3 - Hypokalemia or Hyperkalemia
Editors: Schrier, Robert W.
Title: Manual of Nephrology, 6th Edition
Copyright 2005 Lippincott Williams & Wilkins
> Table of Contents > 3 - The Patient with Hypokalemia or Hyperkalemia
3
The Patient with Hypokalemia or Hyperkalemia
Catherine L. Kelleher
Stuart L. Linas
Potassium is the most abundant cation in the human body. It regulates intracellular enzyme function and helps determine neuromuscular and cardiovascular tissue excitability. Ninety percent of total body potassium is located in intracellular fluid (ICF; primarily in muscle), 10% in the extracellular fluid (ECF), and less than 1% in plasma. The ratio of extracellular potassium to intracellular potassium determines membrane potential. The acuity of changes in serum potassium concentration and membrane potential determines the severity of clinical symptoms and underlies the clinical findings caused by disorders of potassium metabolism.
Overview of potassium physiology. The typical Western diet contains between 40 and 120 mEq of potassium a day. Tight contol of the serum potassium between 3.5 and 5.5 mEq per day is primarily accomplished by the kidney, where secretion varies between 40 and 120 mEq per day. Potassium losses in stool and sweat are small (5 to 10 mEq). Although total body potassium declines with aging, and the rate of decline appears to be influenced by sex and race, the clinical significance of these observations is not clear. The internal regulation of potassium involves the interplay of several hormonal systems as well as the internal acid-base enviornment.
Internal balance. After a meal, potassium is rapidly redistributed between the plasma and intracellular compartments. Several systems interact to regulate the transcellular movement of potassium:
Insulin. The binding of the insulin hormone to insulin receptors causes a hyperpolarization of cell membranes that facilitates potassium uptake. Insulin also activates sodium-potassium adenosine triphosphatase (ATPase) pumps and causes the cellular uptake of potassium.
Catecholamines. Activation of the beta2 adrenoreceptor results in cellular potassium uptake. This effect is transduced by cyclic adenosine monophosphate (cAMP) activation of sodium-potassium ATPase pumps, causing an influx of potassium in exchange for sodium. Therapeutic agents such as theophylline potentiate beta2 adrenoreceptor-mediated potassium uptake by inhibiting the degradation of cAMP.
Aldosterone. Although this mineralocorticoid facilitates potassium uptake into muscle, clear proof of the clinical significance of this effect is lacking.
Acid-base. Inorganic acidosis (e.g., hydrochloric acid) facilitates potassium movement from ICF to ECF. Protons enter cells, whereas impermeant inorganic ions do not. The resulting increases in ICF positive charge favor the outward movement of potassium. Because organic ions (lactate, ketoacids) are less restricted from entering cells, increases in serum potassium may not occur in organic acidosis.
Tonicity. Hyperglycemia causes potassium-rich fluid to leave the cell, thus increasing ECF potassium. Under most conditions, increases in insulin modulate and reverse the effect of increased extracellular tonicity. However, when insulin cannot be increased (as in diabetes mellitus) or hyperglycemia occurs rapidly (as with the administration of 50% glucose), hyperkalemia occurs. Rapid infusions of mannitol also may cause hyperkalemia.
External balance. Urinary potassium excretion is the result of a difference between the potassium secreted and potassium reabsorbed in the distal
P.38
nephron. Potassium is freely filtered at the glomerulus. More than 50% of filtered potassium is reabsorbed in the proximal convoluted tubule. In the descending limb of Henle's loop, especially in deep nephrons, potassium concentration increases. In the ascending limb of the loop of Henle, the Na-K-2Cl cotransporter leads to the reabsorption of potassium. When the tubular fluid reaches the early distal convoluted tubule, only 10% to 15% of filtered potassium remains. Potassium is secreted by the principal cells of the cortical collecting and outer-medullary collecting duct. Potassium is reabsorbed in the collecting tubule, an effect mediated by intercalated cells. A fall in glomerular filtration rate (GFR) is not generally associated with decreased potassium excretion and hyperkalemia until the GFR is less than 20 mL per minute. The major factors regulating potassium excretion follow.Distal nephron flow rate and sodium delivery. Under normal conditions, sodium delivered to the cortical collecting tubule is reabsorbed through apical sodium channels in the principal cells. This increases cell sodium concentration and basolateral sodium-potassium-ATPase activity. The resultant increases in cellular potassium lead to an increased apical potassium transport through specific potassium channels. Increases in sodium delivery facilitate this mechanism of potassium secretion. In addition, increases in tubular flow rate help maintain a low urinary potassium concentration, which favors the movement of potassium from cells into tubular fluid.
Mineralocorticoids. Aldosterone is the major mineralocorticoid; it increases potassium secretion into the tubular fluid by:
Increasing the number and activity of apical sodium channels
Increasing basolateral sodium-potassium ATPase activity
Increasing the number of apical potassium channels
Increases or decreases in dietary potassium increase or decrease urinary potassium, respectively. Renal adaptation to high potassium intake is mediated by a potassium-induced increase in aldosterone secretion and by an increase in distal nephron sodium-potassium ATPase activity. In response to potassium restriction, mineralocorticoid activity decreases, thus causing a decline in potassium secretion.
Increases in relatively nonresorbable anions (e.g., bicarbonate, penicillins) trap secreted potassium in the tubular lumen and limit potassium reabsorption in the medullary collecting duct. The resulting renal potassium losses may lead to severe potassium depletion.
Hypokalemia
Diagnosis. The initial approach to hypokalemia is to determine whether it is spurious, secondary to a shift of potassium from the extracellular to intracellular compartments, or a result of a true decreases in total body potassium (Fig. 3-1).
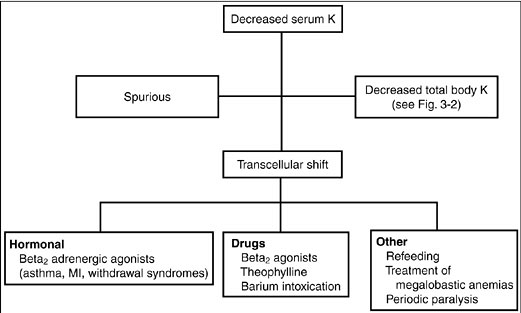
Figure 3-1. Diagnostic approach to hypokalemia. (K, potassium; MI, myocardial infarction.)
Spurious hypokalemia occurs in the setting of extreme leukocytosis (in vitro white blood cells uptake potassium in the test tube) and is not associated with changes in either internal or external potassium balance.
Potassium shifts into cells may occur acutely in conditions associated with increases in endogenous insulin or catecholamines. For example, catecholamine release associated with shortness of breath (asthma, chronic obstructive pulmonary disease exacerbations, heart failure, chest pain syndromes including myocardial infarction or angina) or catecholamine release from a drug withdrawal syndrome (alcohol, narcotics, or barbiturates) shifts potassium to inside cells thereby decreasing the serum potassium concentration. Hypokalemia may also be caused by insulin administration (correction of diabetic ketoacidosis, postresuscitation for hyperkalemia) or beta2-adrenoreceptor mimetics (beta2 agonists, theophylline). Other common causes of decreases in serum potassium without decreases in total body potassium include hypokalemic periodic paralysis (familial and hyperthyroid types), treatment of megaloblastic anemias, and refeeding syndromes
P.39
(probably insulin mediated). The refeeding syndrome, in which severely malnourished patients are begun on nasogastric feeding, is also seen in older adults in whom the clinical manifestations of malnutrition are less clinically apparent.Decreases in total body potassium (Fig. 3-2) are caused by either inadequate potassium intake or by excessive renal or extrarenal potassium losses. The measurement of urinary potassium excretion (by 24-hour measurements or spot potassium concentrations) is used to distinguish renal versus extrarenal potassium loss. Urinary potassium concentrations less than 20 mEq per L suggest poor potassium intake or extrarenal potassium loss. Serum acid-base status is also helpful in evaluating the site of potassium loss. Metabolic acidosis may suggest gastrointestinal (GI) losses (diarrhea of any cause, e.g., infectious, toxic, laxative abuse). A normal serum pH is less helpful, because hypokalemia can be secondary to both decreases in intake and GI losses. Metabolic alkalosis with urinary potassium of less than 20 mEq per L, although rare, is associated with laxative abuse, villous adenoma, or congenital chloride-losing diarrhea. Hypokalemia with a urinary potassium excretion of greater than 20 mEq per L suggests renal potassium wasting. The serum pH also is used to further evaluate etiologies. Metabolic acidosis suggests renal tubular acidosis (type 1 or type 2), diabetic ketoacidosis (osmotic diuresis), ureterosigmoidostomy, or carbonic anhydrase inhibitor use.
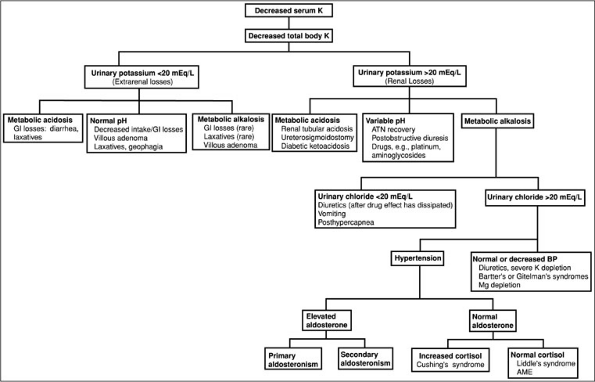
Figure 3-2. Diagnostic approach to hypokalemia. (AME, aseptic meningo-encephalitis; ATN, acute tubular necroses; BP, blood pressure; GI, gastrointestinal; K, potassium.)
More commonly, renal potassium losses are associated with metabolic alkalosis. In this clinical setting, the urinary chloride concentration is helpful. A low urinary chloride concentration (less than 20 mEq per L) suggests potassium losses from upper GI losses, recent diuretic use, or a post-hypercapnic syndrome. Hypokalemia with a high urinary chloride concentration is further distinguished on the basis of the presence or absence of hypertension. In normotensive individuals, hypokalemia with metabolic alkalosis and a high urinary chloride occurs with diuretic use (loop or distal convoluted tubule acting diuretics), in Bartter's and Gitelman's syndrome, and with severe decreases in total body magnesium or potassium.
P.40
P.41
Hypokalemia with renal potassium wasting, renal chloride wasting, and hypertension is further evaluated by urinary aldosterone concentrations. An elevated aldosterone level suggests either primary aldosteronism (adenoma, hyperplasia, glucocorticoid remedial) or secondary aldosteronism (renovascular or accelerated hypertension, diuretic use). Conversely, normal aldosterone levels with increases in serum cortisol suggest Cushing's syndrome or exogenous steroid use. Normal cortisol levels indicate Liddle's syndrome (caused by increases in the activity of the cortical collecting tubule sodium channel) or apparent mineralocorticoid excess syndrome [decreases in 11 hydroxy-steroid dehydrogenase activity in kidney tissue (congenital, licorice ingestion) causing the mineralocorticoid receptor to respond to glucocorticoid]. Increases in urinary potassium excretion without a significant acid-base disorder are seen during the recovery phase of acute tubular necrosis, postobstructive diuresis, and magnesium depletion associated with drugs such as aminoglycosides and cis-platinum, or in myelomonocytic leukemia (secondary to lysozymuria).Finally, hypokalemia is frequently associated with chronic alcolohism. The mechanism behind this electrolyte abnormality is not well defined but is probably multifactorial secondary to poor intake, diarrhea, alcohol withdrawl with respiratory alkalosis, and kaliuresis associated with hypomagnesemia.
Although cell-shift hypokalemia and decreases in total body potassium do occur as isolated problems, they frequently occur simultaneously. Decreases in total body potassium potentiate the effects of drugs and hormones to shift potassium into cells. For example, small changes in potassium during insulin therapy may not cause hypokalemia if total body potassium is normal, but in the setting of total body potassium depletion (e.g., during the treatment of diabetic ketoacidosis or with diuretic use), cellular shifts of potassium during insulin therapy can result in profound hypokalemia.
The manifestations of hypokalemia are mainly cardiac and neuromuscular (Table 3-1). The most dramatic neuromuscular symptoms are paresis, paralysis, and respiratory failure. Potassium depletion causes supraventricular and ventricular arrhythmias, especially in patients on digitalis therapy. Although severe hypokalemia is more likely to cause complications, even minimal decreases in serum or total body potassium can be arrhythmogenic in patients with underlying heart disease or who are on digitalis therapy.
Table 3-1. Clinical Manifestations of Hypokalemia
Cardiovascular Electrocardiographic abnormalities: U waves, QT prolongation, ST depression Predisposition to digitalis toxicity Atrial/ventricular arrhythmias Neuromuscular Skeletal muscle Weakness Cramps Tetany Paralysis flaccid Rhabdomyolysis Smooth muscle Constipation Ileus Urinary retention Endocrine Carbohydrate intolerance Diabetes mellitus Decreased aldosterone Growth retardation Renal/electrolyte Decreased renal blood flow, glomerular filtration rate Nephrogenic diabetes insipidus Increased ammoniagenesis (hepatic encephalopathy) Chloride wasting/metabolic alkalosis Cyst formation Interstitial nephritis Tubular vacuolization The treatment of hypokalemia depends on the underlying cause, the degree of potassium depletion, and the risk of potassium depletion to the patient. In general, hypokalemia secondary to cell shift is managed by treating underlying conditions. For example, hypokalemia in the setting of catecholamine increases, as in chest pain syndromes, is managed with appropriate treatments for the pain. However, when cell-shift hypokalemia is associated with life-threatening conditions such as paresis, paralysis, or hypokalemia in the setting of myocardial infarction, the administration of potassium is indicated. With potassium depletion, replacement therapy depends on the estimated degree of decreases in total body potassium. For example, decreases in total body potassium accompanied by a fall in serum potassium to 3.5 to 3.0 mEq per L are associated with a potassium deficit of 150 to 200 mEq. Decreases in serum potassium from 3 to 2 mEq per L are associated with 200- to 400-mEq additional decreases in total body potassium. Potassium can be administered intravenously, but in limited quantities (10 mEq per hour into a peripheral vein; 15 to 20 mEq per hour into a central vein). Larger potassium requirements can only be accomplished by oral therapy or with dialysis.
Hyperkalemia
The approach to hyperkalemia (Fig. 3-3) is to determine whether increases in serum potassium are spurious, caused by shifts of potassium from cellular to extracellular spaces, or represent a true increase in total body potassium.
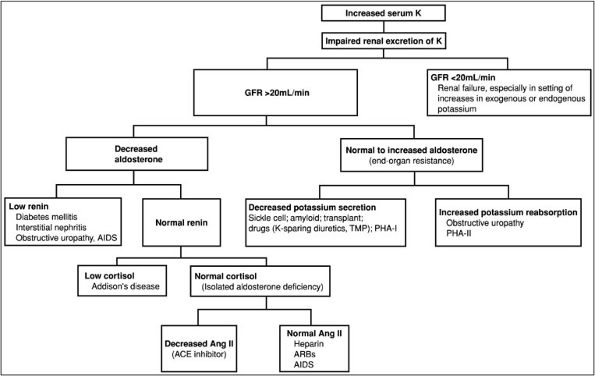
Figure 3-3. Diagnostic approach to hypokalemia. (ACE, angiotensin-converting enzyme; AIDS, acquired immunodeficiency syndrome; ARBs, adrenergic receptor binders; GFR, glomerular filtration rate; K, potassium; PHA, pseudohypoaldosteronism; TMP, trimethoprim-sulfa.)
P.42
Spurious hyperkalemia is caused by red blood cell hemolysis in vitro, ischemic blood draws, extreme thrombocytosis (greater than 1 million mL), or leukocytosis (greater than 50,000 mL). Spurious hyperkalemia is distinguished from true hyperkalemia by the absence of electrocardiographic (ECG) abnormalities. Hyperkalemia caused by cell shifts of potassium occurs acutely and results from decreased potassium transfer into cells (with decreases in insulin or beta-adrenergic blocker therapy), increased potassium movement from cells to the extracellular space (with metabolic acidosis), hypertonicity (with hyperglycemia or the administration of mannitol), exercise, muscle breakdown (with rhabdomyolysis), or drug intoxications from digitalis or succinylcholine.
Sustained hyperkalemia is caused by decreases in renal potassium excretion. This usually is not seen until the glomerular filtration rate is less than 20 mL per minute. However, it may be seen with less severe decreases in GFR when the kidney is challenged with a potassium load from potassium ingestion (e.g., diet, salt substitutes, or drugs, including potassium chloride and potassium citrate) and from increases in endogenous potassium production (e.g., GI bleed, resolving hematoma, rhabdomyolysis, catabolic states, tumor lysis). Hyperkalemia with less severe decreases in renal function is also associated with reductions in the distal nephron flow rate or low serum aldosterone levels as, for example, with hyporenin-hypoaldosteronism. Last, hyperkalemia is also associated with less severe decreases in GFR when drugs that alter potassium excretion [e.g., potassium-sparing diuretics, beta-adrenegic blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, nonsteroidal anti-inflammatory drugs (NSAIDs)] are
P.43
P.44
administered. Clinical studies also suggest that older adults are at increased risk for hyperkalemia. Although no clear explanation exists for this observation, it may be related to an age-associated decline in aldosterone synthesis or possibly a decline in tubular sensitivity to its action. Commonly used medications causing hyperkalemia are shown in Table 3-2.Table 3-2. Commonly Used Medications Causing Hyperkalemia
Medication Mechanism Digitalis overdose Inhibition of the Na-K-ATPase pump Angiotensin II inhibitors Decreased aldosterone excretion NSAIDs Blocks prostaglandin stimulation of renin Trimethoprim A cationic agent that decreases the number of open sodium channels in the luminal membrane of cortical collecting ducts Pentamidine Same mechanism as trimethoprim blocks distal potassium excretion Spirolactone Competes for aldosterone receptor in collecting tubule Amiloride Blocks sodium channel Heparin Decrease aldosterone Salt substitutes Contain potassium Succinylcholine Moves potassium from intracellular to extracellular fluid Cyclosporine Multifactorial, including hyporenin hypoaldosteronism and interference with aldosterone action in the potassium-secreting cells of the cortical collecting duct Pentamidine Blocks distal potassium secretion Hyperkalemia also occurs in the setting of a relatively well preserved GFR. The causes of hyperkalemia in this setting are distinguished on the basis of plasma or urinary aldosterone levels. Decreases in aldosterone occur in the setting of normal, increased, or decreased plasma renin activity. Decreased plasma renin activity (hyporeninemic hypoaldosteronism) tends to occur in older adults and is associated with a number of renal diseases, including diabetes, interstitial nephritis (e.g., sickle cell anemia, analgesic use, heavy metal toxicity), obstructive uropathy, systemic lupus erythematosus, and amyloidosis. Decreases in plasma renin activity also are associated with acquired immunodeficiency syndrome (AIDS) nephropathy, transplantation, and medications including cyclosporine and NSAIDs. Hyperreninemic hypoaldosteronism also occurs both with decreases in cortisol production (Addison's disease) and with normal cortisol production when medications such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and heparin are used. Finally, increases in serum potassium can be associated with normal to high levels of aldosterone and end-organ resistance to aldosterone. Aldosterone resistance is caused by decreases in potassium secretion (drugs such as potassium-sparing diuretics, trimethoprim, and pentamidine), interstitial renal diseases (systemic lupus erythematosus, sickle cell anemia), obstructive uropathy, or transplantation. It also occurs in an unusual hereditary disease called pseudohypoaldosteronism type 1, in which the etiology is either a decrease in aldosterone receptor number or increases in the activity of the sodium chloride (NaCl) cotransporter in the distal convoluted tubule. With decreased activity of the cotransporter, sodium delivery to the distal tubule is decreased. Hyperkalemia in association with normal potassium secretion and increased potassium reabsorption occurs with obstructive uropathy.
Diagnosis. The urinary potassium excretion rate or transtubular potassium gradient (TTKG) (urine potassium divided by serum potassium divided by the ratio of urine osmolality to plasma osmolality) is used to distinguish renal from extrarenal causes of hyperkalemia (Table 3-2). Extrarenal causes of hyperkalemia (e.g., diet) are associated with increases in potassium excretion (24-hour urinary potassium excretion greater than 80 mEq per day and TTKG greater than 10). In contrast, renal causes of hyperkalemia (for example, Addison's disease) are associated with decreases in urinary potassium excretion (less than 20 mEq per day) and TTKG (less than 3). The administration of a mineralocorticoid (0.05 mg fludrocortisone) results in increases in urinary potassium excretion (greater than 40 mEq per day) and TTKG (greater than 7) in patients with aldosterone deficiency (Addison's disease) but no increases in urinary potassium excretion or in TTKG in patients with aldosterone resistance (e.g., sickle cell anemia).
The clinical manifestations of hyperkalemia are predominantly cardiac and neuromuscular. It is important to note that patients with hyperkalemia often present with vague GI complaints and nonspecific unwell feelings. Electrocardiogram (ECG) abnormalities include peak T waves, widened QRS interval, and asystole. Neuromuscular abnormalities include weakness, constipation, and paralysis.
The treatment of hyperkalemia (Fig. 3-4) depends on the presence or absence of ECG and neuromuscular abnormalities. In the absence of symptoms or ECG abnormalities, hyperkalemia is treated conservatively for example, by decreasing dietary potassium or withdrawing offending drugs.
P.46
In the presence of ECG abnormalities or symptoms, the goal of therapy is to stabalize cell membranes. First-line therapy includes calcium gluconate, 10 to 30 mL as a 10% solution (onset of action 1 or 2 minutes). Although the mechanism remains undefined, calcium antagonizes the membranes and depolarizes effects of hyperkalemia. Other therapies include sodium bicarbonate, 50 to 150 mEq (onset 15 to 30 minutes) and insulin 5 to 10 units intravenously (onset 5 to 10 minutes), which increases the activity of the Na-K-ATPase pump in skeletal muscle and drives the potassium into cells. Glucose, 25 grams intravenously, is given simultanously to prevent hypoglycemia. Blood sugars should be monitored for approximately 6 hours to identify and treat hypoglycemia from the insulin. Albuterol nebulizer, 20 mg in 4 mL normal saline (onset 15 to 30 minutes), also activates the Na-K-ATPase and drives potassium into cells. Potassium driven intracellularly generally begins to move extracellularly again after approximately 6 hours, increasing the serum potassium concentration. Therefore, therapy to remove potassium from the body should be started simultaneously. Reductions in total body potassium may be achieved through a potassium exchange resin. The primary potassium resin used is sodium polystyrene sulfonate. One gram of this medication binds approximately 1 meq of potassium and releases 1 to 2 meq of sodium back into the circulation. This medication may be given orally (onset 2 hours) or by enema (onset 30 to 60 minutes). Finally, if indicated, hemodialysis is initiated, removing 25 to 30 mEq of potassium per hour.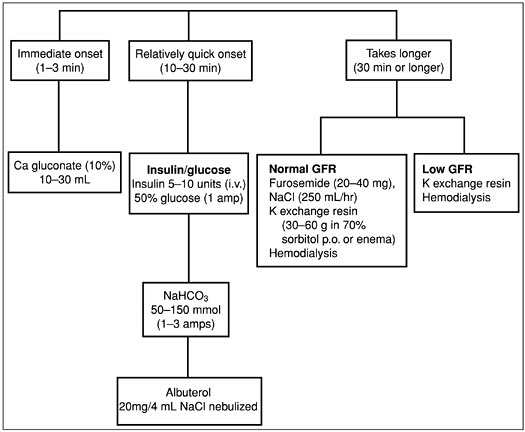
Figure 3-4. Treatment of hyperkalemia. (GFR, glomerular filtration rate; K, potassium.)
P.45
Suggested Readings
Allon M. Hyperkalemia in end stage renal disease: mechanisms and management. J Am Soc Nephrol 1995;6:1134 1142.
Ethier JH, Kanel KS, Magner PO, Lemann J, Halperin ML. The transtubular potassium concentration in patients with hypokalemia and hyperkalemia. Am J Kidney Dis 1990;15:309 315.
Gennari FJ. Hypokalemia. N Engl J Med 1998;339:451 458.
Halperin ML, Kamel SK. Electrolyte quintet: potassium. Lancet 1998;352:135 140.
Kamel KS, Halperin ML. Treatment of hypokalemia and hyperkalemia. In: Brady HR, Wilcox CS, eds. Therapy in nephrology and hypertension. Philadelphia: WB Saunders, 1999:270 278.
Kellerman PS, Linas SL. Disorders of potassium metabolism. In: Feehally J, Johnson R, eds. Comprehensive clinical nephrology. London: Mosby International, 1999.
Osorio FV, Linas SL. Disorders of potassium metabolism. In: Schrier RW, ed. Atlas of diseases of the kidney, vol 1. Philadelphia: Blackwell Science, 1998:sec 1.
Oster JR, Singer I, Fishman LM. Heparin-induced aldosterone suppression and hyperkalemia. Am J Med 1995;98:575 586.
Perazella MA, Mahnensmith RL. Hyperkalemia in the elderly. JGIM. 1997;10:646 656.
Perazella M, Rastegar Asghar. Disorders of potassium and acid base metabolism in association with renal disease. In Schrier RW, ed. Diseases of the kidney and urinary tract, 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2001:2577 2606.
Peterson L, Levi M. Disorders of potassium metabolism. In: Schrier RW, ed. Renal and electrolyte disorders, 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2003:171 215.
Moonseong QH, Heshka S, Wang J, et al. Total body potassium differs by sex and age across the adult like span. Am J Clin Nutri 2003;78:72 77.
Weiner ID, Linas SL, Wingo CS. Disorders of potassium metabolism. In: Johnson RJ, Feehally JF, ed. Comprehensive clinical nephrology, 2nd ed. St. Louis: Mosby, 2003;109 121.
Weiner ID, Wingo CS. Hyperkalemia: a potential silent killer. J Am Soc Nephrol 1998;9:1535 1543.
Weiner ID, Wingo CS. Hypokalemia: consequences, causes, and correction. J Am Soc Nephrol 1997;8:1179 1188.