease are related to the amount and location of the resulting scars. The condition can become chronic because of the poor cell-mediated immunity in HIV-positive patients. Although central nervous system toxoplasmosis is a common complication of AIDS, intraocular infections are somewhat rare.
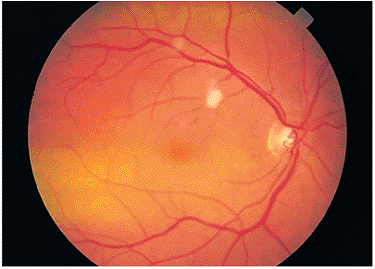 |
Figure 8.48. An asymptomatic young individual was found to have cotton-wool patches and subtle microangiopathy. Serologic testing revealed human immunodeficiency virus as the cause. |
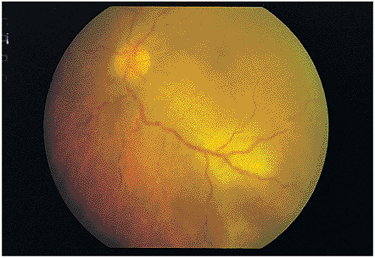 |
Figure 8.49. Toxoplasma gondii retinochoroiditis in a patient with acquired immunodeficiency syndrome (AIDS). The findings of precipitates along peripheral vessels, low-grade vasculitis, and deep retinochoroidal lesions scattered throughout the fundus are characteristic of the immunocompromising effects of AIDS in a patient with ocular toxoplasmosis. The immunocompromising events create multifocal lesions. |
AIDS-Related Kaposi's Sarcoma
Clinical Features
Kaposi's sarcoma is one of the more common cancers associated with AIDS. Ocular manifestations consist of small, reddish lesions and tumorous areas on the conjunctiva or contiguous ocular adnexa (Fig. 8.50). These lesions may initially be misdiagnosed as subconjunctival hemorrhages, but the localized elevation of the area and the lack of spontaneous resolution differentiate the lesions. Biopsy is definitive, and antineoplastic agents are used for treatment.
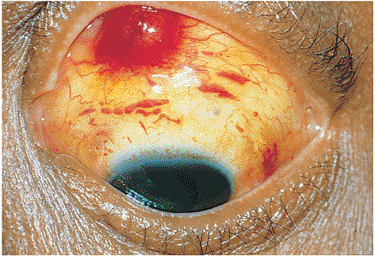 |
Figure 8.50. Kaposi's sarcoma. The conjunctival nodule was originally misdiagnosed as a subconjunctival hemorrhage. Biopsy revealed Kaposi's sarcoma. (Courtesy of Jerry Shields, M.D., Philadelphia, PA.) |
P.310
AIDS-Related Progressive Outer Retinal Necrosis
Herpes zoster infection in the immunocompromised HIV-positive individual can result in a severe, progressive outer retinal necrosis (PORN). It is a severe, potentially blinding disease that can cause irreversible loss of vision within days to weeks if not diagnosed and treated aggressively.
Clinical Features
Patients present with blurred vision and occasionally with ocular pain in one or both eyes. The hallmark of the disease is seen ophthalmoscopically in the form of small, punctate, opaque foci representing inflammation in the outer retina, occurring initially in the periphery (Fig. 8.51). Within days, the small spots coalesce to form a sheet of retinal necrosis that often extends 360 degrees around the globe. Cells in the vitreous are common. If the condition affects the posterior pole, optic nerve, or macular area, the necrosis may cause severe and irreversible loss of vision. Sometimes, systemic herpes zoster infection is detected several weeks before the eye involvement. In most cases, intense systemic immunosuppression has occurred, associated with CD4 counts below 50.
Management
The management of progressive outer retinal necrosis is difficult. The use of foscarnet, ganciclovir, and acyclovir, alone or in combination, has been advocated. Prompt evaluation and treatment may preserve vision. Retinal detachments are common in this condition, and the injection of silicone oil to tamponade the retina may preserve sight.
AIDS-Related Cytomegalovirus Retinitis
CMV retinitis is the most common ocular infection with the greatest ocular morbidity in AIDS patients. CMV retinitis was discussed earlier in this chapter and is reemphasized here because of the frequency with which this opportunistic virus affects the AIDS patient.
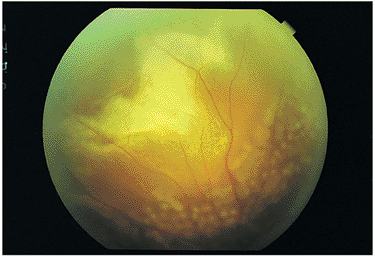 |
Figure 8.51. Progressive outer retinal necrosis (PORN). The PORN syndrome is associated with acute peripheral retinal necrosis, indicated here by the whitish areas located peripherally in the superior retina. Laser burns had been placed inferiorly to help wall off the area. |
Clinical Features
The presenting symptom is a painless decrease in vision in one or both eyes. No significant anterior segment reaction occurs, and ophthalmoscopic examination reveals patchy areas of whitish or grayish-white full-thickness necrotizing retinitis (Fig. 8.52). The necrotizing retinitis extends through contiguous spread into adjacent noninvolved retina, and progressive retinal destruction can lead to blindness. The more the immune system is compromised, the greater is the chance for rapid progression. CD4 counts in affected individuals are usually below 50.
Management
Treatment involves the use of anti-CMV drugs such as ganciclovir or foscarnet. Because these agents are not viricidal, treatment must be continued for the duration of the disease. In difficult cases, intravitreal injections have been used, as have ganciclovir intravitreal implants. If the immune system remains severely suppressed, recurrences may develop.
AIDS-Related Pneumocystis carinii Infection
Pneumocystis carinii is a ubiquitous protozoan organism that almost never causes disease in individuals with normal immune systems but becomes an opportunistic pathogen in immunosuppressed patients. P. carinii pneumonia is the most common form of systemic involvement, affecting 80% of patients with AIDS.
Clinical Features
Eye findings are rare, but choroiditis is the most common ocular manifestation (Fig. 8.53). This is seen ophthalmoscopically as multiple, yellow-white subretinal lesions that are 0.5 to 1 disc diameter. They are found more frequently in the posterior pole. There is no significant ocular inflammatory reaction, and patients are usually asymptomatic unless the macula is involved.
Management
These individuals are severely immunosuppressed, and their systemic status often overshadows the
P.311
ocular portion of the disease. P. carinii infection, including the choroiditis, is treated with trimethoprim-sulfamethoxazole (Bactrim) and pentamidine given parenterally or orally. These drugs may have significant side effects and should be prescribed by a physician familiar with their use.
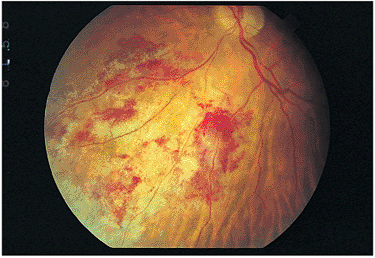 |
Figure 8.52. Cytomegalovirus (CMV) retinitis secondary to acquired immunodeficiency syndrome. The characteristic finding of yellowish CMV retinitis with secondary hemorrhage along the edges is seen off the inferior nasal areas of the human immunodeficiency virus positive individual, who had a CD4 count below 50. |
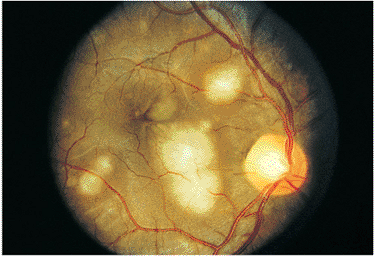 |
Figure 8.53. Pneumocystis carinii choroiditis in a patient with acquired immunodeficiency syndrome. Multifocal, whitish lesions are seen at the level of the choroid. Macular involvement often reduces vision, although the lesions are asymptomatic and clear promptly with appropriate antibiotic therapy. (Courtesy of Trent Wallace, M.D., Nashville, TN.) |
Masquerade Syndromes
Ophthalmologists use the term masquerade syndrome to describe eyes with uveitis (or simulated uveitis) in which the cause of inflammation is either neoplastic or nonuveitic in origin (Fig. 8.54). It is important to be aware of these conditions so that they do not escape clinical recognition and proper therapy. With a careful history and physical examination and a high index of suspicion, most masquerade syndromes can be discovered and treated correctly.
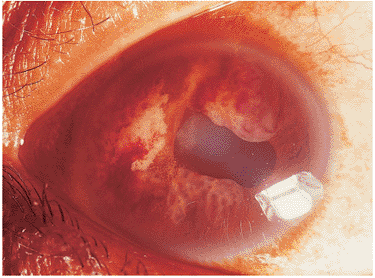 |
Figure 8.54. Anterior segment photograph of an adult male with what was believed to be uveal granulomas. Notice that there are no signs of active inflammation and the lesions turned out to be metastatic carcinoma. |
 |
Figure 8.55. Intraocular foreign body. The computed tomography scan reveals a small pellet in the inferior pars plana of the left eye that was caused by an undiagnosed injury 6 months earlier. Courtesy of Trent Wallace, M.D., Nashville, TN.) |
Intraocular Foreign Body
Clinical Features
A foreign body may penetrate the eye in such an unobtrusive manner that the seriousness of the event is not appreciated. A piece of metal from a lawn mower or a hammer may be tolerated in the eye for several months (Fig. 8.55). If iron is part of the metallic makeup, it can generate a condition called siderosis, which is manifested by low-grade inflammation around the site, cataractous changes, and chronic inflammation (Fig. 8.56).
Management
If a careful history reveals any possibility of an intraocular foreign body, a CT scan is needed to look for the metallic fragments. If siderosis has involved the retina, the electroretinogram may show a diminished b wave, and this may be helpful in making the diagnosis. Heterochromia of the iris can also be seen in chronic cases of siderosis. Surgical extraction of the foreign body is indicated and curative.
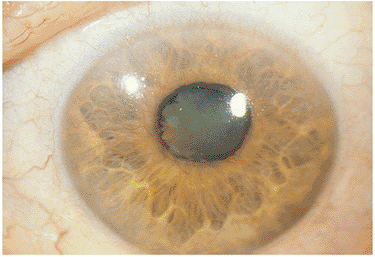 |
Figure 8.56. Siderosis secondary to an intraocular foreign body. Low-grade iridocyclitis, synechiae formation, cataract, and a change in iris coloration are characteristic of an occult intraocular metallic foreign body. |
P.312
Large Cell Lymphoma
Clinical Features
Large cell lymphoma (e.g., reticulum cell sarcoma, non-Hodgkin's lymphoma) is the most common and feared nonmelanoma cancer of the eye among older persons. The characteristic symptoms are chronic, bilateral spots and floaters from infiltration of the vitreous cavity (Fig. 8.57). Other findings include yellow-white, patchy subretinal infiltrates of lymphoma cells (Fig. 8.58), occasional vasculitis, and rarely, anterior segment involvement. Neurologic symptoms can occur. Large cell lymphomas often arise primarily within the eye, without systemic or central nervous system involvement. Systemic lymphoma primarily infiltrates the choroid and manifests as a thickening of the choroidal tissues.
Management
The diagnosis of intraocular large cell lymphoma depends on a high index of suspicion. Investigation includes CT scans, MRI, and a spinal tap, looking for central nervous system involvement. An intraocular vitreous biopsy may help in diagnosing large cell lymphoma (Fig. 8.59). The treatment of intraocular lymphoma is undergoing reevaluation, but it usually includes irradiation and aggressive chemotherapy.
Leukemia
Clinical Features
In younger individuals, leukemia may present as infiltrative lesions in the eye (Fig. 8.60). Ocular disease may present as hypopyon, vitreous infiltration, optic nerve infiltration, or secondary vasculitis. The ophthalmologist may be the first to diagnose the leukemia because of the ocular findings or to diagnose an acute leukemic exacerbation in a person previously in remission. The cells may masquerade as inflammatory cells, but because they are infiltrative, the condition is usually progressive.
Management
An anterior chamber tap or vitrectomy can aid diagnosis. Consultation with an oncologist should be obtained for further diagnosis and treatment.
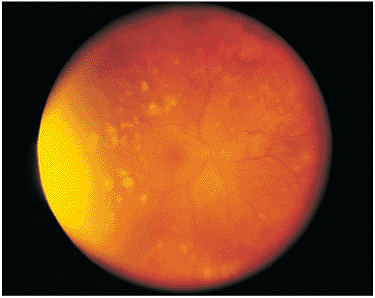 |
Figure 8.57. Reticulum cell sarcoma. A wide-angle photograph shows whitish, multifocal deposits at the level of the pigment epithelium, characteristic of reticulum cell sarcoma. A biopsy specimen revealed the characteristic histopathology. |
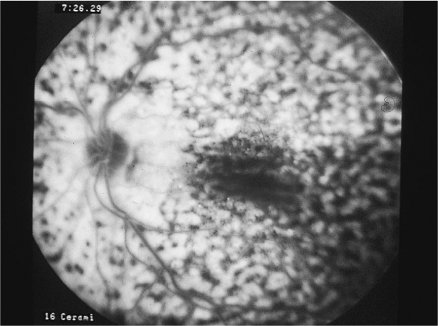 |
Figure 8.58. Fluorescein angiogram demonstrating the leopard spots pattern resulting from patchy subretinal infiltrates in a patient with reticulum cell sarcoma. |
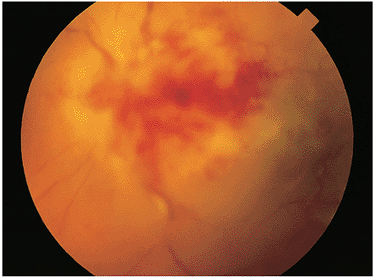 |
Figure 8.59. Disc area of a patient with acquired immunodeficiency syndrome demonstrating infiltration from intraocular lymphoma. A specimen was obtained by needle biopsy. |
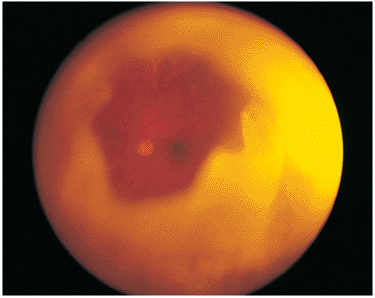 |
Figure 8.60. The wide-angle photograph shows a central core vitrectomy used to sample cells from a progressive opacification of the vitreous, which was thought to be cytomegalovirus retinitis in a leukemic individual in remission. The cells were positive for leukemia, and the patient died of fulminant systemic leukemia soon after the ocular diagnosis was made. (Courtesy of James J. Augsburger, M.D., Philadelphia, PA.) |
P.313
Posterior Scleritis
Posterior scleritis is a disorder in which intense inflammation and swelling primarily involve the posterior sclera. The primary disorder appears to be an immune reaction directed towards scleral collagen (similar to the immune reaction seen in joints of patients with rheumatoid arthritis), causing scleral swelling, dilation of blood vessels within the overlying choroid, and inflammation and other changes in adjacent ocular tissues.
Clinical Features
There is an acute onset of deep-seated and aching pain, usually in one eye only. Blurred vision and pain on eye movement may be present or absent. Fundus examination reveals subretinal edema or thickening that may resemble an ocular tumor, and serous elevation of the overlying retina may also be seen (Fig. 8.61). In less acute cases, choroidal folds may be noted. Depending on the area of involvement, visual acuity may be affected by optic nerve swelling or by elevation and distortion of the macula. Fluorescein angiography reveals a characteristic picture of initial pinpoint hyperfluorescence leading to pooling of fluorescein dye subretinally. The ultrasound scan shows fluid between the layers of the eye and orbital tissues (Fig. 8.62).
Posterior scleritis frequently occurs in the absence of any significant underlying systemic disease, although rheumatoid arthritis, Crohn's disease, and Wegener's granulomatosis have all been associated with posterior scleritis, and must be ruled out. Thorough medical evaluation and laboratory testing for collagen vascular disorders are indicated.
Management
Therapy is focused on controlling the inflammation. Short-term systemic corticosteroid therapy often brings prompt relief, although recurrences are possible. Nonsteroidal antiinflammatory agents are helpful. Posterior scleritis has a variable course, and if inflammatory damage is minimal, visual function can remain excellent.
 |
Figure 8.61. Posterior scleritis. The findings of acute pain and a mass, seen here in the fundus nasally, are characteristic of posterior scleritis. |
 |
Figure 8.62. The A- and B-scan ultrasonograms of the area described in Figure 8.61 show the mass. The characteristic fluid line seen posteriorly is called the T sign. The scan denotes the size of the lesion. (Courtesy of James J. Augsburger, M.D., Philadelphia, PA.) |
Endophthalmitis
Endophthalmitis refers to severe infection involving all the coats of the eye. The infectious process, depending on the underlying cause, may present in different manners. If the infectious nidus is internal and has spread from another infected organ, it is called endogenous. If the infectious process follows intraocular surgery or penetrating ocular trauma, it is called exogenous. Both forms of endophthalmitis are true ophthalmic emergencies that can lead to blindness if not diagnosed and treated appropriately and aggressively.
Endogenous Endophthalmitis
Clinical Features
Endogenous endophthalmitis is usually associated with systemic infection. In immunocompromised individuals, such as those receiving long-term intravenous therapy or hyperalimentation or those debilitated from other causes, an infectious nidus of bacteria or fungi reaches the eye through the choroidal or retinal circulation.
The initial symptoms may be minimal, such as spots and floaters. Severe visual loss may result from opacification of the vitreous; full-thickness inflammation of the retina, choroid, and sclera; and opacification in the anterior segment. Initially, the findings may mimic acute nongranulomatous anterior uveitis, but the rapid progression and the nature of the clinical picture are usually characteristic of endogenous endophthalmitis (Fig. 8.63).
Management
Immediate vitrectomy surgery or vitreous biopsy is needed to identify the offending organism. Intravenous and intraocular broad-spectrum antibiotic treatment should be initiated promptly until the specific organism is isolated. If caught early, endogenous endophthalmitis has a good visual prognosis, but severe cases can lead to severe scarring, blindness, phthisis bulbi, or loss of the eye.
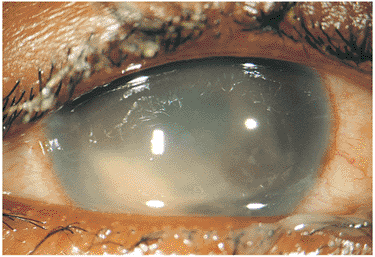 |
Figure 8.63. Subacute bacterial endocarditis (SBE) and endogenous endophthalmitis. Severe vitritis and an acute-onset fibrinoid and exudative reaction were detected in the anterior chamber of a patient with SBE. Culture-proven streptococci were recovered. |
P.314
Exogenous Endophthalmitis
Clinical Features
Exogenous endophthalmitis refers to intraocular infection, usually bacterial, which occurs after intraocular surgery or penetrating ocular trauma. The offending agents can occur from many sources, although the usual source in postsurgical endophthalmitis is patient flora or contamination of instruments or fluids, yielding an infectious culture in the eye. Depending on the series reported, the incidence of endophthalmitis after cataract surgery is one in 500 to 10,000 surgical cases. The technique involved, the degree of intraocular manipulation, and the duration and complexity of surgery may influence the infection rate.
The clinical features include postoperative pain out of proportion to normal postoperative healing within 24 hours to 1 week after surgery, a steamy inflammation of the cornea with a fibrinoid anterior chamber reaction, and hypopyon (Fig. 8.64). The size of the hypopyon often reflects the bacterial load or aggressiveness of the offending organism. Gram-positive bacteria, such as Staphylococcus epidermidis, are some of the more common causes of postoperative endophthalmitis and generally produce milder disease. Staphylococcus aureus, Streptococcus pneumoniae, and Gram-negative organisms usually produce much more severe clinical findings. The onset of pain, redness, and hypopyon and, sometimes, complete opacification of the cornea with intraocular pus formation can occur within 8 hours (Fig. 8.65). Endophthalmitis after penetrating ocular trauma can be caused by multiple organisms, including Bacillus cereus, a particularly virulent organism, which can lead to rapid destruction of the eye.
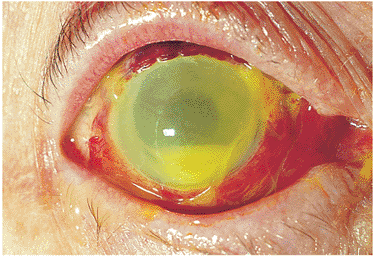 |
Figure 8.64. Acute postoperative endophthalmitis. One day after cataract surgery, the characteristic findings of redness, pain, discomfort, and hypopyon were exhibited by this elderly individual with -hemolysin streptococcal endophthalmitis. Fluorescein staining shows severe superficial punctate keratopathy of the cornea. |
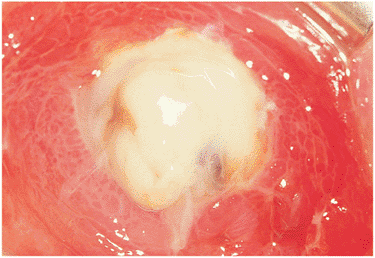 |
Figure 8.65. Severe postoperative endophthalmitis. Severe coagulative necrosis of the cornea from Serratia endophthalmitis is evident 3 days after routine cataract surgery. Despite aggressive therapy, vision in this eye was lost. |
Bleb-associated endophthalmitis is a special case of postsurgical endophthalmitis. Symptoms may develop months to years after the procedure. Unlike chronic endophthalmitis, the onset is sudden. Haemophilus influenzae and S. pneumoniae are common pathogens.
Chronic postoperative endophthalmitis can arise after extracapsular cataract surgery. Usually developing 2 weeks
P.315
to 2 months postoperatively, low-grade granulomatous inflammation causes mild discomfort, occasional hypopyon, vitritis, and blurred vision. Occult infection with Propionibacterium acnes often is the cause, although S. aureus can also result in an identical clinical picture. The infecting organism becomes sequestered in the capsular bag and causes low-grade inflammation.
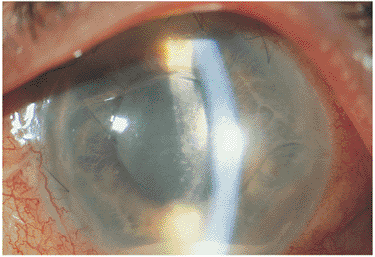 |
Figure 8.66. Propionibacterium acnes endophthalmitis is characteristically indolent, producing minimal pain and photophobia. Granulomatous keratic precipitates and a small hypopyon are seen. |
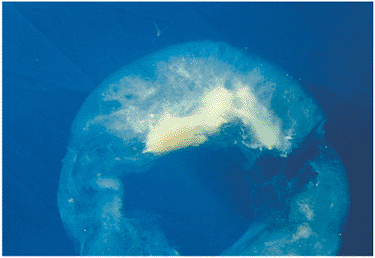 |
Figure 8.67. A biopsy specimen of a peripheral capsular bag shows the whitish lesions characteristic of Propionibacterium acnes infection. These can often be seen clinically and help to make the diagnosis. |
Management
The secret to successful treatment is early recognition and appropriate therapy. Depending on the amount of scarring of important tissues, vision may be quite good, especially if the organism is not virulent and is caught early. Unfortunately, the loss of ciliary body function, corneal opacification, retinal obliteration due to toxins or severe uncontrolled inflammation, and loss of optic nerve function can cause blindness. In chronic endophthalmitis due to P. acnes, removal of the lens and capsular bag or intracameral antibiotics to eradicate the infection usually ensures the return of good vision (Figs. 8.66 and 8.67).
Endogenous or exogenous endophthalmitis can result in complete loss of vision or loss of the eye. The secret to preservation of ocular integrity and vision is prompt recognition and appropriate and aggressive treatment directed to the offending organism. Visual outcome can be good if the infection is caught early.
Bibliography
Michaelson SB. Color atlas of uveitis diagnosis. 2nd ed. St. Louis: Mosby; 1992.
Nussenblatt RB, Whitcup SM, Palestine AG. Uveitis. Fundamentals in clinical practice. 2nd ed. St. Louis: Mosby; 1996.
Opremcak EM. Uveitis: A clinical manual for ocular inflammation. New York: Springer-Verlag; 1995.
Pepose JS, Holland GN, Wilhelmus KR. Ocular infection and immunity. St. Louis: Mosby; 1996.
Rao NA, Augsburger JJ, Forster DJ. The uvea: Uveitis and intraocular neoplasms. New York: Gower; 1992.
Roitt IM. Essential immunology. 3rd ed. Malden, MA: Blackwell Science; 1997.
EAN: 2147483647
Pages: 17