29 - Drug Interactions in Psychopharmacology
Editors: Shader, Richard I.
Title: Manual of Psychiatric Therapeutics, 3rd Edition
Copyright 2003 Lippincott Williams & Wilkins
> Table of Contents > 29 - Drug Interactions in Psychopharmacology
function show_scrollbar() {}
29
Drug Interactions in Psychopharmacology1
Karthik Venkatakrishnan
Richard I. Shader
Lisa L. von Moltke
David J. Greenblatt
A clinically significant drug interaction occurs when the therapeutic or toxic effects of a medication are altered by coadministration with another drug. Likely possibilities include diminished efficacy or enhanced efficacy or toxicity. Conceptually, to think of these interactions as happening between an inhibiting or inducing agent, which one can think of as the perpetrator, and the affected agent, which one can think of as the victim, can be helpful (Fig. 29.1). Drug interactions are particularly notable when the victim drug has a relatively narrow therapeutic index (e.g., tricyclic antidepressants [TCAs], lithium, mood-stabilizing anticonvulsants). Some interactions are predictable, and they are easily understood because they involve the action of two or more separate agents working through different receptors or different sites on the same receptor to enhance the same pharmacodynamic (see below) outcome (e.g., increased central nervous system [CNS] depression when a sedative-hypnotic agent, such as diazepam, is taken with alcohol). Some are easily understood pharmacokinetically (e.g., when two drugs are substrates for the same catabolic enzyme and compete for it). Others are unpredictable; these are discovered by chance or through large-scale epidemiologic studies. An example of the latter is the potentiation of oral anticoagulants by the sedative-hypnotic chloral hydrate. Regrettably, unexpected interactions may sometimes result in serious or fatal drug toxicity.
I. Classification of Drug Drug Interactions: A Mechanistic Perspective
Drug interactions may be categorized as pharmacodynamic (i.e., additive, antagonistic, or synergistic effects at the level of mechanism of action at the target or effect site) or pharmacokinetic (i.e., modulation of the time course or magnitude of drug concentrations in body compartments and thus of drug action at the effect or target site). Drug antagonism can occur when one drug prevents another from reaching or acting at its receptor or binding site. A classic example of such an interaction is that between the TCAs and the antihypertensive agent guanethidine. The mechanism of action of TCAs partly involves the inhibition of neurotransmitter reuptake at the noradrenergic synapse. The site of action of guanethidine is the presynaptic adrenergic neuron where it works as a substitute or false neurotransmitter and depletes catecholamine-containing vesicles of their native neurotransmitter, thereby producing an antihypertensive effect. Guanethidine reaches its site of action by active transport into the neuron, a process mediated by the same transporter that is responsible for norepinephrine reuptake. In patients receiving TCAs, the function of this transporter is blocked by the antidepressant. As a result, guanethidine cannot reach its site of action, making it ineffective as an antihypertensive agent.
Pharmacokinetic interactions can result from a variety of mechanisms, including the modulation of hepatic drug biotransformation, renal clearance, drug transport, distribution, and plasma protein binding. One drug may alter the bioavailability of another by changing its dissolution characteristics, altering the ambient pH, causing variations in gastrointestinal motility, inhibiting presystemic extraction at the level of the small intestine or liver, or modulating drug transport in the intestine. Some drugs are excreted in significant amounts
P.442
P.443
by the kidney, and their excretion may be modified by agents that alter glomerular filtration, tubular function, or urinary pH. For example, thiazide diuretics increase the elimination of sodium and inhibit the renal clearance of lithium; coadministration may result in lithium toxicity, unless compensatory dosage reductions of lithium are made.
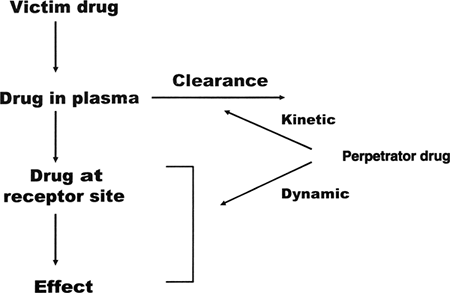 |
FIG. 29.1. A model for drug interactions. |
Many pharmacokinetic drug interactions result from the modulation of drug biotransformation or metabolism. The liver is the major site of drug metabolism, with the small intestine playing a secondary, yet important, role in the oral clearance of certain therapeutic agents. A number of psychotropic drugs can inhibit or induce hepatic drug metabolizing enzymes, thereby impairing or enhancing, respectively, the liver's ability to detoxify coadministered drugs. Metabolic drug drug interactions in psychopharmacology can occur in two ways. First, the metabolic clearance of the psychotropic agent may be altered by another coadministered psychotropic or nonpsychotropic agent. Second, the metabolic clearance of a nonpsychotropic agent may be altered by a psychotropic agent. Several examples of both kinds of interactions have been noted in the clinical literature, and representative examples follow. For example, the detoxification of the TCA nortriptyline requires hepatic biotransformation via hydroxylation, a reaction catalyzed by the hepatic microsomal enzyme cytochrome P-450 (CYP) 2D6 (see section II). The antifungal agent terbinafine is a potent inhibitor of CYP 2D6. Consider a patient receiving long-term terbinafine for onychomycosis. Initiation of antidepressant treatment in this patient with usual therapeutic doses of nortriptyline may result in nortriptyline-related toxicity because nortriptyline concentrations in the plasma, and hence at the sites of pharmacologic or toxicologic action (e.g., brain and heart, respectively), can increase beyond the upper limit of the therapeutic range due to impaired hepatic clearance. In the above example, modulation of the clearance of a psychotropic agent by a coadministered drug belonging to an unrelated therapeutic category is evident. The reverse is illustrated in the interaction between the antimalarial prodrug proguanil and the selective serotonin reuptake inhibitor (SSRI) antidepressant fluvoxamine, a potent inhibitor of CYP 2C19. Proguanil actions require hepatic biotransformation to its active metabolite cycloguanil, a step mediated by CYP 2C19. As a result, proguanil's antimalarial efficacy may be compromised in a patient receiving fluvoxamine through its impairment of CYP 2C19-mediated metabolite formation.
During the last few decades, a dramatic increase has occurred in the understanding of the molecular biology and biochemistry of the CYP enzymes, enabling a better understanding and improved prediction of metabolic drug drug interactions in psychopharmacology. Although the widespread use of SSRIs has greatly improved the prognosis of the depressed patient, it has also become evident that the SSRIs constitute a group of drugs with a high risk of drug interactions as a result of CYP inhibition. Thus, a large fraction of drug interactions in psychopharmacology is mediated by metabolic mechanisms, especially at the level of the CYP enzymes. The following discussion of human CYP enzymes and their interactions with psychopharmacologics will hopefully not only allow the psychiatrist to be aware of the reported interactions (e.g., in a package insert or a computerized drug interaction program) but also will provide a mechanistic basis that will allow their management and the forecasting of unidentified interactions.
II. Human Cytochrome P-450 Isozymes and Psychopharmacologic Agents
CYP enzymes are estimated to account for the biotransformation of approximately 60% of the commonly prescribed drugs in the United States. Although the human drug-metabolizing CYPs are expressed in several tissues, they are concentrated in the smooth endoplasmic reticulum of the zone III hepatocytes in the liver, with lower levels of expression in the intestine, lungs, kidneys, and brain. The multiple CYP enzymes are classified into families, subfamilies, and isoforms based on a systematic nomenclature. The major human drug-metabolizing CYPs belong to families 1, 2, and 3, with the specific isoforms of interest to drug interactions in psychopharmacology being 1A2, 2B6, 2C9, 2C19, 2D6, and 3A4.
P.444
CYP 1A2 is the primary enzyme responsible for the human hepatic metabolism of phenacetin, tacrine, caffeine, and theophylline, and it also plays a role in the metabolism of clozapine and imipramine. Clinically significant CYP 1A2 inhibitors include the SSRI antidepressant fluvoxamine, the antihelminthic agent thiabendazole, and the fluoroquinolone antibiotics ciprofloxacin and enoxacin.
CYP B6 is the sole mediator of the hydroxylation of the antidepressant bupropion, and it contributes significantly to the metabolism of the anticonvulsant S-mephenytoin, the barbiturate S-mephobarbital, the alkylating anticancer agent cyclophosphamide, and the intravenous anesthetic propofol. CYP 2B6 activity is inhibited by the SSRI antidepressants fluvoxamine, sertraline, and paroxetine and by norfluoxetine, the active metabolite of fluoxetine, based on in vitro studies. Based on in vitro studies, the antiretroviral agents ritonavir and efavirenz are also potent CYP 2B6 inhibitors.
CYP 2C8 plays a major role in the biotransformation of the antineoplastic agents paclitaxel and all trans-retinoic acid. This enzyme is also partially responsible for the metabolism of the nonbenzodiazepine (BZ) hypnotic zopiclone, based on in vitro studies. The clinical implications of CYP 2C8-mediated drug metabolism remain to be understood, and insufficient information is available on the clinical consequences of inhibition or induction of this enzyme.
CYP 2C9 is the primary enzyme responsible for the metabolism of the oral hypoglycemic agent tolbutamide, the anticoagulant S-warfarin, the anticonvulsant phenytoin, the antihypertensive agent losartan, and several nonsteroidal antiinflammatory drugs. Clinically significant CYP 2C9 inhibitors include fluoxetine and fluvoxamine, the sulfonamide antimicrobials sulfamethoxazole and sulfinpyrazone, and the azole antifungal agents miconazole and fluconazole. Clinically significant inducers of CYP 2C9 include barbiturates, carbamazepine, and rifampin.
CYP 2C19 is polymorphically expressed, and it contributes significantly to the clearance of the monoamine oxidase inhibitor (MAOI) moclobemide, the BZ diazepam, the antiulcer drug omeprazole, the antimalarial agent proguanil, and the antidepressants citalopram and amitriptyline. Clinically significant CYP 2C19 inhibitors include fluvoxamine, omeprazole, fluconazole, and the antithrombotic agent ticlopidine. The genetic polymorphisms associated with this isoform result in a poor metabolizer phenotype at a frequency of less than 5% in whites and at a greater frequency of 12% to 20% in Asian populations.
CYP 2D6 is partly or entirely responsible for the metabolism of a variety of psychopharmacologic and cardiovascular drugs, including thioridazine, perphenazine, desipramine, nortriptyline, paroxetine, venlafaxine, codeine, metoprolol, encainide, flecainide, propafenone, propofol, and mexiletine. Clinically significant CYP 2D6 inhibitors include fluoxetine and paroxetine; the conventional antipsychotic agents haloperidol, thioridazine, chlorpromazine, and perphenazine; the allylamine antifungal agent terbinafine; the antithrombotic drug ticlopidine; the antiretroviral agent ritonavir; and the antiarrhythmic agent quinidine. CYP 2D6 is polymorphically expressed with a frequency of poor metabolizers of approximately 5% to 10% in the white population. Table 29.1 lists some substrates and inhibitors for CYP 2D6.
CYP 3A4 is the most abundant CYP enzyme in both the human liver and small intestine. Most oxidatively biotransformed therapeutic agents are metabolized at least in part by this enzyme. Examples of drugs that are primarily metabolized by CYP 3A4 include immunosuppressants, such as cyclosporin A and tacrolimus; sedative-hypnotic agents, such as midazolam, triazolam, and alprazolam; antidepressants, such as trazodone and nefazodone; the conventional antipsychotic agent haloperidol; calcium channel blockers such as nifedipine, felodipine, and diltiazem; antiarrhythmic agents, such as amiodarone, quinidine, and lidocaine; antiinfective agents, such as erythromycin, quinine, ritonavir, saquinavir, and amprenavir; antineoplastic agents, such as etoposide, ifosfamide, tamoxifen, and vinblastine; synthetic opioids, such as fentanyl,
P.445
alfentanil, and sufentanil; and the nonsedating antihistaminic agents terfenadine, loratadine, and astemizole. Table 29.2 lists some substrates for CYP 3A.
TABLE 29.1. SELECTED EXAMPLES OF CYTOCHROME P-450 2D6 SUBSTRATES AND INHIBITORS | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Examples of clinically significant CYP 3A4 inhibitors include the macrolide antibiotic erythromycin, the azole antifungal agents ketoconazole and itraconazole, the antiretroviral drug ritonavir, the calcium channel blocking agent diltiazem, the progestin gestodene, and the active metabolite of fluoxetine norfluoxetine. Inducers of CYP 3A4 include rifampin, dexamethasone, phenytoin, carbamazepine, phenobarbital, ritonavir, and nevirapine, as well as the herbal antidepressant St. John's wort. Table 29.3 lists some inhibitors and inducers of CYP 3A.
The various CYP enzymes have distinct yet overlapping substrate specificities. Thus, in many cases, multiple enzymes can mediate the metabolism of a single drug. The consequence of CYP inhibition for a pure substrate of a single CYP isoform will be different from that for a drug that can be metabolized by multiple enzymes. This is evident upon examination of the drug interactions of the sedative-hypnotic agents triazolam and zolpidem with the CYP 3A4-inhibitory antifungal agent ketoconazole. Triazolam is a pure CYP 3A4 substrate; zolpidem is metabolized by multiple CYP isoforms, including CYP 3A4. Coadministration of ketoconazole with triazolam results in a clinically significant interaction due to a greater than 90% inhibition of triazolam clearance. The pharmacodynamic consequences of this interaction can be profound, and they manifest as enhanced CNS depression and prolonged sleep time when compared with the short-lived hypnotic effects of triazolam taken by itself. On the other hand, coadministration of zolpidem with ketoconazole results in
P.446
only a 40% decrement in zolpidem's oral clearance. This is because CYP 3A4 accounts only for 40% to 60% of zolpidem's catabolism. CYPs 2C9 and 1A2 also metabolize zolpidem and can thus compensate when CYP 3A4 is inhibited. Thus, an understanding of the relative contributions of CYP enzymes mediating the biotransformation of a drug is useful for predicting the magnitude and clinical consequences of an interaction resulting from metabolic inhibition.
TABLE 29.2. SELECTED EXAMPLES OF CYTOCHROME P-450 3A SUBSTRATES | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
TABLE 29.3. SELECTED EXAMPLES OF CYTOCHROME P-450 3A INHIBITORS AND INDUCERS | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
Although the inhibition of a CYP isoform that plays a relatively minor role in the overall metabolism of a drug is generally clinically unimportant, the consequences of induction of such relatively minor contributors should not be overlooked. This is because, when induced, the relative contribution of low-affinity CYP isoforms will increase due to increased hepatic enzyme content, which can result in an acceleration of overall drug clearance.
Another important consideration in predicting the outcome of a metabolic drug interaction is the pharmacokinetic properties of the victim drug. This can be explained by considering the interactions between the BZs triazolam or alprazolam and the CYP 3A4 inhibitor ketoconazole. As was discussed above, the clinical consequences of the inhibition of triazolam clearance by ketoconazole are significant; the interaction results in a large increase in the peak plasma concentration that is attained after an oral dose of triazolam, as well as an exaggerated pharmacodynamic response to the drug. On the other hand, the consequences of metabolic inhibition by ketoconazole on alprazolam, another pure CYP 3A4 substrate, pharmacokinetics and pharmacodynamics are much more modest; a significant prolongation of its elimination half-life is seen, coupled with an insignificant increase in its peak plasma concentrations. Alprazolam is a poorly extracted drug having high oral bioavailability. Conversely, triazolam undergoes extensive presystemic extraction by CYP 3A4-mediated metabolism both in the liver and the small intestine. Thus, inhibition of metabolism of triazolam will result in a large increase in peak plasma concentrations after an oral dose and in enhanced pharmacodynamic activity. On the contrary, the predominant effect of metabolic inhibition for a poorly extracted victim drug will be a prolongation of the elimination half-life, which is less likely to affect the pharmacodynamic effects after single doses.
Worth emphasizing is that, although CYPs are the most important drug metabolizing enzymes in humans and are generally the focus of drug interaction mechanisms, other non-CYP enzymes (e.g., glucuronosyl transferases, N-acetyl- transferase, epoxide hydrolase) are also important for some psychopharmacologic agents. For example, lorazepam undergoes glucuronidation that can be inhibited by the uricosuric agent probenecid, resulting in a decrease in lorazepam clearance. Acetylation steps involve the enzyme N-acetyltransferase.
P.447
This enzyme reveals a genetic polymorphism; that is, a single gene yields two or more alleles, and the least common allele is present in at least 1% of the population. Slow acetylator status is found in approximately one-half of white and African-American cohorts. Some Oriental and Eskimo cohorts contain about 10% and 5% slow acetylators, respectively. The most common slow allele that is found in whites is not present in Japanese cohorts. The metabolism of phenelzine involves an acetylation step. Carbamazepine undergoes CYP 3A4-mediated oxidation to a toxic epoxide metabolite that undergoes hydrolysis by epoxide hydrolase to yield a nontoxic diol metabolite. Valproic acid (VPA) can inhibit epoxide hydrolase, and coadministration with carbamazepine can result in neurotoxicity due to elevated levels of carbamazepine epoxide.
In addition to metabolic biotransformation, other processes that influence the pharmacokinetics of a drug include protein binding and drug transport. Modulation of active (i.e., energy-dependent) drug transport by P-glycoprotein is being increasingly recognized as a mechanism of drug drug interactions. P-glycoprotein is an important molecular determinant of the oral bioavailability, tissue distribution, and the clearance of several therapeutically used drugs (e.g., cyclosporin, digoxin, anthracycline antineoplastic agents, vinca alkaloids, opioids, human immunodeficiency virus [HIV] protease inhibitors). Fortunately, most commonly used prescription psychotropic agents are not clinically important P-glycoprotein substrates. In fact, avid P-glycoprotein substrates are generally excluded from the CNS due to the barrier function of this transporter in the blood brain barrier. Interestingly though, the over-the-counter herbal antidepressant St. John's wort is a clinically significant inducer of P-glycoprotein; studies demonstrate a decrease in the oral bioavailability and plasma concentrations of P-glycoprotein substrates, such as digoxin, indinavir, and cyclosporin, when they are coadministered with this herbal preparation. (Note: The net cyclosporin effect is mediated additionally via the induction of CYP 3A4.)
III. Overview of Drug Interactions With Psychopharmacologic Agents
Valuable data on drug interactions have been developed in the last several decades. Misinformation has also been published and perpetuated. Interactions are sometimes suspected on the basis of clinical observation of one or two cases. Unfortunately, when these appear in the medical literature as case reports, many readers and drug-interaction databases interpret them as depicting established fact. Systematic study often reveals such apparent interactions to be coincidental or spurious. Animal studies are another potential source of misinformation. Drug interactions that are strikingly evident in animal investigations may have no clinical relevance. Finally, biochemical or pharmacokinetic theory may predict certain interactions. Regrettably, these theoretical or in vitro interactions may be depicted as fact, even when clinical experience does not substantiate them. Uncritical interpretation of case reports, animal studies, in vitro data, or theoretical models can generate and perpetuate facts that are not really facts.
In the remainder of this chapter, a limited overview of drug interactions relevant to clinical psychopharmacology is presented. Every effort has been made to evaluate the strength of the evidence and to cull out published speculation. Examples of representative agents are presented. For some classes of agents, the interaction may generalize to the whole class. For others, the interaction may be specific to the noted drugs. This chapter is intended to raise clinicians' awareness of drug interactions; it cannot substitute for reading current product labeling on a specific medication or for consulting an up-to-date drug interaction text or computer program. Each major grouping of psychopharmacologic agents is presented separately.
A. Conventional Antipsychotic Agents (Neuroleptics)
These classes of agents (e.g., phenothiazines, butyrophenones) generally have a wide therapeutic index, so an alteration in their own metabolism is rarely a clinical problem in either their reducing efficacy or producing toxicity. However, given the understandable trend toward using the lowest possible doses of these agents, any interaction that further lowers a drug's concentration could have clinical significance. Some antipsychotic agents
P.448
affect the metabolism of other drugs (e.g., TCAs, phenytoin), and some of these interactions have important clinical implications. Most significant interactions reflect toxicity as a consequence of pharmacodynamic interactions. However, recent in vitro studies indicate that perphenazine and thioridazine and, to a lesser extent, chlorpromazine and haloperidol are clinically significant CYP 2D6 inhibitors and that they can produce decrements in the clearance of drugs that are in large part metabolized by this enzyme.
Anticholinergic agents. Anticholinergic antiparkinsonian agents (e.g., benztropine, biperiden, trihexyphenidyl) are commonly given with antipsychotic agents to reduce the extrapyramidal effects of the latter (see Chapter 20). When they are used with the so-called low potency antipsychotic agents (e.g., chlorpromazine, thioridazine), additive anticholinergic effects may lead to toxicity (e.g., delirium, paralytic ileus). Whether anticholinergic agents affect the efficacy of antipsychotic agents remains unresolved; most studies suggest that they do not.
Mood-stabilizing anticonvulsant agents. Carbamazepine induces microsomal enzymes, and it may lead to lower systemic levels of antipsychotic medications. This may result in clinical deterioration, but the consequences are highly variable depending on the doses of the specific antipsychotic agent, whether it has active or toxic metabolites, the disorder(s) being treated, and other concurrent drug therapies. VPA may inhibit the metabolism of chlorpromazine but not that of haloperidol. Chlorpromazine may inhibit the metabolism of phenytoin. Prochlorperazine and thioridazine may similarly interact with phenytoin, but often no change in metabolism is seen with these combinations. Haloperidol does not affect phenytoin levels; phenytoin, however, may reduce haloperidol or clozapine levels. Antipsychotic agents (e.g., loxapine) may lower the seizure threshold and may antagonize the actions of phenytoin.
Antidepressants. TCAs increase the plasma levels of some antipsychotic drugs. Imipramine and nortriptyline increase chlorpromazine levels. Fluoxetine impairs haloperidol metabolism; it likely interferes with the metabolism of other antipsychotic agents as well, because oxidative metabolism is characteristic for this group. Fluoxetine may also have indirect additive or synergistic effects in reducing dopamine activity, which may lead to extrapyramidal symptoms. When perphenazine is combined with paroxetine, an increase in perphenazine plasma levels and CNS side effects, including extrapyramidal effects, can occur. Both fluoxetine and paroxetine are potent CYP 2D6 inhibitors, explaining at least in part their interactions with a number of conventional antipsychotic agents. Bupropion as an inhibitor of CYP 2D6 also has the potential to inhibit the clearance of agents, such as haloperidol and thioridazine.
Haloperidol, thiothixene, chlorpromazine, perphenazine, and fluoxetine inhibit the metabolism of imipramine, desipramine, and nortriptyline. Thioridazine increases desipramine levels. These effects are mediated via the inhibition of CYP 2D6-mediated metabolism of the TCAs. Increased levels of many TCAs may lead to anticholinergic problems (e.g., constipation, paralytic ileus) or cardiac toxicity (e.g., prolonged PR or QTc intervals); monitoring of electrocardiograms is strongly advised.
Antihypertensives. The two major types of interactions of clinical concern between antipsychotic agents and antihypertensives are those that cause delirium (see Chapter 4) and those with an enhanced or blocked hypotensive effect, depending on the specific antihypertensive agent. Captopril and perhaps other angiotensin-converting enzyme inhibitors, propranolol, and methyldopa may have enhanced hypotensive effects when they are coadministered with chlorpromazine. Although this effect
P.449
seems predictable from chlorpromazine's strong 1-adrenergic receptor antagonist properties, even haloperidol may interact with propranolol to produce serious hypotensive episodes. Atenolol, a drug that is predominantly cleared renally, may be a useful alternative to propanolol and metoprolol when CYP 2D6-inhibitory antipsychotic agents, such as thioridazine or perphenazine, are used. Chlorpromazine coadministration with methyldopa may be associated with both hypotension and hypertension. The antihypertensive effect of guanethidine may be reduced by the concomitant use of chlorpromazine, because the latter inhibits noradrenergic reuptake. The antagonism is weaker with or thiothixene; it does not occur, however, with molindone. Bethanidine, debrisoquin, and guanadrel are similar to guanethidine in their mechanisms of action, and they would be expected to interact with antipsychotic agents in a similar manner.Benzodiazepines. The most serious interaction between BZs and antipsychotic agents causes respiratory depression that may occur when clozapine is concurrently used with a BZ. Other antipsychotic agents do not appear to present problems when they are given together with BZs, and combinations are frequently used. Patients taking alprazolam with fluphenazine or haloperidol may have higher antipsychotic agent blood levels. When additional sedation is required, adding a BZ is often preferable to using higher doses of antipsychotic agents.
Lithium. Encephalopathic syndromes characterized by lethargy, fever, confusion, extrapyramidal symptoms, and cerebellar dysfunction have been reported when lithium is coprescribed with some antipsychotic agents. Although most of these reports have involved a haloperidol lithium combination, similar reports are found with thioridazine, perphenazine, and thiothixene. The greater reporting frequency for the haloperidol lithium interaction likely reflects the greater clinical use of this combination. Many believe that these reports reflect patients with idiosyncratic manifestations of lithium toxicity (i.e., these are not true drug interactions). Nevertheless, caution should be used when prescribing these combinations, even though most patients tolerate them well.
Antiretroviral agents. The coadministration of the HIV-1 protease inhibitor ritonavir and pimozide is contraindicated. This interaction may involve the inhibition of drug transport, as well as the inhibition of CYP 3A4. Ritonavir also is a potent inhibitor of CYP 2D6, and it may thus impair the clearance of perphenazine, risperidone, and thioridazine.
Antiarrhythmic agents. The clearance of propafenone and encainide is CYP 2D6 mediated. Coadministration with thioridazine or perphenazine can potentially result in the impairment of clearance.
Other medications. Antacids, cimetidine, and antidiarrheals may impair the absorption of some antipsychotic agents. Cimetidine may impair the metabolism of clozapine and may lead to toxicity. Buspirone may increase haloperidol levels. When alcohol is ingested during the use of some antipsychotic agents, it may enhance sedation. Alcohol use may also worsen extrapyramidal symptoms. The coadministration of amantadine and thioridazine can worsen the tremor in elderly patients with Parkinson disease. However, whether amantadine combined with other antipsychotic agents produces a similar response is not known. Erythromycin and clarithromycin impair the metabolism of pimozide via the inhibition of CYP 3A4, resulting in cardiac toxicity (e.g., bradycardia); thus, coadministration is contraindicated. Ketoconazole has been shown to reduce the clearance of quetiapine. Azithromycin
P.450
may be a safe alternative macrolide antibiotic because it does not inhibit CYP activity.
B. Atypical (novel) antipsychotic agents
Unlike the conventional antipsychotic agents, several of which are inhibitors of CYP 2D6, the atypical agents clozapine, olanzapine, quetiapine, and ziprasidone do not perpetrate drug interactions as CYP inhibitors.
Clozapine is associated with a significant risk for agranulocytosis, the mechanism of which remains unclear. Therefore, clozapine should not be used with other agents having a known potential to suppress marrow function (i.e., a potential pharmacodynamic interaction). In addition, the oxidation metabolism of clozapine, which is mediated by multiple CYP isoforms, including 2C19, 3A4, 1A2, and 2D6, is impaired by the coadministration of any SSRIs that inhibit these CYPs. The most notable of these is fluvoxamine. The resulting increase in serum concentrations of clozapine may increase the likelihood of clozapine's unwanted effects. (Note: Although many of these unwanted effects are concentration dependent, concentration per se is not.) Such combinations should be approached with caution, and patients may require close monitoring.
Olanzapine's metabolism is mediated by CYPs 1A2 and 2D6, flavin-containing monooxygenase 3, and glucuronidation. Because of the contribution of CYP 1A2 to the metabolism of both olanzapine and clozapine, the clearance of these two agents is higher and, therefore, the steady-state concentrations are lower in smokers. Even so, routine dosage adjustments are not usually recommended.
Quetiapine undergoes hepatic clearance that is largely mediated by CYP 3A4. Coadministration of CYP 3A4 inhibitors can therefore result in impaired clearance and elevated concentrations, as has been shown with the CYP 3A-inhibiting azole antifungal agent ketoconazole. CYP 3A inducers may exert the reverse effect. Increased amounts of quetiapine may be required for patients taking this along with phenytoin or other CYP 3A inducers (e.g., carbamazepine, barbiturates, rifampin, glucocorticoids). Coadministration with thioridazine produces an approximately 70% increase in the apparent oral clearance of quetiapine and an associated decrease in the steady-state concentrations of quetiapine. The mechanism of this interaction is unclear because thioridazine has not been shown to produce clinically significant induction of CYP 3A in humans.
Ziprasidone's clearance is mediated by CYP 3A4 and by the non-CYP cytosolic enzyme aldehyde oxidase2. Two-thirds of ziprasidone's clearance in humans is mediated by aldehyde oxidase, whereas the remaining one-third is mediated largely by CYP 3A4. Thus, the magnitude of any pharmacokinetic interaction resulting from CYP 3A4 inhibition or induction should be small. No clinically significant inhibitors or inducers of aldehyde oxidase have been identified at the present time. Based on this and published findings to date, ziprasidone is not expected to be a victim of clinically significant drug interaction resulting from CYP inhibition or induction, although carbamazepine may cause a small increase in ziprasidone clearance. However, because of the infrequent but potentially life-threatening significance of QTc interval prolongation (see Chapter 20), ziprasidone should not be used with other agents known to have this effect, and it should be used with caution and close monitoring when combined with potent CYP 3A inhibitors.
Aripiprazole's literature is too limited at this time to comment on its drug interaction liability.
P.451
C. Monamine Oxidase Inhibitors
All MAOIs currently marketed in the United States (e.g., phenelzine, tranylcypromine, isocarboxazid, selegiline), when they are used in dosages that are effective in depression, are nonselective inhibitors of the enzymes monoamine oxidase A and B. (Note: Selegiline becomes nonselective at dosages of greater than 10 mg per day.) This effect has to be considered when these agents are coadministered with other medications that affect biogenic amine metabolism.
Although most clinicians immediately associate monoamine oxidase inhibition with antidepressant agents, remembering that other compounds have potent inhibitory actions on monoamine oxidase is important (e.g., the cancer chemotherapy agent procarbazine, the antihypertensive agent pargyline, the antibiotic linezolid).
Antidepressants. The combined use of any MAOI with another type of antidepressant should either be avoided or should be prescribed only with caution and careful monitoring in patients refractory to more conventional approaches. Some combinations are more likely to be problematic than others, and some patients are more susceptible to harmful complications. Unfortunately, which will cause this cannot always be determined in advance. When an interaction does occur, it usually involves some combination of central excitation, restlessness, rigidity, hyperpyrexia, respiratory depression, coma, or cardiovascular shock. Death may ensue. This unfortunate clinical outcome is most often a form of serotonin syndrome. Therefore, antidepressants with strong or selective proserotonergic activity are of special concern. These include fluoxetine, sertraline, paroxetine, fluvoxamine, and clomipramine, as well as some other TCAs. One exception in this category may be trazodone; some reports have suggested that concurrent use with phenelzine is safe. Combinations of amitriptyline with tranylcypromine or phenelzine and of trimipramine with phenelzine or isocarboxazid have also been used safely. That nortriptyline, in modest dosages, can also be used safely in combination with MAOIs seems probable. Some of the most serious interactions have involved clomipramine or imipramine; combinations of MAOIs with these two TCAs are best avoided. Some clinicians also avoid the use of MAOIs with desipramine.
Combinations of moclobemide with nortriptyline or desipramine may result in impaired clearance of these antidepressants and in toxicity because studies of sparteine and dextromethorphan metabolism in healthy volunteers have shown moclobemide to be a CYP 2D6 inhibitor.
Although the safety of selected combinations seems reasonably clear, any greater efficacy for combination therapy has not been established. Despite this, some clinical experience supports cautious trials of appropriate combinations in patients refractory to other treatment regimens. When combination oral therapy is chosen, initiating both medications at the same time and at lower than usual dosages is safest. Another possible role for combination therapy is to provide sedation with, for example, low doses of trazodone or trimipramine for patients on MAOIs who are having difficulty with sleep-onset insomnia. No data on the use of mirtazapine for this adjunctive role could be located.
Antihypertensive agents. Reserpine, when it is administered with an MAOI, may result in autonomic arousal, delirium, agitation, or hypertension. The antihypertensive action of guanethidine, and possibly also of clonidine, may be blocked when it is given with an MAOI. Some clinicians recommend avoiding propranolol altogether or using only low doses of the drug to prevent hypertensive reactions in patients also taking an MAOI. The hypotensive action of thiazide diuretics may be
P.452
potentiated by MAOIs. One case report suggests that hallucinosis may be seen with pargyline, an MAOI used in the treatment of hypertension, and methyldopa; however, some patients tolerate this combination without difficulty. Nonetheless, in animal models, a central excitatory syndrome occurs with this combination.Antipsychotic agents. Droperidol may potentiate hypotension in patients taking an MAOI. (Note: Droperidol is not available in some countries because of its association with lengthened QTc intervals.) Chlorpromazine and other antipsychotic agents may block the pressor response to tyramine in patients taking MAOIs.
Sedative-hypnotics
Barbiturates. MAOIs may enhance and prolong barbiturate sedation and other barbiturate effects due to an inhibition of metabolism of the barbiturate.
Benzodiazepines and other hypnotic agents binding to the benzodiazepine receptor. Chorea has been reported in one patient and edema in two others. Given the widespread clinical use of these combinations, concomitant use seems unlikely to be problematic.
Buspirone. Elevated blood pressure has been reported in a few patients already receiving MAOIs when buspirone was started in them; other monitored patients have not shown blood-pressure changes while taking buspirone and MAOIs. Nevertheless, conservative clinical practice should include periodic blood-pressure monitoring. The package insert for buspirone recommends that it not be given with MAOIs.
Alcohol. Pargyline may cause a disulfiram-like reaction when alcohol is coingested. Tyramine-free alcoholic beverages probably do not interact with other MAOIs.
Ginseng. Ginseng, which is found in many herbal health remedies, may cause insomnia, headache, tremor, and even hypomania when it is ingested by patients taking MAOIs.
Hypoglycemic agents. An enhanced hypoglycemic response may occur in patients taking MAOIs and insulin or oral hypoglycemics.
Opioids. Two types of interactions may occur between opioids and MAOIs. The first is a syndrome characterized by hypotension or, less commonly, hypertension; excitement; sweating; rigidity; and hyperreflexia that can progress to hyperthermia, coma, and death. This syndrome may occur after meperidine or dextromethorphan (or a structurally close analogue) is administered to patients who are taking an MAOI. These opioid analgesics should be avoided in patients receiving MAOIs.
The second type of interaction is the potentiation of opioid effects. Some clinicians recommend one-fifth to one-half of the typical opioid dose when analgesia is accomplished with fentanyl, morphine, or codeine. However, the latter agents do not appear to cause the dangerous interactions noted with meperidine-like agents.
Succinylcholine. Phenelzine may reduce pseudocholinesterase levels; prolonged apnea after succinylcholine administration has been reported in one phenelzine-treated patient. In general, mivacurium or rapacuronium can be safely substituted for succinylcholine (see Chapter 24).
Sympathomimetic amines. Direct-acting sympathomimetic amines, such as epinephrine, norepinephrine, and isoproterenol, may have enhanced pressor effects when they are administered to patients taking MAOIs; hypertensive crises may occur. Hypertensive reactions and crises are more common, however, after the use of indirect-acting sympathomimetics (e.g., dextroamphetamine, ephedrine, phenylpropanolamine, pseudoephedrine, reserpine, tyramine) in patients taking MAOIs. Levodopa (L-dopa) should also be avoided.
P.453
D. Tricyclic Antidepressants and Other Heterocyclic or Atypical Antidepressants
Analgesics. Proserotonergic antidepressants (e.g., amitriptyline, imipramine, trazodone) are sometimes prescribed to relieve migraine headaches and temporomandibular joint pain or to treat a variety of other chronic pain syndromes. In these and other conditions, they may be coadministered with opioid analgesics. Animal studies and clinical experience suggest that antidepressants may enhance opioid analgesia. Some evidence implies that proserotonergic antidepressants may be the most effective; desipramine, however, potentiates morphine analgesia in some animal models.
Anesthetic agents. Halothane and pancuronium may induce cardiac arrhythmias in patients on antidepressants that have anticholinergic properties (e.g., amitriptyline).
Anticoagulants. A single case report describes a 30% reduction in prothrombin time in a patient taking trazodone and warfarin.
Antihypertensive agents. The antihypertensive effects of guanethidine, bethanidine, and debrisoquin are blocked by antidepressants that inhibit the presynaptic transporter of norepinephrine (see above). Virtually all available TCAs have this action to some degree; doxepin and trimipramine are the weakest. Newer antidepressants, such as fluoxetine, sertraline, paroxetine, nefazodone, bupropion, and trazodone, have even weaker norepinephrine-transporter blocking properties, and they thus should not antagonize the antihypertensive action of guanethidine or its related compounds. Venlafaxine has norepinephrine-transporter blocking properties.
As an inhibitor of CYP 2D6, bupropion could increase the hypotensive effects of -adrenergic receptor antagonists, such as metoprolol.
The antihypertensive action of clonidine also may be blocked by some antidepressants. Desipramine, imipramine, and amitriptyline reverse the antihypertensive effects of clonidine in humans, and animal studies suggest other TCAs act similarly. Bupropion does not interact with clonidine.
Barbiturates. Barbiturates induce hepatic microsomal enzymes and enhance the metabolism of oxidatively metabolized antidepressants.
Carbamazepine. A growing number of reports note that carbamazepine may induce the metabolism of imipramine, desipramine, nortriptyline, and bupropion. As with the barbiturates, carbamazepine is likely to induce the metabolism of all oxidatively metabolized antidepressants. Additive or synergistic cardiotoxicity has also been reported. Some evidence from animal studies and one case report suggests that imipramine and desipramine may inhibit carbamazepine metabolism, thus leading to carbamazepine toxicity, as levels of the 10,11-epoxide metabolite of carbamazepine are elevated. Although some authorities caution against combining carbamazepine and MAOIs, clinical experience suggests that the combination of phenelzine or tranylcypromine with carbamazepine is safe and that it does not affect carbamazepine blood levels.
Cimetidine. Cimetidine impairs the metabolism of desipramine, doxepin, amitriptyline, nortriptyline, and other oxidatively metabolized antidepressants; ranitidine and famotidine do not.
Digoxin. Trazodone may elevate digoxin levels.
Disulfiram. This drug inhibits the metabolism of imipramine, desipramine, and other oxidatively metabolized antidepressants. Delirium has been reported with the combination of disulfiram and amitriptyline. It has been suggested to potentiate the effects of amitriptyline by an unknown mechanism.
Estrogens. Women on long-term low-dose estrogen-containing oral contraceptive treatment and those on estrogen replacement may have a
P.454
decreased clearance of imipramine. This can lead to toxicity unless appropriate dosage reductions are made.Alcohol. With the exception of the selective serotonin reuptake inhibiting antidepressants (see section III.E) and bupropion, antidepressants enhance the sedative and psychomotor effects of alcohol. Acute alcohol consumption inhibits the metabolism of most antidepressants; chronic alcohol use, in the absence of cirrhosis, leads to enhanced drug metabolism.
Levodopa. The anticholinergic properties of TCAs may impair the absorption of L-dopa. Whether this occurs with paroxetine is not known.
Triiodothyronine (T3). T3 augmentation of antidepressants may be useful in some treatment-refractory depressions, particularly in women.
Methylphenidate. Methylphenidate may inhibit the metabolism of imipramine and other antidepressants having a CYP 2D6-mediated pathway. In some patients with treatment-refractory depressions, the addition of a stimulant, such as methylphenidate or pemoline, may be beneficial.
Phenytoin. Some reports suggest that imipramine, nortriptyline, and trazodone may increase phenytoin levels.
Reserpine. Reserpine has been used to potentiate antidepressant response, and it may be associated with a greater propensity for medication-induced switching into mania. Significant adverse effects may be associated with these combinations.
Antiarrhythmic agents. Bupropion, through its inhibition of CYP 2D6, can increase concentrations of type 1C antiarrhythmic agents, such as propafenone and flecainide.
Sulfonylureas. Hypoglycemia has been reported when sulfonylureas have been administered with doxepin and nortriptyline. This likely could occur with other TCAs.
Sympathomimetic amines. The effects of direct-acting sympathomimetic amines may be enhanced in patients taking antidepressants. As long as intravenous administration is avoided, this rarely presents clinical problems. Pressor effects of indirect-acting sympathomimetics (ephedrine, pseudoephedrine, tyramine, and others) may be antagonized by the TCAs, venlafaxine, mirtazapine, and maprotiline, because norepinephrine reuptake is reduced or blocked.
Antiretroviral agents. The HIV-1 protease inhibitor ritonavir is a potent CYP 2D6 inhibitor, and it produces an extremely large mean increase in the area under the curve of desipramine. An interaction with nortriptyline is also highly likely because of the similar mechanism of clearance (CYP 2D6 mediated). Clinically significant interactions with the tertiary amines imipramine and amitriptyline are also likely due to the broad-spectrum CYP inhibitory effects of ritonavir and the inhibition of downstream metabolism of the demethylated active metabolites of these drugs. CYP 2D6 is the primary molecular determinant of the clearance (via O-demethylation) of the serotonin and norepinephrine reuptake inhibitor antidepressant venlafaxine, and thus ritonavir can be expected to impair its clearance. Nefazodone and trazodone are CYP 3A4 substrates based on in vitro studies, and ritonavir is a highly potent CYP 3A4 inhibitor. (Note: The net effect of ritonavir on CYP 3A4 substrates is difficult to predict because ritonavir produces concurrent inhibition and induction of this enzyme.) The exact nature of the interaction will depend on the dose of treatment with ritonavir and the time of ingestion before the addition of the CYP 3A4 substrate.
Antifungal agents. The allylamine antifungal agent terbinafine is a potent inhibitor of CYP 2D6 based on in vitro and in vivo studies. Case reports of nortriptyline toxicity and elevated plasma concentrations in terbinafine-treated patients are available. A clinically significant interaction with desipramine is also likely, given the importance of CYP 2D6 to desipramine clearance. The CYP 3A4-inhibitory antifungal
P.455
agent ketoconazole inhibits reboxetine clearance, which is reflected as a 1.5-fold prolongation of the elimination half-life of single dose reboxetine. An increase in steady-state reboxetine concentrations is thus predicted to result from CYP 3A4 inhibition. Ketoconazole and itraconazole may impair the clearance of trazodone and nefazodone, given the importance of CYP 3A4 in their metabolism.
E. Specific Serotonin Reuptake Inhibiting Antidepressants
The SSRI antidepressants fluoxetine, fluvoxamine, sertraline, paroxetine, and citalopram are all CYP enzyme inhibitors with varying specificities and potencies toward the multiple isoforms. Citalopram is the only SSRI without any clinically significant CYP inhibitory activity, and thus it is superior to the other agents from a drug interaction perspective. CYP 3A4 is inhibited by norfluoxetine, the active metabolite of fluoxetine. Given the high plasma concentrations of norfluoxetine that are attained during multiple-dose administration of fluoxetine, significant drug interactions occur between fluoxetine and CYP 3A4 substrates. Because of the long half-life of norfluoxetine (7 to 15 days), a significant risk of interaction exists for weeks after cessation of fluoxetine therapy. Thus, the inhibitory effects of fluoxetine take a sustained interval to reach maximum effect, and they persist for an extended period of time. Fluoxetine and paroxetine are potent CYP 2D6 inhibitors. Sertraline and desmethylsertraline are weak CYP 2D6 inhibitors. Fluoxetine and fluvoxamine inhibit CYP 2C9, and clinically significant interactions are likely with CYP 2C9 substrate drugs with a narrow therapeutic index, such as phenytoin or warfarin, necessitating an adjustment of dosage and emphasizing the importance of therapeutic drug monitoring in patients receiving these drug combinations. The same is true for sertraline as well, although it is a much weaker CYP 2C9 inhibitor and interactions with CYP 2C9 substrates are less likely and are probably variable. Paroxetine, sertraline, fluvoxamine, and norfluoxetine are all inhibitors of CYP 2B6 activity in vitro, and they can potentially impair the clearance of the antidepressant and antismoking agent bupropion. Fluvoxamine is a potent inhibitor of CYP 1A2 and CYP 2C19 and a less potent, yet clinically significant, inhibitor of CYPs 2C9 and 3A4.
Antidepressants. Fluoxetine inhibits the metabolism of all TCAs to varying degrees. Concomitant administration of fluoxetine and desipramine, for example, may lead to a 10-fold reduction in clearance as a result of CYP 2D6 inhibition and to 4-fold prolongation of the elimination half-life. When imipramine and fluoxetine are coadministered, clearance may be reduced fourfold. The effects on imipramine and amitriptyline clearance are of a smaller magnitude because multiple CYP isoforms metabolize these tertiary amine TCAs, and CYP 2D6 and 3A4, the main targets of fluoxetine and norfluoxetine, respectively, only partially contribute to their metabolic clearance. Nevertheless, the interactions are clinically meaningful because of the inhibition of the downstream metabolism of the pharmacologically active N-demethylated metabolites of the TCA by the SSRI.
Fluoxetine impairs bupropion clearance. If this interaction is not considered, the coadministration of fluoxetine and bupropion may result in a clinical syndrome resembling stimulant-induced psychosis or a bupropion-induced lowering of seizure threshold, even at doses of bupropion below 400 mg per day. This is explained at least in part by the norfluoxetine-mediated inhibition of CYP 2B6, the major molecular determinant of bupropion clearance in humans. Similar interactions of the CYP 2B6-inhibitory SSRIs paroxetine, sertraline, and fluvoxamine with bupropion can be predicted based on in vitro studies. The addition of bupropion to patients receiving SSRI antidepressants is not uncommon. Such combination treatments may potentially increase the risk of bupropion-related seizures, and an appropriate dosage adjustment may be necessary.
P.456
Combination therapy with TCAs and fluoxetine is sometimes tried to improve response rates in refractory depression, to reverse tolerance to fluoxetine's antidepressant response, or to induce hypnotic effects in patients with insomnia as a prominent symptom of depression or secondary to fluoxetine. Trazodone may be used for hypnotic effects in patients receiving fluoxetine, and the potential inhibition of CYP 3A4-mediated metabolism of trazodone by norfluoxetine may need to be considered.
Antipsychotic agents. Fluoxetine may impair the metabolism of some antipsychotic agents, resulting in elevated serum levels of the latter. Because of the wide therapeutic index of antipsychotic agents, this is usually not of clinical significance. In some cases, however, enhanced dopaminergic activity may lead to extrapyramidal symptoms, some of which may be due to a pharmacodynamic interaction.
Barbiturates and benzodiazepines. Concomitant administration of fluoxetine may impair the metabolism of barbiturates and triazolobenzodiazepines, thus enhancing their sedative and psychomotor effects. The effect on barbiturates is CYP 2D6 mediated; the triazolobenzodiazepine effect is CYP 3A4 mediated. Inhibition of the clearance of triazolam, alprazolam, estazolam, and midazolam by fluoxetine is mainly explained by the potent CYP 3A4-inhibitory effects of norfluoxetine. Fluvoxamine decreases the clearance of intravenous midazolam by 33%. The effects on oral midazolam are expected to be greater given the large contribution of the small intestine in the presystemic extraction of oral midazolam.
Buspirone. The antianxiety effect of buspirone may be reversed by fluoxetine. One uncontrolled study suggested that buspirone may augment fluoxetine's effects in obsessive-compulsive disorder; this was not confirmed by a subsequent controlled trial. Fluvoxamine was shown to produce a small decrease in buspirone clearance in a controlled study with healthy volunteers. Although the interaction may be minor from a pharmacokinetic standpoint, a pharmacodynamic interaction is possible when combining any proserotonergic agent with an SSRI.
Mood-stabilizing anticonvulsants. Elevated levels of carbamazepine and toxicity have resulted with concurrent fluoxetine administration. Increases in VPA blood levels without clinical consequences have also been reported.
Cyproheptadine. Cyproheptadine, when used to treat anorgasmia in patients taking fluoxetine, may antagonize the antidepressant and anorectic effects of fluoxetine.
Alcohol. Fluoxetine does not affect the psychomotor performance, subjective effects, or metabolism of alcohol. Similarly, other SSRIs do not appear to affect or to be affected by alcohol. Some studies suggest that SSRI use may decrease ad libitum alcohol intake in both animals and humans.
Lithium. Lithium may augment the antidepressant response to fluoxetine, but a potential for toxicity also exists. Fluvoxamine may produce seizures and hyperpyrexia when combined with lithium. This may be a form of serotonin syndrome, in which case it could occur when lithium and any SSRI are combined.
L-Tryptophan. The combination of L-tryptophan and fluoxetine, sertraline, or paroxetine may result in toxicity when high doses are administered. Symptoms of agitation, restlessness, insomnia, aggressive behavior, headaches, chills, nausea, abdominal cramps, and diarrhea have been reported. This may be a variant of a serotonin syndrome.
Opioids. Fluoxetine may potentiate the analgesic effects of opioids in animal models of pain, but clinical experience with the combination is limited. The analgesic and narcotic effects of codeine can be expected to
P.457
be diminished in patients receiving antidepressant therapy with fluoxetine or paroxetine due to the inhibition of the CYP 2D6-mediated O-demethylation process that produces the pharmacologically active metabolite morphine. Whether bupropion would also inhibit the biotransformation of codeine to morphine has not been established. The synthetic opioids fentanyl, sufentanil, alfentanil, and methadone are all primarily metabolized by CYP 3A4. The apparent oral clearance of these agents may be impaired by fluoxetine due to the inhibition of CYP 3A4 by norfluoxetine. In principle, the pharmacokinetics of intravenous fentanyl and sufentanil should not be affected by fluoxetine because of their high extraction ratio, which makes their clearance flow dependent.Calcium channel blocking agents. Three patients in whom concurrent use of fluoxetine and calcium channel blocking agents led to unwanted effects have been described; these effects were presumed to be secondary to increased concentrations of calcium channel blocking agents. Verapamil was associated with edema in one patient and with an increase in headaches in another. Nifedipine was associated with nausea and flushing in the third patient. These interactions are most likely explained by the inhibition of CYP 3A4-mediated clearance of these calcium channel blocking agents by norfluoxetine. To minimize the risk of an interaction, paroxetine, sertraline, or citalopram may be suitable alternatives for patients receiving calcium channel blocking agents.
Antiarrhythmic agents. The clearance of propafenone and encainide is CYP 2D6 mediated. Coadministration with fluoxetine or paroxetine can result in the impairment of clearance. Because propafenone's major metabolite 5-hydroxy propafenone is pharmacologically active, the predominant effect of clearance impairment is enhanced -adrenergic receptor antagonism, a side effect of propafenone that is not shared by the metabolite. Fluvoxamine coadministration results in an approximately 40% decrement in the clearance of quinidine, a CYP 3A4 substrate, which is consistent with the moderate inhibition of CYP 3A4 by this SSRI.
-Adrenergic receptor antagonists. The clearance of metoprolol is CYP 2D6 mediated. Paroxetine is a potent inhibitor of metoprolol metabolism in vitro, and it produces an 83% decrement in oral metoprolol clearance and enhanced pharmacodynamic effects as measured by reductions in exercise heart rate and exercise systolic blood pressure. Case reports of severe bradycardia when metoprolol was coadministered with fluoxetine are available and are consistent with the in vitro data. Dosage reductions of metoprolol may be required when it is coadministered with paroxetine or fluoxetine. Fluvoxamine is reported to increase the plasma concentrations and effects of propranolol, an effect that is probably explained by the inhibition of CYP 1A2.
Tacrine. Fluvoxamine (100 mg per day) causes an 88% decrease in the apparent oral clearance of tacrine, and it may potentially modulate the hepatotoxicity of tacrine, depending on the relative contribution of tacrine and its reactive metabolites to this toxicity. This interaction results from potent CYP 1A2 inhibition by fluvoxamine. The prevalence of tacrine use has diminished since the introduction of other agents for Alzheimer disease that are not CYP 1A2 substrates (see Chapter 5).
Melatonin. Fluvoxamine is a clinically significant inhibitor of melatonin clearance, and plasma melatonin levels are increased upon coadministration. The interaction is characterized by a large increase in the peak plasma melatonin concentrations without an alteration of the elimination half-life; it is explained by the inhibition of CYP 1A2-mediated metabolism of melatonin.
P.458
F. Lithium
Because lithium is not metabolized by the liver, enzyme induction and inhibition have no relevance for the understanding of lithium's drug interactions. Renal excretion is primarily responsible for lithium clearance; drugs that affect renal function, especially sodium reabsorption, may have clinically important effects on lithium levels. The therapeutic index of lithium is narrow; small increments in therapeutic lithium levels may lead to toxicity. Pharmacodynamic interactions may also be important in lithium therapy, and some CNS toxicity (e.g., confusion, memory loss, fever, seizures) may occur with haloperidol and lithium in the absence of elevated serum lithium levels (intracellular levels may be elevated as reflected by increased red blood cell concentrations; see Chapters 19 and 20).
Aminophylline and theophylline. These increase lithium excretion, and they have been used to treat lithium toxicity, although osmotic diuretics, intravenous sodium bicarbonate, and dialysis are preferred for serious lithium intoxication (see Chapter 3). Patients with pulmonary diseases who take lithium together with these drugs should have lithium levels monitored and the dosage increased when necessary.
Antimicrobials. Several antimicrobials (e.g., spectinomycin, tetracycline, metronidazole) have been associated with increased lithium levels in case reports. The interactions have not been systematically studied in humans. In animal studies, lithium clearance was not affected by tetracycline, ampicillin, or metronidazole.
Antidepressants. Lithium may potentiate the therapeutic effects of MAOIs, TCAs, atypical antidepressants, and SSRIs. As with many drug combinations that enhance the response, a risk of increased toxicity is also seen. Caution should also be exercised when the combination is used to treat depressed bipolar patients because lithium may not provide prophylaxis against the ability of antidepressants to increase cycling.
Antihypertensive agents. See section III.F.11 for a review of the diuretics. Angiotensin-converting enzyme inhibitors may elevate serum lithium levels and thus may lead to toxicity. The glomerular filtration rate in some patients taking lithium may depend on angiotensin II, thus putting these patients at risk for lithium toxicity. Enalapril, captopril, and any related agents should be used with caution in lithium-treated patients. Alternative antihypertensive medications are preferable.
In patients taking lithium, methyldopa may cause a toxic syndrome that is characterized by confusion, tremors, slurred speech, blurred vision, sedation, and dysphoria. It may occur even in the presence of therapeutic lithium levels.
Patients with bipolar disorder have smaller decrements in blood pressure when they are given clonidine while taking lithium, compared with patients taking clonidine alone.
-Adrenergic receptor antagonists and prazosin do not appear to interact with lithium. They may be the preferred antihypertensive agents for patients taking lithium.
Antiinflammatory agents. Increased lithium levels and toxicity have been reported when patients taking lithium have also taken indomethacin, diclofenac, clomethacin, phenylbutazone, or ibuprofen. Aspirin and sulindac do not elevate serum lithium levels.
Antipsychotic agents (see section III.A.6).
Antithyroid agents. Lithium enhances the action of thyroid-suppressing agents, such as methimazole and carbimazole; it has been used as an adjunct to radioactive iodine in the treatment of thyrotoxicosis. Lithium use may expose or aggravate hypothyroidism in patients with Hashimoto (autoimmune) hypothyroidism.
Benzodiazepines. A single case report describes hypothermia (30 C [86 F]) and a comatose state with reduced reflexes, dilated pupils, hypotension,
P.459
bradycardia, and absent piloerector response that were produced by the combination of lithium and diazepam. No subsequent reports have appeared; this may best be understood as an idiosyncratic reaction. In another report, steady-state serum lithium concentrations were increased by 3% when alprazolam was given concurrently, a change that was clinically insignificant.The combination of lithium with a BZ (e.g., clonazepam, lorazepam) is a common practice to control agitation in bipolar patients in the acute stages of mania. Based on the extensive clinical use of these combinations, BZs appear to be a safe and useful adjunct to lithium therapy. Animal studies do suggest the possibility of an interaction between lithium and BZs, although the relevance of these results to clinical practice is not apparent. For example, some studies have shown decreases in BZ receptor density in the rat frontal cortex after 4 weeks of lithium administration.
Digitalis glycosides. In a single case report, the combination of lithium and digoxin resulted in confusion and nodal bradycardia alternating with atrial fibrillation in the presence of therapeutic lithium levels. Some clinicians believe that, with proper clinical monitoring, the combination can be used safely.
Disulfiram. Rats have increased mortality when disulfiram and lithium are administered concurrently, but no clinically significant interactions have been reported in humans.
Diuretics. Diuretics are generally divided into the following seven classes: osmotic diuretics, carbonic anhydrase inhibitors, thiazide diuretics, loop diuretics, aldosterone antagonists, potassium-sparing diuretics, and methylxanthines. Osmotic diuretics, such as urea, and carbonic anhydrase inhibitors, such as acetazolamide, increase lithium excretion. The thiazides consist of the thiazide diuretics plus other agents that are chemically dissimilar yet work through similar mechanisms of action (e.g., the sulfonamide derivatives chlorthalidone, quinethazone, metolazone, methyclothiazide, and indapamide). All agents in this class impair the excretion of lithium, and many case reports of severe toxicity have appeared that describe patients in whom appropriate decreases in lithium dosage were not made. Most case reports suggest that symptom onset occurs rapidly, within 2 to 10 days after adding the diuretic and lithium. With a 25 mg daily dose of hydroflumethiazide or a 2.5 mg daily dose of bendrofluazide, an average reduction in renal lithium clearance of 24% occurred; some patients showed no change. Other studies with chlorothiazide report average reductions in lithium clearance of 40%, 58%, and 68%, for daily chlorothiazide doses of 500, 750, and 1,000 mg, respectively. Therefore, a reduction of approximately 40% in the lithium dose may be required when a daily dose of 500 mg chlorothiazide is coadministered. Similarly, a 35% increase in the mean serum lithium level was found in normal volunteers taking 900 mg lithium carbonate and 50 mg of hydrochlorothiazide daily. Most clinicians reduce the lithium dosage by approximately 50% when this combination is used. In addition to the treatment of hypertension in bipolar patients, the benzothiadiazides are sometimes used to treat lithium-induced diabetes insipidus.
Loop diuretics, such as furosemide and bumatanide, have been associated with lithium toxicity in case reports, but carefully designed studies have not demonstrated a clinically significant effect. One study found an 11% decrease in lithium clearance after single doses of 40 or 80 mg of furosemide. In another study, 40 mg of furosemide added to lithium carbonate (900 mg per day) resulted in no significant change in the serum lithium levels in five of six volunteers; in the sixth volunteer, however, the level increased from 0.44 to 0.72 mEq per L, coincident with her premenstrual period. The consequences of the addition of
P.460
furosemide to lithium appear to be less predictable than those for the addition of thiazides. In part, this may reflect differences in usage furosemide is often used in single doses, whereas thiazides are more often used chronically, and potassium to lithium alterations may be less critical. Frequent serum monitoring is required when the combination is used.Ethacrynic acid does not have a clinically significant effect on lithium clearance. Single doses of the aldosterone antagonist spironolactone do not have a clinically important effect on lithium clearance; daily doses of 100 mg per day, however, may cause increases in serum lithium concentration.
The potassium-sparing diuretics triamterene and amiloride have not been extensively studied in combination with lithium. One case report with triamterene describes an elevation in the lithium level from 0.65 to 0.95 mEq per L. In a study of the effects of amiloride on lithium-induced polyuria, the mean lithium levels did not change significantly, although one patient had an increase from 0.8 to 2 mEq per L. When potassium-sparing diuretics lead to large volume contractions and reduced glomerular filtration, they appear possibly to reduce lithium clearance. As was discussed earlier, methylxanthines, such as aminophylline, theophyline, and caffeine, may increase lithium clearance and may thereby lower the serum levels.
Alcohol. Lithium may block the alcohol-induced subjective high, may decrease the desire to continue drinking, and may reduce the cognitive dysfunction associated with intoxication. Lithium, however, is not an effective agent to treat alcohol withdrawal; its role in promoting abstinence after the withdrawal period is uncertain.
Anticonvulsants (e.g., phenytoin, carbamazepine, and VPA). The combination of lithium and phenytoin may produce a toxic state that is characterized by coarse tremor, drowsiness, ataxia, gastrointestinal symptoms, and coma. In two of three reported cases, lithium levels were within the therapeutic range. Preexisting brain injury or damage may predispose a patient to this toxic interaction. In most cases, these drugs can be coadministered safely, as long as close supervision and monitoring occur.
Carbamazepine is often coadministered with lithium in refractory bipolar patients (see Chapter 19). Although occasional reports of additive toxicity are seen, these are rare and they are thought by some to reflect too rapid dosage increases of carbamazepine. Both drugs reduce thyroid activity, which should be monitored during combination therapy. Lithium may obscure carbamazepine-induced decreases in leukocytes and neutrophils; this outcome is not prophylactic against carbamazepine-induced myelosuppression. Carbamazepine has an antidiuretic effect; this, however, is not an effective treatment for lithium-induced diabetes insipidus. Lithium may reduce the hyponatremia caused by carbamazepine. VPA added to lithium may improve the response in some patients with refractory bipolar disorder.
Electroconvulsive therapy (ECT) and anesthesia (see also Chapter 24). When patients taking lithium must undergo general anesthesia for surgery or ECT, the potential for several drug interactions exists. Methoxyflurane, an inhalational anesthetic, should be avoided because it can cause renal toxicity. Because renal excretion is the major mechanism of lithium elimination, the two agents are rarely used together.
Some clinicians recommend that barbiturate dosages are reduced or that their use be avoided in patients taking lithium based on animal data that demonstrate increased barbiturate sleeping times in lithium-treated mice. This appears to be an acute effect of lithium that is not seen during chronic treatment in mice; no interactions in humans have been reported. In another animal study, one strain of rat became oliguric
P.461
with decreased renal clearance after the use of amobarbital anesthesia. Ketamine, an injectable anesthetic agent that produces dissociative anesthesia, should be avoided in patients taking lithium because it may prolong seizure duration during ECT.With respect to ECT, an initial case report suggested that delayed recovery from anesthesia could occur after ECT in patients taking lithium. A subsequent retrospective chart review in 17 patients who had a total of 78 treatments indicated that lithium use did not prolong anesthesia duration. Further complicating the issue of lithium and ECT are reports that patients may be more likely to develop greater memory loss, confusion, and atypical neurologic findings if they are receiving lithium while being treated with ECT.
The use of certain neuromuscular blocking agents must be avoided in patients taking lithium. One report describes a woman taking lithium who required an emergency cesarean section. Anesthesia was induced by pancuronium, thiopental, and succinylcholine. Postoperatively, apnea requiring mechanical ventilation occurred for a 4-hour period. In another case, a woman receiving lithium required an elective thoracotomy; anesthesia was induced by thiamylal, succinylcholine, and pancuronium bromide. Reversal of her neuromuscular blockade required atropine and neostigmine.
A number of animal studies support an interaction between lithium and neuromuscular blocking agents. Several studies indicate that lithium prolongs the duration of the blockade produced by succinylcholine, decamethonium, or pancuronium. These data, taken together with the case reports of surgical complications, suggest that lithium should be avoided during elective anesthesia.
G. Benzodiazepines and Nonbenzodiazepine g-Aminobutyric Acid-A Receptor Agonists
Aminophylline, theophylline, and caffeine. These may antagonize the action of diazepam, lorazepam, and probably all BZs. This interaction may be secondary to reduced -aminobutyric acid (GABA) transmission by competition at the BZ receptor site or by effects at the adenosine receptor.
Anesthetic agents. Case reports have suggested that diazepam can increase the magnitude of the neuromuscular blockade produced by gallamine and that it can decrease that of succinylcholine. Other reports have not supported these findings. Midazolam does not affect the neuromuscular blockade produced by succinylcholine or pancuronium. The intravenous anesthetic agent propofol acts synergistically with the BZs when this combination is used in the induction of general anesthesia. The mechanism of this interaction is the apparent increased affinity of propofol to GABAA receptors in the presence of the BZ agonists.
Antacids. The transformation of clorazepate to desmethyldiazepam is dependent on gastric pH, and concomitant administration of an antacid may reduce the rate and amount of desmethyldiazepam that is formed. In chronic dosing of clorazepate, however, steady-state levels are not significantly affected by antacids. Antacids may also reduce the rate, but not the extent, of diazepam or chlordiazepoxide absorption. When single doses of BZs are administered with antacids, the subjective relief of anxiety may be delayed.
Anticonvulsants. VPA may increase the unbound fraction of diazepam twofold due to the displacement of diazepam from plasma albumin, theoretically causing a temporary increase in BZ effects. However, no evidence exists to indicate that a clinically important interaction occurs in vivo. Some reports suggest VPA may also inhibit diazepam metabolism. VPA does not affect clonazepam metabolism, although case reports of oversedation and the appearance of absence seizures with the combination do exist.
P.462
Several reports have indicated that diazepam may increase phenytoin levels; the opposite effect has also been reported. Chlordiazepoxide and clonazepam may also increase phenytoin serum levels. Clearance of diazepam, clonazepam, and oxazepam and possibly of other BZs may be enhanced by phenytoin.
Carbamazepine induces the metabolism of clonazepam and alprazolam and probably of other oxidatively metabolized BZs. Tiagabine does not alter triazolam pharmacokinetics or pharmacodynamics.
Antituberculosis agents. Isoniazid decreases the clearance of diazepam, triazolam, and probably all BZs metabolized by demethylation or hydroxylation. Rifampin is an enzyme inducer that increases the clearance of diazepam, triazolam, and midazolam and probably that of all other oxidatively metabolized BZs. Triazolam may be ineffective in patients taking rifampin; temazepam may be a useful alternative. The clearance of the imidazopyridine BZ receptor agonist zolpidem is also induced by rifampin, leading to a considerable reduction in hypnotic effect.
Barbiturates. These have additive sedative effects when they are given with BZs; they may also induce the metabolism of BZs.
Cigarette (tobacco) smoking. Although a retrospective study indicated that the sedative effect of diazepam was reduced in smokers, carefully designed studies have found no difference in the metabolism of chlordiazepoxide, lorazepam, or triazolam between smokers and nonsmokers. The half-life of estazolam is lower in smokers compared with nonsmokers. Most, but not all, studies have found no difference in diazepam metabolism in smokers versus nonsmokers. Conflicting reports exist concerning clorazepate. That any interaction between BZs and cigarette smoking would present clinical problems is unlikely. Curiously, nicotine patches contain a product warning for an interaction between nicotine and oxazepam, the basis for which is not obvious especially since the patches are free of the inhaled aryl hydrocarbons that were presumed to account for any of the interactions observed between cigarette smoking and oxidatively metabolized BZs.
Clozapine. See section III.B.
Digoxin. A small increase in the digoxin half-life and increased serum levels of digoxin may occur in the presence of diazepam. One case report describes an increase in digoxin levels with alprazolam, but a larger study found no interaction.
Disulfiram. Disulfiram impairs the metabolism of chlordiazepoxide and diazepam and probably of other BZs metabolized by demethylation or hydroxylation. It does not affect the metabolism of lorazepam or oxazepam.
Alcohol. The combination of alcohol and BZs enhances sedation, respiratory depression, and psychomotor impairment. Some studies suggest that temazepam may produce a less intense interaction with alcohol. The pharmacokinetic interactions between alcohol and the BZs are complex, and studies are contradictory, depending in part on whether single (inhibiting) or chronic (inducing) alcohol ingestion is involved and on the BZ's metabolic pathway.
Histamine (H2) receptor antagonists. Cimetidine impairs the metabolism of chlordiazepoxide; diazepam; dealkylflurazepam, the active metabolite of flurazepam; alprazolam; and triazolam. Evidence for an interaction with midazolam is contradictory. Cimetidine does not alter the metabolism of oxazepam, lorazepam, or temazepam, although lorazepam absorption may be increased in the presence of cimetidine. In patients who require therapy with BZs and H2 blockers, famotidine or ranitidine may be more suitable. When cimetidine is used, BZs that primarily undergo glucuronidation (e.g., lorazepam) may be less likely to produce a clinically significant
P.463
interaction. An interesting and as yet unexplained interaction is the reduction in the clearance of triazolam by ranitidine.Heparin. In healthy nonfasting subjects, heparin can cause a rise in the free fraction of diazepam, chlordiazepoxide, and oxazepam but not in lorazepam. In principle, this would not present clinical problems because the increased BZ effects would be transient.
Levodopa. Anecdotal reports suggest that BZs may decrease the antiparkinsonian effects of L-dopa.
Oral contraceptives. Oral contraceptives containing ethinyl estradiol reduce the clearance of both diazepam and chlordiazepoxide. In one study of women receiving oral contraceptives, the mean clearances of triazolam and alprazolam were not altered; however, the half-life of alprazolam was increased by 29%. Data for BZs metabolized by glucuronidation (e.g., lorazepam, oxazepam, temazepam) are contradictory; the reasons for this are not yet established.
Tricyclic antidepressants. Imipramine plasma levels may increase during concurrent alprazolam treatment. The effects on other antidepressants are unknown.
Propranolol and other -adrenergic receptor antagonists. Propranolol may reduce the clearance of diazepam but not that of alprazolam or lorazepam. Metoprolol may also impair diazepam clearance, but atenolol does not. In general, BZs metabolized by N-demethylation or N-dealkylation (e.g., diazepam, flurazepam) are most likely to interact with -adrenergic receptor antagonists.
Calcium channel blockers. The coadministration of the CYP 3A4 inhibitors mibefradil, verapamil, and diltiazem with triazolam or midazolam is contraindicated. Large increases in plasma concentrations and CNS depressive effects have been demonstrated, and interactions of a smaller magnitude may occur with alprazolam as well.
Antibiotics. Erythromycin and clarithromycin inhibit the CYP 3A4-mediated metabolism of triazolam, alprazolam, and midazolam. Coadministration results in decreased BZ clearance and increased CNS depression. Interactions with flunitrazepam are also likely based on in vitro studies. Azithromycin does not inhibit CYP 3A4, and it may be a suitable alternative. When erythromycin or clarithromycin is used, BZs that primarily undergo glucuronidation (e.g., lorazepam) may be less likely to produce a clinically significant interaction.
Antiretroviral agents. The HIV-1 protease ritonavir is a highly potent inhibitor of CYP 3A4, the primary enzyme that mediates the clearance of the oxidatively metabolized BZs triazolam, alprazolam, and midazolam. Short-term low dose ritonavir inhibits triazolam and alprazolam clearance to a clinically significant extent. However, long-term treatment with ritonavir for 10 days did not significantly alter alprazolam clearance. This is because ritonavir treatment results in an acute inhibitory effect, while continued or chronic treatment causes an induction of CYP 3A4. Concurrent inhibition and induction of CYP 3A4 makes the prediction of the outcome of a drug interaction difficult. If a patient is on long-term ritonavir and a BZ is added, the net effect may be difficult to predict because the net effects of induction and inhibition may balance out, resulting in a minor interaction. If, however, antiretroviral treatment with ritonavir is initiated in a patient receiving a BZ, such as triazolam or alprazolam, the acute effects of inhibition of CYP 3A4 will produce a clinically significant increase in the plasma concentrations of the BZ and thus increased CNS depression. Based on in vitro studies, the HIV-1 protease inhibitor nelfinavir is also a CYP 3A4 inhibitor, and coadministration with triazolam or midazolam is contraindicated.
Antifungal agents. The azole antifungal agents ketoconazole, itraconazole, and voriconazole are potent CYP 3A4 inhibitors, and they
P.464
impair the clearance of triazolam, alprazolam, and midazolam. In vitro studies predict clinically significant effects on flunitrazepam clearance as well. Coadministration of ketoconazole, itraconazole, or voriconazole with these BZs results in greatly enhanced CNS depression, and thus it is contraindicated. In patients taking these antifungal agents, temazepam may be chosen as a hypnotic in place of triazolam. In patients taking triazolam, alprazolam, or midazolam, terbinafine, rather than itraconazole, may be chosen for the treatment of dermatophytic infections. Although fluconazole is a less potent inhibitor of CYP 3A4, it does produce a significant impairment of triazolam and midazolam clearance, and coadministration requires a dose reduction of the BZ. The clearance of zolpidem is impaired by only 40% by ketoconazole, and the interaction with itraconazole and fluconazole is clinically unimportant. Itraconazole does not produce a clinically significant interaction with zopiclone.Selective serotonin reuptake inhibitors. See section III.E.3.
H. Carbamazepine
Carbamazepine has several properties that predispose it to drug interactions. It is an inducer of the cytochrome P-450 system, and it is subject to autoinduction. Carbamazepine metabolism is exclusively hepatic; it can be affected by other drugs that induce or impair hepatic metabolism. It also has an active metabolite, carbamazepine-10,11-epoxide. Any evaluation of drug interactions must take into account the effects on both the parent compound and this potentially more toxic metabolite.
Anesthetic and neuromuscular blocking agents. One case report describes tachyarrhythmias after halothane anesthesia in an 8-year-old girl receiving carbamazepine. Recovery from the neuromuscular blocking effects of pancuronium and vocuronium is faster in patients taking carbamazepine.
Antianxiety agents. See section III.G.4.
Antimicrobial agents. Erythromycin and triacetyloleandomycin inhibit the metabolism of carbamazepine. Macrolide antibiotics, with the exception of azithromycin, should be used with caution in patients taking carbamazepine. Clearance of the tetracyclic antibiotic doxycycline is enhanced with concurrent carbamazepine treatment. Apparently, carbamazepine has no effect on the metabolism of tetracycline, methacycline, oxytetracycline, demeclocycline, or chlortetracycline. However, demeclocycline and other tetracyclines inhibit antidiuretic hormone-sensitive adenylate cyclase activity, and they may reduce the hyponatremia that is sometimes produced by carbamazepine. No data are found concerning an interaction between carbamazepine and minocycline, a tetracyclic antibiotic that is partially metabolized in the liver. Carbamazepine metabolism is impaired with the coadministration of isoniazid; cases of neurotoxicity have been reported.
Anticoagulants. The metabolism of warfarin is increased, and its anticoagulant effect is reduced in the presence of carbamazepine. Frequent monitoring of prothrombin times is necessary when carbamazepine is added to or withdrawn from a therapeutic regimen.
Other anticonvulsants. VPA impairs the metabolism of the epoxide metabolite of carbamazepine by inhibiting the enzyme epoxide hydrolase. It also displaces carbamazepine from plasma proteins. Neurotoxicity may result from this combination, and plasma levels may not appear to be elevated if the epoxide level is not separately analyzed and reported. Carbamazepine lowers the levels of VPA and increases the levels of its 2-propyl-4-pentanoic acid metabolite, which is hepatotoxic and teratogenic. Despite potential problems, this drug combination is widely and usually safely used in the treatment of epilepsy.
P.465
Phenytoin induces the metabolism of carbamazepine and, to a lesser extent, of its epoxide metabolite. The effects of carbamazepine on phenytoin levels are variable. One case report describes sinoatrial block in an elderly woman taking the combination, thus suggesting an increased risk of cardiotoxicity with concurrent use.
Carbamazepine does not alter phenobarbital levels, but phenobarbital does induce the metabolism of carbamazepine and, to a lesser extent, of the epoxide. Carbamazepine reduces primidone levels while increasing the level of its metabolite phenobarbital. Primidone decreases carbamazepine levels.
Population pharmacokinetic studies indicate that tiagabine clearance is 60% greater in patients taking carbamazepine, whether with or without other enzyme-inducing antiepileptic drugs. This is most likely explained by the induction of CYP 3A4. Tiagabine does not affect the steady-state plasma concentrations of carbamazepine or its epoxide metabolite in patients with epilepsy.
Antidepressants. Carbamazepine induces the metabolism of imipramine, desipramine, nortriptyline, and bupropion and perhaps that of other antidepressants. Additive or synergistic cardiotoxicity has also been reported. Some evidence from both animal studies and one case report suggests that imipramine and desipramine may inhibit carbamazepine metabolism, leading to toxicity. Increased carbamazepine blood levels and clinical toxicity have been noted with the concurrent use of fluoxetine.
Although some authorities caution against the combination of carbamazepine and MAOI antidepressants, clinical experience suggests that the combination of phenelzine or tranylcypromine with carbamazepine is safe and that it does not alter carbamazepine plasma levels.
Calcium channel blocking agents. Diltiazem and verapamil, but not nifedipine or nimodipine, inhibit the metabolism of carbamazepine, which may lead to increased levels and possible neurotoxicity.
Cyclosporine. Carbamazepine induces the metabolism of cyclosporine; it may reduce its clinical effectiveness when dosage adjustments are not made.
Digoxin. Bradycardia has been reported with the combination of digoxin and carbamazepine. In addition, anecdotal evidence suggests that carbamazepine may reduce digoxin levels and that digoxin may increase carbamazepine levels; this is not well established.
Danazol. Danazol, a synthetic androgen used in the treatment of fibrocystic breast disease and endometriosis, impairs the clearance of carbamazepine; it may lead to toxicity.
Glucocorticoids. Carbamazepine induces the metabolism of dexamethasone, prednisolone, and methylprednisolone. It has also been associated with false-positive dexamethasone suppression tests, probably from the increased metabolism of dexamethasone or from direct effects on the hypothalamic-pituitary-adrenal axis.
Histamine receptor antagonists. Cimetidine may cause transient increases in carbamazepine levels, but these usually are not clinically significant. Ranitidine does not interact with carbamazepine; famotidine also probably does not.
Lithium. See section III.F.13.
Opioids. Carbamazepine may induce the metabolism of methadone and may reduce its levels enough to lead to mild withdrawal symptoms. Propoxyphene and dextropropoxyphene may inhibit the metabolism of carbamazepine, leading to increased levels and neurotoxicity. Carbamazepine has been used to potentiate the analgesic effects of hydromorphone in pain syndromes. Higher doses than usual are required to maintain fentanyl anesthesia in patients taking carbamazepine.
Oral contraceptives. Carbamazepine may be associated with breakthrough bleeding and pregnancy in women taking oral contraceptives. Proposed mechanisms of the interaction are the increased metabolism of ethinyl estradiol and levonorgestrel or an increase in the binding globulins for the sex hormones. A larger than typical dose of ethinyl estradiol may be required. Some authorities recommend starting the treatment with a preparation containing 50 g of ethinyl estradiol and then increasing this when breakthrough bleeding occurs.
Theophylline. Carbamazepine may induce the metabolism of theophylline, leading to subtherapeutic levels and the exacerbation of asthma symptoms.
Conventional antipsychotic agents. See section III.A.2.
P.466
I. Valproic Acid
The metabolism of VPA occurs via glucuronidation and -oxidation to yield 2-E VPA, 3-hydroxy VPA, and 3-keto VPA. Thus, CYP inhibitors do not affect VPA clearance. Other anticonvulsants, such as phenytoin, carbamazepine, and phenobarbital, can induce VPA metabolism. Patients on VPA monotherapy will generally have longer half-lives than will those receiving polytherapy with other anticonvulsants.
Antacids. An aluminum hydroxide-magnesium hydroxide antacid was found to increase the extent of absorption of a single 500 mg dose of VPA. The clinical significance of this finding is unknown, but antacids are often administered with VPA to counter its gastrointestinal effects.
Antidepressants. Increases in the VPA elimination half-life were seen after doses of 100 mg per day of amitriptyline for 3 weeks. The clearance and the extent of absorption, however, were not affected. The clinical significance of this interaction is not known. Increases in VPA blood levels that were without clinical consequences have been observed with the concurrent use of fluoxetine.
Antipsychotic agents. Chlorpromazine, but not haloperidol, may be associated with increases in the steady-state levels of VPA when it is coadministered. Chlorpromazine may inhibit the metabolism of VPA (see section III.A).
Aspirin. The steady-state free fraction of VPA may increase from 12% to 43% during concurrent aspirin administration. Aspirin may displace VPA from protein-binding sites. Aspirin decreases VPA clearance by the inhibition of -oxidation. The dosage of VPA may need to be reduced when aspirin is added.
Carbamazepine. See section III.H.5.
Clonazepam and diazepam. See section III.G.4.
Ethosuximide. VPA inhibits the metabolism of ethosuximide. Dosage reduction is necessary and plasma concentration monitoring of both drugs is desirable when this combination is used.
Barbiturates. VPA impairs phenobarbital metabolism; decreased clearance and the prolonged elimination half-life of phenobarbital in the presence of VPA have been consistently demonstrated. Without an appropriate adjustment of the phenobarbital dose, this combination can result in increased CNS depression. The combination may also result in lowered VPA levels due to phenobarbital-mediated enzyme induction. Dosage adjustments based on careful monitoring of serum concentrations are necessary. Data with primidone and VPA are contradictory. Mephobarbital may also decrease the serum levels of VPA. The unbound fraction of thiopental may be increased in patients taking VPA. This could enhance its anesthetic effects, but the clinical significance of this interaction is unknown.
Phenytoin. VPA inhibits the metabolism of phenytoin and displaces it from its protein-binding sites. Some reports have indicated that, when VPA is added to phenytoin, total serum phenytoin levels decline whereas the free fraction increases. The magnitude of this change is
P.467
highly variable, and some studies actually report declines in both measures. In most cases the interaction will not result in serious problems, although toxicity characterized by delirium and increased seizure frequency has been observed. The susceptible patient appears to be one who has high levels of phenytoin before the introduction of VPA; the transient increase in unbound phenytoin leads to toxicity.Felbamate. The coadministration of VPA and felbamate (1,200 to 2,400 mg per day) produces up to a 50% increase in peak plasma concentrations of VPA. Use of this combination generally requires a reduction in the dose of VPA.
Zidovudine. VPA (250 or 500 mg every 8 hours) has been shown to decrease zidovudine clearance by 38% without altering its elimination half-life in HIV-seropositive patients.
Rifampin. Treatment with 600-mg daily doses of rifampin for 5 days resulted in a 40% increase in the apparent oral clearance of VPA. Patients taking rifampin may need an adjustment of their VPA dose.
Other interactions. VPA displaces warfarin and tolbutamide from plasma proteins, although the clinical significance of this effect is not known.
J. Central Nervous System Stimulants, Especially Agents Used in the Treatment of Attention Deficit Hyperactivity Disorder (see Chapter 22)
A decreased seizure threshold has been reported in patients receiving pemoline concomitantly with antiepileptic medications. Insulin requirements in diabetes mellitus may be altered in association with the use of methamphetamine and the concomitant dietary regimen. Methamphetamine may decrease the hypotensive effect of guanethidine. Methamphetamine should not be used concurrently with MAOIs. Literature reports suggest that methamphetamines may be associated with significant elevation of plasma corticosteroids. This should be considered if the determination of plasma corticosteroid levels is desired in a patient taking methamphetamine. The combination of methamphetamine with ethanol results in increased cardiac work, which may produce cardiac toxicity.
K. Buspirone
Buspirone has a high presystemic extraction, making it particularly susceptible to metabolic drug interactions. Buspirone clearance is significantly impaired and plasma concentrations are elevated upon coadministration with CYP 3A4 inhibitory drugs, such as the calcium channel blockers diltiazem and verapamil, the azole antifungal agent itraconazole, and the macrolide antibiotic erythromycin. These interactions are also characterized by increasing the pharmacodynamic effects of buspirone. Rifampin treatment causes a significant induction of buspirone clearance, and the pharmacodynamic effects are reduced. Induction by phenytoin and carbamazepine is also likely, and this requires investigation.
IV. Herbals and Natural Products
A. St. John's Wort
The over-the-counter herbal antidepressant St. John's wort is associated with a high risk of pharmacokinetic drug interactions that stem from the modulation of CYP-mediated metabolism and P-glycoprotein mediated transport of coadministered substrates of these proteins. Some of the constituents of St. John's wort (e.g., hyperforin, I3, II8-biapigenin, and hypericin) are all highly potent inhibitors of CYP 3A4 with submicromolar inhibition constants in vitro. No in vivo evidence indicates that St. John's wort is a CYP 3A4 inhibitor. Furthermore, potent inhibition of CYPs 2C9, 2D6, and 1A2 has also been described in in vitro experiments. In contrast to the results of these in vitro studies performed in liver microsomal systems that do not support the biosynthesis of the metabolic enzymes, other in vitro studies using human hepatocytes have described the induction of CYP 3A4 by St. John's wort, an effect that is mediated by the activation of the pregnane X
P.468
receptor. Thus, although St. John's wort is an inhibitor of CYP activity in vitro, its inductive effects, which are generally observable only after multiple dose administration of the herbal, form the basis of several clinically important interactions with CYP 3A substrates, such as cyclosporin and oral contraceptive agents. Such drug combinations result in subtherapeutic plasma concentrations of the CYP 3A substrate with the potential consequences of transplant rejection (cyclosporin) or ineffective contraception (oral contraceptives). In addition to its effect on CYP 3A substrates, St. John's wort also decreases the plasma concentrations of digoxin, probably via induction of P-glycoprotein, an efflux transporter in the small intestine; thus, it potentially reduces the absorption of P-glycoprotein substrates, such as cyclosporin and digoxin. In addition, tannic acid present in St. John's wort may inhibit the absorption of iron.
B. Grapefruit Juice
Certain furanocoumarin constituents of grapefruit juice are potent irreversible inhibitors of CYP 3A4. Intake of grapefruit juice results in an increase in oral bioavailability of some CYP 3A substrates, with a decrease in the apparent oral clearance. However, intravenous clearance is generally unaffected, demonstrating that the effect of grapefruit juice is to inhibit the CYP 3A-mediated metabolism in the small intestine, without affecting hepatic drug extraction. Several psychopharmacologic agents, such as the oxidatively metabolized BZs triazolam and alprazolam, and antidepressants, such as trazodone and nefazodone, are metabolized almost entirely by CYP 3A4, and they are also administered by the oral route. Clinically significant interactions with grapefruit juice have been described for triazolam, a CYP 3A substrate with high presystemic extraction. Interactions with drugs, like alprazolam, with low to moderate extraction ratios are less significant.
V. Additional Information
Many medical facilities and pharmacies maintain and use drug interaction software programs. Many clinicians have similar programs on their personal computers or personal digital assistants (PDAs). Although they provide useful information and they are a good starting place, many of the programs are not up to date, and many include interactions from case reports in the literature where causality has not been established. Useful information is available on the following websites:
http://www.medicine.iupui.edu/flockhart
http://www.gentest.com/human_p450_database/srchh450.asp
http://www.drugdigest.org/DD
http://www.healthatoz.com/Atoz/Jsp/drugdb/drugSearch.jsp
http://www.drugsearch.doctorsnetaccess.com
http://www.personalmd.com/drugdatabase.shtml
http://www.mhc.com/Cytochromes/
ADDITIONAL READING
Blackwell B. Monoamine oxidase inhibitor interactions with other drugs. J Clin Psychopharmacol 1991;11:55 59.
Ciraulo DA, Shader RI. Fluoxetine drug-drug interactions. I. Antidepressants and antipsychotics. J Clin Psychopharmacol 1990;10:48 50.
Ciraulo DA, Shader RI. Fluoxetine drug-drug interactions. II. J Clin Psychopharmacol 1990;10:213 217.
Ciraulo DA, Shader RI, Greenblatt DJ, Creelman W, eds. Drug interactions in psychiatry. Baltimore: Williams & Wilkins, 1989.
Goff DC, Baldessarini RJ. Drug interactions with antipsychotic agents. J Clin Psychopharmacol 1993;13:55 70.
Greenblatt DJ, Patki KC, von Moltke LL, et al. Drug interactions with grapefruit juice: an update. J Clin Psychopharmacol 2001;21:357 359.
P.469
Greenblatt DJ, von Moltke LL. Sedative-hypnotic and anxiolytic agents. In: Levy RH, Thummel KE, Trager WF, et al., eds. Metabolic drug interactions. Philadelphia: Lippincott Williams & Wilkins, 2000:259 270.
Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Human cytochromes and some newer antidepressants: kinetics, metabolism, and drug interactions. J Clin Psychopharmacol 1999;19:23S 35S.
Hansten PD, Horn JR. Drug interactions, 6th ed. Philadelphia: Lea & Febiger, 1989.
Johnson FN, ed. Lithium combination treatment. Basel, Switzerland: Karger, 1987.
Ketter TA, Post RM, Worthington K. Principles of clinically important drug interactions with carbamazepine. Part I. J Clin Psychopharmacol 1991;11:198 203.
Ketter TA, Post RM, Worthington K. Principles of clinically important drug interactions with carbamazepine. Part II. J Clin Psychopharmacol 1991;11:306 313.
Ragheb M. The clinical significance of lithium-nonsteroidal antiinflammatory drug interactions. J Clin Psychopharmacol 1990;10:350 354.
Shinn AF, ed. Evaluations of drug interactions. New York: Macmillan, 1988.
Small JG, Milstein V. Lithium interactions: lithium and electroconvulsive treatment. J Clin Psychopharmacol 1990;10:346 349.
Spina E, Pisani F, Perucca E. Clinically significant pharmacokinetic drug interactions with carbamazepine. Clin Pharmacokinet 1996;31:198 214.
Sproule BA, Naranjo CA, Bremner KE, et al. Selective serotonin reuptake inhibitors and CNS drug interactions: a critical review of the evidence. Clin Pharmacokinet 1997;33:454 471.
Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism in humans: clinical relevance. Clin Pharmacokinet 2000;38:111 180.
Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Human drug metabolism and the cytochromes P450: application and relevance of in vitro models. J Clin Pharmacol 2001;41:1149 1179.
von Moltke LL, Greenblatt DJ. Drug transporters in psychopharmacology are they important? J Clin Psychopharmacol 2000;20:291 294.
Yuan R, Flockhart DA, Balian JD. Pharmacokinetic and pharmacodynamic consequences of metabolism-based drug interactions with alprazolam, midazolam, and triazolam. J Clin Pharmacol 1999;39:1109 1125.
1This chapter is deliberately long. This decision was made to address the relatively sparse background that many students and clinicians have about drug metabolism and drug interactions.
2Aldehyde oxidase is a molybdenum-containing cytosolic redox enzyme that catalyzes the oxidation of azaheterocylic compounds, such as zaleplon and famciclovir, and the reduction of zonisamide. Menadione, a vitamin K analogue, is an in vitro antagonist.
EAN: 2147483647
Pages: 37