10 - Heart
Editors: McPhee, Stephen J.; Papadakis, Maxine A.; Tierney, Lawrence M.
Title: Current Medical Diagnosis & Treatment, 46th Edition
Copyright 2007 McGraw-Hill
> Table of Contents > 13 - Blood
function show_scrollbar() {}
13
Blood
Charles A. Linker MD
Anemias
General Approach to Anemias
Anemia is present in adults if the hematocrit is less than 41% (hemoglobin < 13.5 g/dL) in males or less than 37% (hemoglobin < 12 g/dL) in females. Congenital anemia is suggested by the patient's personal and family history. Poor diet results in folic acid deficiency and contributes to iron deficiency, but bleeding is much more commonly the cause of iron deficiency in adults. Physical examination includes attention to signs of primary hematologic diseases (lymphadenopathy, hepatosplenomegaly, or bone tenderness). Mucosal changes such as a smooth tongue suggest megaloblastic anemia.
Anemias are classified according to their pathophysiologic basis, ie, whether related to diminished production or accelerated loss of red blood cells (Table 13-1), or according to cell size (Table 13-2). The diagnostic possibilities in microcytic anemia are iron deficiency, thalassemia, and anemia of chronic disease. A severely microcytic anemia (mean cell volume [MCV] < 70 fL) is due either to iron deficiency or thalassemia. Macrocytic anemia may be due to megaloblastic (folate or vitamin B12 deficiency) or nonmegaloblastic causes, in particular myelodysplasia and the use of antiretroviral drugs. A severely macrocytic anemia (MCV > 125 fL) is almost always megaloblastic; exceptions are the myelodysplastic syndromes.
Iron Deficiency Anemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Serum ferritin < 12 mcg/L.
Caused by bleeding in adults unless proved otherwise.
Responds to iron therapy.
General Considerations
Iron deficiency is the most common cause of anemia worldwide. The causes are listed in Table 13-3. Iron is necessary for the formation of heme and other enzymes. Total body iron ranges between 2 and 4 g: approximately 50 mg/kg in men and 35 mg/kg in women. Most (70 95%) of the iron is present in hemoglobin in circulating red blood cells. One milliliter of packed red blood cells (not whole blood) contains approximately 1 mg of iron. In men, red blood cell volume is approximately 30 mL/kg. A 70-kg man will therefore have approximately 2100 mL of packed red blood cells and consequently 2100 mg of iron in his circulating blood. In women, the red cell volume is about 27 mL/kg; a 50-kg woman will thus have 1350 mg of iron circulating in her red blood cells. Only 200 400 mg of iron is present in myoglobin and nonheme enzymes. Aside from circulating red blood cells, the major location of iron in the body is the storage pool, as ferritin or as hemosiderin and in macrophages. The range for storage iron is wide (0.5 2 g); approximately 25% of women in the United States have none.
The average American diet contains 10 15 mg of iron per day. About 10% of this amount is absorbed. Absorption occurs in the stomach, duodenum, and upper jejunum. Dietary iron present as heme is efficiently absorbed (10 20%) but nonheme iron less so (1 5%), largely because of interference by phosphates, tannins, and other food constituents. Small amounts of iron approximately 1 mg/d are normally lost though exfoliation of skin and mucosal cells. There is no physiologic mechanism for increasing normal body iron losses.
Menstrual blood loss in women plays a major role in iron metabolism. The average monthly menstrual blood loss is approximately 50 mL, or about 0.7 mg/d. However, menstrual blood loss may be five times the average. To maintain adequate iron stores, women with heavy menstrual losses must absorb 3 4 mg of iron from the diet each day. This strains the upper limit of what may reasonably be absorbed, and women with menorrhagia of this degree will almost always become iron deficient without iron supplementation.
In general, iron metabolism is balanced between absorption of 1 mg/d and loss of 1 mg/d. Pregnancy may also upset the iron balance, since requirements increase to 2 5 mg of iron per day during pregnancy and lactation.
P.494
Normal dietary iron cannot supply these requirements, and medicinal iron is needed during pregnancy and lactation. Repeated pregnancy (especially with breast-feeding) may cause iron deficiency if increased requirements are not met with supplemental medicinal iron. Decreased iron absorption can on very rare occasions cause iron deficiency and usually occurs after gastric surgery, though concomitant bleeding is frequent.
Table 13-1. Classification of anemias by pathophysiology. | |
|---|---|
|
By far the most important cause of iron deficiency anemia is blood loss, especially gastrointestinal blood loss. Chronic aspirin use may cause it even without a documented structural lesion. Iron deficiency demands a search for a source of gastrointestinal bleeding if other sites of blood loss (menorrhagia, other uterine bleeding, and repeated blood donations) are excluded.
Table 13-2. Classification of anemias by mean cell volume. | |
|---|---|
|
Table 13-3. Causes of iron deficiency. | |
|---|---|
|
Chronic hemoglobinuria may lead to iron deficiency since iron is lost in the urine; traumatic hemolysis due to a prosthetic cardiac valve and other causes of intravascular hemolysis (eg, paroxysmal nocturnal hemoglobinuria) should also be considered.
Clinical Findings
A. Symptoms and Signs
As a rule, the only symptoms of iron deficiency anemia are those of the anemia itself (easy fatigability, tachycardia, palpitations and tachypnea on exertion). Severe deficiency causes skin and mucosal changes, including a smooth tongue, brittle nails, and cheilosis. Dysphagia because of the formation of esophageal webs (Plummer-Vinson syndrome) also occurs. Many iron-deficient patients develop pica, craving for specific foods (ice chips, etc) often not rich in iron.
B. Laboratory Findings
Iron deficiency develops in stages. The first is depletion of iron stores. At this point, there is anemia and no change in red blood cell size. The serum ferritin will become abnormally low. A ferritin value less than 30 mcg/L is a highly reliable indicator of iron deficiency. The serum total iron-binding capacity (TIBC) rises. Bone marrow biopsy for evaluation of iron stores is now rarely performed because of intraobserver variation in its interpretation.
After iron stores have been depleted, red blood cell formation will continue with deficient supplies of iron. Serum iron values decline to less than 30 mcg/dL and transferrin saturation to less than 15%.
In the early stages, the MCV remains normal. Subsequently, the MCV falls and the blood smear shows hypochromic microcytic cells. With further progression, anisocytosis (variations in red blood cell size) and poikilocytosis (variation in shape of red cells) develop. Severe iron deficiency will produce a bizarre peripheral blood smear, with severely hypochromic cells, target cells, hypochromic pencil-shaped cells, and occasionally small numbers of nucleated red blood cells. The platelet count is commonly increased.
P.495
Differential Diagnosis
Other causes of microcytic anemia include anemia of chronic disease, thalassemia, and sideroblastic anemia. Anemia of chronic disease is characterized by normal or increased iron stores in the bone marrow and a normal or elevated ferritin level; the serum iron is low, often drastically so, and the TIBC is either normal or low. Thalassemia produces a greater degree of microcytosis for any given level of anemia than does iron deficiency. Red blood cell morphology on the peripheral smear is abnormal earlier in the course of thalassemia.
Treatment
The diagnosis of iron deficiency anemia can be made either by demonstrating an iron-deficient state or by evaluating the response to a therapeutic trial of iron replacement.
Since the anemia itself is rarely life-threatening, the most important part of treatment is identification of the cause especially a source of occult blood loss.
A. Oral Iron
Ferrous sulfate, 325 mg three times daily, which provides 180 mg of iron daily of which up to 10 mg is absorbed (though absorption may exceed this amount in cases of severe deficiency), is the preferred therapy. Compliance is improved by introducing the medicine more slowly in a gradually escalating dose with food. Alternatively, in cases of poor tolerance, one pill of ferrous sulfate can be taken at bedtime on an empty stomach. It is preferable to prescribe a lower dose of iron or to allow ingestion concurrent with food than to insist on a more rigorous schedule that will not be followed. An appropriate response is a return of the hematocrit level halfway toward normal within 3 weeks with full return to baseline after 2 months. Iron therapy should continue for 3 6 months after restoration of normal hematologic values to replenish iron stores. Failure of response to iron therapy is usually due to noncompliance, although occasional patients may absorb iron poorly. Other reasons for failure to respond include incorrect diagnosis (anemia of chronic disease, thalassemia) and ongoing gastrointestinal blood loss that exceeds the rate of new erythropoiesis.
B. Parenteral Iron
The indications are intolerance to oral iron, refractoriness to oral iron, gastrointestinal disease (usually inflammatory bowel disease) precluding the use of oral iron, and continued blood loss that cannot be corrected. Because of the possibility of anaphylactic reactions, parenteral iron therapy should be used only in cases of persistent anemia after a reasonable course of oral therapy. Until recently, iron dextran had been the only form of parenteral iron available in the United States. Now, sodium ferric gluconate is available and has been shown to result in a lower incidence of severe anaphylaxis. To date, no deaths have been reported with the use of this preparation.
The dose (total 1.5 2 g) may be calculated by estimating the decrease in volume of red blood cell mass and then supplying 1 mg of iron for each milliliter of volume of red blood cells below normal. Approximately 1 g should then be added for storage iron. The entire dose may be given as an intravenous infusion over 4 6 hours. A test dose of a dilute solution is given first, and the patient should be observed during the entire infusion for anaphylaxis.
Capurso G et al: Can patient characteristics predict the outcome of endoscopic evaluation of iron deficiency anemia: a multiple logistic regression analysis. Gastrointest Endosc 2004; 59:766.
Cook JD et al: The quantitative assessment of body iron. Blood 2003;101:3359.
Eichbaum Q et al: Is iron gluconate really safer than iron dextran? Blood 2003;101:3756.
Makrides M et al: Efficacy and tolerability of low-dose iron supplements during pregnancy: a randomized controlled trial. Am J Clin Nutr 2003;78:145.
Yates JM et al: Iron deficiency anaemia in general practice: clinical outcomes over three years and factors influencing diagnostic investigations. Postgrad Med J 2004;80:405.
Anemia of Chronic Disease
Many chronic systemic diseases are associated with mild or moderate anemia. Common causes include chronic infection or inflammation, cancer, and liver disease. The anemia of chronic renal failure is somewhat different in pathophysiology and is usually more severe.
Red blood cell survival is modestly reduced, and the bone marrow fails to compensate adequately by increasing red blood cell production. Failure to increase red cell production is largely due to sequestration of iron within the reticuloendothelial system. Decrease in erythropoietin is rarely an important cause of underproduction of red cells except in renal failure.
Clinical Findings
A. Symptoms and Signs
The clinical features are those of the causative condition. The diagnosis should be suspected in patients with known chronic diseases; it is confirmed by the findings of low serum iron, low TIBC, and normal or increased serum ferritin (or normal or increased bone marrow iron stores). In cases of significant anemia, coexistent iron deficiency or folic acid deficiency should be suspected. Decreased dietary intake of folate or iron is common in these ill patients, and many will also have ongoing gastrointestinal blood losses. Patients undergoing hemodialysis regularly lose both iron and folate during dialysis.
B. Laboratory Findings
The hematocrit rarely falls below 60% of baseline (except in renal failure). The MCV is usually normal or slightly reduced. Red blood cell morphology is nondiagnostic, and the reticulocyte count is neither strikingly
P.496
reduced nor increased. Serum iron values may be unmeasurable, and transferrin saturation may be extremely low, leading to an erroneous diagnosis of iron deficiency. In contrast to iron deficiency, serum ferritin values should be normal or increased. A serum ferritin value of less than 30 mcg/L should suggest coexistent iron deficiency.
Treatment
In most cases, no treatment is necessary. Purified recombinant erythropoietin (epoetin alfa) is effective for treatment of the anemia of renal failure and other secondary anemias, such as anemia related to cancer or inflammatory disorders (eg, rheumatoid arthritis). In renal failure, optimal response to epoetin alfa requires adequate intensity of dialysis. Epoetin alfa must be injected subcutaneously and is very expensive. One effective schedule is 30,000 units once weekly. This agent is used only when the patient is transfusion dependent or when the quality of life is clearly improved by the hematologic response.
Ganz T: Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003;102:783.
Smith RE Jr et al: A dose- and schedule-finding study of darbepoetin alpha for the treatment of chronic anaemia of cancer. Br J Cancer 2003;88:1851.
Weiss G et al: Anemia of chronic disease. N Engl J Med 2005; 352:1011.
Weiss G et al: Possible role of cytokine-induced tryptophan degradation in anaemia of inflammation. Eur J Haematol 2004; 72:130.
The Thalassemias
![]() Essentials of Diagnosis
Essentials of Diagnosis
Microcytosis out of proportion to the degree of anemia.
Positive family history or lifelong personal history of microcytic anemia.
Abnormal red blood cell morphology with microcytes, acanthocytes, and target cells.
In -thalassemia, elevated levels of hemoglobin A2 or F.
General Considerations
The thalassemias are hereditary disorders characterized by reduction in the synthesis of globin chains ( or ). Reduced globin chain synthesis causes reduced hemoglobin synthesis and eventually produces a hypochromic microcytic anemia because of defective hemoglobinization of red blood cells. Thalassemias can be considered among the hypoproliferative anemias, the hemolytic anemias, and the anemias related to abnormal hemoglobin, since all of these factors play a role in pathogenesis.
Normal adult hemoglobin is primarily hemoglobin A, which represents approximately 98% of circulating hemoglobin. Hemoglobin A is formed from a tetramer two chains and two chains and can be designated 2 2. Two copies of the -globin gene are located on chromosome 16, and there is no substitute for -globin in the formation of hemoglobin. The -globin gene resides on chromosome 11 adjacent to genes encoding the -like globin chains, and . The tetramer of 2 2 forms hemoglobin A2, which normally comprises 1 2% of adult hemoglobin. The tetramer 2 2 forms hemoglobin F, which is the major hemoglobin of fetal life but which comprises less than 1% of normal adult hemoglobin.
-Thalassemia is due primarily to gene deletion causing reduced -globin chain synthesis (Table 13-4). Since all adult hemoglobins are containing, -thalassemia produces no change in the percentage distribution of hemoglobins A, A2, and F. In severe forms of -thalassemia, excess chains may form a 4 tetramer called hemoglobin H.
-Thalassemias are usually caused by point mutations rather than deletions (Table 13-5). These mutations result in premature chain termination or in problems with transcription of RNA and ultimately result in reduced or absent -globin chain synthesis. The molecular defects leading to -thalassemia are numerous and heterogeneous. Defects that result in absent globin chain expression are termed 0, whereas those causing reduced synthesis are termed +. The reduced -globin chain synthesis in -thalassemia results in a relative increase in the percentages of hemoglobins A2 and F compared to hemoglobin A, as the -like globins ( and ) substitute for the missing chains. In the presence of reduced chains, the excess chains are unstable and precipitate, leading to damage of red blood cell membranes. This leads to intramedullary and peripheral hemolysis. The bone marrow becomes hyperplastic under the drive of anemia and ineffective erythropoiesis resulting from the intramedullary destruction of the developing erythroid
P.497
cells. In cases of severe thalassemia, the marked expansion of the erythroid element in the bone marrow may cause severe bony deformities, osteopenia, and pathologic fractures.
Table 13-4. -Thalassemia syndromes. | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
Table 13-5. -Thalassemia syndromes. | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
Clinical Findings
A. Symptoms and Signs
The -thalassemia syndromes are seen primarily in persons from southeast Asia and China and, less commonly, in blacks. Normally, adults have four copies of the -globin chain. When three -globin genes are present, the patient is hematologically normal (silent carrier). When two -globin genes are present, the patient is said to have -thalassemia trait, one form of thalassemia minor. These patients are clinically normal and have a normal life expectancy and performance status, with a mild microcytic anemia. When only one -globin chain is present, the patient has hemoglobin H disease. This is a chronic hemolytic anemia of variable severity (thalassemia minor or intermedia). Physical examination will reveal pallor and splenomegaly. Although affected individuals do not usually require transfusions, they may do so during periods of hemolytic exacerbation caused by infection or other stresses. When all four -globin genes are deleted, the affected fetus is stillborn as a result of hydrops fetalis.
-Thalassemia primarily affects persons of Mediterranean origin (Italian, Greek) and to a lesser extent Chinese, other Asians, and blacks. Patients homozygous for -thalassemia have thalassemia major. Affected children are normal at birth, but after 6 months, when hemoglobin synthesis switches from hemoglobin F to hemoglobin A, severe anemia requiring transfusion develops. Numerous clinical problems ensue, including growth failure, bony deformities (abnormal facial structure, pathologic fractures), hepatosplenomegaly, and jaundice. The clinical course is modified significantly by transfusion therapy, but the transfusional iron overload (hemosiderosis) results in a clinical picture similar to hemochromatosis, with heart failure, cirrhosis, and endocrinopathies, usually after more than 100 units of red blood cells. These problems develop because of the body's inability to excrete the iron (see above) from transfused red cells. Before the application of allogeneic stem cell transplantation and the development of more effective forms of iron chelation, death from cardiac failure usually occurred between the ages of 20 and 30 years.
Patients homozygous for a milder form of -thalassemia (allowing a higher rate of globin gene synthesis) have thalassemia intermedia. These patients have chronic hemolytic anemia but do not require transfusions except under periods of stress. Iron overload because of periodic transfusion may also develop. They survive into adult life but with hepatosplenomegaly and bony deformities. Patients heterozygous for -thalassemia have thalassemia minor and a clinically insignificant microcytic anemia.
Prenatal diagnosis is available, and genetic counseling should be offered and the opportunity for prenatal diagnosis discussed.
B. Laboratory Findings
1. -Thalassemia trait
Patients with two -globin genes have mild anemia, with hematocrits between 28% and 40%. The MCV is strikingly low (60 75 fL) despite the modest anemia, and the red blood count is normal or increased. The peripheral blood smear shows microcytes, hypochromia, occasional target cells, and acanthocytes (cells with irregularly spaced bulbous projections). The reticulocyte count and iron parameters are normal. Hemoglobin electrophoresis will show no increase in the percentage of hemoglobins A2 or F and no hemoglobin H. -Thalassemia trait is thus usually diagnosed by exclusion. Genetic testing to demonstrate -globin gene deletion is available in research laboratories.
2. Hemoglobin H disease
These patients have a more marked hemolytic anemia, with hematocrits between 22% and 32%. The MCV is remarkably low (60 70 fL) and the peripheral blood smear is markedly abnormal, with hypochromia, microcytosis, target cells, and poikilocytosis. The reticulocyte count is elevated. Hemoglobin electrophoresis will show the presence of a fast migrating hemoglobin (hemoglobin H), which comprises 10 40% of the hemoglobin. A peripheral blood smear can be stained with supravital dyes to demonstrate the presence of hemoglobin H.
P.498
3. -Thalassemia minor
As in -thalassemia trait, these patients have a modest anemia with hematocrit between 28% and 40%. The MCV ranges from 55 to 75 fL, and the red blood cell count is normal or increased. The peripheral blood smear is mildly abnormal, with hypochromia, microcytosis, and target cells. In contrast to -thalassemia, basophilic stippling may be present. The reticulocyte count is normal or slightly elevated. Hemoglobin electrophoresis (using quantitative techniques) may show an elevation of hemoglobin A2 to 4 8% and occasional elevations of hemoglobin F to 1 5%.
4. -Thalassemia major
-Thalassemia major produces severe anemia, and without transfusion the hematocrit may fall to less than 10%. The peripheral blood smear is bizarre, showing severe poikilocytosis, hypochromia, microcytosis, target cells, basophilic stippling, and nucleated red blood cells. Little or no hemoglobin A is present. Variable amounts of hemoglobin A2 are seen, and the major hemoglobin present is hemoglobin F.
Differential Diagnosis
Mild forms of thalassemia must be differentiated from iron deficiency. Compared to iron deficiency anemia, patients with thalassemia have a lower MCV, a more normal red blood count, and a more abnormal peripheral blood smear at modest levels of anemia. Iron studies are normal. Severe forms of thalassemia may be confused with other hemoglobinopathies. The diagnosis is made by hemoglobin electrophoresis.
Treatment
Patients with mild thalassemia ( -thalassemia trait or -thalassemia minor) require no treatment and should be identified so that they will not be subjected to repeated evaluations and treatment for iron deficiency. Patients with hemoglobin H disease should take folate supplementation and avoid medicinal iron and oxidative drugs such as sulfonamides. Patients with severe thalassemia are maintained on a regular transfusion schedule and receive folate supplementation. Splenectomy is performed if hypersplenism causes a marked increase in the transfusion requirement. Deferoxamine is routinely given as an iron-chelating agent to avoid or postpone hemosiderosis. Deferipone is a new oral iron chelator that has been approved for clinical use in Europe, but is not currently available in the United States. A diet low in iron may help in all of these patients.
Allogeneic bone marrow transplantation has become the treatment of choice for -thalassemia major. Children who have not yet experienced iron overload and chronic organ toxicity do well, with long-term survival in more than 80% of cases.
Chaidos A et al: Treatment of beta-thalassemia patients with recombinant human erythropoietin: effect on transfusion requirements and soluble adhesion molecules. Acta Haematol 2004;111:189.
Chui D et al: Hemoglobin H disease: not necessarily a benign disorder. Blood 2003;101:791.
Cunningham MJ et al: Thalassemia Clinical Research Network. Complications of beta-thalassemia major in North America. Blood 2004;104:34.
Hoffbrand AV et al: Role of deferipone in chelation therapy for transfusional iron overload. Blood 2003;102:17.
Rund D et al: -Thalassemia. N Engl J Med 2005;353:1135.
Sideroblastic Anemia
The sideroblastic anemias are a heterogeneous group of disorders in which hemoglobin synthesis is reduced because of failure to incorporate heme into protoporphyrin to form hemoglobin. Iron accumulates, particularly in the mitochondria. A Prussian blue stain of the bone marrow will reveal ringed sideroblasts, cells with iron deposits encircling the red cell nucleus. The disorder is usually acquired. Sometimes it represents a stage in evolution of a generalized bone marrow disorder (myelodysplasia) that may ultimately terminate in acute leukemia. Other causes include chronic alcoholism and lead poisoning.
Patients have no specific clinical features other than those related to anemia. The anemia is usually moderate, with hematocrits of 20 30%, but transfusions may occasionally be required. Although the MCV is usually normal or slightly increased, it may occasionally be low, leading to confusion with iron deficiency. The peripheral blood smear characteristically shows a dimorphic population of red blood cells, one normal and one hypochromic. In cases of lead poisoning, coarse basophilic stippling of the red cells is seen.
The diagnosis is made by examination of the bone marrow. Characteristically, there is marked erythroid hyperplasia, a sign of ineffective erythropoiesis (expansion of the erythroid compartment of the bone marrow that does not result in the production of reticulocytes in the peripheral blood). The iron stain of the bone marrow shows a generalized increase in iron stores and the presence of ringed sideroblasts. Other characteristic laboratory features include a high serum iron and a high transferrin saturation. In lead poisoning, serum lead levels will be elevated.
Occasionally, the anemia is so severe that support with transfusion is required. These patients usually do not respond to erythropoietin therapy.
Germing U et al: Two types of acquired idiopathic sideroblastic anaemia (AISA): a time-tested distinction. Br J Haematol 2000;108:724.
Vitamin B12 Deficiency
![]() Essentials of Diagnosis
Essentials of Diagnosis
Macrocytic anemia.
Macro-ovalocytes and hypersegmented neutrophils on peripheral blood smear.
Serum vitamin B12 level less than 100 pg/mL.
P.499
General Considerations
Vitamin B12 belongs to the family of cobalamins and serves as a cofactor for two important reactions in humans. As methylcobalamin, it is a cofactor for methionine synthetase in the conversion of homocysteine to methionine, and as adenosylcobalamin for the conversion of methylmalonyl-coenzyme A (CoA) to succinyl-CoA. All vitamin B12 comes from the diet and is present in all foods of animal origin. The daily absorption of vitamin B12 is 5 mcg.
After being ingested, vitamin B12 is bound to intrinsic factor, a protein secreted by gastric parietal cells. Other cobalamin-binding proteins (called R factors) compete with intrinsic factor for vitamin B12. Vitamin B12 bound to R factors cannot be absorbed. The vitamin B12-intrinsic factor complex travels through the intestine and is absorbed in the terminal ileum by cells with specific receptors for the complex. It is then transported through plasma and stored in the liver. Three plasma transport proteins have been identified. Transcobalamins I and III (differing only in carbohydrate structure) are secreted by white blood cells. Although approximately 90% of plasma vitamin B12 circulates bind to these proteins, only transcobalamin II is capable of transporting vitamin B12 into cells. The liver contains 2000 5000 mcg of stored vitamin B12. Since daily losses are 3 5 mcg/d, the body usually has sufficient stores of vitamin B12 so that vitamin B12 deficiency develops more than 3 years after vitamin B12 absorption ceases.
Since vitamin B12 is present in all foods of animal origin, dietary vitamin B12 deficiency is extremely rare and is seen only in vegans strict vegetarians who avoid all dairy products as well as meat and fish (Table 13-6). Abdominal surgery may lead to vitamin B12 deficiency in several ways. Gastrectomy will eliminate that site of intrinsic factor production; blind loop syndrome will cause competition for vitamin B12 by bacterial overgrowth in the lumen of the intestine; and surgical resection of the ileum will eliminate the site of vitamin B12 absorption. Rare causes of vitamin B12 deficiency include fish tapeworm (Diphyllobothrium latum) infection, in which the parasite uses luminal vitamin B12, pancreatic insufficiency (with failure to inactivate competing cobalamin-binding proteins), and severe Crohn's disease, causing sufficient destruction of the ileum to impair vitamin B12 absorption.
Table 13-6. Causes of vitamin B12 deficiency. | |
|---|---|
|
The most common cause of vitamin B12 deficiency is associated with pernicious anemia. Although the disease is hereditary, it is rare clinically before age 35 years. Pernicious anemia produces a number of clinical findings in addition to vitamin B12 deficiency. Atrophic gastritis is invariably present and results in histamine-fast achlorhydria. These patients may also have a number of other autoimmune diseases, including immunoglobulin (Ig) A deficiency, as well as polyglandular endocrine insufficiency. The atrophic gastritis is associated with an increased risk of gastric carcinoma.
Clinical Findings
A. Symptoms and Signs
The hallmark of symptomatic vitamin B12 deficiency is megaloblastic anemia. However, subclinical cobalamin deficiency is an increasingly recognized condition, especially in those with predisposing conditions such as ileal disease or gastric surgery. In advanced cases, the anemia may be severe, with hematocrits as low as 10 15%, and may be accompanied by leukopenia and thrombocytopenia. The megaloblastic state also produces changes in mucosal cells, leading to glossitis, as well as other vague gastrointestinal disturbances such as anorexia and diarrhea. Vitamin B12 deficiency also leads to a complex neurologic syndrome. Peripheral nerves are usually affected first, and patients complain initially of paresthesias. The posterior columns next become impaired, and patients complain of difficulty with balance. In more advanced cases, cerebral function may be altered as well, and on occasion dementia and other neuropsychiatric changes may precede hematologic changes.
Patients are usually pale and may be mildly icteric. Neurologic examination may reveal decreased vibration and position sense but is more commonly normal in early stages of the disease.
B. Laboratory Findings
The megaloblastic state produces an anemia of variable severity that on occasion may be very severe. The MCV is usually strikingly elevated, between 110 and 140 fL. However, it is possible to have vitamin B12 deficiency with a normal MCV. Occasionally, the normal MCV may be explained by coexistent thalassemia or iron deficiency, but in other cases the reason is obscure. Patients with neurologic symptoms and signs that suggest possible
P.500
vitamin B12 deficiency should be evaluated for that deficiency despite a normal MCV and the absence of anemia. The peripheral blood smear is usually strikingly abnormal, with anisocytosis and poikilocytosis. A characteristic finding is the macro-ovalocyte, but numerous other abnormal shapes are usually seen. The neutrophils are hypersegmented. Typical features include a mean lobe count greater than four or the finding of six-lobed neutrophils. The reticulocyte count is reduced. Because vitamin B12 deficiency affects all hematopoietic cell lines, in severe cases the white blood cell count and the platelet count are reduced, and pancytopenia is present.
Bone marrow morphology is characteristically abnormal. Marked erythroid hyperplasia is present as a response to defective red blood cell production (ineffective erythropoiesis). Megaloblastic changes in the erythroid series include abnormally large cell size and asynchronous maturation of the nucleus and cytoplasm ie, cytoplasmic maturation continues while impaired DNA synthesis causes retarded nuclear development. In the myeloid series, giant metamyelocytes are characteristically seen.
Other laboratory abnormalities include elevated serum lactate dehydrogenase (LDH) and a modest increase in indirect bilirubin. These two findings are a reflection of intramedullary destruction of developing abnormal erythroid cells and are similar to those observed in peripheral hemolytic anemias.
The diagnosis of vitamin B12 deficiency is made by finding an abnormally low vitamin B12 (cobalamin) serum level. Whereas the normal vitamin B12 level is > 240 pg/mL, most patients with overt vitamin B12 deficiency will have serum levels < 170 pg/mL, with symptomatic patients usually having levels < 100 pg/mL. A level of 170 240 pg/mL is borderline. When the serum level of vitamin B12 is borderline, the diagnosis is best confirmed by finding an elevated level of serum methylmalonic acid (> 1000 nmol/L). However, elevated levels of serum methylmalonic acid can be due to renal insufficiency. The Schilling test is now rarely used.
Differential Diagnosis
Vitamin B12 deficiency should be differentiated from folic acid deficiency, the other common cause of megaloblastic anemia, in which red blood cell folate is low while vitamin B12 levels are normal. The distinction between vitamin B12 deficiency and myelodysplasia (the other common cause of macrocytic anemia with abnormal morphology) is based on the characteristic morphology and the low vitamin B12 level.
Treatment
Patients with pernicious anemia have historically been treated with parenteral therapy. Intramuscular injections of 100 mcg of vitamin B12 are adequate for each dose. Replacement is usually given daily for the first week, weekly for the first month, and then monthly for life. It is a lifelong disorder, and if patients discontinue their monthly therapy the vitamin deficiency will recur. Oral cobalamin may be used instead of parenteral therapy and can provide equivalent results. The dose is 1000 mcg/d and must be continued indefinitely.
Patients respond to therapy with an immediate improvement in their sense of well-being. Hypokalemia may complicate the first several days of therapy, particularly if the anemia is severe. A brisk reticulocytosis occurs in 5 7 days, and the hematologic picture normalizes in 2 months. Central nervous system symptoms and signs are reversible if they are of relatively short duration (less than 6 months) but become permanent if treatment is not initiated promptly.
Andres E et al: Vitamin B12 (cobalamin) deficiency in elderly patients. CMAJ 2004;171:251.
Bolaman Z et al: Oral versus intramuscular cobalamin treatment in megaloblastic anemia: a single-center, prospective, randomized, open-label study. Clin Ther 2003;25:3124.
Carmel R: Current concepts in cobalamin deficiency. Annu Rev Med 2000;51:357.
Folic Acid Deficiency
![]() Essentials of Diagnosis
Essentials of Diagnosis
Macrocytic anemia.
Macro-ovalocytes and hypersegmented neutrophils on peripheral blood smear.
Normal serum vitamin B12 levels.
Reduced folate levels in red blood cells or serum.
General Considerations
Folic acid is the term commonly used for pteroylmonoglutamic acid. In its reduced form of tetrahydrofolate, it serves as an important mediator of many reactions involving one-carbon transfers. Important reactions include the conversion of homocysteine to methionine and of deoxyuridylate to thymidylate, an important step in DNA synthesis.
Folic acid is present in most fruits and vegetables (especially citrus fruits and green leafy vegetables) and daily requirements of 50 100 mcg/d are usually met in the diet. Total body stores of folate are approximately 5000 mcg, enough to supply requirements for 2 3 months.
By far the most common cause of folate deficiency is inadequate dietary intake (Table 13-7). Alcoholics, anorectic patients, persons who do not eat fresh fruits and vegetables, and those who overcook their food are candidates for folate deficiency. Reduced folate absorption is rarely seen, since absorption occurs from the entire gastrointestinal tract. However, drugs such as phenytoin,
P.501
trimethoprim-sulfamethoxazole, or sulfasalazine may interfere with folate absorption. Folic acid requirements are increased in pregnancy, hemolytic anemia, and exfoliative skin disease, and in these cases the increased requirements (five to ten times normal) may not be met by a normal diet. Patients with increased folate requirements should receive supplementation with 1 mg/d of folic acid.
Table 13-7. Causes of folate deficiency. | |
|---|---|
|
Clinical Findings
A. Symptoms and Signs
The features are similar to those of vitamin B12 deficiency, with megaloblastic anemia and megaloblastic changes in mucosa. However, there are none of the neurologic abnormalities associated with vitamin B12 deficiency.
B. Laboratory Findings
Megaloblastic anemia is identical to anemia resulting from vitamin B12 deficiency (see above). However, the serum vitamin B12 level is normal. A red blood cell folate level of less than 150 ng/mL is diagnostic of folate deficiency.
Differential Diagnosis
The megaloblastic anemia of folate deficiency should be differentiated from vitamin B12 deficiency by the finding of a normal vitamin B12 level and a reduced red blood cell folate or serum folate level. Alcoholics, who often have folate deficiency, may also have anemia of liver disease. This latter macrocytic anemia does not cause megaloblastic morphologic changes but rather produces target cells in the peripheral blood. Hypothyroidism is associated with mild macrocytosis but also with pernicious anemia.
Treatment
Folic acid deficiency is treated with folic acid, 1 mg/d orally. The response is similar to that seen in the treatment of vitamin B12 deficiency, with rapid improvement and a sense of well-being, reticulocytosis in 5 7 days, and total correction of hematologic abnormalities within 2 months. Large doses of folic acid may produce hematologic responses in cases of vitamin B12 deficiency but will allow neurologic damage to progress.
Clarke R et al: Vitamin B12 and folate deficiency in later life. Age Ageing 2004;33:34.
Pure Red Cell Aplasia
Adult acquired pure red cell aplasia is rare. It appears to be an autoimmune disease mediated either by T lymphocytes or (rarely) by an IgG antibody against erythroid precursors. In adults, the disease is usually idiopathic. However, cases have been seen in association with systemic lupus erythematosus, chronic lymphocytic leukemia (CLL), lymphomas, or thymoma. Some drugs (phenytoin, chloramphenicol) may cause red cell aplasia. Transient episodes of red cell aplasia are probably common in response to viral infections, especially parvovirus infections. However, these acute episodes will go unrecognized unless the patient has a chronic hemolytic disorder, in which case the hematocrit may fall precipitously.
The only signs are those of anemia unless the patient has an associated autoimmune or lymphoproliferative disorder. The anemia is often severe and normochromic, with low or absent reticulocytes. Red blood cell morphology is normal, and the myeloid and platelet lines are unaffected. The bone marrow is normocellular. All elements present are normal, but erythroid precursors are markedly reduced or absent. In some cases, chest imaging studies will reveal a thymoma.
The disorder is distinguished from aplastic anemia (in which the marrow is hypocellular and all cell lines are affected) and from myelodysplasia. This latter disorder is recognized by the presence of morphologic abnormalities that should not be present in pure red cell aplasia.
Possible offending drugs should be stopped. With thymoma, resection results in amelioration of anemia in some instances. High-dose intravenous immune globulin has produced excellent responses in a small number of cases, especially in parvovirus-related cases. For most cases, the treatment of choice is immunosuppressive therapy with a combination of antithymocyte globulin and cyclosporine (or tacrolimus) similar to therapy of aplastic anemia. Anti-CD20 monoclonal antibody (rituximab) has also had some success.
Casadevall N et al: Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med 2002;346:469.
Zecca M et al: Anti-CD20 monoclonal antibody for the treatment of severe immune-mediated pure red cell aplasia and hemolytic anemia. Blood 2001;97:3995.
Hemolytic Anemias
The hemolytic anemias are a group of disorders in which red blood cell survival is reduced, either episodically
P.502
or continuously. The bone marrow has the ability to increase erythroid production up to eightfold in response to reduced red cell survival, so anemia will be present only when the ability of the bone marrow to compensate is outstripped. This will occur when red cell survival is extremely short or when the ability of the bone marrow to compensate is impaired for some second reason.
Since red blood cell survival is normally 120 days, in the absence of red cell production the hematocrit will fall at the rate of approximately 1/100 of the hematocrit per day, which translates to a decrease in the hematocrit reading of approximately 3% per week. For example, a fall of hematocrit from 45% to 36% over 3 weeks need not indicate hemolysis, since this rate of fall would result simply from cessation of red blood cell production. If the hematocrit is falling at a rate faster than that due to decreased production, blood loss or hemolysis is the cause.
Reticulocytosis is an important clue to the presence of hemolysis, since in most hemolytic disorders the bone marrow will respond with increased red blood cell production. However, hemolysis can be present without reticulocytosis when a second disorder (infection, folate deficiency) is superimposed on hemolysis; in these circumstances, the hematocrit will fall rapidly. However, reticulocytosis also occurs during recovery from hypoproliferative anemia or bleeding. Hemolysis is correctly diagnosed (when bleeding is excluded) if the hematocrit is either falling or stable despite reticulocytosis.
Hemolytic disorders are generally classified according to whether the defect is intrinsic to the red cell or due to some external factor (Table 13-8). Intrinsic defects have been described in all components of the red blood cell, including the membrane, enzyme systems, and hemoglobin; most of these disorders are hereditary. Hemolytic anemias due to external factors are the immune and microangiopathic hemolytic anemias.
Table 13-8. Classification of hemolytic anemias. | |
|---|---|
|
Certain laboratory features are common to all the hemolytic anemias. Haptoglobin, a normal plasma protein that binds and clears hemoglobin released into plasma, may be depressed in hemolytic disorders. However, haptoglobin levels are influenced by many factors and, by themselves, are not a reliable indicator of hemolysis. When intravascular hemolysis occurs, transient hemoglobinemia occurs. Hemoglobin is filtered through the glomerulus and is usually reabsorbed by tubular cells. Hemoglobinuria will be present only when the capacity for reabsorption of hemoglobin by these cells is exceeded. In its absence, evidence for prior intravascular hemolysis is the presence of hemosiderin in shed renal tubular cells (positive urine hemosiderin). With severe intravascular hemolysis, hemoglobinemia and methemalbuminemia may be present. Hemolysis increases the indirect bilirubin, and the total bilirubin may rise to 4 mg/dL. Bilirubin levels higher than this may indicate some degree of hepatic dysfunction. Serum LDH levels are strikingly elevated in cases of microangiopathic hemolysis (thrombotic thrombocytopenic purpura [TTP], hemolytic-uremic syndrome [HUS]) and may be elevated in other hemolytic anemias.
Hereditary Spherocytosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Positive family history.
Splenomegaly.
Spherocytes and increased reticulocytes on peripheral blood smear.
Microcytic, hyperchromic indices.
General Considerations
Hereditary spherocytosis is a disorder of the red blood cell membrane, leading to chronic hemolytic anemia. Normally, the red blood cell is a biconcave disk with a diameter of 7 8 mcm. The red blood cells must be both strong and deformable strong to withstand the stress of circulating for 120 days and deformable so as to pass through capillaries 3 mcm in diameter and splenic fenestrations in the cords of the red pulp of approximately 2 mcm. The red blood cell skeleton, made up primarily of the proteins spectrin and actin, gives the red cells these characteristics of strength and deformability.
The membrane defect in hereditary spherocytosis is an abnormality in spectrin, the protein providing most of the scaffolding for the red blood cell membranes. The result is a decrease in surface-to-volume ratio that results in a spherical shape of the cell. These spherical cells are less deformable and unable to pass through 2-mcm fenestrations in the splenic red pulp. Hemolysis
P.503
takes place because of trapping of red blood cells within the spleen.
Clinical Findings
A. Symptoms and Signs
Hereditary spherocytosis is an autosomal dominant disease of variable severity. It is often diagnosed during childhood, but milder cases may be discovered incidentally late in adult life. Anemia may or may not be present, since the bone marrow may be able to compensate for shortened red cell survival. Severe anemia (aplastic crisis) may occur in folic acid deficiency or when bone marrow compensation is temporarily impaired by infection. Chronic hemolysis causes jaundice and pigment (calcium bilirubinate) gallstones, leading to attacks of cholecystitis. Examination may reveal icterus and a palpable spleen.
B. Laboratory Findings
The anemia is of variable severity, and the hematocrit may be normal. Reticulocytosis is always present. The peripheral blood smear shows the presence of spherocytes, small cells that have lost their central pallor. Spherocytes usually make up only a small percentage of red blood cells on the peripheral smear. Hereditary spherocytosis is the only important disorder associated with microcytosis and an increased mean corpuscular hemoglobin concentration (MCHC), often greater than 36 g/dL. As with other hemolytic disorders, there may be an increase in indirect bilirubin. The Coombs test is negative.
Because spherocytes are red cells that have lost some membrane surface, they are abnormally vulnerable to swelling induced by hypotonic media. Increased osmotic fragility merely reflects the presence of spherocytes and does not distinguish hereditary spherocytosis from other spherocytic hemolytic disorders such as autoimmune hemolytic anemia. In some laboratories, the osmotic fragility test has been supplanted by ektacytometry, which has the advantages of better reliability and the ability to distinguish spherocytes from other red blood cell abnormalities such as elliptocytosis.
Treatment
These patients should receive uninterrupted supplementation with folic acid, 1 mg/d. The treatment of choice is splenectomy, which will correct neither the membrane defect nor the spherocytosis but will eliminate the site of hemolysis. In very mild cases discovered late in adult life, splenectomy may not be necessary.
Kimura F et al: Partial splenic embolization for the treatment of hereditary spherocytosis. AJR Am J Roentgenol 2003;181: 1021.
Paroxysmal Nocturnal Hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is an acquired clonal stem cell disorder that results in abnormal sensitivity of the red blood cell membrane to lysis by complement. The underlying cause is a defect in the gene for phosphatidylinositol class A (PICA), which results in a deficiency of the glycosylphosphatidylinositol (GPI) anchor for cellular membrane proteins. In particular, the complement-regulating proteins CD55 and CD59 are deficient. Paroxysmal nocturnal hemoglobinuria should be suspected in confusing cases of hemolytic anemia or pancytopenia. The best screening test is flow cytometry to demonstrate deficiency of CD59 on red blood cells. This test has largely replaced the classic sucrose hemolysis test.
Clinical Findings
A. Symptoms and Signs
Classically, patients report episodic hemoglobinuria resulting in reddish brown urine. Hemoglobinuria is most often noticed in the first morning urine, probably because of its increased concentration. In addition to being prone to anemia, these patients are prone to thrombosis, especially mesenteric and hepatic vein thromboses. Other common sites of thrombosis include the central nervous system (saggital vein) and the skin, with formation of painful nodules. This hypercoagulopathy may be related to platelet activation by complement. As this is a stem cell disorder, paroxysmal nocturnal hemoglobinuria may progress either to aplastic anemia, to myelodysplasia, or to acute myelogenous leukemia (AML).
B. Laboratory Findings
Anemia is of variable severity, and reticulocytosis may or may not be present. Abnormalities on the blood smear are nondiagnostic and may include macro-ovalocytes. Since the episodic hemolysis in paroxysmal nocturnal hemoglobinuria is intravascular, the finding of urine hemosiderin is a useful test. Serum LDH is characteristically elevated. Iron deficiency is commonly present and is related to chronic iron loss from hemoglobinuria, since hemolysis is primarily intravascular.
The white blood cell count and platelet count may be decreased. A decreased leukocyte alkaline phosphatase evidence for a qualitative abnormality in the myeloid series may be seen. Bone marrow morphology is variable and may show either generalized hypoplasia or erythroid hyperplasia. Flow cytometric assays may confirm the diagnosis by demonstrating the absence of CD59.
Treatment
Iron replacement is often indicated for treatment of iron deficiency. This may improve the anemia but may also cause a transient increase in hemolysis. For unclear reasons, prednisone is effective in decreasing hemolysis, and some patients can be managed effectively with alternate-day corticosteroids. In severe cases and cases of transformation to myelodysplasia, allogeneic bone marrow transplantation has been used to treat the disorder.
P.504
Hillmen P et al: Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med 2004;350:552.
Moyo VM et al: Natural history of paroxysmal nocturnal haemoglobinuria using modern diagnostic assays. Br J Haematol 2004;126:133.
Parker C et al; International PNH Interest Group: Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005;106:3699.
Rosse WF et al: Clinical manifestations of paroxysmal nocturnal hemoglobinuria: present state and future problems. Int J Hematol 2003;77:113.
Glucose-6-Phosphate Dehydrogenase Deficiency
![]() Essentials of Diagnosis
Essentials of Diagnosis
X-linked recessive disorder seen commonly in American black men.
Episodic hemolysis in response to oxidant drugs or infection.
Minimally abnormal peripheral blood smear.
Reduced levels of glucose-6-phosphate dehydrogenase between hemolytic episodes.
General Considerations
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is a hereditary enzyme defect that causes episodic hemolytic anemia because of the decreased ability of red blood cells to deal with oxidative stresses. The hexose monophosphate shunt is not an important source of energy in red cells but is important in generating reduced glutathione, which protects hemoglobin from oxidative denaturation. The first step in this pathway is the production of nicotinamide adenine dinucleotide phosphate (reduced) (NADPH) by the action of G6PD on glucose 6-phosphate. NADPH serves as a cofactor for glutathione reductase in generating reduced glutathione, which detoxifies hydrogen peroxide. In the absence of reduced glutathione, hemoglobin may become oxidized. Oxidized hemoglobin denatures and forms precipitants called Heinz bodies. These Heinz bodies cause membrane damage, which leads to removal of these cells by the spleen.
Numerous types of G6PD enzymes have been described. The normal type found in whites is designated G6PD-B. Most American blacks have G6PD-A, which is normal in function. Ten to 15 percent of American blacks have the variant G6PD designated A , in which there is only 15% of normal enzyme activity, and enzyme activity declines rapidly as the red blood cell ages past 40 days, a fact that explains many of the clinical findings in this disorder. Many other G6PD variants have been described, including some Mediterranean variants with extremely low enzyme activity.
Clinical Findings
G6PD deficiency is an X-linked recessive disorder affecting 10 15% of American black males. Female carriers are rarely affected only when an unusually high percentage of cells producing the normal enzyme is inactivated.
A. Symptoms and Signs
Patients are usually healthy, without chronic hemolytic anemia or splenomegaly. Hemolysis occurs as a result of oxidative stress on the red blood cells, generated either by infection or exposure to certain drugs. Common drugs initiating hemolysis include dapsone, primaquine, quinidine, quinine, sulfonamides, and nitrofurantoin. Even with continuous use of the offending drug, the hemolytic episode is self-limited because older red blood cells (with low enzyme activity) are removed and replaced with a population of young red blood cells with adequate functional levels of G6PD. Severe G6PD deficiency (as in Mediterranean variants) may produce a chronic hemolytic anemia.
B. Laboratory Findings
Between hemolytic episodes, the blood is normal. During episodes of hemolysis, there is reticulocytosis and increased serum indirect bilirubin. The red blood cell smear is not diagnostic but may reveal a small number of bite cells cells that appear to have had a bite taken out of their periphery. This indicates pitting of hemoglobin aggregates by the spleen. Heinz bodies may be demonstrated by staining a peripheral blood smear with cresyl violet; they are not visible on the usual Wright-stained blood smear. Specific enzyme assays for G6PD may reveal a low level but may be misleading if they are performed shortly after a hemolytic episode when the enzyme-deficient cohort of cells has been removed. In these cases, the enzyme assays should be repeated weeks after hemolysis has resolved. In severe cases of G6PD deficiency, enzyme levels are always low.
Treatment
No treatment is necessary except to avoid known oxidant drugs.
Mehta A et al: Glucose-6-phosphate dehydrogenase deficiency. Baillieres Best Pract Res Clin Haematol 2000;13:21.
Sickle Cell Anemia & Related Syndromes
![]() Essentials of Diagnosis
Essentials of Diagnosis
Irreversibly sickled cells on peripheral blood smear.
Positive family history and lifelong history of hemolytic anemia.
Recurrent painful episodes.
Hemoglobin S is the major hemoglobin seen on electrophoresis.
P.505
General Considerations
Sickle cell anemia is an autosomal recessive disorder in which an abnormal hemoglobin leads to chronic hemolytic anemia with numerous clinical consequences. A single DNA base change leads to an amino acid substitution of valine for glutamine in the sixth position on the -globin chain. The abnormal chain is designated s and the tetramer of 2 s2 is designated hemoglobin S.
When in the deoxy form, hemoglobin S forms polymers that damage the red blood cell membrane. Both polymer formation and early membrane damage are reversible. However, red blood cells that have undergone repeated sickling are damaged beyond repair and become irreversibly sickled.
The rate of sickling is influenced by a number of factors, most importantly by the concentration of hemoglobin S in the individual red blood cell. Red cell dehydration makes the cell quite vulnerable to sickling. Sickling is also strongly influenced by the presence of other hemoglobins within the cell. Hemoglobin F cannot participate in polymer formation, and its presence markedly retards sickling. Other factors that increase sickling are those that lead to formation of deoxyhemoglobin S, eg, acidosis and hypoxemia, either systemic or locally in tissues.
Prenatal diagnosis is now available for couples at risk of producing a child with sickle cell anemia. DNA from fetal cells can be directly examined, and the presence of the sickle cell mutation can be accurately and definitively diagnosed. Genetic counseling should be made available to such couples.
Clinical Findings
A. Symptoms and Signs
The hemoglobin S gene is carried in 8% of American blacks, and one birth out of 400 in American blacks will produce a child with sickle cell anemia. The disorder has its onset during the first year of life, when hemoglobin F levels fall as a signal is sent to switch from production of -globin to -globin.
Chronic hemolytic anemia produces jaundice, pigment (calcium bilirubinate) gallstones, splenomegaly, and poorly healing ulcers over the lower tibia. The chronic anemia may become life-threatening when severe anemia is produced by hemolytic or aplastic crises. The latter occur when the ability of the bone marrow to compensate is reduced by viral or other infection or by folate deficiency. Hemolytic crises may be related to splenic sequestration of sickled cells (primarily in childhood, before the spleen has been infarcted as a result of repeated sickling) or with coexistent disorders such as G6PD deficiency.
Acute painful episodes due to acute vaso-occlusion may occur spontaneously or be provoked by infection, dehydration, or hypoxia. Clusters of sickled red cells occlude the microvasculature of the organs involved. These episodes last hours to days and produce acute pain and low-grade fever. Common sites of acute painful episodes include the bones (especially the back and long bones) and the chest. Acute vaso-occlusion may also cause strokes due to sinus thrombosis and priapism. Vaso-occlusive episodes are not associated with increased hemolysis.
Repeated episodes of vascular occlusion affect a large number of organs, especially the heart and liver. Ischemic necrosis of bone occurs, rendering the bone susceptible to osteomyelitis due to staphylococci or (less commonly) salmonellae. Infarction of the papillae of the renal medulla causes renal tubular concentrating defects and gross hematuria, more often encountered in sickle cell trait than in sickle cell anemia. Retinopathy similar to that noted in diabetes is often present and may lead to blindness.
These patients are prone to delayed puberty. An increased incidence of infection is related to hyposplenism as well as to defects in the alternative pathway of complement.
On examination, patients are often chronically ill and jaundiced. There is hepatomegaly, but the spleen is not palpable in adult life. The heart is enlarged, with a hyperdynamic precordium and systolic murmurs. Nonhealing ulcers of the lower leg and retinopathy may be present.
Sickle cell anemia becomes a chronic multisystem disease, with death from organ failure. With improved supportive care, average life expectancy is now between 40 and 50 years of age.
B. Laboratory Findings
Chronic hemolytic anemia is present. The hematocrit is usually 20 30%. The peripheral blood smear is characteristically abnormal, with irreversibly sickled cells comprising 5 50% of red cells. Other findings include reticulocytosis (10 25%), nucleated red blood cells, and hallmarks of hyposplenism such as Howell-Jolly bodies and target cells. The white blood cell count is characteristically elevated to 12,000 15,000/mcL, and thrombocytosis may occur. Indirect bilirubin levels are high.
Most clinical laboratories offer a screening test for sickle cell hemoglobin, and the diagnosis of sickle cell anemia is then confirmed by hemoglobin electrophoresis (Table 13-9). Hemoglobin S has an abnormal migration pattern on electrophoresis and will usually comprise 85 98% of hemoglobin. In homozygous S disease, no hemoglobin A will be present. Hemoglobin F levels are variably increased, and high hemoglobin F levels are associated with a more benign clinical course.
Table 13-9. Hemoglobin distribution in sickle cell syndromes. | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
Treatment
No specific treatment is available for the primary disease. Patients are maintained on folic acid supplementation and given transfusions for aplastic or hemolytic
P.506
crises. Pneumococcal vaccination reduces the incidence of infections with this pathogen.
When acute painful episodes occur, precipitating factors should be identified and infections treated if present. The patient should be kept well hydrated, and oxygen should be given if the patient is hypoxic.
Acute vaso-occlusive crises can be treated with exchange transfusion. These are primarily indicated for the treatment of intractable pain crises, priapism, and stroke.
Cytotoxic agents increase hemoglobin F levels by stimulating erythropoiesis in more primitive erythroid precursors. Hydroxyurea (500 750 mg/d) reduces the frequency of painful crises in patients whose quality of life is disrupted by frequent pain crises. Long-term safety is uncertain, and concern remains about the potential of secondary malignancies. Allogeneic bone marrow transplantation is being studied as a possible curative option for severely affected young patients.
Adams RJ et al; The Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP 2) Trial Investigators: Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med 2005;353:2769.
Alexander N et al: Are there clinical phenotypes of homozygous sickle cell disease? Br J Haematol 2004;126:606.
Hankins JS et al: Long-term hydroxyurea therapy for infants with sickle cell anemia: the HUSOFT extension study. Blood 2005; 106:2269.
Steinberg MH et al: Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA 2003;289:1645.
Stuart MJ et al: Sickle-cell disease. Lancet 2004;364:1343.
Sickle Cell Trait
Patients with the heterozygous genotype (AS) have sickle cell trait. These persons are clinically normal and have acute painful episodes only under extreme conditions such as vigorous exertion at high altitudes (or in unpressurized aircraft). The patients are hematologically normal, with no anemia and normal red blood cells on peripheral blood smear. They may, however, have a defect in renal tubular function, causing an inability to concentrate the urine, and experience episodes of gross hematuria. This appears to be caused by many years of sickling in the sluggish circulation of the renal medulla. A screening test for sickle hemoglobin will be positive, and hemoglobin electrophoresis will reveal that approximately 40% of hemoglobin is hemoglobin S (Table 13-9).
No treatment is necessary. Genetic counseling is a reasonable strategy.
Sickle Thalassemia
Patients with homozygous sickle cell anemia and -thalassemia have a somewhat milder form of hemolysis because of a slower rate of sickling related to reduced MCHC within the red blood cell.
Patients who are double heterozygotes for sickle cell anemia and -thalassemia are clinically affected with sickle cell syndromes. Sickle 0-thalassemia is clinically very similar to homozygous SS disease. Vaso-occlusive crises may be somewhat less severe, and the spleen is usually not infarcted. Hematologically, the MCV is usually low, in contrast to the normal MCV of sickle cell anemia. Hemoglobin electrophoresis (Table 13-9) reveals no hemoglobin A but will show an increase in hemoglobin A2, which is not present in sickle cell anemia.
Sickle +-thalassemia is a milder disorder than homozygous SS disease, with fewer crises. The spleen is usually palpable. The hemolytic anemia is less severe, and the hematocrit is usually 30 38%, with reticulocytes of 5 10%. Hemoglobin electrophoresis shows the presence of some hemoglobin A.
Hemoglobin C Disorders
Hemoglobin C is formed by a single amino acid substitution at the same site of substitution as in sickle hemoglobin but with lysine instead of valine substituted for glutamine at the 6 position. Hemoglobin C is nonsickling but may participate in polymer formation in association with hemoglobin S. Homozygous hemoglobin C disease produces a mild hemolytic anemia with splenomegaly, mild jaundice, and pigment (calcium bilirubinate) gallstones. The peripheral blood smear shows generalized red cell targeting and occasional cells with rectangular crystals of hemoglobin C. Persons heterozygous for hemoglobin C are clinically normal.
P.507
Patients with hemoglobin SC disease are double heterozygotes for S and C. These patients, like those with sickle +-thalassemia, have a milder hemolytic anemia and milder clinical course than those with homozygous SS disease. There are fewer vaso-occlusive events, and the spleen remains palpable in adult life. However, persons with hemoglobin SC disease have more retinopathy and more ischemic necrosis of bone than those with SS disease. The hematocrit is usually 30 38%, with 5 10% reticulocytes and few irreversibly sickled cells on the blood smear. Target cells are more numerous than in SS disease. Hemoglobin electrophoresis will show approximately 50% hemoglobin C, 50% hemoglobin S, and no increase in hemoglobin F levels.
Nagel RL et al: The paradox of hemoglobin SC disease. Blood Rev 2003;17:167.
Unstable Hemoglobins
Unstable hemoglobins are prone to oxidative denaturation even in the presence of a normal G6PD system. The disorder is autosomal dominant and of variable severity. Most patients have a mild chronic hemolytic anemia with splenomegaly, mild jaundice, and pigment (calcium bilirubinate) gallstones. Less severely affected patients are not anemic except under conditions of oxidative stress.
The diagnosis is made by the finding of Heinz bodies and a normal G6PD level. Hemoglobin electrophoresis is usually normal, since these hemoglobins characteristically do not have a change in their migration pattern. These hemoglobins precipitate in isopropanol. Usually no treatment is necessary. Patients with chronic hemolytic anemia should receive folate supplementation and avoid known oxidative drugs. In rare cases, splenectomy may be required.
Autoimmune Hemolytic Anemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Acquired anemia caused by IgG autoantibody.
Spherocytes and reticulocytosis on peripheral blood smear.
Positive Coombs' test.
General Considerations
Autoimmune hemolytic anemia is an acquired disorder in which an IgG autoantibody is formed that binds to the red blood cell membrane. The antibody is most commonly directed against a basic component of the Rh system present on most human red blood cells. When IgG antibodies coat the red blood cell, the Fc portion of the antibody is recognized by macrophages present in the spleen and other portions of the reticuloendothelial system. The interaction between splenic macrophage and the antibody-coated red blood cell results in removal of red blood cell membrane and the formation of a spherocyte because of the decrease in surface-to-volume ratio of the red blood cell. These spherocytic cells have decreased deformability and become trapped in the red pulp of the spleen because of their inability to squeeze through the 2-mcm fenestrations. When large amounts of IgG are present on red blood cells, complement may be fixed. Direct lysis of cells is rare, but the presence of C3b on the surface of red blood cells allows Kupffer cells in the liver to participate in the hemolytic process because of the presence of C3b receptors on Kupffer cells.
Approximately 50% of all cases of autoimmune hemolytic anemia are idiopathic. The disorder may also be seen in association with systemic lupus erythematosus, CLL, or lymphomas. It must be distinguished from drug-induced hemolytic anemia. Penicillin (and other drugs) coats the red blood cell membrane, and the antibody is directed against the membrane-drug complex.
The Coombs antiglobulin test forms the basis for diagnosis of these immune hemolytic disorders. The Coombs reagent is a rabbit IgM antibody raised against human IgG or human complement. The direct Coombs test is performed by mixing the patient's red blood cells with the Coombs reagent and looking for agglutination, which indicates the presence of antibody on the red blood cell surface. The indirect Coombs test is performed by mixing the patient's serum with a panel of type O red blood cells. After incubation of the test serum and panel red blood cells, the Coombs reagent is added. Agglutination in this system indicates the presence of free antibody in the patient's serum.
Clinical Findings
A. Symptoms and Signs
Autoimmune hemolytic anemia typically produces an anemia of rapid onset that may be life-threatening in severity. Patients complain of fatigue and may present with angina or congestive heart failure. On examination, jaundice and splenomegaly are usually present.
B. Laboratory Findings
The anemia is of variable severity but may be severe, with hematocrit of less than 10%. Reticulocytosis is usually present, and spherocytes are seen on the peripheral blood smear. In cases of severe hemolysis, the stressed bone marrow may also release nucleated red blood cells. As with other hemolytic disorders, indirect bilirubin is increased. Approximately 10% of patients with autoimmune hemolytic anemia have coincident immune thrombocytopenia (Evans's syndrome).
The direct Coombs test is positive, and the indirect Coombs test may or may not be positive. A positive indirect Coombs test indicates the presence of a large amount of autoantibody that has saturated binding
P.508
sites in the red blood cell and consequently appears in the serum. Because the patient's serum usually contains the autoantibody, it may be difficult to obtain a compatible cross-match with donor's cells.
Treatment
Initial treatment consists of prednisone, 1 2 mg/kg/d in divided doses. Most transfused blood will survive similarly to the patient's own red blood cells. Because of difficulty in performing the cross-match, incompatible blood may be given. Decisions regarding transfusions should be made in consultation with a hematologist. If prednisone is ineffective or if the disease recurs on tapering the dose, splenectomy should be performed. Patients with autoimmune hemolytic anemia refractory to prednisone and splenectomy may be treated with a variety of agents. Treatment with rituximab, a monoclonal antibody against the B cell antigen CD20, is effective in some cases. The suggested dose is 375 mg/m2 intravenously weekly for 4 weeks. Danazol, 600 800 mg/d, is less often effective than in immune thrombocytopenia but is well suited for long-term use because of its low toxicity. Immunosuppressive agents, including cyclophosphamide, azathioprine, or cyclosporine, may also be used. High-dose intravenous immune globulin (1 g daily for 1 or 2 days) may be highly effective in controlling hemolysis. The benefit is short-lived (1 3 weeks), and the drug is very expensive. The long-term prognosis for patients with this disorder is good, especially if there is no underlying autoimmune disorder or lymphoma. Splenectomy is often successful in controlling the disorder.
Petz LD: A physician's guide to transfusion in autoimmune haemolytic anaemia. Br J Haematol 2004;124:712.
Robak T: Monoclonal antibodies in the treatment of autoimmune cytopenias. Eur J Haematol 2004;72:79.
Cold Agglutinin Disease
![]() Essentials of Diagnosis
Essentials of Diagnosis
Increased reticulocytes and spherocytes on peripheral blood smear.
Coombs' test positive only for complement.
Positive cold agglutinin test.
General Considerations
Cold agglutinin disease is an acquired hemolytic anemia due to an IgM autoantibody usually directed against the I antigen on red blood cells. These IgM autoantibodies characteristically will not react with cells at 37 C but only at lower temperatures. Since the blood temperature (even in the most peripheral parts of the body) rarely goes lower than 20 C, only antibodies active at higher temperatures will produce clinical effects. Hemolysis results indirectly from attachment of IgM, which in the cooler parts of the circulation (fingers, nose, ears) binds and fixes complement. When the red blood cell returns to a warmer temperature, the IgM antibody dissociates, leaving complement on the cell. Lysis of cells rarely occurs. Rather, C3b present on the red cells is recognized by Kupffer cells (which have receptors for C3b), and red blood cell sequestration ensues.
Most cases of chronic cold agglutinin disease are idiopathic. Others occur in association with Waldenstr m's macroglobulinemia, in which a monoclonal IgM paraprotein is produced. Acute postinfectious cold agglutinin disease occurs following mycoplasmal pneumonia or infectious mononucleosis (with antibody directed against antigen i rather than I).
Clinical Findings
A. Symptoms and Signs
In chronic cold agglutinin disease, symptoms related to red blood cell agglutination occur on exposure to cold, and patients may complain of mottled or numb fingers or toes. Hemolytic anemia is rarely severe, but episodic hemoglobinuria may occur on exposure to cold. The hemolytic anemia in acute postinfectious syndromes is rarely severe.
B. Laboratory Findings
Mild anemia is present with reticulocytosis and spherocytes. The direct Coombs test will be positive for complement only. Occasionally, a micro-Coombs test is necessary to reveal bound complement (low-titer cold agglutinin disease). A bedside cold agglutinin test may be performed by placing a glass slide in ice and then putting a few drops of heparinized blood on it. Inspection may reveal small clumps of agglutinated blood.
Treatment
Treatment is largely symptomatic, based on avoiding exposure to cold. Patients with severe involvement may be treated with alkylating agents such as cyclophosphamide or with immunosuppressive agents such as cyclosporine. Splenectomy and prednisone are usually ineffective since hemolysis takes place in the liver. High-dose intravenous immunoglobulin (2 g/kg) may be effective temporarily, but is rarely used because of the extreme cost and short duration of benefit. Rituximab, a monoclonal antibody directed against the CD20 antigen on B lymphocytes, is emerging as the treatment of choice. The dose is 375 mg/m2 intravenously weekly for 4 weeks.
Berentsen S et al: Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood 2004;103:2925.
P.509
Microangiopathic Hemolytic Anemias
The microangiopathic hemolytic anemias are a group of disorders in which red blood cell fragmentation takes place. The anemia is intravascular, producing hemoglobinemia, hemoglobinuria, and, in severe cases, methemalbuminemia. The hallmark of the disorder is the finding of fragmented red blood cells (schistocytes, helmet cells) on the peripheral blood smear.
These fragmentation syndromes can be caused by a variety of disorders (Table 13-8). TTP is the most important of these and is discussed below. Clinical features are variable and depend on the underlying disorder. Coagulopathy and thrombocytopenia are variably present.
Chronic microangiopathic hemolytic anemia (such as is present with a malfunctioning cardiac valve prosthesis) may cause iron deficiency anemia because of continuous low-grade hemoglobinuria.
Aplastic Anemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Pancytopenia.
No abnormal cells seen.
Hypocellular bone marrow.
General Considerations
All hematopoietic cells are derived from a pluripotent stem cell that gives rise to precursors of erythroid, myeloid, and platelet forms. Injury to or suppression of this hematopoietic stem cell will result in pancytopenia. Aplastic anemia is a condition of bone marrow failure that arises from injury to or abnormal expression of the stem cell. The bone marrow becomes hypoplastic, and pancytopenia develops.
There are a number of causes of aplastic anemia (Table 13-10). Direct stem cell injury may be caused by radiation, chemotherapy, toxins, or pharmacologic agents. Systemic lupus erythematosus may rarely cause suppression of the hematopoietic stem cell by an IgG autoantibody directed against the stem cell. However, the most common pathogenesis of aplastic anemia appears to be autoimmune suppression of hematopoiesis by a T cell-mediated cellular mechanism.
Table 13-10. Causes of aplastic anemia. | |
|---|---|
|
Clinical Findings
A. Symptoms and Signs
Patients come to medical attention because of the consequences of bone marrow failure. Anemia leads to symptoms of weakness and fatigue, neutropenia causes vulnerability to bacterial infections, and thrombocytopenia results in mucosal and skin bleeding. Physical examination may reveal signs of pallor, purpura, and petechiae. Other abnormalities such as hepatosplenomegaly, lymphadenopathy, or bone tenderness should not be present, and their presence should lead to questioning the diagnosis.
B. Laboratory Findings
The hallmark of aplastic anemia is pancytopenia. However, early in the evolution of aplastic anemia, only one or two cell lines may be reduced.
Anemia may be severe and is always associated with decreased reticulocytes. Red blood cell morphology is unremarkable. The MCV is usually normal but occasionally may be increased. Neutrophils and platelets are reduced in number, and no immature or abnormal forms are seen. The bone marrow aspirate and the bone marrow biopsy appear hypocellular, with only scant amounts of normal hematopoietic progenitors. No abnormal cells are seen.
Differential Diagnosis
The diagnosis of aplastic anemia is made in cases of pancytopenia with a hypocellular marrow biopsy containing no abnormal cells. Aplastic anemia must be differentiated from other causes of pancytopenia (Table 13-11). Myelodysplastic disorders, especially hypocellular
P.510
forms of myelodysplasia, or acute leukemia may occasionally be confused with aplastic anemia. These are differentiated by the presence of morphologic abnormalities or increased blasts, or by the presence of abnormal cytogenetics in bone marrow cells. Hairy cell leukemia has been misdiagnosed as aplastic anemia and should be recognized by the presence of splenomegaly and by abnormal lymphoid cells on the bone marrow biopsy. Pancytopenia with a normocellular bone marrow is usually due to systemic lupus erythematosus, disseminated infection, or hypersplenism. Isolated thrombocytopenia may occur early as aplastic anemia develops and be confused with immune thrombocytopenia.
Table 13-11. Causes of pancytopenia. | |
|---|---|
|
Treatment
Mild cases of aplastic anemia may be treated with supportive care. Red blood cell transfusions and platelet transfusions are given as necessary, and antibiotics are used to treat infections.
Severe aplastic anemia is defined by a neutrophil count of less than 500/mcL, platelets less than 20,000/mcL, reticulocytes less than 1%, and bone marrow cellularity less than 20%. When this constellation of features is present (or three of the four), the median survival without treatment is approximately 3 months, and only 20% of patients survive for 1 year. The treatment of choice for young adults (under age 50) who have HLA-matched siblings is allogeneic bone marrow transplantation. Children or young adults may also benefit from allogeneic transplantation using an unrelated donor. The use of reduced-intensity preparative regimens for allogeneic transplantation has reduced the toxicity of transplantation. Because of the increased risks associated with unrelated-donor transplantation, this treatment is usually reserved for patients who have not benefited from immunosuppressive therapy.
For adults over age 50 years or those without HLA-matched siblings, the treatment of choice for severe aplastic anemia is immunosuppression with antithymocyte globulin (ATG) plus cyclosporine. ATG is given in the hospital in conjunction with transfusion and antibiotic support. A useful regimen is 40 mg/kg/d for 4 days in combination with cyclosporine, 6 mg/kg orally twice daily. ATG must be used in combination with corticosteroids (prednisone 1 2 mg/kg/d initially, followed by a rapid taper) to avoid complications of serum sickness. Responses usually occur in 4 12 weeks and are usually only partial, but the blood counts rise high enough to give patients a safe and transfusion-free life.
High-dose immunosuppression with cyclophosphamide, 200 mg/kg, has produced remissions in refractory cases and should be considered for patients without suitable bone marrow donors. Androgens have been widely used in the past, with a low response rate. However, a few patients can be maintained successfully with this form of treatment. One regimen is oxymetholone, 2 3 mg/kg orally daily.
Course & Prognosis
Patients with severe aplastic anemia have a rapidly fatal illness if left untreated. Allogeneic bone marrow transplantation is highly successful in children and young adults, especially with HLA-matched siblings. For this group of patients, the durable complete response rate exceeds 80%. Advances in the field of unrelated donor transplantation have made this a more attractive option than in the past. ATG treatment leads to partial response in approximately 60% of adults, and the long-term prognosis of responders appears to be good. After many years of follow-up, there is increasing evidence that clonal hematologic disorders, such as paroxysmal nocturnal hemoglobinuria or myelodysplasia, may develop in some fraction (as many as 25%) of these nontransplanted patients.
Ades L et al: Long-term outcome after bone marrow transplantation for severe aplastic anemia. Blood 2004;103:2490.
Frickhofen N et al: Antithymocyte globulin with or without cyclosporin A: 11-year follow-up of a randomized trial comparing treatments of aplastic anemia. Blood 2003;101:1236.
Yamaguchi H et al: Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med 2005; 352:1413.
Young NS: Acquired aplastic anemia. Ann Intern Med 2002; 136:534.
Neutropenia
Neutropenia is present when the neutrophil count is below 1500/mcL, though blacks and other specific population groups may normally have neutrophil counts as low as 1200/mcL. The neutropenic patient is increasingly vulnerable to infection by gram-positive and gram-negative bacteria and by fungi. The risk of infection is related to the severity of neutropenia. Patients with chronic benign neutropenia are free of infection for years despite very low neutrophil levels.
A variety of bone marrow disorders and nonmarrow conditions may cause neutropenia (Table 13-12). All the causes of aplastic anemia (Table 13-10) and pancytopenia (Table 13-11) may cause neutropenia. Isolated neutropenia is often due to an idiosyncratic reaction to a drug, and agranulocytosis (complete absence of neutrophils in the peripheral blood) is almost always due to a drug reaction. In these cases, examination of the bone marrow shows an almost complete absence of myeloid precursors, with other cell lines undisturbed. Felty's syndrome immune neutropenia associated with seropositive nodular rheumatoid arthritis and splenomegaly is another cause. Neutropenia in the presence of a normal bone marrow may be due to immunologic peripheral destruction, sepsis,
P.511
or hypersplenism. Severe neutropenia may be associated with clonal disorders of T lymphocytes, often with the morphology of large granular lymphocytes.
Table 13-12. Causes of neutropenia. | |
|---|---|
|
Clinical Findings
Neutropenia results in stomatitis and in infections due to gram-positive or gram-negative aerobic bacteria or to fungi such as Candida or Aspergillus. The most common infections are septicemia, cellulitis, and pneumonia. In the presence of severe neutropenia, the usual signs of inflammatory response to infection may be absent. Nevertheless, fever in the neutropenic patient should always be assumed to be of infectious origin.
Treatment
Potential causative drugs are discontinued. Infections are treated with broad-spectrum antibiotics, but particular attention should be paid to enteric gram-negative bacteria. Effective antibiotics include the quinolones such as levofloxacin, 500 mg orally or intravenously daily, or new cephalosporins such as cefepime, 2 g intravenously every 8 hours. New antifungal agents such as voriconazole and caspofungin can provide both better efficacy and reduced toxicity compared to amphotericin.
Many cases of idiopathic or autoimmune neutropenia respond to myeloid growth factors such as granulocyte colony-stimulating factor (G-CSF). Once-weekly or twice-weekly dosage will often be sufficient to produce a protective neutrophil count.
When Felty's syndrome leads to repeated bacterial infections, splenectomy has been the treatment of choice, but it now appears that sustained use of G-CSF is effective and provides a nonsurgical alternative. The prognosis of patients with neutropenia depends on the underlying cause. Most patients with drug-induced agranulocytosis can be supported with broad-spectrum antibiotics and will recover completely. The myeloid growth factors G-CSF (filgrastim) and GM-CSF (sargramostim) may be useful in shortening the duration of neutropenia associated with chemotherapy. The neutropenia associated with large granular lymphocytes may respond to therapy with either cyclosporin or low-dose methotrexate.
Cullen M et al; Simple Investigation in Neutropenic Individuals of the Frequency of Infection after Chemotherapy +/- Antibiotic in a Number of Tumours (SIGNIFICANT) Trial Group: Antibacterial prophylaxis after chemotherapy for solid tumors and lymphomas. N Engl J Med 2005;353:988.
Walsh TJ et al: Caspofungin versus liposomal amphotericin B for empirical antifungal therapy in patients with persistent fever and neutropenia. N Engl J Med 2004;351:1391.
Leukemias & Other Myeloproliferative Disorders
Myeloproliferative disorders are due to acquired clonal abnormalities of the hematopoietic stem cell. Since the stem cell gives rise to myeloid, erythroid, and platelet cells, qualitative and quantitative changes are seen in all these cell lines. In some disorders (chronic myelogenous leukemia [CML]), specific characteristic chromosomal changes are seen. In others, no characteristic cytogenetic abnormalities are seen.
Classically, the myeloproliferative disorders produce characteristic syndromes with well-defined clinical and laboratory features (Tables 13-13 and 13-14). However, these disorders are grouped together because the disease may evolve from one form into another and because hybrid disorders are commonly seen. All of the myeloproliferative disorders may progress to AML.
Polycythemia Vera
![]() Essentials of Diagnosis
Essentials of Diagnosis
Increased red blood cell mass.
Splenomegaly.
Normal arterial oxygen saturation.
Usually elevated white blood count and platelet count.
P.512
Table 13-13. Classification of myeloproliferative disorders. | |
|---|---|
|
Table 13-14. Laboratory features of myeloproliferative disorders. | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
General Considerations
Polycythemia vera is an acquired myeloproliferative disorder that causes overproduction of all three hematopoietic cell lines, most prominently the red blood cells. The hematocrit is elevated (at sea level) when values exceed 54% in males or 51% in females (Table 13-15).
When the hematocrit is elevated, the red blood cell mass should be measured to determine whether true polycythemia or relative polycythemia exists. Normal values for red blood cell mass are 26 34 mL/kg in men and 21 29 mL/kg in women. Relative ( spurious ) polycythemia presents in middle-aged men who are overweight and hypertensive (often on diuretic therapy); the hematocrit is almost always less than 60%, and they have a high-normal red cell mass and a low-normal plasma volume.
If the red blood cell mass is increased, it is necessary to determine whether the increase is primary or secondary. Primary polycythemia (polycythemia vera) is a bone marrow disorder characterized by autonomous overproduction of erythroid cells. Erythroid production is independent of erythropoietin, and the serum erythropoietin level is low. In vitro, erythroid progenitor cells grow without added erythropoietin, a finding not seen in normal individuals. A mutation in JAK2, a signaling molecule, has been demonstrated in most cases and is likely involved in the pathogenesis.
Clinical Findings
A. Symptoms and Signs
Most presenting symptoms are related to expanded blood volume and increased blood viscosity. Common complaints include headache, dizziness, tinnitus, blurred vision, and fatigue. Generalized pruritus, especially following a warm shower or bath, may be a striking symptom and is related to histamine release from the increased number of basophils present. Patients may also initially complain of epistaxis. This is probably related to engorgement of mucosal blood vessels in combination with abnormal hemostasis due to qualitative abnormalities in platelet function. Sixty percent of patients are men, and the median age at presentation is 60 years. Polycythemia rarely occurs in persons under age 40 years.
Physical examination reveals plethora and engorged retinal veins. The spleen is palpable in 75% of cases but is nearly always enlarged when imaged.
Thrombosis is the most common complication of polycythemia vera and the major cause of morbidity and death in this disorder. Thrombosis appears to be related to increased blood viscosity and abnormal platelet function. Uncontrolled polycythemia leads to a very high incidence of thrombotic complications of surgery, and elective surgery should be deferred until the condition has been treated. Paradoxically, in addition to thrombosis, increased bleeding also occurs. There is a high incidence of peptic ulcer disease.
B. Laboratory Findings
The hallmark of polycythemia vera is a hematocrit above normal, at times greater than 60%. Red blood cell morphology is normal. By definition, the red blood cell mass is elevated, but this is rarely measured. The white blood count is elevated to 10,000 20,000/mcL and the platelet count is variably increased, sometimes to counts exceeding 1,000,000/mcL. Platelet morphology is usually normal. White blood cells are usually normal, but basophilia and eosinophilia are frequently present.
The bone marrow is hypercellular, with panhyperplasia of all hematopoietic elements. Iron stores are usually absent from the bone marrow, having been transferred to the increased circulating red blood cell mass. Iron deficiency may also result from chronic gastrointestinal blood
P.513
loss. Bleeding may lower the hematocrit to the normal range (or lower), creating diagnostic confusion.
Table 13-15. Causes of polycythemia. | |
|---|---|
|
Vitamin B12 levels are strikingly elevated because of increased levels of transcobalamin III (secreted by white blood cells). Overproduction of uric acid may lead to hyperuricemia.
Although red blood cell morphology is usually normal at presentation, microcytosis, hypochromia, and poikilocytosis may result from iron deficiency following treatment by phlebotomy (see below). Progressive hypersplenism may also lead to elliptocytosis.
Differential Diagnosis
Spurious polycythemia, in which an elevated hematocrit is due to contracted plasma volume rather than increased red cell mass, may be related to diuretic use or may occur without obvious cause.
A secondary cause of polycythemia should be suspected if splenomegaly is absent and the high hematocrit is not accompanied by increases in other cell lines. Arterial oxygen saturation should be measured to determine if hypoxia is the cause. A smoking history should be taken; carboxyhemoglobin levels may be elevated in smokers. A renal CT scan or sonogram may be considered to look for an erythropoietin-secreting cyst or tumor (see Figure 13-1). A positive family history should lead to investigation for congenital high-oxygen-affinity hemoglobin.
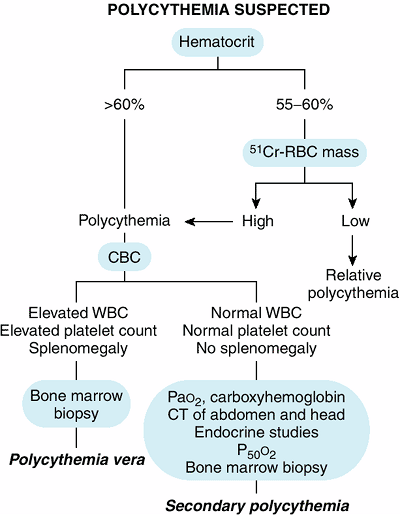 |
Figure 13-1. Diagnostic evaluation of patients with suspected polycythemia. (RBC, red blood count; CBC, complete blood count; WBC, white blood count; PaO2, partial pressure of oxygen in arterial blood; P50O2, partial pressure of oxygen at which hemoglobin is 50% saturated.) (From Nicoll D, McPhee SJ, Pignone M [editors]: Pocket Guide to Diagnostic Tests, 4th ed. McGraw-Hill, 2004 . Modified from:Stein JH [editor]: Internal Medicine, 5th ed. 1998 with permission from Elsevier.) |
Polycythemia vera should be differentiated from other myeloproliferative disorders (Table 13-14). Marked elevation of the white blood count (above 30,000/mcL) suggests CML. This disorder is confirmed by the presence of the Philadelphia chromosome or the bcr/abl fusion gene. Abnormal red blood cell morphology and nucleated red blood cells in the peripheral blood are seen in myelofibrosis. This condition is diagnosed by bone marrow biopsy showing fibrosis of the marrow. Essential thrombocytosis is diagnosed when the platelet count is strikingly elevated and the red blood cell mass is normal.
Treatment
The treatment of choice is phlebotomy. One unit of blood (approximately 500 mL) is removed weekly until the hematocrit is less than 45%; the hematocrit is maintained at less than 45% by repeated phlebotomy as necessary. Because repeated phlebotomy intentionally produces iron deficiency, the requirement for phlebotomy should gradually decrease. It is important to avoid medicinal iron supplementation, as this can thwart the goals of a phlebotomy program. Maintaining the hematocrit at normal levels has been shown to decrease the incidence of thrombotic complications. A diet low in iron may also increase the intervals between phlebotomies.
Occasionally, myelosuppressive therapy is indicated. Indications include a high phlebotomy requirement, thrombocytosis, and intractable pruritus. There is evidence that reduction of the platelet count to less than 600,000/mcL will reduce the risk of thrombotic complications. Alkylating agents have been shown to increase the risk of conversion of this disease to acute leukemia and should be avoided. Hydroxyurea is now being widely used when myelosuppressive therapy is indicated. The usual dose is 500 1500 mg/d orally, adjusted to keep platelets < 500,000/mcL without reducing the neutrophil count to < 2000/mcL. Anagrelide may be substituted or added when hydroxyurea is not well tolerated but is not the preferred initial agent. Low-dose aspirin (75 81 mg daily) has been shown to reduce the risk of thrombosis without excessive bleeding, and should be part of therapy for all patients without contraindications to aspirin.
Allopurinol may be indicated for hyperuricemia. Antihistamine therapy with diphenhydramine or other H1-blockers may be helpful for control of pruritus, and some reports suggest the efficacy of selective serotonin reuptake inhibitors in refractory cases.
Prognosis
Polycythemia is an indolent disease with median survival of 11 15 years. The major cause of morbidity and mortality is arterial thrombosis. Over time, polycythemia
P.514
vera may convert to myelofibrosis or to CML. In approximately 5% of cases, the disorder progresses to AML, which is usually refractory to therapy.
Finazzi G et al; ECLAP Investigators: Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664.
Landolfi R et al: Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004;350:114.
Marchioli R et al: Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23: 2224.
Tefferi A: Polycythemia vera: a comprehensive review and clinical recommendations. Mayo Clin Proc 2003;78:174.
Essential Thrombocytosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Elevated platelet count in absence of other causes.
Normal red blood cell mass.
Absence of Philadelphia chromosome.
General Considerations
Essential thrombocytosis is an uncommon myeloproliferative disorder of unknown cause in which marked proliferation of the megakaryocytes in the bone marrow leads to elevation of the platelet count. As with polycythemia vera, the recent finding of a high frequency of mutations of JAK2 in these patients promises to advance the understanding of this disorder.
Clinical Findings
A. Symptoms and Signs
The median age at presentation is 50 60 years, and there is a slightly increased incidence in women. The disorder is often suspected when an elevated platelet count is found. Less frequently, the first sign is thrombosis, which is the most common clinical problem. The risk of thrombosis rises with age. Venous thromboses may occur in unusual sites such as the mesenteric, hepatic, or portal vein. Some patients experience erythromelalgia, painful burning of the hands accompanied by erythema; this symptom is reliably relieved by aspirin. Bleeding, typically mucosal, is less common and is related to a concomitant qualitative platelet defect. Splenomegaly is present in at least 25% of patients.
B. Laboratory Findings
An elevated platelet count is the hallmark of this disorder, and may be over 2,000,000/mcL. The white blood cell count is often mildly elevated, usually not above 30,000/mcL, but with some immature myeloid forms. The hematocrit is normal. The peripheral blood smear reveals large platelets, but giant degranulated forms seen in myelofibrosis are not observed. Red blood cell morphology is normal. The bleeding time is prolonged in 20% of patients.
The bone marrow shows increased numbers of megakaryocytes but no other morphologic abnormalities. The Philadelphia chromosome is absent but should be assayed by molecular testing of peripheral blood for the bcr/abl fusion gene in all suspected cases to differentiate the disorder from chronic myeloid leukemia.
Differential Diagnosis
Essential thrombocytosis must be distinguished from secondary causes of an elevated platelet count. In reactive thrombocytosis, the platelet count seldom exceeds 1,000,000/mcL. Inflammatory disorders such as rheumatoid arthritis and ulcerative colitis cause significant elevations of the platelet count, as may chronic infection. The thrombocytosis of iron deficiency is observed only when anemia is significant. The platelet count is temporarily elevated after splenectomy.
Regarding other myeloproliferative disorders, the lack of elevated hematocrit and red blood cell mass distinguishes it from polycythemia vera. Unlike myelofibrosis, red blood cell morphology is normal, nucleated red blood cells are absent, and giant degranulated platelets are not seen. In chronic myeloid leukemia, the Philadelphia chromosome (or bcr/abl by molecular testing) establishes the diagnosis.
Treatment
The risk of thrombosis can be reduced by control of the platelet count, which should be kept at less than 500,000/mcL. The treatment of choice is hydroxyurea in a dose of 0.5 2 g/d. Hydroxyurea has been shown to be more effective than anagrilide in preventing thrombotic events, with no increase in toxicity. In cases in which hydroxyurea is not well tolerated because of anemia, low doses of anagrilide, 1 2 mg/d, may be added. Higher doses of anagrilide are often complicated by headache, peripheral edema, and congestive heart failure.
Vasomotor symptoms such as erythromelalgia and paresthesias respond rapidly to aspirin and eventually to control of the platelet count. The role of chronic low-dose aspirin therapy to reduce the risk of thrombosis remains unsettled. In the unusual event of severe bleeding, the platelet count can be lowered rapidly with plateletpheresis.
Course & Prognosis
Essential thrombocytosis is an indolent disorder and allows long-term survival. Average survival is longer than 15 years from diagnosis, and the survival of patients younger than 50 years does not appear different from matched controls. The major source of morbidity thrombosis can be reduced by appropriate platelet
P.515
control. Late in the course of the disease, the bone marrow may become fibrotic, and massive splenomegaly may occur, sometimes with splenic infarction. There is a 10 15% risk of progression to myelofibrosis after 15 years, and a 1 5% risk of transformation to acute leukemia over 20 years.
Barbui T et al: Practice guidelines for the therapy of essential thrombocythemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and the Italian Group for Bone Marrow Transplantation. Haematologica 2004;89:215.
Harrison CN et al; United Kingdom Medical Research Council Primary Thrombocythemia 1 Study: Hydroxyurea Compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33.
Harrison CN et al: Essential thrombocythemia. Hematol Oncol Clin North Am 2003;17:1175.
Myelofibrosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Striking splenomegaly.
Teardrop poikilocytosis on peripheral smear.
Leukoerythroblastic blood picture; giant abnormal platelets.
Hypercellular bone marrow with reticulin or collagen fibrosis.
General Considerations
Myelofibrosis (myelofibrosis with myeloid metaplasia, agnogenic myeloid metaplasia) is a myeloproliferative disorder characterized by fibrosis of the bone marrow, splenomegaly, and a leukoerythroblastic peripheral blood picture with teardrop poikilocytosis. It is widely believed that fibrosis occurs in response to increased secretion of platelet-derived growth factor (PDGF) and possibly other cytokines. In response to bone marrow fibrosis, extramedullary hematopoiesis takes place in the liver, spleen, and lymph nodes. In these sites, mesenchymal cells responsible for fetal hematopoiesis can be reactivated. As with other myeloproliferative diseases, abnormalities of JAK2 signalling pathways may be involved in the pathogenesis.
Clinical Findings
A. Symptoms and Signs
Myelofibrosis develops in adults over age 50 years and is usually insidious in onset. Patients most commonly present with fatigue due to anemia or abdominal fullness related to splenomegaly. Uncommon presentations include bleeding and bone pain. On examination, splenomegaly is almost invariably present and is commonly massive. The liver is enlarged in more than 50% of cases.
Later in the course of the disease, progressive bone marrow failure takes place as it becomes increasingly more fibrotic. Anemia becomes severe, requiring transfusion. Progressive thrombocytopenia leads to bleeding. The spleen continues to enlarge, which leads to early satiety. Painful episodes of splenic infarction may occur. Late in the course, the patient becomes cachectic and may experience severe bone pain, especially in the upper legs. Hematopoiesis in the liver leads to portal hypertension with ascites, esophageal varices, and occasionally transverse myelitis caused by myelopoiesis in the epidural space.
B. Laboratory Findings
Patients are almost invariably anemic at presentation. The white blood count is variable either low, normal, or elevated and may be increased to 50,000/mcL. The platelet count is variable. The peripheral blood smear is dramatic, with significant poikilocytosis and numerous teardrop forms in the red cell line. Nucleated red blood cells are present and the myeloid series is shifted, with immature forms including a small percentage of promyelocytes or myeloblasts. Platelet morphology may be bizarre, and giant degranulated platelet forms (megakaryocyte fragments) may be seen. The triad of teardrop poikilocytosis, leukoerythroblastic blood, and giant abnormal platelets is highly suggestive of myelofibrosis.
The bone marrow usually cannot be aspirated (dry tap), though early in the course of the disease it is hypercellular, with a marked increase in megakaryocytes. Fibrosis at this stage is detected by a silver stain demonstrating increased reticulin fibers. Later, biopsy reveals more severe fibrosis, with eventual replacement of hematopoietic precursors by collagen. There is no characteristic chromosomal abnormality.
Differential Diagnosis
A leukoerythroblastic blood picture from other causes may be seen in response to severe infection, inflammation, or infiltrative bone marrow processes. However, teardrop poikilocytosis and giant abnormal platelet forms will not be present. Bone marrow fibrosis may be seen in metastatic carcinoma, Hodgkin's disease, and hairy cell leukemia. These disorders are diagnosed by characteristic morphology of involved tissues.
Concerning other myeloproliferative disorders, CML is diagnosed when there is marked leukocytosis, normal red blood cell morphology, and the presence of the Philadelphia chromosome or bcr/abl fusion gene. Polycythemia vera is characterized by an elevated red blood cell mass. Essential thrombocytosis shows predominant and consistent platelet count elevations.
Treatment
Patients with mild forms of the disease may require no therapy or occasional transfusion support. Recently,
P.516
biologic agents have shown some benefit. Thalidomide has produced definite responses with acceptable toxicity, and the investigational agent lenalidomide (Revlimid) may have equivalent efficacy with less toxicity. Allogeneic bone marrow transplantation has been performed successfully with 50% long-term survival and should be considered in younger patients. The use of less toxic, nonmyeloablative regimens for allogeneic transplantation has produced encouraging results. Anemic patients are supported with transfusion. Erythropoietin may increase red blood cell production and decrease transfusion requirements. Splenectomy is not routinely performed but is indicated for splenic enlargement causing recurrent painful episodes, severe thrombocytopenia, or an unacceptable transfusion requirement.
Course & Prognosis
It is difficult to date the onset of myelofibrosis, but the median survival from time of diagnosis is approximately 5 years. New therapies with biologic agents, such as thalidomide and lenalidomide, and the application of reduced-intensity allogeneic stem cell transplantation now appear to offer the possibility of improving the outcome for many patients. End-stage myelofibrosis is characterized by generalized debility, liver failure, and bleeding from thrombocytopenia, with some cases terminating in AML.
Deeg HJ et al: Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood 2003;102:3912.
Dingli D et al: Myelofibrosis with myeloid metaplasia: new developments in pathogenesis and treatment. Intern Med 2004; 43:540.
Kralovics R et al: A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005;352:1779.
Mesa RA et al: Durable responses to thalidomide-based drug therapy for myelofibrosis with myeloid metaplasia. Mayo Clin Proc 2004;79:883.
Chronic Myelogenous Leukemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Strikingly elevated white blood count.
Markedly left-shifted myeloid series but with a low percentage of promyelocytes and blasts.
Presence of Philadelphia chromosome or bcr/abl gene.
General Considerations
CML is a myeloproliferative disorder characterized by overproduction of myeloid cells. These myeloid cells retain the capacity for differentiation, and normal bone marrow function is retained during the early phases. The disease usually remains stable for years and then transforms to a more overtly malignant disease.
CML is characterized by a specific chromosomal abnormality, the Philadelphia chromosome, a reciprocal translocation between the long arms of chromosomes 9 and 22. A large portion of 22q is translocated to 9q, and a smaller piece of 9q is moved to 22q. The portion of 9q that is translocated contains abl, a protooncogene that is the cellular homolog of the Ableson murine leukemia virus. The abl gene is received at a specific site on 22q, the break point cluster (bcr). The fusion gene bcr/abl produces a novel protein that differs from the normal transcript of the abl gene in that it possesses tyrosine kinase activity (a characteristic activity of transforming genes). Evidence that the bcr/abl fusion gene is pathogenic is provided by transgenic mouse models in which introduction of the gene almost invariably leads to leukemia. Furthermore, targeted therapy with imatinib, which inhibits the tyrosine kinase activity of the bcr/abl protein, is remarkably effective as treatment.
Approximately 5% of cases of CML are Philadelphia chromosome negative at the level of light microscope cytogenetics, though molecular studies demonstrate the bcr/abl fusion gene, and these patients appear to have the same clinical outcome as those with the overt cytogenetic finding. The entity formerly known as Philadelphia chromosome-negative CML is now recognized as chronic myelomonocytic leukemia, a subtype of myelodysplasia.
Early CML ( chronic phase ) does not behave like a malignant disease. Normal bone marrow function is retained, white blood cells differentiate, and, despite some qualitative abnormalities (low leukocyte alkaline phosphatase), the neutrophils combat infection normally. However, CML is inherently unstable, and the disease progresses to an accelerated phase and finally after several years, to blast crisis. This progression of the disease is often associated with added chromosomal defects superimposed on the Philadelphia chromosome. Blast crisis CML is morphologically indistinguishable from acute leukemia.
Clinical Findings
A. Symptoms and Signs
CML is a disorder of middle age (median age at presentation is 55 years). Patients usually present with fatigue, night sweats, and low-grade fever related to the hypermetabolic state caused by overproduction of white blood cells. At other times, the patient complains of abdominal fullness related to splenomegaly. In many cases, an elevated white blood count is discovered incidentally. Rarely, the patient will present with a clinical syndrome related to leukostasis with blurred vision, respiratory distress, or priapism. The white blood count in these cases is usually greater than 500,000/mcL.
On examination, the spleen is enlarged (often markedly so), and sternal tenderness may be present as a sign of marrow overexpansion. In cases discovered during
P.517
routine laboratory monitoring, these findings are often absent.
Acceleration of the disease is often associated with fever in the absence of infection, bone pain, and splenomegaly. In blast crisis, patients may experience bleeding and infection related to bone marrow failure.
B. Laboratory Findings
The hallmark of CML is an elevated white blood count; the median white blood count at diagnosis is 150,000/mcL, although in some cases the white blood cell count is only modestly increased. The peripheral blood is characteristic. The myeloid series is left shifted, with mature forms dominating and with cells usually present in proportion to their degree of maturation. Blasts are usually less than 5%. Basophilia and eosinophilia of granulocytes may be present. At presentation, the patient is usually not anemic. Red blood cell morphology is normal, and nucleated red blood cells are rarely seen. The platelet count may be normal or elevated (sometimes to strikingly high levels). Platelet morphology is usually normal, but abnormally large forms may be seen.
The bone marrow is hypercellular, with left-shifted myelopoiesis. Myeloblasts comprise less than 5% of marrow cells.
The hallmark of the disease is that the bcr/abl gene is detected in the peripheral blood. This is best done by the polymerase chain reaction (PCR) test, which has now supplanted cytogenetics in looking for the Philadelphia chromosome. A bone marrow examination is not necessary for diagnosis, although it is useful for prognosis and for detecting additional chromosomal abnormalities in addition to the Philadelphia chromosome.
With progression to the accelerated and blast phases, progressive anemia and thrombocytopenia occur, and the percentage of blasts in the blood and bone marrow increases. Blast phase CML is diagnosed when blasts comprise more than 30% of bone marrow cells.
Differential Diagnosis
Early CML must be differentiated from the reactive leukocytosis associated with infection. In such cases, the white blood count is usually less than 50,000/mcL, splenomegaly is absent, and the bcr/abl gene is not present.
CML must be distinguished from other myeloproliferative disease (Table 13-14). The hematocrit should not be elevated, the red blood cell morphology is normal, and nucleated red blood cells are rare or absent. Definitive diagnosis is made by finding the bcr/abl gene.
Treatment
Treatment is usually not emergent even with white blood counts over 200,000/mcL, since the majority of circulating cells are mature myeloid cells that are smaller and more deformable than primitive leukemic blasts. In the rare instances in which symptoms result from extreme hyperleukocytosis (priapism, respiratory distress, visual blurring, altered mental status), emergent leukapheresis is performed in conjunction with myelosuppressive therapy.
The treatment of CML has been transformed by the introduction of imatinib mesylate. This drug is a specifically designed inhibitor of the tyrosine kinase activity of the bcr/abl oncogene. It is well tolerated and results in nearly universal (98%) hematologic control of chronic phase disease. It has now replaced both interferon and hydroxyurea as standard therapy. For patients with the chronic phase of CML, the standard dose is 400 mg orally daily. Higher doses, such as 600 800 mg daily, may overcome some degree of resistance and may produce more rapid initial responses, but side effects are more prominent with these doses. The most common toxicities are nausea, periorbital swelling, edema, rash, and myalgia, but most of these are modest. Fewer than 5% of patients discontinue the drug due to unacceptable side effects.
Response is assessed in several ways. First, the patient should enter hematologic complete remission, with normalization of blood counts and splenomegaly. This usually occurs within several weeks, but should occur within 3 months. Second, cytogenetic remission should be achieved, ideally within 6 months but certainly within 12 months. A major cytogenetic response is identified when < 35% of metaphases contain the Philadelphia chromosome, and a complete cytogenetic response indicates the absence of the abnormal chromosome. More recently, quantitative assessment of the bcr/abl gene using PCR assays has become the standard method of assessment. At this time, with lead patients out 5 years from start of imatinib, 100% of patients with the best response (complete cytogenetic response and > 3 log reduction in bcr/abl) remain free of progression. Others with lesser degrees of response have increased risk of early progression, and these patients are best treated with allogeneic stem cell transplantation. Although imatinib has been a remarkable new treatment, since almost all clinical experience with imatinib began in early 2000, the long-term outcome is uncertain.
Current clinical trials focus on new molecular targeted agents that can overcome resistance to imatinib. Dasatinib has been shown to produce responses in a high proportion of patients whose disease has become resistant to imatinib and is being evaluated as initial therapy. Other investigational agents are similarly being evaluated. It is possible that combinations of molecular targeted agents will be used in the future.
Hydroxyurea was formerly the standard treatment for this disease and can be used for patients who do not tolerate imatinib. Hydroxyurea is oral and very well tolerated. The usual dose is 0.5 2.5 g/d, adjusted to keep the white blood cell count ideally near 5000/mcL but in any case above 2000/mcL. It is given without interruption because of rapid white blood count rebound.
The only available curative therapy for CML is allogeneic bone marrow transplantation. The best results (80% cure rate) are obtained in patients who are under 40 years of age and transplanted within 1 year
P.518
after diagnosis from HLA-matched siblings. The introduction of imatinib has changed the approach to allogeneic transplant for CML. Patients with the best transplant outcomes (under 40 years of age with matched sibling donors) may be offered allogeneic transplant as initial therapy. An alternative approach is to initiate imatinib and to recommend transplant if there is a suboptimal response, either lack of a complete cytogenetic response, a suboptimal molecular response, or an increasing level of bcr/abl transcripts. These recommendations are in flux and may change as long-term experience with imatinib and other agents accumulates. For patients without sibling donors whose disease is not well controlled by imatinib, HLA-matched unrelated donors may be located through registries such as the National Marrow Donors Program. Results are somewhat inferior to those achieved with matched sibling transplants but offer a cure rate of 40 60% in an otherwise invariably fatal disease.
Allogeneic transplantation cures CML by initial cytoreduction followed by long-term immunologic control mediated by the donor's immune system. This alloimmune phenomenon has been called the graft-versus-leukemia effect. The most compelling evidence for its importance is that chronic phase disease that has recurred after allogeneic transplantation can usually be reversed without additional chemotherapy by the infusion of T lymphocytes from the initial bone marrow donor. This donor lymphocyte infusion can lead to long-term remission in 50 70% of cases. In response to appreciation of the importance of the graft-versus-leukemia effect, less toxic forms of allogeneic transplantation (nonmyeloablative) have been developed that require much less initial cytoreductive therapy and rely exclusively on the immune effect for long-term disease control. This approach has been encouraging and is likely to further expand the role of allogeneic transplant for CML, allowing transplant in selected patients up to age 70 years.
Course & Prognosis
In the past, median survival was 3 4 years. In the era of imatinib therapy, and with the recent development of new molecular targeted agents, more than 80% of patients remain alive and in remission at 4 years. It is impossible to predict at this time to what extent long-term survival will be impacted. At the current time, allogeneic stem cell transplantation remains the only proven curative treatment.
Crossman LC et al: Imatinib therapy in chronic myeloid leukemia. Hematol Oncol Clin North Am 2004;18:605.
Deininger M et al: The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005;105:2640.
Kantarjian HM et al: Long-term survival benefit and improved complete cytogenetic and molecular response rates with imatinib mesylate in Philadelphia chromosome-positive chronic-phase chronic myeloid leukemia after failure of interferon-alpha. Blood 2004;104:1979.
Or R et al: Nonmyeloablative allogeneic stem cell transplantation for the treatment of chronic myeloid leukemia in first chronic phase. Blood 2003;101:441.
Radich JP et al: HLA-matched related hematopoietic cell transplantation for chronic-phase CML using a targeted busulfan and cyclophosphamide preparative regimen. Blood 2003; 102:31.
Shah NP et al: Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004;305:399.
Myelodysplastic Syndromes
![]() Essentials of Diagnosis
Essentials of Diagnosis
Cytopenias with a hypercellular bone marrow.
Morphologic abnormalities in two or more hematopoietic cell lines.
General Considerations
The myelodysplastic syndromes are a group of acquired clonal disorders of the hematopoietic stem cell. They are characterized by the constellation of cytopenias, a hypercellular marrow, and a number of morphologic and cytogenetic abnormalities. The disorders are usually idiopathic but may be seen after cytotoxic chemotherapy.
Despite the presence of adequate numbers of hematopoietic progenitor cells, ineffective hematopoiesis occurs, resulting in various cytopenias. Ultimately, the disorder may evolve into AML, and the term preleukemia has been used in the past to describe these disorders. Although no specific chromosomal abnormality is seen in myelodysplasia, there are frequently abnormalities involving the long arm of chromosome 5 (which contains a number of genes encoding both growth factors and receptors involved in myelopoiesis) as well as deletions of chromosomes 5 and 7.
Myelodysplasia encompasses several heterogeneous syndromes. Those without excess bone marrow blasts are termed refractory anemia, with or without ringed sideroblasts. Syndromes with excess blasts are diagnosed as refractory anemia with excess blasts (RAEB 5 19% blasts). Those with a proliferative syndrome including peripheral blood monocytosis greater than 1000/mcL are termed chronic myelomonocytic leukemia (CMML). An International Prognostic Scoring System (IPSS) has been developed that classifies patients by risk status based on the percentage of bone marrow blasts, cytogenetics, and the severity of cytopenias.
Clinical Findings
A. Symptoms and Signs
Patients are usually over age 60 years. Many are diagnosed while asymptomatic because of the finding of abnormal blood counts. Patients usually present with fatigue, infection, or bleeding related to bone marrow
P.519
failure. The course may be indolent, and the disease may present as a wasting illness with fever, weight loss, and general debility. On examination, splenomegaly may be present in combination with pallor, bleeding, and various signs of infection.
B. Laboratory Findings
Anemia may be marked and may require transfusion support. The MCV is normal or increased, and macro-ovalocytes may be seen on the peripheral blood smear. The reticulocyte count is usually reduced. The white blood cell count is usually normal or reduced, and neutropenia is common. The neutrophils may exhibit morphologic abnormalities, including deficient numbers of granules or a bilobed nucleus (Pelger-Huet). The myeloid series may be left shifted, and small numbers of promyelocytes or blasts may be seen. The platelet count is normal or reduced, and hypogranular platelets may be present.
The bone marrow is characteristically hypercellular, but may be hypocellular. Erythroid hyperplasia is common, and signs of abnormal erythropoiesis include megaloblastic features, nuclear budding, or multinucleated erythroid precursors. The Prussian blue stain may demonstrate ringed sideroblasts. The myeloid series is often left shifted, with variable increases in blasts. Deficient or abnormal granules may be seen. A characteristic abnormality is the presence of dwarf megakaryocytes with a unilobed nucleus. A variety of cytogenetic abnormalities in the bone marrow are characteristic of myelodysplasia. Some patients with an indolent form of the disease have an isolated partial deletion of chromosome 5 (5q- syndrome). The presence of other abnormalities such as monosomy 7 is associated with more aggressive disease.
Differential Diagnosis
In subtle cases, cytogenetic evaluation of the bone marrow may help distinguish this clonal disorder from other causes of cytopenias. As the number of blasts increases in the bone marrow, myelodysplasia is arbitrarily separated from AML by the presence of less than 20% blasts.
Treatment
Historically, there has been no effective therapy for this disorder, and patients have been managed with supportive care. However, this is now changing. Patients affected primarily by anemia may be supported with red blood cell transfusions. Erythropoietin (epoetin alfa), 30,000 units subcutaneously weekly, reduces the red cell transfusion requirement in some patients. The response rate is 20%, but a 4-week trial of epoetin alfa is reasonable since it will be of benefit and cost-effective for the subgroup of responders. The combination of myeloid growth factors and high doses of epoetin alfa produces a higher response rate, but the cost is very high. Lenalidomide has recently been approved for the treatment of transfusion-dependent anemia due to myelodysplasia. It is remarkably effective in patients with the 5q- cytogenetic abnormality, with significant responses in 70% of patients. The recommended initial dose is 10 mg daily. The most common side effects are neutropenia and thrombocytopenia, but venous thrombosis is also seen. The cost of this new agent is extremely high. Patients affected primarily with severe neutropenia may benefit from the use of myeloid growth factors such as G-CSF or GM-CSF.
Azacitidine (5-azacytidine) has been approved as an effective treatment based on its ability to improve both symptoms and blood counts and to prolong the time to conversion to acute leukemia. It is now the treatment of choice for many patients, especially those with higher risk disease based on increased blasts in the bone marrow. Occasional patients can benefit from immunosuppressive therapy including ATG. Patients with hypocellular bone marrows and those with HLA A15 have an increased chance of response. Allogeneic stem cell transplantation is the only curative therapy for myelodysplasia, but its role is limited by the advanced age of many patients and the indolent course of disease in some subsets of patients. The optimal use and timing of allogeneic transplantation are controversial, but the use of reduced-intensity preparative regimens for transplantation has expanded the role of this therapy, using both family and matched unrelated donors.
Course & Prognosis
Myelodysplasia is an ultimately fatal disease, and allogeneic transplantation is the only curative therapy, with cure rates of 30 60% depending primarily on the risk status of the disease. Patients most commonly die of infections or bleeding. The risk of transformation to AML depends on the percentage of blasts in the bone marrow. Patients with refractory anemia may survive many years, and the risk of leukemia is low (< 10%). Those with excess blasts or CMML have short survivals (usually < 2 years) and have a higher (20 50%) risk of developing acute leukemia. The finding of full deletions of chromosomes 5 and 7 is associated with a poor prognosis.
Cortes J et al: Phase I Study of BMS-214662, a farnesyl transferase inhibitor in patients with acute leukemias and high-risk myelodysplastic syndromes. J Clin Oncol 2005;23:2805.
Ho AY et al: Reduced-intensity allogeneic hematopoietic stem cell transplantation for myelodysplastic syndrome and acute myeloid leukemia with multilineage dysplasia using fludarabine, busulphan, and alemtuzumab (FBC) conditioning. Blood 2004;104:1616.
J dersten M et al: Long-term outcome of treatment of anemia in MDS with erythropoietin and G-CSF. Blood 2005;106:803.
List A et al: Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med 2005;352:549.
Silverman LR et al: Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 2002;20:2429.
P.520
Acute Leukemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Short duration of symptoms, including fatigue, fever, and bleeding.
Cytopenias or pancytopenia.
More than 20% blasts in the bone marrow.
Blasts in peripheral blood in 90% of patients.
General Considerations
Acute leukemia is a malignancy of the hematopoietic progenitor cell. The malignant cell loses its ability to mature and differentiate. These cells proliferate in an uncontrolled fashion and replace normal bone marrow elements. Most cases arise with no clear cause. However, radiation and some toxins (benzene) are leukemogenic. In addition, a number of chemotherapeutic agents (especially procarbazine, melphalan, other alkylating agents, and etoposide) may cause leukemia. The leukemias seen after toxin or chemotherapy exposure often develop from a myelodysplastic prodrome and are associated with abnormalities in chromosomes 5 and 7.
Much has been learned about the molecular biology of the leukemias. One subtype, acute promyelocytic leukemia, is characterized by chromosomal translocation t(15;17), which produces the fusion gene PML-RAR . This change in the retinoic acid receptor produces a block in differentiation that can be overcome with pharmacologic doses of retinoic acid (see below).
Most of the clinical findings in acute leukemia are due to replacement of normal bone marrow elements by the malignant cell. Less common manifestations result from organ infiltration (skin, gastrointestinal tract, meninges). Acute leukemia is potentially curable with combination chemotherapy.
Acute lymphoblastic leukemia (ALL) comprises 80% of the acute leukemias of childhood. The peak incidence is between 3 and 7 years of age. It is also seen in adults, causing approximately 20% of adult acute leukemias. AML is primarily an adult disease with a median age at presentation of 60 years and an increasing incidence with advanced age.
Clinical Findings
A. Symptoms and Signs
Most patients have been ill only for days or weeks. Bleeding (usually due to thrombocytopenia) occurs in the skin and mucosal surfaces, with gingival bleeding, epistaxis, or menorrhagia. Less commonly, widespread bleeding is seen in patients with disseminated intravascular coagulation (DIC) (in acute promyelocytic leukemia and monocytic leukemia). Infection is due to neutropenia, with the risk of infection rising as the neutrophil count falls below 500/mcL; with neutrophil counts less than 100/mcL, infection within days is the rule. The most common pathogens are gram-negative bacteria (Escherichia coli, Klebsiella, Pseudomonas) or fungi (Candida, Aspergillus). Common presentations include cellulitis, pneumonia, and perirectal infections; death within a few hours may occur if treatment with appropriate antibiotics is delayed.
Patients may also seek medical attention because of gum hypertrophy and bone and joint pain. The most dramatic presentation is hyperleukocytosis, in which a markedly elevated circulating blast count (usually > 200,000/mcL) leads to impaired circulation, presenting as headache, confusion, and dyspnea. Such patients require emergent leukapheresis and chemotherapy.
On examination, patients appear pale and have purpura and petechiae; signs of infection may not be present. Stomatitis and gum hypertrophy may be seen in patients with monocytic leukemia, as may rectal fissures. There is variable enlargement of the liver, spleen, and lymph nodes. Bone tenderness may be present, particularly in the sternum, tibia, and femur.
B. Laboratory Findings
The hallmark of acute leukemia is the combination of pancytopenia with circulating blasts. However, blasts may be absent from the peripheral smear in as many as 10% of cases ( aleukemic leukemia ). The bone marrow is usually hypercellular and dominated by blasts. More than 20% blasts are required to make a diagnosis of acute leukemia.
A number of other laboratory abnormalities are noted. Hyperuricemia may be seen. If DIC is present, the fibrinogen level will be reduced, the prothrombin time prolonged, and fibrin degradation products or fibrin D-dimers present. Patients with ALL (especially T cell) may have a mediastinal mass visible on chest radiograph. Meningeal leukemia will have blasts present in the spinal fluid, seen in approximately 5% of cases at diagnosis; it is more common in monocytic types of AML.
Acute leukemia should be classified as either ALL or AML. The Auer rod, an eosinophilic needle-like inclusion in the cytoplasm, is pathognomonic of AML. To confirm the myeloid nature of the cells, histochemical stains demonstrating myeloid enzymes such as peroxidase may be useful. Monocytic lineage can be established by the finding of butyrate esterase. ALL is considered when there is no morphologic or histochemical evidence of myeloid or monocytic lineage. The diagnosis is confirmed by demonstrating surface markers characteristic of primitive lymphoid cells, typically by flow cytometry; terminal deoxynucleotidyl transferase (TdT) is present in 95% of cases of ALL. A variety of monoclonal antibodies have been used to define other phenotypes of ALL. Primitive B lymphocyte antigens include CD19 and sometimes CD10. T cell ALL is diagnosed by the finding of CD2, CD5, and CD7.
P.521
AML is usually categorized on the basis of morphology and histochemistry as follows: acute undifferentiated leukemia (M0), acute myeloblastic leukemia (M1), acute myeloblastic leukemia with differentiation (M2), acute promyelocytic leukemia (M3), acute myelomonocytic leukemia (M4), acute monoblastic leukemia (M5), erythroleukemia (M6), and megakaryoblastic leukemia (M7). The World Health Organization (WHO) has sponsored a new classification of the leukemias and other hematologic malignancies that incorporates cytogenetic, molecular, and immunophenotype information.
ALL is most usefully classified by immunologic phenotype as follows: common, early B lineage, and T cell.
Cytogenetic studies are the most powerful prognostic factors in the acute leukemias. Favorable cytogenetics in AML include t(8;21), t(15;17), and inv(16)(p13;q22). These patients have a higher chance of achieving both short- and long-term disease control. In ALL, the hyperdiploid states are associated with a better prognosis. Unfavorable cytogenetics in AML are monosomy 5 and 7 and complex abnormalities; unfavorable cytogenetics in ALL are the Philadelphia chromosomes t(9;22) and t(4;11).
Differential Diagnosis
AML must be distinguished from other myeloproliferative disorders, CML, and myelodysplastic syndromes. Acute leukemia also resembles a left-shifted bone marrow recovering from a previous toxic insult. If the question is in doubt, a bone marrow study should be repeated in several days to see if maturation has taken place. ALL must be separated from other lymphoproliferative disease such as CLL, lymphomas, and hairy cell leukemia. It may also be confused with the atypical lymphocytosis of mononucleosis and pertussis.
Treatment
Most young patients with acute leukemia are treated with the objective of effecting a cure. The first step in treatment is to obtain complete remission, defined as normal peripheral blood with resolution of cytopenias, normal bone marrow with no excess blasts, and normal clinical status. The type of initial chemotherapy depends on the subtype of leukemia. Most patients with AML are treated with a combination of an anthracycline (daunorubicin or idarubicin) plus cytarabine, either alone or in combination with other agents. This therapy will produce complete remissions in 70 80% of patients under age 60 years and in 40 60% of older patients. Acute promyelocytic leukemia is treated differently from other forms of AML. Induction therapy includes an anthracycline plus all-trans-retinoic acid. This agent is an analog of vitamin A that leads to terminal differentiation of acute promyelocytic leukemia cells through an interaction with the abnormal retinoic acid receptor created by a specific chromosomal translocation, which is the hallmark of the subtype of leukemia. With this approach 90 95% of patients will achieve complete remission. Adults with ALL are treated with combination chemotherapy, including daunorubicin, vincristine, prednisone, and asparaginase. This treatment produces complete remissions in 80 90% of patients. Those patients with Philadelphia chromsome-positive ALL (or bcr-abl plus ALL) should have imatinib added to their initial chemotherapy.
Once a patient has entered remission, postremission therapy is given with curative intent. Options include standard chemotherapy and autologous and allogeneic transplantation. The optimal treatment strategy depends on the patient's age and clinical status and the risk factor profile of the leukemia. Acute promyelocytic leukemia is generally treated with chemotherapy plus retinoic acid, and 70 80% of patients remain in long-term remission. Arsenic trioxide has been approved for treatment of relapsed disease, and is under investigation during primary therapy in the hope of increasing cure rates. For average-risk patients with AML, cure rates for postremission therapy are 35 40% for chemotherapy, 50% for autologous transplantation, and 50 60% for allogeneic transplantation. Some types of AML whose cytogenetics involved core-binding factors have a more favorable prognosis, with cure rates of 40 60% with chemotherapy and 70% with autologous transplantation. Patients who do not enter remission or who have high-risk cytogenetics (such as monosomy 7 and complex cytogenetics) do far more poorly and are rarely cured with chemotherapy. Allogeneic transplantation is the treatment of choice, but cure rates are only 20 30%.
Once leukemia has recurred after initial chemotherapy, the prognosis is much more guarded. For patients in second remission, transplantation (autologous or allogeneic) offers a 20 40% chance of cure. For those patients with acute promyelocytic leukemia who relapse, arsenic trioxide is a novel therapy that can produce second remissions, and autologous transplant in second remission produces cure rates of 60 70%.
ALL is treated initially with combination chemotherapy, including daunorubicin, vincristine, prednisone, and asparaginase. Remission induction therapy for ALL is less myelosuppressive than treatment for AML and does not necessarily produce marrow aplasia. After achieving complete remission, patients receive central nervous system prophylaxis so that meningeal sequestration of leukemic cells does not develop. As with AML, patients may be treated with either chemotherapy or high-dose chemotherapy plus bone marrow transplantation. Treatment decisions are made based on patient age and risk factors of the disease. High-risk patients with adverse cytogenetics or poor responses to chemotherapy are best treated with allogeneic transplantation. Autologous transplantation is a possibility in high-risk patients who lack a suitable donor.
Prognosis
Approximately 70 80% of adults with AML under age 60 years achieve complete remission. High-dose postremission chemotherapy leads to cure in 35 40%
P.522
of these patients, and high-dose cytarabine has been shown to be superior to therapy with lower doses. Allogeneic bone marrow transplantation (for younger adults with HLA-matched siblings) is curative in 50 60% of cases. Autologous bone marrow transplantation may be superior to nonablative chemotherapy. Older adults with AML achieve complete remission in up to 50% of instances. The cure rates for older patients with AML have been very low (approximately 10 15%), even if they achieve remission and are able to receive postremission chemotherapy. The use of reduced-intensity allogeneic transplantation is being explored in order to improve on these outcomes.
Berg SL et al; Children's Oncology Group: Phase II Study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children's Oncology Group. J Clin Oncol 2005;23:3376.
Breems DA et al: Prognostic index for adult patients with acute myeloid leukemia in first relapse. J Clin Oncol 2005;23: 1969.
Farag S et al: Outcome if induction and postremission therapy in younger adults with acute myeloid leukemia with normal karyotype: A cancer and leukemia group b study. J Clin Oncol 2005;23:482.
Lee S et al: The effect of first-line imatinib interim therapy on the outcome of allogeneic stem cell transplantation in adults with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 2005;105:3449.
Mancini M et al: A comprehensive genetic classification of adult acute lymphoblastic leukemia (ALL): analysis of the GIMEMA 0496 protocol. Blood 2005;105:3434.
Pui CH et al: Treatment of acute lymphoblastic leukemia. N Engl J Med 2006;354:166.
Sanz MA et al: Tricks of the trade for the appropriate management of newly diagnosed acute promyelocytic leukemia. Blood 2005;105:3019.
Chronic Lymphocytic Leukemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Most patients asymptomatic at presentation.
Splenomegaly typical.
Lymphocytosis > 5000/mcL.
Mature appearance of lymphocytes.
Coexpression of CD19, CD5.
General Considerations
Chronic lymphocytic leukemia (CLL) is a clonal malignancy of B lymphocytes. The disease is usually indolent, with slowly progressive accumulation of long-lived small lymphocytes. These cells are immunoincompetent and respond poorly to antigenic stimulation.
CLL is manifested clinically by immunosuppression, bone marrow failure, and organ infiltration with lymphocytes. Immunodeficiency is also related to inadequate antibody production by the abnormal B cells. With advanced disease, CLL may cause damage by direct tissue infiltration.
Information about CLL is now evolving rapidly, with new findings in biology and new treatment options.
Clinical Findings
A. Symptoms and Signs
CLL is a disease of older patients, with 90% of cases occurring after age 50 years and a median age at presentation of 65 years. Many patients will be incidentally discovered to have lymphocytosis. Others present with fatigue or lymphadenopathy. On examination, 80% of patients will have lymphadenopathy and 50% will have enlargement of the liver or spleen.
A prognostically useful staging system (Rai system) has been developed as follows: stage 0, lymphocytosis only; stage I, lymphocytosis plus lymphadenopathy; stage II, organomegaly; stage III, anemia; stage IV, thrombocytopenia.
CLL usually pursues an indolent course; a variant, prolymphocytic leukemia, is more aggressive. The morphology of the latter is different, characterized by larger and more immature cells. In 5 10% of cases, CLL may be complicated by autoimmune hemolytic anemia or autoimmune thrombocytopenia. In approximately 5% of cases, while the systemic disease remains stable, an isolated lymph node transforms into an aggressive large cell lymphoma (Richter's syndrome).
B. Laboratory Findings
The hallmark of CLL is isolated lymphocytosis. The white blood count is usually greater than 20,000/mcL and may be markedly elevated to several hundred thousand. Usually 75 98% of the circulating cells are lymphocytes. Lymphocytes appear small and mature, with condensed nuclear chromatin, and are morphologically indistinguishable from normal small lymphocytes, but smaller numbers of larger and activated lymphocytes may be seen. The hematocrit and platelet count are usually normal at presentation. The bone marrow is variably infiltrated with small lymphocytes. The immunophenotype of CLL demonstrates coexpression of the B lymphocyte lineage marker CD19 with the T lymphocyte marker CD5; this finding is commonly observed only in CLL and mantle cell lymphoma. CLL is distinguished from mantle cell lymphoma by the expression of CD23 and the typical low expression of surface immunoglobulin and CD20. Patients whose CLL cells have mutated forms of the immunoglobulin gene (which can currently be tested only in research laboratories) appear to have a more indolent form of disease; these cells typically express low levels of the surface antigen CD38 and do not express the zeta-associated protein (ZAP-70). Conversely, patients whose cells have
P.523
unmutated IgV genes and high levels of ZAP-70 expression do less well. The assessment of genomic changes by fluorescence in-situ hybridization (FISH) provides important prognostic information. The findings of deletions of chromosome 17p or 11q have a poor prognosis, whereas those whose only genomic change is deletion of 13q have a very favorable outcome.
Hypogammaglobulinemia is present in 50% of patients and becomes more common with advanced disease. In some, a small amount of IgM paraprotein is present in the serum. Pathologic changes in lymph nodes are the same as in diffuse small cell lymphocytic lymphoma.
Differential Diagnosis
Few syndromes can be confused with CLL. Viral infections producing lymphocytosis should be obvious from the presence of fever and other clinical findings; however, fever may occur in CLL from concomitant bacterial infection. Pertussis may cause a particularly high total lymphocyte count. Other lymphoproliferative diseases such as Waldenstr m's macroglobulinemia, hairy cell leukemia, or lymphoma (especially mantle cell) in the leukemic phase are distinguished on the basis of the morphology and immunophenotype of circulating lymphocytes and bone marrow.
Treatment
Most cases of early indolent CLL require no specific therapy, and the standard of care for early stage disease has been observation. However, with advances in therapy and with no information on biologic prognostic factors, new clinical trials will investigate whether there is a role for early intervention in subsets of patients with early stage disease. At the present time, indications for treatment include progressive fatigue, symptomatic lymphadenopathy, or anemia or thrombocytopenia. These patients have either symptomatic and progressive stage II disease or stage III/IV disease. The current treatment of choice is the combination of the chemotherapeutic drug fludarabine plus the antibody rituximab. Treatment is usually given monthly for 6 months and then stopped. Other combinations including fludarabine plus cyclophosphamide (and the three-drug combination adding rituximab) also produce high response rates and are being studied but produce somewhat more toxicity. Chlorambucil, 0.6 1 mg/kg orally every 3 weeks for approximately 6 months, was the standard treatment prior to the development of fludarabine. This treatment is convenient, well tolerated, and remains a reasonable first choice for elderly patients for whom frequent trips to the physician's office is a hardship. The monoclonal antibody alemtuzumab has been approved for treatment of refractory CLL and has been shown to reduce minimal residual disease during primary therapy. However, it produces significant immunosuppression, and its role in primary therapy remains to be determined.
Associated autoimmune hemolytic anemia or immune thrombocytopenia may require treatment with rituximab, prednisone, or splenectomy. Fludarabine should be avoided in patients with autoimmune hemolytic anemia since it may exacerbate this condition. Patients with recurrent bacterial infections and hypogammaglobulinemia benefit from prophylactic infusions of gamma globulin (0.4 g/kg/month), but this treatment is very expensive and can be justified only when these infections are severe.
Allogeneic transplantation offers potentially curative treatment for patients with CLL, but it should be used only in patients whose disease cannot be controlled by standard therapies. Nonmyeloablative allogeneic transplant has produced encouraging results and may expand the role of transplant in CLL.
Prognosis
New therapies appear to be changing the prognosis of CLL. In the past, median survival was approximately 6 years, and only 25% of patients lived more than 10 years. Patients with stage 0 or stage I disease have a median survival of 10 15 years, and these patients may be reassured that they can live a normal life for many years. Patients with stage III or stage IV disease had a median survival of less than 2 years in the past, but with new fludarabine-based combination therapies, 2-year survival is now greater than 90% and the long-term outlook appears to be substantially changed. Biologic markers, such as IgV gene mutation status, ZAP-70 expression, and genomic abnormalities, will better predict outcomes of subsets of patients with CLL and will help direct appropriate patients to reduced-intensity allogeneic transplant.
Byrd JC et al: Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood 2005;105:49.
Chiorazzi N et al: Chronic lymphocytic leukemia. N Engl J Med 2005;352:804.
Keating MJ et al: Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005;23:4079.
Montserrat E et al: How I treat refractory CLL. Blood 2006;107: 1276.
Moreno C et al: Allogeneic stem-cell transplantation may overcome the adverse prognosis of unmutated VH gene in patients with chronic lymphocytic leukemia. J Clin Oncol 2005;23:3433.
Moreton P et al: Eradication of minimal residual disease in b-cell chronic lymphocytic leukemia after alemtuzumab therapy is associated with prolonged survival. J Clin Oncol 2005;23:2971.
Hairy Cell Leukemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Pancytopenia.
Splenomegaly, often massive.
Hairy cells present on blood smear and especially in bone marrow biopsy.
P.524
General Considerations
Hairy cell leukemia, an uncommon form of leukemia, is an indolent cancer of B lymphocytes.
Clinical Findings
A. Symptoms and Signs
The disease characteristically presents in middle-aged men. The median age at presentation is 55 years, and there is a striking 5:1 male predominance. Most patients present with gradual onset of fatigue, others complain of symptoms related to markedly enlarged spleen, and some come to attention because of infection.
Splenomegaly is almost invariably present and may be massive. The liver is enlarged in 50% of cases; lymphadenopathy is uncommon.
Hairy cell leukemia is usually an indolent disorder whose course is dominated by pancytopenia and recurrent infections, including mycobacterial infections.
B. Laboratory Findings
The hallmark of hairy cell leukemia is pancytopenia. Anemia is nearly universal, and 75% of patients have thrombocytopenia and neutropenia. Nearly all patients have striking monocytopenia, which is encountered in almost no other condition. The hairy cells are usually present in small numbers on the peripheral blood smear and have a characteristic appearance with numerous cytoplasmic projections. The bone marrow is usually inaspirable (dry tap), and the diagnosis is made by characteristic morphology on bone marrow biopsy. The hairy cells have a characteristic histochemical staining pattern, with tartrate-resistant acid phosphatase (TRAP). On immunophenotyping, the cells coexpress the antigens CD11c and CD22. Pathologic examination of the spleen shows marked infiltration of the red pulp with hairy cells. This is in contrast to the usual predilection of lymphomas to involve the white pulp of the spleen.
Differential Diagnosis
Hairy cell leukemia should be distinguished from other lymphoproliferative diseases such as Waldenstr m's macroglobulinemia and non-Hodgkin's lymphomas. It also may be confused with other causes of pancytopenia, including hypersplenism due to any cause, aplastic anemia, and paroxysmal nocturnal hemoglobinuria.
Treatment
The treatment of choice is cladribine (2-chlorodeoxyadenosine; CdA), 0.14 mg/kg daily for 7 days. This is a relatively nontoxic drug that produces benefit in 95% of cases and complete remission in more than 80%. Responses are long-lasting, with few patients relapsing in the first few years. Treatment with pentostatin produces similar results, but that drug is more cumbersome to administer.
Course & Prognosis
The development of new therapies has changed the prognosis of this disease. Formerly, median survival was 6 years, and only one-third of patients survived longer than 10 years. It now appears that more than 90% of patients with hairy cell leukemia will live longer than 10 years.
Chadha P et al: Treatment of hairy cell leukemia with 2-chlorodeoxyadenosine (2-CdA): long-term follow-up of the Northwestern University experience. Blood 2005;106:241.
Jehn U et al: An update: 12-year follow-up of patients with hairy cell leukemia following treatment with 2-chlorodeoxyadenosine. Leukemia 2004;18:1476.
Robak T: Monoclonal antibodies in the treatment of chronic lymphoid leukemias. Leuk Lymphoma 2004;45:205.
Lymphomas
Non-Hodgkin'S Lymphomas
The non-Hodgkin's lymphomas are a heterogeneous group of cancers of lymphocytes. The disorders vary in clinical presentation and course from indolent to rapidly progressive.
Molecular biology has provided clues to the pathogenesis of these disorders. The best-studied example is Burkitt's lymphoma, in which a characteristic cytogenetic abnormality of translocation between the long arms of chromosomes 8 and 14 has been identified. The protooncogene c-myc is translocated from its normal position on chromosome 8 to the heavy chain locus on chromosome 14. Cells committed to B cell differentiation are likely to have enhanced expression of this heavy chain locus, and it is likely that overexpression of c-myc (in its new anomalous position) is related to malignant transformation. In the follicular lymphomas, the t(14,18) translocation is characteristic and results in overexpression of bcl-2, resulting in protection against apoptosis, the usual mechanism of cell death.
Classification of the lymphomas is a controversial area still undergoing evolution. The most recent grouping (see Table 13-16) separates diseases based on both clinical and pathologic features.
Clinical Findings
A. Symptoms and Signs
Patients with indolent lymphomas usually present with painless lymphadenopathy, which may be isolated
P.525
or widespread. Involved lymph nodes may be present in the retroperitoneum, mesentery, and pelvis. The indolent lymphomas are usually disseminated at the time of diagnosis, and bone marrow involvement is frequent. Patients with intermediate and high-grade lymphomas also have constitutional symptoms such as fever, drenching night sweats, or weight loss.
Table 13-16. World Health Organization proposed classification of non-Hodgkin's lymphomas. | |
|---|---|
|
On examination, lymphadenopathy may be isolated, or extranodal sites of disease (skin, gastrointestinal tract) may be found. Patients with Burkitt's lymphoma are noted to have abdominal pain or abdominal fullness because of the predilection of the disease for the abdomen.
Once a pathologic diagnosis is established, the patient is staged. Chest radiograph and CT scan of the abdomen and pelvis, bone marrow biopsy, and lumbar puncture (in selected cases with high-risk morphology) are performed.
B. Laboratory Findings
The peripheral blood is usually normal, but a number of lymphomas may present in a leukemic phase.
Bone marrow involvement is manifested as paratrabecular lymphoid aggregates. In some high-grade lymphomas, the meninges are involved and malignant cells are found with cerebrospinal fluid cytology. The chest radiograph may show a mediastinal mass in lymphoblastic lymphoma. The serum LDH has been shown to be a useful prognostic marker and is now incorporated in risk stratification of treatment.
The diagnosis of lymphoma is made by tissue biopsy. Needle aspiration may yield suspicious results, but a lymph node biopsy (or biopsy of involved extranodal tissue) is required for diagnosis and staging.
Molecular profiling based on the examination of gene expression may lead to a new classification of the lymphomas.
Treatment
The treatment of indolent lymphoma depends on the stage of disease and the clinical status of the patient. A small number of patients have limited disease with only one abnormal lymph node and may be treated with localized irradiation with curative intent. Most patients with indolent lymphoma have disseminated disease at the time of diagnosis. If the disease is not bulky and the patient not symptomatic, no initial therapy may be required. Some patients will have spontaneous remissions and may defer treatment for 1 3 years. There are an increasing number of reasonable treatment options for low-grade lymphomas, but no clear consensus has emerged on the best strategy. Treatment with the anti-CD20 antibody rituximab is a commonly used treatment because of its very low toxicity and avoidance of chemotherapy. Combinations of rituximab with chemotherapy may also be used. Radioimmunoconjugates that fuse anti-B cell antibodies with radiation may produce improved results with modest increases in toxicity compared with antibody alone, and one such agent (yttrium-90 ibritumomab tiuxetan) is in use. Some patients with clinically aggressive low-grade lymphomas may be appropriate candidates for allogeneic transplantation. As in other hematologic malignancies, the use of less toxic nonmyeloablative regimens for allogeneic transplant may expand the role of transplant in this disease. The role of autologous transplantation for follicular lymphoma remains uncertain, but some patients with recurrent disease appear to have prolonged remissions.
Patients with intermediate-grade lymphomas such as diffuse large cell lymphoma are treated with curative intent. Those with localized disease receive either short-course chemo-immunotherapy (such as three courses of rituximab, cyclophosphamide, doxorubicin [hydroxydaunomycin; Adriamycin], vincristine [Oncovin], and prednisone [R-CHOP]) plus localized radiation or six courses of chemotherapy without radiation. Most patients who have more advanced disease are treated with six to eight cycles of chemotherapy such as R-CHOP. Individuals with very high-risk lymphoma are best treated with autologous stem cell transplantation early in the course. Patients with intermediate-grade lymphoma who relapse after initial chemotherapy may still be cured by autologous stem cell transplantation if their disease remains responsive to chemotherapy.
Persons with special forms of lymphoma require individualized therapy. Burkitt's lymphoma is treated with intensive regimens specifically tailored for this histologic type. Those with lymphoblastic lymphoma receive regimens similar to those used for T cell ALL. Mantle cell lymphoma is not effectively treated with standard chemotherapy regimens. Intensive initial
P.526
therapy including autologous stem cell transplantation has been shown to improve outcomes and is now the standard of care. Patients with mucosal associated lymphoid tumors (MALT lymphomas) of the stomach may be appropriately treated with combination antibiotics directed against Helicobacter pylori but require frequent endoscopic monitoring.
Prognosis
The median survival of patients with indolent lymphomas has been 6 8 years, but this appears to be changing for the better. These diseases ultimately become refractory to chemotherapy. This often occurs at the time of histologic progression of the disease to a more aggressive form of lymphoma.
The International Prognostic Index is now widely used to categorize patients with intermediate-grade lymphoma into risk groups. Factors that confer adverse prognosis are age over 60 years, elevated serum LDH, stage III or stage IV disease, and poor performance status. Patients with no risk factors or one risk factor have high complete response rates (80%) to standard chemotherapy, and most responses (80%) are durable. Patients with two risk factors have a 70% complete response rate, 70% being long-lasting. Patients with higher-risk disease have lower response rates and poor survival with standard regimens, and alternative treatments are needed. Early treatment with high-dose therapy and autologous stem cell transplantation improves the outcome.
For patients who relapse after initial chemotherapy, the prognosis depends on whether the lymphoma is still partially sensitive to chemotherapy. If it is, autologous transplantation offers a 50% chance of long-term salvage.
The treatment of older patients with lymphoma has been difficult because of poorer tolerance of aggressive chemotherapy. The use of myeloid growth factors and prophylactic antibiotics to reduce neutropenic complications may improve outcomes.
New techniques of molecular profiling using gene array technology are being studied to better define subsets of lymphomas with different biologic features and prognoses.
Abramson JS et al: Advances in the biology and therapy of diffuse large B-cell lymphoma: moving toward a molecularly targeted approach. Blood 2005;106:1164.
Dreyling M et al: Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 2005;105:2677.
Escalon MP et al: Nonmyeloablative allogeneic hematopoietic transplantation: a promising salvage therapy for patients with non-Hodgkin's lymphoma whose disease has failed a prior autologous transplantation. J Clin Oncol 2004;22:2419.
Feugier P et al: Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study for the Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 2005;23:4117.
Fisher RI et al: New treatment options have changed the survival of patients with follicular lymphoma. J Clin Oncol 2005; 23:8447.
Marcus R et al: CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood 2005;105:1417.
Milpied N et al; Groupe Ouest-Est des Leucemies et des Autres Maladies du Sang: Initial treatment of aggressive lymphoma with high-dose chemotherapy and autologous stem-cell support. N Engl J Med 2004;350:1287.
Sehn LH et al: Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. J Clin Oncol 2005; 23:5027.
Hodgkin's Disease
![]() Essentials of Diagnosis
Essentials of Diagnosis
Painless lymphadenopathy.
Constitutional symptoms may or may not be present.
Pathologic diagnosis by lymph node biopsy.
General Considerations
Hodgkin's disease is a group of cancers characterized by Reed-Sternberg cells in an appropriate reactive cellular background. The nature of the malignant cell is a subject of controversy.
Clinical Findings
There is a bimodal age distribution, with one peak in the 20s and a second over age 50 years. Most patients present because of a painless mass, commonly in the neck. Others may seek medical attention because of constitutional symptoms such as fever, weight loss, or drenching night sweats, or because of generalized pruritus. An unusual symptom of Hodgkin's disease is pain in an involved lymph node following alcohol ingestion.
An important feature of Hodgkin's disease is its tendency to arise within single lymph node areas and spread in an orderly fashion to contiguous areas of lymph nodes. Only late in the course of the disease will vascular invasion lead to widespread hematogenous dissemination.
Hodgkin's disease is divided into several subtypes: lymphocyte predominance, nodular sclerosis, mixed cellularity, and lymphocyte depletion. Hodgkin's disease should be distinguished pathologically from other malignant lymphomas and may occasionally be confused with reactive lymph nodes seen in infectious mononucleosis, cat-scratch disease, or drug reactions (eg, phenytoin).
P.527
Patients undergo a staging evaluation to determine the extent of disease. The staging nomenclature (Ann Arbor) is as follows: stage I, one lymph node region involved; stage II, involvement of two lymph node areas on one side of the diaphragm; stage III, lymph node regions involved on both sides of the diaphragm; and stage IV, disseminated disease with bone marrow or liver involvement. In addition, patients are designated stage A if they lack constitutional symptoms and stage B if 10% weight loss over 6 months, fever, or night sweats are present. If symptoms indicate careful evaluation for higher numerical stage, clinical stage IB (for example) is highly likely to emerge as stage II or stage IIIB.
Treatment
The treatment of Hodgkin's disease has evolved, with radiation therapy used as initial treatment only for patients with low-risk stage IA and IIA disease. Staging is usually clinical, and laparotomy is no longer routinely performed. The addition of limited chemotherapy for some patients treated with radiation appears promising.
Most patients with Hodgkin's disease (including all with stage IIIB and IV disease) are best treated with combination chemotherapy using doxorubicin (Adriamycin), bleomycin, vincristine, and dacarbazine (ABVD). New shorter and more intensive regimens have produced promising results and may supplant ABVD in the treatment of advanced disease.
Prognosis
All patients with both localized and disseminated disease should be treated with curative intent. The prognosis of patients with stage IA or IIA disease treated by radiotherapy is excellent, with 10-year survival rates in excess of 80%. Patients with disseminated disease (IIIB, IV) have 5-year survival rates of 50 60%. Poorer results are seen in patients who are older, those who have bulky disease, and those with lymphocyte depletion or mixed cellularity on histologic examination. Others whose disease recurs after initial radiotherapy treatment may still be curable with chemotherapy. The treatment of choice for patients who relapse after initial chemotherapy is high-dose chemotherapy with autologous stem cell transplantation. This offers a 35 50% chance of cure when disease is still chemotherapy sensitive.
Bonadonna G et al: ABVD plus subtotal nodal versus involved-field radiotherapy in early-stage Hodgkin's disease: long-term results. J Clin Oncol 2004;22:2835.
Engert A et al; German Hodgkin's Study Group: Hodgkin's lymphoma in elderly patients: a comprehensive retrospective analysis from the German Hodgkin's Study Group. J Clin Oncol 2005;23:5052.
Friedberg JW et al: FDG-PET is superior to gallium scintigraphy in staging and more sensitive in the follow-up of patients with de novo Hodgkin lymphoma: a blinded comparison. Leuk Lymphoma 2004;45:85.
le Maignan C et al: Three cycles of adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD) or epirubicin, bleomycin, vinblastine, and methotrexate (EBVM) plus extended field radiation therapy in early and intermediate Hodgkin disease: 10-year results of a randomized trial. Blood 2004;103:58.
Re D et al: From Hodgkin disease to Hodgkin lymphoma: biologic insights and therapeutic potential. Blood 2005;105:4553.
Multiple Myeloma
![]() Essentials of Diagnosis
Essentials of Diagnosis
Bone pain, often in the lower back.
Monoclonal paraprotein by serum and urine protein electrophoresis or immunoelectrophoresis.
Replacement of bone marrow by malignant plasma cells.
General Considerations
Multiple myeloma is a malignancy of plasma cells characterized by replacement of the bone marrow, bone destruction, and paraprotein formation. Myeloma causes clinical symptoms and signs through a variety of mechanisms.
Replacement of the bone marrow (and perhaps humoral suppression of myelopoiesis) leads initially to anemia and later to general bone marrow failure. Malignant plasma cells can form tumors (plasmacytomas) that may cause spinal cord compression. Bone involvement causes bone pain, osteoporosis, lytic lesions, pathologic fractures, and hypercalcemia. The pathogenesis of osteoclast activation in myeloma appears to involve osteoprotegerin ligand, and the decoy receptor osteoprotegerin may be able to interfere with this pathway.
The paraproteins secreted by the malignant plasma cells may cause problems in their own right. Very high paraprotein levels (either IgG or IgA) may cause hyperviscosity, though this is more often caused by IgM in Waldenstr m's macroglobulinemia. The light chain component of the immunoglobulin often leads to renal failure (often aggravated by hypercalcemia). Light chain components may be deposited in tissues as amyloid, worsening renal failure with albuminuria and causing a vast array of systemic symptoms.
Myeloma patients are prone to recurrent infections for a number of reasons, including neutropenia and the immunosuppressive effects of chemotherapy. More often, there is a failure of antibody production in response to antigen challenge, and myeloma patients are especially prone to infections with encapsulated organisms such as Streptococcus pneumoniae and Haemophilus influenzae.
Clinical Findings
A. Symptoms and Signs
Myeloma is a disease of older adults (median age at presentation, 65 years). The most common presenting
P.528
complaints are those related to anemia, bone pain, and infection. Bone pain is most common in the back or ribs or may present as a pathologic fracture, especially of the femoral neck. Patients may also come to medical attention because of renal failure, spinal cord compression, or the hyperviscosity syndrome (mucosal bleeding, vertigo, nausea, visual disturbances, alterations in mental status). Equally as often, patients are diagnosed because of laboratory findings of hypercalcemia, proteinuria, elevated sedimentation rate, or abnormalities on serum protein electrophoresis obtained for symptoms or in routine screening studies. A few patients come to medical attention because of amyloidosis.
Examination may reveal pallor, bone tenderness, and soft tissue masses. Patients may have neurologic signs related to neuropathy and spinal cord compression. Patients with amyloidosis may have an enlarged tongue, neuropathy, congestive heart failure, or hepatomegaly. Splenomegaly is absent unless amyloidosis is present. Fever occurs only with infection.
B. Laboratory Findings
Anemia is nearly universal. Red blood cell morphology is normal, but rouleau formation is common and may be marked. The neutrophil and platelet counts are usually normal at presentation. Only rarely will plasma cells be visible on peripheral smear (plasma cell leukemia).
The hallmark of myeloma is the finding of a paraprotein on serum protein electrophoresis (SPEP). The majority of patients will have a monoclonal spike visible in the - or -globulin region. Immunofixation (IF) will reveal this to be a monoclonal protein. Approximately 15% of patients will have no demonstrable paraprotein in the serum. In these, IF of the urine will reveal either complete immunoglobulin or light chains. Overall, approximately 60% of myeloma patients will have an IgG paraprotein, 25% an IgA, and 15% light chains only. In sporadic cases, no paraprotein is present ( nonsecretory myeloma ); these patients have particularly aggressive disease.
The bone marrow will be infiltrated by variable numbers of plasma cells ranging from 5% to 100%. The plasma cells will occasionally appear normal but more commonly are morphologically abnormal. Many benign processes can result in plasmacytosis, but the presence of highly atypical plasma cells or effacement of normal bone marrow elements helps to distinguish myeloma. Bone radiographs are important in establishing the diagnosis of myeloma. Lytic lesions are most commonly seen in the axial skeleton: skull, spine, proximal long bones, and ribs. At other times, only generalized osteoporosis is seen. The radionuclide bone scan is not useful in detecting bone lesions in myeloma, as there is usually no osteoblastic component. Positron emission tomography (PET) scans are being evaluated in the staging of myeloma, and may become routine.
The level of 2-microglobulin has strong prognostic significance in myeloma, with levels > 3 mg/L associated with poor survival. Bone marrow cytogenetic characteristics have greater prognostic significance, with deletions of chromosome 13q associated with a dismal outcome. Other laboratory features include hypercalcemia, renal failure, and an elevated erythrocyte sedimentation rate; alkaline phosphatase is not elevated despite extensive bony involvement. Some patients have proximal renal tubular acidosis, with phosphaturia, glycosuria, uricosuria, and aminoaciduria. The urinalysis may reveal proteinuria, but the dipstick test (which detects primarily albumin) is unreliable for light chains. Often there is a narrow anion gap when the paraprotein is cationic (70% of cases).
The standard staging system for multiple myeloma, the Salmon-Durie system, has been based on the level of paraprotein, blood counts, bone radiographs, and serum calcium. A new International Staging System has been proposed based on serum albumin and 2-microglobulin.
Differential Diagnosis
When a patient is discovered to have a monoclonal paraprotein, the distinction between myeloma and monoclonal gammopathy of unknown significance (MGUS) must be made. MGUS is present in 1% of all adults and 3% of adults over age 70 years. Thus, among all patients with paraproteins, MGUS is far more common than myeloma. Most commonly, patients with MGUS will have a monoclonal IgG spike less than 2.5 g/dL, and the height of the spike remains stable. In approximately 25% of cases, MGUS progresses to overt malignant disease, but this may take many years.
Myeloma is distinguished from MGUS by findings of replacement of the bone marrow, bone destruction, and progression. Although the height of the paraprotein spike should not be used by itself to distinguish benign from malignant disease, nearly all patients with IgG spikes greater than 3.5 g/dL prove to have myeloma; an IgA spike of > 2 g/dL is similarly suggestive. If there is doubt about whether paraproteinemia is benign or malignant, the patient should be observed without therapy, since there is no advantage to early treatment of asymptomatic multiple myeloma.
Myeloma must be distinguished from reactive polyclonal hypergammaglobulinemia. Myeloma may also be similar to other malignant lymphoproliferative diseases such as Waldenstr m's macroglobulinemia, lymphomas, and primary amyloidosis (with which it is commonly associated).
Treatment
Patients with minimal disease or in whom the diagnosis of malignancy is in doubt should be observed without treatment. Most commonly, patients require treatment at diagnosis because of bone pain or other symptoms related to the disease. The treatment of myeloma is rapidly changing, and the optimal initial treatment regimen is in flux. The most commonly used initial regimen is the combination of thalidomide
P.529
plus dexamethasone. This combination has been shown to be more effective than vincristine-doxorubicin-dexamethasone (VAD) chemotherapy for initial disease control and is more convenient since the agents are given orally, although prophylaxis against deep venous thrombosis is warranted and not all patients tolerate thalidomide. Other combinations including the nonchemotherapeutic agents bortezomib and lenalidomide are being explored. Bortezomib, available only intravenously, is a proteosome inhibitor and has significant activity in myeloma, either as a single agent or in combination. Lenalidomide, an oral agent, is a derivative of thalidomide with both improved efficacy and greatly reduced toxicity. However, both of these new agents are extremely expensive.
After initial disease control ( induction therapy ), the optimal consolidation therapy for patients under age 70 years with myeloma is autologous stem cell transplantation. Early aggressive treatment prolongs both duration of remission and overall survival. Clinical trials are now evaluating the role of posttransplant maintenance therapy with agents such as lenalidomide. For young patients with aggressive disease, autologous transplant followed by a reduced-intensity allogeneic transplant is under active investigation.
Allogeneic transplantation is potentially curative in myeloma, but its role has been limited because of the unusually high mortality rate (40 50%) in myeloma patients. Newer and less toxic forms of allogeneic transplantation using nonmyeloablative regimens have produced encouraging results, especially when performed early in the course of disease, such as the minimal disease state following autologous stem cell transplantation.
Localized radiotherapy may be useful for palliation of bone pain or for eradicating tumor at the site of pathologic fracture. Hypercalcemia should be treated aggressively and immobilization and dehydration avoided. The bisphosphonates (pamidronate 90 mg or zoledronic acid 4 mg intravenously monthly) reduce pathologic fractures in patients with significant bony disease and are an important adjunct in this subset of patients.
Prognosis
The median survival of patients with myeloma has been 3 years, but the outlook appears to be changing with new treatment approaches. The prognosis is markedly affected by a number of prognostic features, with shorter survivals in those with high paraprotein spikes, renal failure, hypercalcemia, or extensive bony disease. Patients are said to have a low tumor burden (stage I) if the IgG spike is less than 5 g/dL, there is no more than one lytic bone lesion, and there is no evidence of hypercalcemia or renal failure. Such patients have a median survival of 5 6 years. Conversely, patients with a high tumor burden (stage III) have an IgG spike greater than 7 g/dL, hematocrit less than 25%, calcium greater than 12 mg/dL, or more than three lytic bone lesions. Median survival for this group was formerly 1 2 years, but early intervention with autologous stem cell transplantation prolongs median survival to 5 6 years in this group. A new staging system that incorporates 2-microglobulin, serum albumin, and bone marrow cytogenetics has been proposed, and appears to predict outcome better than the traditional staging system. Broader use of immunotherapy with allogeneic transplantation may improve the outlook further, and it is likely that the new biologic agents will lead to additional therapeutic gains.
Barlogie B et al: Treatment of multiple myeloma. Blood 2004;103:20.
Cavo M et al: Superiority of thalidomide and dexamethasone over vincristine-doxorubicin-dexamethasone (VAD) as primary therapy in preparation for autologous transplantation for multiple myeloma. Blood 2005;106:35.
Crawley C et al: Outcome for reduced-intensity allogeneic transplantation for multiple myeloma: an analysis of prognostic factors from the Chronic Leukaemia Working Party of the EBMT. Blood 2005;105:4532.
Greipp PR et al: International staging system for multiple myeloma. J Clin Oncol 2005;23:3412.
Rajkumar SV et al: Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 2005;106:4050.
Richardson PG et al: Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005;352:2487.
Waldenstr M'S Macroglobulinemia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Symptoms nonspecific; splenomegaly common on examination.
Monoclonal IgM paraprotein.
Infiltration of bone marrow by plasmacytic lymphocytes.
Absence of lytic bone disease.
General Considerations
Waldenstr m's macroglobulinemia is a malignant disease of B cells that appear to be a hybrid of lymphocytes and plasma cells. These cells characteristically secrete an IgM paraprotein, and many clinical manifestations of the disease are related to this macroglobulin.
Clinical Findings
A. Symptoms and Signs
This disease characteristically develops insidiously in patients in their 60s or 70s. Patients usually present with fatigue related to anemia. Hyperviscosity of serum may be
P.530
manifested in a number of ways. Mucosal and gastrointestinal bleeding are related to engorged blood vessels and platelet dysfunction. Other complaints include nausea, vertigo, and visual disturbances. Alterations in consciousness vary from mild lethargy to stupor and coma. The IgM paraprotein may also cause symptoms of cold agglutinin disease or peripheral neuropathy.
On examination, there may be hepatosplenomegaly or lymphadenopathy. The retinal veins are engorged. Purpura may be present. There should be no bone tenderness.
B. Laboratory Findings
Anemia is nearly universal, and rouleau formation is common. The anemia is related in part to expansion of the plasma volume by 50 100% due to the presence of the paraprotein. Other blood counts are usually normal. The abnormal plasmacytic lymphocytes usually appear in small numbers on the peripheral blood smear. The bone marrow is characteristically infiltrated by the plasmacytic lymphocytes.
The hallmark of macroglobulinemia is the presence of a monoclonal IgM spike seen on SPEP in the - or -globulin region. The serum viscosity is usually increased above the normal of 1.4 1.8 times that of water. Symptoms of hyperviscosity usually develop when the serum viscosity is over four times that of water, and marked symptoms usually arise when the viscosity is over six times that of water. Because paraproteins vary in their physicochemical properties, there is no strict correlation between the concentration of paraprotein and serum viscosity.
The IgM paraprotein may cause a positive Coombs test or have cold agglutinin or cryoglobulin properties. If macroglobulinemia is suspected but the SPEP shows only hypogammaglobulinemia, the test should be repeated while taking special measures to maintain the blood at 37 C, since the paraprotein may precipitate out at room temperature if it is cryoprecipitable.
Bone radiographs are normal, and there is no evidence of renal failure.
Differential Diagnosis
Waldenstr m's macroglobulinemia is differentiated from monoclonal gammopathy of unknown significance by the finding of bone marrow infiltration. It is distinguished from chronic lymphocytic leukemia and multiple myeloma by bone marrow morphology and the finding of the characteristic IgM spike, and also on clinical grounds.
Treatment
Patients with marked hyperviscosity syndrome (stupor or coma) should be treated on an emergency basis with plasmapheresis. On a chronic basis, some patients can be managed with periodic plasmapheresis alone. As with other indolent lymphoid diseases, fludarabine and rituximab have replaced alkylator-based chemotherapy for initial therapy.
As with multiple myeloma, autologous stem cell transplantation is playing a more important role in management and is considered in younger patients with more aggressive disease.
Prognosis
Waldenstr m's macroglobulinemia is an indolent disease with a median survival rate of 3 5 years. However, patients may survive 10 years or longer.
Dimopoulos MA et al: Diagnosis and management of Waldenstr m's macroglobulinemia. J Clin Oncol 2005;23:1564.
Kyle RA et al: Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood 2003;102: 3759.
Mitsiades CS et al: Novel biologically based therapies for Waldenstr m's macroglobulinemia. Semin Oncol 2003;30: 309.
Munshi NC et al: Role for high-dose therapy with autologous hematopoietic stem cell support in Waldenstrom's macroglobulinemia. Semin Oncol 2003;30:282.
Disorders Of Hemostasis
Disorders of hemostasis may be due to defects in either platelet number or function or to problems in formation of a fibrin clot (coagulation). Bleeding due to platelet disorders is typically mucosal or dermatologic. Common problems include epistaxis, gum bleeding, menorrhagia, gastrointestinal bleeding, purpura, and petechiae. Petechiae are seen almost exclusively in conditions of thrombocytopenia and not platelet dysfunction. Bleeding due to coagulopathy may occur as deep muscle hematomas as well as skin bleeding. Spontaneous hemarthroses are seen only in severe hemophilia.
Idiopathic (Autoimmune) Thrombocytopenic Purpura
![]() Essentials of Diagnosis
Essentials of Diagnosis
Isolated thrombocytopenia.
Other hematopoietic cell lines normal.
No systemic illness.
Spleen not palpable.
Normal bone marrow with normal or increased megakaryocytes.
General Considerations
Idiopathic thrombocytopenic purpura is an autoimmune disorder in which an IgG autoantibody is formed that binds to platelets. It is not clear which antigen
P.531
on the platelet surface is involved. Although the antiplatelet antibody may bind complement, platelets are not destroyed by direct lysis. Rather, destruction takes place in the spleen, where splenic macrophages with Fc receptors bind to antibody-coated platelets. Since the spleen is the major site both of antibody production and platelet sequestration, splenectomy is highly effective therapy.
Clinical Findings
A. Symptoms and Signs
Idiopathic thrombocytopenic purpura occurs commonly in childhood, frequently precipitated by viral infection and usually self-limited. In contrast, the adult form is usually a chronic disease and only infrequently follows a viral infection. It is a disease of young persons, with peak incidence between ages 20 and 50 years, and there is a 2:1 female predominance.
Patients are systemically well and usually not febrile. The presenting complaint is mucosal or skin bleeding. Common types of bleeding are epistaxis, oral bleeding, menorrhagia, purpura, and petechiae.
On examination, the patient appears well, and there are no abnormal findings other than those related to bleeding. An enlarged spleen should lead one to doubt the diagnosis. Common signs of bleeding are purpura, petechiae, and hemorrhagic bullae in the mouth.
B. Laboratory Findings
The hallmark of the disease is thrombocytopenia, with platelet counts that may be less than 10,000/mcL. Other counts are usually normal except for occasional mild anemia, which can be explained by bleeding or associated hemolysis. Peripheral blood cell morphology is normal except that platelets are slightly enlarged (megathrombocytes). These larger platelets are young platelets produced in response to enhanced platelet destruction. Approximately 10% of patients will have coexistent autoimmune hemolytic anemia (Evans's syndrome), and in these cases anemia, reticulocytosis, and spherocytes are seen on peripheral smear. Red blood cell fragmentation should not be seen.
The bone marrow will appear normal, with a normal or increased number of megakaryocytes. Coagulation studies will be entirely normal.
Differential Diagnosis
Thrombocytopenia may be produced either by abnormal bone marrow function or by peripheral destruction (Table 13-17). Although most bone marrow disorders produce abnormalities in addition to isolated thrombocytopenia, diagnoses such as myelodysplasia can be excluded only by examining the bone marrow. Most causes of thrombocytopenia resulting from peripheral destruction can be ruled out by initial evaluation. Disorders such as DIC, TTP, HUS, hypersplenism, and sepsis are easily excluded by the absence of systemic illness. Thus, patients with isolated thrombocytopenia with no other abnormal findings almost certainly have immune thrombocytopenia. Patients should be questioned regarding drug use, especially sulfonamides, quinine, thiazides, cimetidine, gold, and heparin. Heparin is now the most common cause of drug-induced thrombocytopenia in hospitalized patients. Systemic lupus erythematosus and CLL are common causes of secondary thrombocytopenic purpura, hematologically identical to idiopathic thrombocytopenic purpura.
Table 13-17. Causes of thrombocytopenia. | ||
|---|---|---|
|
Treatment
Few adults with idiopathic thrombocytopenic purpura will have spontaneous remissions, and most will require treatment. Initial treatment is with prednisone, 1 2 mg/kg/d. Prednisone works primarily by decreasing the affinity of splenic macrophages for antibody-coated platelets. High-dose prednisone therapy also reduces the binding of antibody to the platelet surface, and long-term therapy may decrease antibody production. Bleeding will often diminish within 1 day after beginning prednisone even before the platelet count begins to rise. This effect has been attributed to enhanced vascular stability. The platelet count will usually begin to rise within a week, and responses are almost always seen within 3 weeks. About 80% of patients will respond, and the platelet count will usually return to normal. High-dose therapy should be continued until the platelet count is normal, and the dose should then be gradually tapered. In most, thrombocytopenia will recur if prednisone is completely withdrawn, and the aim is to find a dose that
P.532
will maintain an adequate platelet count. It is not necessary for the platelet count to be entirely normal; the risk of bleeding is small with platelet counts above 50,000/mcL. An alternative steroid regimen is the use of high-dose dexamethasone, 40 mg/d for 4 days.
Splenectomy is the most definitive treatment for idiopathic thrombocytopenic purpura, and most adult patients will ultimately undergo splenectomy. High-dose prednisone therapy should not be continued indefinitely in an attempt to avoid surgery. Splenectomy is indicated if patients do not respond to prednisone initially or require unacceptably high doses to maintain an adequate platelet count. Other patients may be intolerant of prednisone or may simply prefer the surgical alternative. Splenectomy can be performed safely even with platelet counts less than 10,000/mcL. Eighty percent of patients benefit from splenectomy with either complete or partial remission.
High-dose intravenous immunoglobulin, 1 g/kg for 1 or 2 days, is highly effective in rapidly raising the platelet count. The response rate is 90%, and the platelet count rises within 1 5 days. However, this treatment is expensive, and the beneficial effect lasts only 1 2 weeks. Immunoglobulin treatment should be reserved for bleeding emergencies or situations such as preparing a severely thrombocytopenic patient for surgery.
For patients who fail to respond to prednisone and splenectomy, danazol, 600 mg/d, has been used, with responses obtained in about 50% of cases. Immunosuppressive agents employed in refractory cases include vincristine, azathioprine, cyclosporine, and cyclophosphamide. Rituximab can produce good responses in some patients with refractory disease. Rare patients with severe and refractory disease are now being treated with high-dose immunosuppression and autologous stem cell transplantation.
Platelet transfusions are rarely used in the treatment of idiopathic thrombocytopenic purpura, since exogenous platelets will survive no better than the patient's own and will usually survive less than a few hours. Platelet transfusion should be reserved for cases of life-threatening bleeding in which even fleeting hemostasis may be of benefit.
Prognosis
The prognosis for remission is good. In most cases, the disease is initially controlled with prednisone, and splenectomy offers definitive therapy. The major concern during the initial phases is cerebral hemorrhage, which becomes a risk when the platelet count is less than 5000/mcL. These patients usually exhibit warning signs of mucosal bleeding. However, even at these very low platelet counts, fatal bleeding is rare.
Cines DB et al: How I treat idiopathic thrombocytopenic purpura (ITP). Blood 2005;106:2244.
Kojouri K et al: Splenectomy for adult patients with idiopathic thrombocytopenia purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004;104:2623.
Vesely SK et al: Management of adult patients with persistent idiopathic thrombocytopenic purpura following splenectomy: a systematic review. Ann Intern Med 2004;140:112.
Heparin-Induced Thrombocytopenia
![]() Essentials of Diagnosis
Essentials of Diagnosis
Thrombocytopenia occurring during heparin use.
Positive test for antibodies to platelet factor 4, usually in a complex with heparin.
Arterial and venous thrombosis.
General Considerations
Heparin-induced thrombocytopenia (HIT) is the most common type of drug-induced thrombocytopenia, and one of the most common causes of thrombocytopenia in hospitalized patients. It is caused by an IgG autoantibody that reacts with platelet factor 4 (PF4) on the platelet surface, usually in a complex with heparin. The risk of HIT is significantly higher with the use of unfractionated heparin than with low-molecular-weight heparin and, for unclear reasons, is higher in surgical patients. The interaction of the antibody with PF4 is profoundly prothrombotic, possibly because of the release of platelet microparticles into the circulation.
Clinical Findings
HIT typically develops during heparin use, the risk rising after the first 4 days. Most often, the first finding is asymptomatic thrombocytopenia. However, both arterial and venous thromboses may occur. Laboratory testing can confirm the diagnosis, with the finding of HIT antibodies based on assays using either washed platelets or PF4 antigen.
Differential Diagnosis
HIT must be distinguished from other types of immune thrombocytopenia, including other drug-induced types. The HIT antibody assay is helpful in making this distinction.
Treatment
Once the HIT syndrome has been recognized, the heparin must be stopped. Because of the high risk of associated thrombosis and the high mortality rate of these events, alternative forms of anticoagulation are indicated. The direct thrombin inhibitors such as argatroban and lepirudin are effective.
P.533
Prognosis
The HIT syndrome is self-limited, and there is no anamnestic response to distant challenge with heparin. The key is the recognition of the syndrome and the discontinuation of heparin. However, the syndrome is serious, with a mortality rate of approximately 5% due to thrombosis.
Alving BM: How I treat heparin-induced thrombocytopenia and thrombosis. Blood 2003;101:31.
Di Nisio M et al: Direct thrombin inhibitors. N Engl J Med 2005;353:1028.
Hirsh J et al: Treatment of heparin-induced thrombocytopenia: a critical review. Arch Intern Med 2004;164:361.
Warkentin TE et al: Heparin-induced thrombocytopenia: recognition, treatment, and prevention: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004;126(3 Suppl):311S.
Thrombotic Thrombocytopenic Purpura
![]() Essentials of Diagnosis
Essentials of Diagnosis
Thrombocytopenia.
Microangiopathic hemolytic anemia.
Neurologic and renal abnormalities, fever.
Reduced level of ADAMTS13.
Normal coagulation tests.
Elevated serum LDH.
General Considerations
TTP is an uncommon syndrome with microangiopathic hemolytic anemia, thrombocytopenia, and a markedly elevated serum LDH. Noninfectious fever, neurologic disorders, and renal abnormalities are less commonly seen. The pathogenesis of sporadic adult TTP appears to involve a deficiency of a von Willebrand factor-cleaving protease, ADAMTS13, in some cases due to an antibody directed against the protease. In the absence of appropriate cleavage, ultralarge multimers of von Willebrand factor accumulate and lead to platelet agglutination and adhesion to endothelium. It is likely that the full expression of the syndrome involves an additional inciting factor.
TTP is seen primarily in young adults between ages 20 and 50 years, and there is a slight female predominance. The syndrome is occasionally precipitated by estrogen use, pregnancy, drugs, or infections. The most common drugs implicated are quinine and ticlopidine. The syndrome may also occur as a complication of bone marrow transplantation or the use of cyclosporine or tacrolimus. Familial cases occur, but are rare.
Clinical Findings
A. Symptoms and Signs
Patients come to medical attention because of anemia, bleeding, or neurologic abnormalities. The neurologic symptoms and signs are unusual in that they may wax and wane over minutes. Neurologic symptoms include headache, confusion, aphasia, and alterations in consciousness from lethargy to coma. With more advanced disease, hemiparesis and seizures may occur.
On examination, the patient appears acutely ill and is usually febrile. Pallor, purpura, petechiae, and signs of neurologic dysfunction may be detected. Patients may have abdominal pain and tenderness due to pancreatitis.
B. Laboratory Findings
Anemia is universal and may be marked. There is usually marked reticulocytosis and occasional circulating nucleated red blood cells. The hallmark is a microangiopathic blood picture with fragmented red blood cells (schistocytes, helmet cells, triangle forms) on the smear. The diagnosis cannot be made without significant red blood cell fragmentation. Thrombocytopenia is invariably present and may be severe.
Hemolysis may be manifested by increasing indirect bilirubin and occasionally hemoglobinemia and hemoglobinuria; methemalbuminemia may impart a brown color to the plasma. The LDH is markedly elevated in proportion to the severity of hemolysis; the Coombs test is negative.
Coagulation tests (prothrombin time, partial thromboplastin time, fibrinogen) are normal unless ischemic tissue damage causes secondary DIC. Elevated fibrin degradation products may be seen, as in other acutely ill patients. Renal insufficiency may be present, with an abnormal urinalysis. ADAMTS13 is usually absent during active disease. However, its absence should not deter diagnosis (and treatment) in a clinically compelling case, and its presence does not, by itself, define the syndrome.
Pathologically, there may be thrombi in capillaries and small arteries, with no evidence of inflammation.
Differential Diagnosis
The normal values of coagulation tests differentiate TTP from DIC. Other conditions causing microangiopathic hemolysis (Table 13-18) should be excluded. Evans's
P.534
syndrome is the combination of autoimmune thrombocytopenia and autoimmune hemolytic anemia, but the peripheral smear will show spherocytes and not red blood cell fragments. Skin biopsy is usually not necessary for diagnosis but may be helpful when vasculitis is a consideration. TTP and HUS are not distinct disease entities rather, there is a spectrum of disease, with TTP characterized by more neurologic findings and more severe thrombocytopenia and HUS with more renal failure.
Table 13-18. Causes of microangiopathic hemolytic anemia. | |
|---|---|
|
Treatment
TTP should be treated emergently with large-volume plasmapheresis. Sixty to 80 mL/kg of plasma should be removed and replaced with fresh-frozen plasma. Treatment should be continued daily until the patient is in complete remission. The optimal duration of plasmapheresis after remission is unknown. Prednisone and antiplatelet agents (aspirin [325 mg three times daily] and dipyridamole [75 mg three times daily]) have been used in addition to plasmapheresis, but their role is unclear.
The management of patients who do not respond to plasmapheresis or who have rapid recurrences is controversial. Increasing the volume or frequency of plasma exchange is often beneficial. The combination of splenectomy, corticosteroids, and dextran has been used with success. Splenectomy performed in remission may prevent subsequent relapses. Immunosuppression with drugs such as cyclophosphamide has also been effective.
Prognosis
With the advent of plasmapheresis, the formerly dismal prognosis of TTP has been dramatically changed; 80 90% of patients now recover completely. Neurologic abnormalities are almost always completely reversed. Most complete responses are durable, but in 20% of cases the disease will be chronic and relapsing.
Ahmad A et al: Rituximab for treatment of refractory/relapsing thrombotic thrombocytopenic purpura (TTP). Am J Hematol 2004;77:171.
Fakhouri F et al: Efficiency of curative and prophylactic treatment with rituximab in ADAMTS 13-deficient thrombotic thrombocytopenic purpura: a study of 11 cases. Blood 2005;106: 1932.
Knovich MA et al: Simplified assay for VWF cleaving protease (ADAMTS13) activity and inhibitor in plasma. Am J Hematol 2004;76:286.
Zheng XL et al: Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood 2004;103:4043.
Hemolytic-Uremic Syndrome
![]() Essentials of Diagnosis
Essentials of Diagnosis
Microangiopathic hemolytic anemia.
Thrombocytopenia and renal failure.
Elevated serum LDH.
Normal coagulation tests.
Absence of neurologic abnormalities.
General Considerations
HUS is an uncommon disorder consisting of microangiopathic hemolytic anemia, thrombocytopenia, and renal failure due to microangiopathy (with decreased glomerular filtration, proteinuria, and hematuria). The cause is unclear. The disease is similar to TTP except that different vascular beds are involved. In fact, the two diseases are probably best considered as part of a spectrum of HUS-TTP disorders. The pathogenesis of the two disorders is probably similar, and a platelet-agglutinating factor found in plasma may be involved. In children, HUS frequently occurs after a diarrheal illness secondary to infections with Shigella, Salmonella, E coli strain O157:H7, or viral agents. The mortality rate of this form is low (< 5%). In adults, this syndrome is often precipitated by estrogen use or by the postpartum state. HUS may be seen as a delayed complication of high-dose corticosteroid therapy and autologous bone marrow or stem cell transplantation, or of the use of cyclosporine or tacrolimus as immunosuppression in allogeneic transplantation. A familial (hereditary) type has been identified in which members of a family have recurrent episodes over several years.
Clinical Findings
A. Symptoms and Signs
Presenting symptoms include anemia, bleeding, or renal failure. The renal failure may or may not be oliguric. In contrast to TTP, there are no neurologic manifestations other than those due to the uremic state.
B. Laboratory Findings
As in TTP, there is microangiopathic hemolytic anemia and thrombocytopenia, but the thrombocytopenia is often less severe. The peripheral blood smear should show striking red blood cell fragmentation, and the diagnosis is untenable without this finding. The LDH is usually elevated out of proportion to the degree of hemolysis, and the Coombs test is negative. Coagulation tests are normal with the exception of elevated fibrin degradation products. As in TTP, levels of ADAMTS13 are usually low.
Kidney biopsy will show endothelial hyaline thrombi in the afferent arterioles and glomeruli. Ischemic necrosis in the renal cortex may occur with obstruction from intravascular coagulation.
Differential Diagnosis
DIC is excluded by normal coagulation results. Other causes of microangiopathic hemolytic anemia (Table 13-18)
P.535
should be entertained. Occasionally, vasculitis or acute glomerulonephritis is considered, and in these cases renal biopsy may be necessary to establish the diagnosis if the platelet count will allow it.
HUS is arbitrarily distinguished from TTP by the consistent presence of renal failure and the lack of neurologic findings.
Treatment
In children, HUS is almost always self-limited and requires only conservative management of acute renal failure. In adults, however, without treatment, there is a high rate of permanent renal insufficiency and death. The treatment of choice (as in TTP) is large-volume plasmapheresis with fresh-frozen replacement (exchange of up to 80 mL/kg), repeated daily until remission is achieved.
Prognosis
The prognosis of HUS in adults remains unclear. Without effective therapy, up to 40% of patients have died, and 80% have had chronic renal insufficiency. Early institution of aggressive therapy with plasmapheresis promises to be beneficial. Survival and correction of hematologic abnormalities are the rule, but restoration of renal function requires that treatment be initiated early.
Garg AX et al: Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: a systematic review, meta-analysis, and meta-regression. JAMA 2003;290:1360.
Vesely SK et al: ADAMTS13 activity in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients. Blood 2003;102:60.
Congenital Qualitative Platelet Disorders
Bleeding disorders characterized by prolonged bleeding times despite a normal platelet count are called qualitative platelet disorders. Patients have a family history or lifelong personal history of the defect. The disorders may be classified as (1) von Willebrand's disease, a congenital disorder of a plasma protein necessary for platelet adhesion, and (2) congenital disorders intrinsic to the platelet (Table 13-19). When an intrinsic qualitative platelet disorder is suspected, platelet aggregation studies should be evaluated to make a specific diagnosis.
Table 13-19. Qualitative platelet disorders. | |
|---|---|
|
1. von Willebrand's Disease
![]() Essentials of Diagnosis
Essentials of Diagnosis
Family history with autosomal dominant pattern of inheritance.
Prolonged bleeding time, either at baseline or after challenge with aspirin.
Reduced levels of factor VIII antigen or ristocetin cofactor.
Reduced levels of factor VIII coagulant activity in some patients.
General Considerations
von Willebrand's disease is the most common congenital disorder of hemostasis. It is transmitted in an autosomal dominant pattern. It is a group of disorders characterized by deficient or defective von Willebrand factor (vWF), a protein that mediates platelet adhesion. Adhesion is a process separate from platelet aggregation. Platelets adhere to the subendothelium via vWF, which is bound to a specific receptor on the platelet composed of glycoprotein Ib (and missing in Bernard-Soulier syndrome). Platelets aggregate via fibrinogen, which binds to a different receptor composed of glycoproteins IIb and IIIa (deficient in Glanzmann's thrombasthenia). The platelet aggregation system is entirely normal in von Willebrand's disease.
vWF is synthesized in megakaryocytes and endothelial cells and circulates in plasma as multimers of varying size. Only the large multimeric forms are functional in mediating platelet adhesion. vWF has a separate function of binding the factor VIII coagulant protein and protecting it from degradation. The factor VIII coagulant protein (factor VIII:C), a protein encoded by a gene on the X chromosome, is the protein deficient in classic hemophilia. Any of the multimeric forms of vWF can bind and protect factor VIII:C. von Willebrand's disease, although primarily a disorder of platelet function, may secondarily cause a coagulation disturbance because of deficient levels of factor VIII:C. However, this coagulopathy is rarely severe.
There are several subtypes of von Willebrand's disease. The most common type (type I, 80% of all cases) is caused by a quantitative decrease in vWF. Type IIa is
P.536
caused by a qualitative abnormality in protein that prevents multimer formation. Only small multimers are present, and both intermediate and large forms that mediate platelet adhesion are missing. Type IIb von Willebrand's disease is caused by a qualitative abnormality in the protein that causes rapid clearance of the large multimeric forms. Type III von Willebrand's disease is a rare autosomal recessive disorder in which vWF is nearly absent. Pseudo-von Willebrand disease is a rare disorder manifested as an abnormal platelet membrane with excessive avidity for the large multimeric forms of vWF, causing their clearance from plasma.
Clinical Findings
A. Symptoms and Signs
von Willebrand's disease is a common disorder affecting both men and women. Most cases are mild. Most bleeding is mucosal (epistaxis, gingival bleeding, menorrhagia), but gastrointestinal bleeding may occur. In most cases, incisional bleeding occurs after surgery or dental extractions. von Willebrand's disease is rarely as severe as hemophilia, and spontaneous hemarthroses do not occur (except in the rare type III). The bleeding tendency is exacerbated by aspirin. Characteristically, bleeding decreases during pregnancy or estrogen use.
B. Laboratory Findings
Platelet number and morphology are normal, and the bleeding time is usually (not always) prolonged. The bleeding time should be ascertained whenever this diagnosis is considered; it correlates most closely with clinical bleeding. When the bleeding time is normal, it is prolonged markedly by aspirin. Normal persons will prolong their bleeding time to a minor extent with aspirin but rarely out of the normal range. In the most common form of von Willebrand's disease (type I), vWF levels in plasma are reduced. This may be measured by factor VIII antigen, which measures the immunologic presence of vWF, or by ristocetin cofactor activity, which measures functional properties of vWF in mediating platelet adhesion.
When factor VIII antigen is reduced, factor VIII coagulant (factor VIII:C) levels may also decrease. When factor VIII:C levels are less than 25%, the partial thromboplastin time (PTT) will be prolonged. Platelet aggregation studies with standard agonists (adenosine diphosphate [ADP], collagen, thrombin) are normal, but platelet aggregation in response to ristocetin may be subnormal.
In difficult cases, it may be helpful to assay directly the multimeric composition of vWF.
Differential Diagnosis
When patients present with a prolonged bleeding time, von Willebrand's disease must be distinguished from other qualitative platelet disorders (Table 13-19). Acquired qualitative disorders are suggested by recent onset of the bleeding tendency. Congenital intrinsic platelet disorders may present with a positive family history and lifelong history of bleeding episodes. von Willebrand's disease is diagnosed by the finding of abnormal measurements of vWF and by normal results of platelet aggregation.
Table 13-20. Causes of prolonged partial thromboplastin time. | |
|---|---|
|
When patients present with a prolonged PTT, measurements of factor VIII:C will distinguish von Willebrand's disease from all disorders except hemophilia (Table 13-20). Hemophilia is diagnosed when factor VIII:C is reduced but all measurements of vWF (factor VIII antigen, ristocetin cofactor activity) are normal.
Patients with a suspicious bleeding history but with normal bleeding time and PTT pose a diagnostic problem. On occasion, the postaspirin bleeding time can be used to unmask a bleeding disorder. At other times, further plasma assays of vWF must be performed to make the diagnosis. von Willebrand's disease waxes and wanes in severity and may be difficult to diagnose, especially in a woman taking estrogens, which raise vWF levels.
It is often useful to distinguish between subtypes of von Willebrand's disease (Table 13-21), because type I usually responds to desmopressin and type IIb may be aggravated by its use.
Treatment
The bleeding disorder is characteristically mild, and no treatment is routinely given other than avoidance of aspirin. However, patients often need to be prepared for surgical or dental procedures. The bleeding time is probably the best indicator of the likelihood of bleeding, and prophylactic therapy may be reasonably withheld if the procedure is minor and the bleeding time is normal.
Desmopressin acetate (DDAVP) is useful for mild type I von Willebrand's disease and should be considered first. The dose is 0.3 mcg/kg, after which vWF levels usually rise twofold to threefold in 30 90 minutes. It can also be given as a nasal spray; levels peak 2 hours after use. DDAVP appears to cause release of stored vWF from endothelial cells. The treatment can be given
P.537
only every 24 hours as stores of vWF become depleted. It is best to give a therapeutic trial of DDAVP before relying on it for effective hemostasis during surgery or procedures. The drug is not effective in type IIa von Willebrand's disease, in which no endothelial stores are present, and may be harmful in type IIb, leading to thrombocytopenia and increased bleeding.
Table 13-21. Types of von Willebrand's disease. | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
Factor VIII concentrates are available that have replaced cryoprecipitate as the treatment of choice for von Willebrand's disease if factor replacement is required. Some (not all) of these products now contain functional vWF and do not transmit HIV or hepatitis. One appropriate product is Humate-P (Armour). The dose is 20 50 units/kg depending on disease severity.
The antifibrinolytic agent -aminocaproic acid (EACA) is useful as adjunctive therapy during dental procedures. After DDAVP, the patient is given 4 g every 4 hours to reduce the likelihood of bleeding.
Prognosis
The prognosis is excellent. In most cases, the bleeding disorder is mild, and in the more serious cases replacement therapy is effective.
Federici AB et al: Biologic response to desmopressin in patients with severe type 1 and type 2 von Willebrand disease: results of a multicenter European study. Blood 2004;103:2032.
Laffan M et al: The diagnosis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors' Organization. Haemophilia 2004;10:199.
Sadler JE et al: Von Willebrand disease type 1: a diagnosis in search of a disease. Blood 2003;101:2089.
Vincentelli A et al: Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med 2003;349:343.
2. Disorders Intrinsic to the Platelets
Glanzmann's Thrombasthenia
This is a rare autosomal recessive intrinsic platelet disorder causing bleeding. Platelets are unable to aggregate because of lack of receptors (containing glycoproteins IIb and IIIa) for fibrinogen, which form the bridges between platelets during aggregation. Clinically, it is manifested chiefly as mucosal (epistaxis, gingival bleeding, menorrhagia) and postoperative bleeding. The defect is of variable severity but may be severe.
P.538
Platelet numbers and morphology are normal, but the bleeding time is markedly prolonged. Platelets fail to aggregate in response to typical agonists (ADP, collagen, thrombin) but aggregate normally in response to ristocetin, which causes platelet clumping by a separate mechanism.
Patients are treated with platelet transfusions when necessary. Platelet transfusion therapy is limited by the tendency of these patients to develop multiple alloantibodies.
Bernard-Soulier Syndrome
This is a rare autosomal recessive intrinsic platelet disorder causing bleeding. Platelets cannot adhere to subendothelium because they lack receptors (composed of glycoprotein Ib) for von Willebrand factor, which mediates platelet adhesion. This is often a severe bleeding disorder with mucosal and postoperative bleeding.
Thrombocytopenia may be present, and platelets on smear are abnormally large. The bleeding time is markedly prolonged. Platelet aggregation is normal in response to standard agonists (collagen, ADP, thrombin), but platelets fail to aggregate in response to ristocetin. Measurements of von Willebrand factor in the plasma are normal. Patients are treated with platelet transfusion when necessary.
Storage Pool Disease
This is a group of mild bleeding disorders characterized by defective secretion of platelet granule contents (especially ADP) that stimulates platelet aggregation. Most patients are mildly affected and have increased bruising and postoperative bleeding.
Platelets are normal in number and morphology, but the bleeding time is slightly prolonged. In some cases, the baseline bleeding time is normal, but it becomes markedly prolonged after aspirin. There are variable abnormalities in platelet aggregation studies.
Most patients do not require treatment but should avoid aspirin. Platelet transfusions transiently correct the bleeding tendency. Some patients respond to DDAVP, 0.3 mcg/kg every 24 hours.
Beguin S et al: Fibrin polymerization is crucial for thrombin generation in platelet-rich plasma in a VWF-GPIb-dependent process, defective in Bernard-Soulier syndrome. J Thromb Haemost 2004;2:170.
Toogeh G et al: Presentation and pattern of symptoms in 382 patients with Glanzmann thrombasthenia in Iran. Am J Hematol 2004;77:198.
Acquired Qualitative Platelet Disorders
A number of acquired disorders lead to abnormal platelet function (Table 13-19).
Uremia
Uremia causes abnormal platelet function by unknown mechanisms. The severity of the bleeding tendency is roughly proportionate to the degree of renal insufficiency. Bleeding is most commonly mucosal and gastrointestinal and may occasionally be severe. Dialysis is effective in reducing the bleeding tendency but may not completely eliminate it. Patients respond to DDAVP, 0.3 mcg/kg every 24 hours.
Myeloproliferative Disorders
All the myeloproliferative disorders can produce abnormalities in platelet function. A number of biochemical abnormalities are present in these platelets, but the cause of the bleeding tendency is unclear. The severity of the bleeding tendency correlates roughly with the height of the platelet count, although conditions causing reactive thrombocytosis of normal platelets are not associated with abnormal function. Bleeding decreases when the platelet count is controlled with myelosuppressive therapy. In cases of life-threatening bleeding with high platelet counts, plateletpheresis may be necessary.
Other Disorders
Aspirin causes a mild bleeding tendency by irreversibly acetylating cyclooxygenase, an enzyme that participates in platelet aggregation. The effect lasts for the life of the platelet and may be manifest for 7 10 days, although the major effect lasts 3 5 days. The effect is not dose dependent, and 65 mg of aspirin is sufficient.
Aspirin by itself does not cause significant bleeding, but it may unmask bleeding disorders such as mild von Willebrand's disease or mild thrombocytopenia. Certain antibiotics (ticarcillin, some cephalosporins) cause a mild bleeding tendency, presumably by coating the surface of platelets. Nonsteroidal anti-inflammatory drugs cause an aspirin-like effect that disappears when the drug leaves the system.
Patients with autoantibodies against platelets may have prolonged bleeding times even in the absence of thrombocytopenia. Platelet-associated IgG levels should be high, and the bleeding tendency responds quickly to modest doses of prednisone, eg, 20 mg/d. Acquired storage pool disease refers to the circulation of exhausted platelets that have been stimulated to release their granule contents and hence are no longer functional. Such granule release occurs in response to cardiopulmonary bypass and severe vasculitis.
Noris M et al: Uremic bleeding: closing the circle after 30 years of controversies? Blood 1999;94:2569.
Hemophilia A
![]() Essentials of Diagnosis
Essentials of Diagnosis
X-linked recessive pattern of inheritance with only males affected.
Low factor VIII coagulant (VIII:C) activity.
Normal factor VIII antigen.
Spontaneous hemarthroses.
General Considerations
Hemophilia A (classic hemophilia, factor VIII deficiency hemophilia) is a hereditary disorder in which bleeding is due to deficiency of the coagulation factor VIII (VIII:C). In most cases, the factor VIII coagulant protein is quantitatively reduced, but in a small number of cases the coagulant protein is present by immunoassay but defective.
Hemophilia is an X-linked recessive disease, and as a rule only males are affected. In rare instances, female carriers are clinically affected if their normal X chromosomes are disproportionately inactivated. Females may also become affected if they are the offspring of a hemophiliac father and carrier mother.
Hemophilia is classified as severe if factor VIII:C levels are less than 1%, moderate if levels are 1 5%, and mild if levels are greater than 5%. Families tend to breed true in the severity of hemophilia produced.
Clinical Findings
A. Symptoms and Signs
Hemophilia A is the most common severe bleeding disorder and after von Willebrand's disease is the most common congenital bleeding disorder overall. Approximately one in 10,000 males is affected. The bleeding tendency is related to factor VIII:C levels. Bleeding may occur anywhere.
P.539
The most common sites of bleeding are into joints (knees, ankles, elbows), into muscles, and from the gastrointestinal tract. Spontaneous hemarthroses are so characteristic of hemophilia that they are almost diagnostic of the disorder. Patients with mild hemophilia bleed only after major trauma or surgery, those with moderately severe hemophilia bleed with mild trauma or surgery, and those with severe disease bleed spontaneously.
Many hemophiliacs are now seropositive for HIV infection transmitted via factor VIII concentrate, and many already have AIDS. HIV-associated immune thrombocytopenia may aggravate the bleeding tendency.
B. Laboratory Findings
The PTT is prolonged, and other measures of coagulation, including prothrombin time, bleeding time, and fibrinogen level, are normal. Levels of factor VIII:C are reduced, but measurements of vWF are normal (Table 13-21).
If plasma from a hemophiliac patient is mixed with normal plasma, the PTT will become normal. Failure of the PTT to normalize in such a mixing test is diagnostic of the presence of a factor VIII inhibitor.
A low platelet count should raise a suspicion of HIV-associated immune thrombocytopenia.
Differential Diagnosis
The finding of a reduced factor VIII:C level will distinguish this disorder from other causes of prolonged PTT (Table 13-20). Clinically, factor VIII hemophilia is indistinguishable from factor IX hemophilia, and only specific factor assays can distinguish these disorders. In cases of mild hemophilia, the disorder needs to be distinguished from von Willebrand's disease by VIII:A assay, which shows normal levels of factor VIII antigen in the former.
An important issue for the families of hemophiliac patients is identifying which females are carriers. They can usually be identified by the presence of low or normal levels of factor VIII:C with normal levels of factor VIII antigen.
Treatment
Standard treatment is based on infusion of factor VIII concentrates, either recombinant or heat treated. The level of factor VIII achieved in plasma depends on the severity of the bleeding problem. In response to minor bleeding, it may be necessary to raise factor VIII:C levels to only 25% with one infusion. For moderate bleeding (such as deep muscle hematomas), it is adequate to raise the level initially to 50% and maintain the level at greater than 25% with repeated infusion for 2 3 days. When major surgery is to be performed, the factor VIII:C level is raised to 100% and then the factor level is maintained at greater than 50% continuously for 10 14 days. Head injuries (with or without neurologic signs) should be emergently treated as though major bleeding were present.
The dose of factor VIII concentrate is calculated assuming that one unit of factor VIII is the amount present in 1 mL of plasma. Plasma volume is 40 mL/kg, and the volume of distribution of factor VIII:C is 1.5 times the plasma volume. Thus, to raise the level 100%, the dose should be 40 1.5 = 60 units/kg, or approximately 4000 units for a 70-kg individual. To raise the levels to 25% would require 1000 units. The half-life of factor VIII:C is approximately 12 hours. Thus, during major surgery, to achieve an initial level of 100% and maintain it continuously at greater than 50%, a dose of 60 units/kg (approximately 4000 units) initially followed by 30 units/kg (approximately 2000 units) every 12 hours should be adequate. During surgery, initially verify that these doses give the anticipated factor VIII levels. If factor VIII levels fail to rise as expected, an inhibitor should be suspected.
For mild hemophiliacs, DDAVP, 0.3 mcg/kg every 24 hours, may be useful in preparing for minor surgical procedures. It causes release of factor VIII:C and will raise the factor VIII:C levels two- to threefold for several hours. In the management of persistent bleeding following use of either desmopressin acetate or factor VIII concentrate, patients may be treated with EACA (Amicar), 4 g orally every 4 hours for several days.
Aspirin should be avoided in these patients.
Prognosis
The prognosis of patients with hemophilia has been transformed by the availability of factor VIII replacement. Gene therapy is currently in the developmental phase, but could further transform the outlook for these patients. The major limiting factor is disability from recurrent joint bleeding. Hepatitis B and C and HIV infection from recurrent transfusion are diminishing in incidence. Approximately 15% of patients develop inhibitors to factor VIII, and these patents cannot be adequately supported with factor VIII.
Goudemand J et al: Influence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood 2006; 107:46.
Plug I et al: Thirty years of hemophilia treatment in the Netherlands, 1972 2001. Blood 2004;104:3494.
Acquired Factor Viii Antibodies
Antibodies to factor VIII may develop either postpartum or with no underlying illness. Factor VIII antibodies also occur in 15 20% of patients with factor VIII hemophilia who have received infusions of plasma concentrates. Inhibitors typically develop in patients with severe disease, and are thought to represent an immune response to a foreign antigen in the form of infused factor VIII. However, some cases of moderate hemophilia caused by mutations are also associated with inhibitor development. In cases of hemophilia A with inhibitors, one treatment approach is
P.540
immune tolerance induction, based on the use of repeated exposure to low levels of antigen. The success rate is approximately 70%. Cases of life-threatening bleeding can be treated by removal of the antibody with plasma exchange combined with infusion of high doses of factor VIII. Another approach is the use of factor VIII bypassing agents or activated factor VIIa.
Factor VIII antibodies can also occur spontaneously, and then usually produce a severe bleeding disorder. The PTT is prolonged, and the fibrinogen level, prothrombin time, and platelet count are not affected. A plasma mixing test will usually reveal the presence of an inhibitor by the failure of normal plasma to correct the prolonged PTT. However, the mixing test may require incubation for 2 4 hours to reveal the inhibitor. Factor VIII coagulant levels are low.
Factor VIII antibodies should be suspected in any acquired severe bleeding disorder associated with a prolonged PTT. Factor VIII antibodies are distinguished from lupus anticoagulants both by the presence of clinical bleeding and more importantly by the reduced factor VIII:C level. The diagnosis is confirmed by mixing tests and in vivo by the failure of factor VIII concentrates to raise the factor VIII:C levels by the expected amount.
The treatment of choice is cyclophosphamide, usually combined with prednisone. In the interim, aggressive factor VIII replacement may be necessary. Products that bypass factor VIII, such as activated factor VII, may be effective but are expensive.
Hind D et al: Recombinant factor VIIa concentrate versus plasma derived concentrates for the treatment of acute bleeding episodes in people with haemophilia A and inhibitors. Cochrane Database Syst Rev 2004;(2):CD004449.
Stasi R et al: Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424.
United Kingdom Haemophelia Centre Doctors' Organisation: Guidelines on the selection and use of therapeutic products to treat haemophilia and other hereditary bleeding disorders. Haemophilia 2003;9:1.
Hemophilia B
![]() Essentials of Diagnosis
Essentials of Diagnosis
X-linked recessive inheritance, with only males affected.
Low levels of factor IX coagulant activity.
Spontaneous hemarthroses.
General Considerations
Hemophilia B (Christmas disease, factor IX hemophilia) is a hereditary bleeding disorder due to deficiency of coagulation factor IX. Most commonly, factor IX is quantitatively reduced, but in one-third of cases an abnormally functioning molecule is immunologically present. Factor IX deficiency is one-seventh as common as factor VIII deficiency hemophilia but is otherwise clinically and genetically identical.
The PTT is prolonged, and factor IX levels are reduced when measured by specific factor assays. Other laboratory features are the same as for factor VIII hemophilia.
Treatment
Factor IX hemophilia is managed with factor IX concentrates. Factor VIII concentrates are ineffective in this type of hemophilia. The same dosing considerations apply as in factor VIII hemophilia, with the exception that the volume of distribution of factor IX is twice the plasma volume, so that 80 units/kg is necessary to achieve a 100% level. In addition, the half-life of factor IX is 18 hours. Thus, to maintain a patient through major surgery, the dosage should be 80 units/kg (approximately 6000 units) initially followed by 40 units/kg (3000 units) every 18 hours. Factor IX levels should be measured to ensure that expected levels are achieved and that an inhibitor is not present.
Unlike factor VIII concentrates, factor IX concentrates contain a number of other proteins, including activated coagulating factors that appear to contribute to a risk of thrombosis with recurrent usage of factor IX concentrates. Because of the risk of thrombosis, more care is needed in deciding to use these concentrates. DDAVP is not useful in this disorder, and patients should be cautioned to avoid aspirin.
Prognosis
The prognosis for these patients is the same as for those with factor VIII hemophilia.
Shapiro AD et al: The safety and efficacy of recombinant human blood coagulation factor IX in previously untreated patients with severe or moderately severe hemophilia B. Blood 2005; 105:518.
Other Congenital Coagulation Disorders
Factor XI Deficiency
This disorder is seen primarily among Ashkenazi Jews and is autosomal recessive. The PTT may be markedly prolonged, and specific assays of factor XI will show reduced levels. This is usually a mild bleeding disorder manifested primarily by postoperative bleeding. Factor replacement is given with fresh-frozen plasma when necessary.
Afibrinogenemia
In this rare disorder, fibrinogen is absent and both prothrombin time and partial thromboplastin time are markedly prolonged. These patients may have a severe
P.541
bleeding disorder similar to hemophilia. Fibrinogen is replaced with cryoprecipitate.
Other Coagulation Disorders
Bleeding disorders due to isolated deficiency of factors II, V, X, or VII are extremely rare. Deficiencies of factor XII and the contact pathway factors cause a markedly prolonged PTT but are not associated with any increased bleeding.
Factor XIII deficiency results in delayed bleeding after trauma or surgery. All coagulation tests are normal. The disorder is diagnosed by showing instability of the fibrin clot in 8 M urea. Factor XIII is replaced with cryoprecipitate or plasma. A rare cause of bleeding is deficiency of the normal inhibitors of fibrinolytic activity: 2-antiplasmin and plasminogen activator inhibitor.
Roberts HR et al: The use of recombinant factor VIIa in the treatment of bleeding disorders. Blood 2004;104:3858.
Coagulopathy Of Liver Disease
The liver is the site of synthesis of all the coagulation factors except factor VIII. As hepatic insufficiency develops, the vitamin K-dependent factors (factors II, VII, IX, X) and factor V are the first to be affected. Because of its rapid turnover (half-life 6 hours), factor VII levels are the first to decline. Conversely, fibrinogen levels are remarkably well conserved, and decreased fibrinogen synthesis does not occur unless liver disease is very severe.
Liver disease has a number of other effects on the hemostatic system. Increased fibrinolysis occurs because the liver synthesizes 2-antiplasmin (the main inhibitor of fibrinolysis), which is responsible for the clearance of plasminogen activator. Biliary tract disease may lead to malabsorption of vitamin K, and congestive splenomegaly may produce mild thrombocytopenia. A variety of chronic liver diseases cause abnormal posttranslation modification of fibrinogen with resultant dysfibrinogenemia. The majority of patients with cirrhosis have very low levels of thrombopoietin, and this may contribute to the thrombocytopenia.
Long-term treatment of hepatic coagulopathy with factor replacement is usually ineffective. Fresh-frozen plasma is the treatment of choice, and volume overload will limit the ability to maintain hemostatic factor levels. For example, to maintain factor levels greater than 25%, the level must initially be raised to 50% with 50% of the plasma volume (20 mL/kg) and then 10 mL/kg must be replaced every 6 hours to maintain adequate factor VII levels. In average-sized persons, this will require transfusion of 1400 mL of plasma initially followed by 700 mL every 6 hours. Factor IX concentrates are contraindicated in liver disease because of their tendency to cause DIC. If thrombocytopenia is present, platelet transfusion may be of some help, but platelet recovery is usually disappointing because of hypersplenism.
The prognosis is that of the underlying liver disease.
Youssef WI et al: Role of fresh frozen plasma infusion in correction of coagulopathy of chronic liver disease: a dual phase study. Am J Gastroenterol 2003;98:1391.
Vitamin K Deficiency
![]() Essentials of Diagnosis
Essentials of Diagnosis
Underlying dietary deficiency or antibiotic use.
Prothrombin time more prolonged than PTT.
Rapid correction with vitamin K replacement.
General Considerations
Vitamin K plays a role in coagulation by acting as a cofactor for the posttranslational -carboxylation of zymogens II, VII, IX, and X. The modified zymogens (with -carboxyglutamic acid residues) are able to bind to platelets in a calcium-dependent reaction and consequently better participate in the complex reactions that activate factors X and II. Without -carboxylation, these reactions on the platelet surface occur slowly and hemostasis is impaired.
Vitamin K is supplied in the diet primarily in leafy vegetables and endogenously from synthesis by intestinal bacteria. Factors that contribute to vitamin K deficiency include poor diet, malabsorption, and broad-spectrum antibiotics suppressing colonic flora. A characteristic setting for vitamin K deficiency is a postoperative patient who is not eating and who is receiving antibiotics. Body stores of vitamin K are small, and deficiency may develop in as little as 1 week.
Clinical Findings
A. Symptoms and Signs
There are no specific clinical features, and bleeding may occur at any site.
B. Laboratory Findings
The prothrombin time (PT) is prolonged to a greater extent than the PTT, and with mild vitamin K deficiency only the PT is defective (Tables 13-22 and 13-23). Fibrinogen
P.542
level, thrombin time, and platelet count are not affected.
Table 13-22. Causes of isolated prolonged prothrombin time. | |
|---|---|
|
Table 13-23. Causes of prolonged prothrombin time and partial thromboplastin time. | |
|---|---|
|
Differential Diagnosis
Vitamin K deficiency can be distinguished from hepatic coagulopathy only by assessing the response to vitamin K therapy. Surreptitious warfarin use will produce laboratory features indistinguishable from those of vitamin K deficiency.
Vitamin K deficiency differs from DIC by normal platelet count and fibrinogen levels in the former.
Treatment
Vitamin K deficiency responds rapidly to subcutaneous vitamin K, and a single dose of 15 mg will completely correct laboratory abnormalities in 12 24 hours.
Prognosis
The prognosis is excellent, as vitamin K deficiency can be completely corrected with replacement.
Crowther MA et al: Oral vitamin K lowers the international normalized ratio more rapidly than subcutaneous vitamin K in the treatment of warfarin-associated coagulopathy. A randomized, controlled trial. Ann Intern Med 2002;137:251.
Disseminated Intravascular Coagulation
![]() Essentials of Diagnosis
Essentials of Diagnosis
Underlying serious illness.
Microangiopathic hemolytic anemia may be present.
Hypofibrinogenemia, thrombocytopenia, fibrin degradation products, and prolonged PT.
General Considerations
Coagulation is usually confined to a localized area by the combination of blood flow and circulating inhibitors of coagulation, especially antithrombin III. If the stimulus to coagulation is too great, these control mechanisms can be overwhelmed, leading to the syndrome of DIC. Antithrombin III levels may be low in DIC due to the combination of increased consumption and decreased synthesis. In pathophysiologic terms, DIC can be thought of as the consequence of the presence of circulating thrombin (normally confined to a localized area). The effects of thrombin are to cleave fibrinogen to fibrin monomer, stimulate platelet aggregation, activate factors V and VIII, and release plasminogen activator, which generates plasmin. Plasmin in turn cleaves fibrin, generating fibrin degradation products, and further inactivates factors V and VIII. Thus, the excess thrombin activity produces hypofibrinogenemia, thrombocytopenia, depletion of coagulation factors, and fibrinolysis.
Other physiologic mechanisms that contribute to the control of thrombin generation may also be disrupted in DIC. Activated protein C inhibits the activated forms of factors V and VIII, and activated protein C levels may be reduced both by consumption and by the downmodulation of thrombomodulin on endothelial cells (necessary for the activation of protein C) induced by high levels of tumor necrosis factor (TNF). Recent evidence demonstrates that much of the stimulus to thrombin activation in DIC comes from the tissue factor/factor VIIa pathway. It is possible that deficiencies in the tissue factor pathway inhibitor also contribute to the pathogenesis of DIC.
DIC can be caused by a number of serious illnesses, including sepsis (especially with gram-negative bacteria but possible with any widespread bacterial or fungal infection), severe tissue injury (especially burns and head injury), obstetric complications (amniotic fluid embolus, septic abortion, retained fetus), cancer (acute promyelocytic leukemia, mucinous adenocarcinomas), and major hemolytic transfusion reactions.
Clinical Findings
A. Symptoms and Signs
DIC leads to both bleeding and thrombosis. Bleeding is far more common than thrombosis, but the latter may dominate if coagulation is activated to a far greater extent than fibrinolysis. Bleeding may occur at any site, but spontaneous bleeding and oozing at venipuncture sites or wounds are important clues to the diagnosis. Thrombosis is most commonly manifested by digital ischemia and gangrene, but catastrophic events such as renal cortical necrosis and hemorrhagic adrenal infarction may occur. DIC may also secondarily produce microangiopathic hemolytic anemia.
Subacute DIC is seen primarily in cancer patients and is manifested primarily as recurrent superficial and deep venous thromboses (Trousseau's syndrome).
B. Laboratory Findings
DIC produces a complex coagulopathy with the characteristic constellation of hypofibrinogenemia, elevated
P.543
fibrin degradation products, thrombocytopenia, and a prolonged PT. Of the fibrin degradation products, the D-dimer is the most sensitive, since its cross-linking implies origin from fibrin in a clot. All fibrin degradation products are cleared by the liver and thus may be elevated in hepatic dysfunction. Hypofibrinogenemia is another important diagnostic laboratory feature, because only a few other disorders (congenital hypofibrinogenemia, severe liver disease) will lower the fibrinogen level. In some cases of DIC, when the patient's baseline fibrinogen level is markedly elevated, the initial fibrinogen level may be normal. However, since the half-life of fibrinogen is approximately 4 days, a declining fibrinogen level will confirm the diagnosis of DIC.
Other laboratory abnormalities are variably present. The PTT may or may not be prolonged. In approximately 25% of cases, a microangiopathic hemolytic anemia is present, and fragmented red blood cells are seen on the peripheral smear. Antithrombin III levels may be markedly depleted. When fibrinolysis is activated, levels of plasminogen and 2-antiplasmin may be low.
Subacute DIC produces a very different laboratory picture. Thrombocytopenia and elevated D-dimer are usually the only abnormalities. Fibrinogen levels are normal, and the PTT may be normal.
Differential Diagnosis
Liver disease may prolong both the PT and PTT, but fibrinogen levels are usually normal, and the platelet count is usually normal or only slightly reduced. However, severe liver disease may be difficult to distinguish from DIC. Vitamin K deficiency will not affect the fibrinogen level or platelet count and will be completely corrected by vitamin K replacement.
Sepsis may produce thrombocytopenia and digital ischemia, and coagulopathy may be present because of vitamin K deficiency. However, in these cases, the fibrinogen level should be normal.
TTP may produce fever and microangiopathic hemolytic anemia. However, fibrinogen levels and other coagulation studies should be normal.
Treatment
The primary focus should be the diagnosis and treatment of the underlying disorder that has given rise to DIC. In many cases, DIC will produce laboratory abnormalities with only mild clinical manifestations, and in these cases no specific therapy is required.
When the underlying cause of DIC is rapidly reversible (such as in obstetric cases), replacement therapy alone may be indicated. The role of heparin in the treatment of DIC is controversial. In some cases, when any increase in bleeding is unacceptable (neurosurgical procedures), heparin therapy is contraindicated. However, when DIC is producing serious clinical consequences and the underlying cause is not rapidly reversible, heparin may be helpful. One clear indication for heparin therapy is the presence of thrombosis or fibrin deposition leading to acral cyanosis. In using heparin, a dose of 500 750 units per hour is usually sufficient. Heparin cannot be effective if antithrombin III levels are markedly depleted. Antithrombin III levels should be measured, and fresh-frozen plasma or concentrates of antithrombin III used to raise levels to greater than 50%. In using heparin, it is not necessary to prolong the PTT. Successful therapy is indicated by a rising fibrinogen level. Fibrin degradation products will decline over 1 2 days. Improvement in the platelet count may lag as much as 1 week behind control of the coagulopathy.
In replacement therapy, platelet transfusion should be used to maintain a platelet count greater than 30,000/mcL, and 50,000/mcL if possible. Fibrinogen is replaced with cryoprecipitate, and the aim is for a plasma fibrinogen level of 150 mg/dL. One unit of cryoprecipitate usually raises the fibrinogen level by 6 8 mg/dL, so that 15 units of cryoprecipitate will raise the level from 50 to 150 mg/dL. Coagulation factor deficiency may require replacement with fresh-frozen plasma.
In some cases, when DIC is complicated by excessive fibrinolysis, even the combination of heparin and replacement therapy may not be adequate to control bleeding. In these cases, EACA, 1 g intravenously per hour, may be added to decrease the rate of fibrinolysis, raise the fibrinogen level, and control bleeding. EACA should not be used without heparin in DIC because of the risk of thrombosis.
Although infusions of large doses of antithrombin III have not reduced the mortality rate of severe sepsis complicated by DIC, one large trial has shown benefit for the use of activated protein C.
Prognosis
The prognosis is that of the underlying disease.
Franchini M et al: Update on the treatment of disseminated intravascular coagulation. Hematology 2004;9:81.
Hoffmann JN et al: Effect of long-term and high-dose antithrombin supplementation on coagulation and fibrinolysis in patients with severe sepsis. Crit Care Med 2004;32:1851.
Hypercoagulable States
Most cases of thromboembolism likely result from a convergence of an underlying genetic predisposition and acquired precipitating events such as immobilization, pregnancy, or surgery (Table 13-24).
Table 13-24. Causes of hypercoagulability. | |
|---|---|
|
Cancer is associated with an increased risk of both venous and arterial thrombosis. In some cases, low-grade DIC appears to be responsible. In unusual cases,
P.544
a unique cancer procoagulant stimulates the clotting system. Myeloproliferative disorders such as polycythemia vera, essential thrombocytosis, and paroxysmal nocturnal hemoglobinuria are associated with a high incidence of thrombosis, caused by qualitative platelet abnormalities. Venous thrombosis may occur in unusual locations such as the mesenteric, hepatic, or splenic venous beds. Arterial thrombosis occurs as well and may be manifested as large vessel occlusion (stroke, myocardial infarction) or as microvascular events with burning in the hands and feet.
A number of congenital biochemical defects have also been associated with hypercoagulability (Table 13-24). A family history is usually present. The thromboses are almost always venous and may occur in the large veins of the abdomen. Thromboses often occur during early adulthood rather than in childhood and are often precipitated by factors such as trauma or pregnancy. The most common of these disorders is an abnormal factor V (factor V Leiden), which is resistant to degradation by activated protein C. Dysfibrinogenemia is diagnosed by a prolonged reptilase time.
The syndrome of warfarin-induced skin necrosis may occur in patients with undiagnosed protein C deficiency. Protein C is vitamin K dependent and has a shorter half-life than the coagulation proteins. Warfarin, by creating a vitamin K-dependent state, will transiently deplete protein C before it leads to anticoagulation. During the period of hypercoagulability due to unopposed protein C depletion, thrombosis of skin vessels may lead to infarction and necrosis. The syndrome can be prevented by the use of heparin for 5 7 days until warfarin induces anticoagulation.
Treatment
If a patient is recognized to be at increased risk of thrombosis, effective prophylactic therapy is usually available. Low-molecular-weight heparin has largely replaced unfractionated heparin because of improved efficacy and ease of monitoring. Warfarin has long been the standard outpatient therapy for prevention of thrombosis, except in cancer-associated thrombosis when low-grade DIC is a pathogenic mechanism. The investigational agent ximelagatran, a direct thrombin inhibitor, may come to replace current anticoagulants based on its ease of use (oral administration, fixed dose, no monitoring) as well as its efficacy and safety (see Chapter 9).
In patients with myeloproliferative disease who have had symptoms of thrombosis, antiplatelet therapy may be helpful. However, such therapy should not be used indiscriminately, because these patients are also at increased risk of bleeding. For patients with erythromelalgia (painful redness and burning of the hands), aspirin, 325 mg daily, is effective.
In congenital biochemical defects such as deficiency of antithrombin III or the vitamin K-dependent proteins C and S, warfarin is effective and is given indefinitely. Family members are screened for the presence of the defect.
Kovalevsky G et al: Evaluation of the association between hereditary thrombophilias and recurrent pregnancy loss: a meta-analysis. Arch Intern Med 2004;164:558.
Mann KG et al: Factor V: a combination of Dr Jekyll and Mr Hyde. Blood 2003;101:20.
Prandoni P: How I treat venous thromboembolism in patients with cancer. Blood 2005;106:4027.
Rieder MJ et al: Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med 2005;352:2285.
Lupus Anticoagulant
The lupus anticoagulant is an IgM or IgG immunoglobulin that produces a prolonged PTT by binding to the phospholipid used in the in vitro PTT assay. As such, it is a laboratory artifact and does not cause a clinical bleeding disorder. The lupus anticoagulant is seen in 5 10% of patients with systemic lupus erythematosus. More commonly, it is seen without an underlying disorder or in patients taking phenothiazines.
There is no bleeding defect unless a second disorder such as thrombocytopenia, hypoprothrombinemia, or a prolonged bleeding time is present. In fact, the lupus anticoagulant has been associated with an increased risk of thrombosis and of recurrent spontaneous abortions.
The PTT is prolonged and fails to correct when the patient's plasma is mixed in a 1:1 dilution with normal plasma. The PT is either normal or slightly prolonged. The fibrinogen level and thrombin time are normal. The Russell viper venom (RVV) time is a more sensitive assay and is specifically designed to demonstrate the presence of a lupus anticoagulant. An antiphospholipid, the lupus anticoagulant will cause a false-positive VDRL test for syphilis. A related autoantibody, anticardiolipin, can be detected by separate assays.
P.545
Lupus anticoagulant should be suspected in cases of a markedly prolonged PTT without clinical bleeding (other causes are factor XII or contact factor deficiency). The plasma mixing test will demonstrate the presence of an inhibitor by the failure of normal plasma to correct the PTT. When acquired factor VIII inhibitors are being considered, a factor VIII:C level may be measured; this will be normal in patients with lupus anticoagulant.
No specific treatment is necessary. Prednisone will usually rapidly eliminate the lupus anticoagulant, and it has been suggested that prednisone therapy reduces spontaneous abortions in this syndrome. It is not clear whether prednisone has any effect on the thrombotic tendency associated with lupus anticoagulant. Patients with thromboses should be treated with anticoagulation in standard doses. Because of the artificially prolonged PTT, heparin therapy is difficult to monitor properly, and low-molecular-weight heparin may be preferred. The dose of warfarin administered may also be inadequate if the baseline PT is prolonged.
Brouwer JL et al: The contribution of inherited and acquired thrombophilic defects, alone or combined with antiphospholipid antibodies, to venous and arterial thromboembolism in patients with systemic lupus erythematosus. Blood 2004;104:143.
Proven A et al: Clinical importance of positive test results for lupus anticoagulant and anticardiolipin antibodies. Mayo Clin Proc 2004;79:467.
Blood Transfusions
Red Blood Cell Transfusions
Red blood cell transfusions are given to raise the hematocrit levels in patients with anemia or to replace losses after acute bleeding episodes.
Sources of Red Cells for Transfusion
Several types of components containing red blood cells are available.
A. Fresh Whole Blood
The advantage of whole blood for transfusion is the simultaneous presence of red blood cells, plasma, and fresh platelets. Fresh whole blood is never absolutely necessary, since all the above components are available separately. The major indications for use of whole blood are cardiac surgery or massive hemorrhage when more than 10 units of blood are required in a 24-hour period.
B. Packed Red Blood Cells
Packed red cells are the component most commonly used to raise the hematocrit. Each unit has a volume of about 300 mL, of which approximately 200 mL consists of red blood cells. One unit of packed red cells will usually raise the hematocrit by approximately 4%. The expected rise in hematocrit can be calculated using an estimated red blood cell volume of 200 mL/unit and a total blood volume of about 70 mL/kg. For example, a 70-kg man will have a total blood volume of 4900 mL, and each unit of packed red blood cells will raise the hematocrit by 200 4900, or 4%.
C. Leukopoor Blood
Patients with severe leukoagglutinin reactions to packed red blood cells may require depletion of white blood cells and platelets from transfused units. White blood cells can be removed either by centrifugation or by washing. Preparation of leukopoor blood is expensive and leads to some loss of red cells.
D. Frozen Blood
Red blood cells can be frozen and stored for up to 3 years, but the technique is cumbersome and expensive, and frozen blood should be used sparingly. The major application is for the purpose of maintaining a supply of rare blood types. Patients with such types may donate units for autologous transfusion should the need arise. Frozen red cells are also occasionally needed for patients with severe leukoagglutinin reactions or anaphylactic reactions to plasma proteins, since frozen blood has essentially all white blood cells and plasma components removed.
E. Autologous Packed Red Blood Cells
Patients scheduled for elective surgery may donate blood for autologous transfusion. These units may be stored for up to 35 days.
Compatibility Testing
Before transfusion, the recipient's and the donor's blood are cross-matched to avoid hemolytic transfusion reactions. Although many antigen systems are present on red blood cells, only the ABO and Rh systems are specifically tested prior to all transfusions. The A and B antigens are the most important, because everyone who lacks one or both red cell antigens has isoantibodies against the missing antigen or antigens in his or her plasma. These antibodies activate complement and can cause rapid intravascular lysis of the incompatible red cells. In emergencies, type O blood can be given to any recipient, but only packed cells should be given to avoid transfusion of donor plasma containing anti-A or anti-B antibodies.
The other important antigen routinely tested for is the D antigen of the Rh system. Approximately 15% of the population lack this antigen. In patients lacking the antigen, anti-D antibodies are not naturally present, but the antigen is highly immunogenic. A recipient whose red cells lack D and who receives D-positive blood may develop anti-D antibodies that can cause severe lysis of subsequent transfusions of D-positive red cells.
P.546
Blood typing includes assay of recipient serum for unusual antibodies by mixing the serum with panels of red cells representing commonly occurring weak antigens. The screening is particularly important if the recipient has had previous transfusions.
Hemolytic Transfusion Reactions
The most severe reactions are those involving mismatches in the ABO system. Most of these cases are due to clerical errors and mislabeled specimens. Hemolysis is rapid and intravascular, releasing free hemoglobin into the plasma. The severity of these reactions depends on the dose of red blood cells given. The most severe reactions are those seen in surgical patients under anesthesia.
Hemolytic transfusion reactions caused by minor antigen systems are typically less severe. The hemolysis usually takes place at a slower rate and is extravascular. Sometimes these transfusion reactions may be delayed for 5 10 days after transfusion. In such cases, the recipient has received blood containing an immunogenic action, and in the time since transfusion, a new alloantibody has been formed. The most common antigens involved in such reactions are Duffy, Kidd, Kell, and C and E loci of the Rh system.
A. Symptoms and Signs
Major hemolytic transfusion reactions cause fever and chills, with backache and headache. In severe cases, there may be apprehension, dyspnea, hypotension, and vascular collapse. The transfusion must be stopped immediately. In severe cases, DIC, acute renal failure from tubular necrosis, or both can occur.
Patients under general anesthesia will not manifest such signs, and the first indication may be generalized bleeding and oliguria.
B. Laboratory Findings
Identification of the recipient and of the blood should be checked. The donor transfusion bag with its pilot tube must be returned to the blood bank, and a fresh sample of the recipient's blood must accompany the donor bag for retyping of donor and recipient blood samples and for repeat of the cross-match.
The hematocrit will fail to rise by the expected amount. Coagulation studies may reveal evidence of renal failure and DIC. Hemoglobinemia will turn the plasma pink and eventually result in hemoglobinuria. In cases of delayed hemolytic reactions, the hematocrit will fall and the indirect bilirubin will rise. In these cases, the new offending alloantibody is easily detected in the patient's serum.
C. Treatment
If a hemolytic transfusion reaction is suspected, the transfusion should be stopped at once. A sample of anticoagulated blood from the recipient should be centrifuged to detect free hemoglobin in the plasma. If hemoglobinemia is present, the patient should be vigorously hydrated to prevent acute tubular necrosis. Forced diuresis with mannitol may help prevent renal damage.
Leukoagglutinin Reactions
Most transfusion reactions are not hemolytic but represent reactions to antigens present on white blood cells in patients who have been sensitized to the antigens through previous transfusions or pregnancy. Most commonly, patients will develop fever and chills within 12 hours after transfusion. In severe cases, cough and dyspnea may occur and the chest x-ray may show transient pulmonary infiltrates. Because no hemolysis is involved, the hematocrit rises by the expected amount despite the reaction.
Leukoagglutinin reactions may respond to acetaminophen and diphenhydramine; corticosteroids are also of value. Removal of leukocytes by filtration before blood storage will reduce the incidence of these reactions.
Anaphylactic Reactions
Rarely, patients will develop urticaria or bronchospasm during a transfusion. These reactions are almost always due to plasma proteins rather than white blood cells. Patients who are IgA deficient may develop these reactions because of antibodies to IgA. Patients with such reactions may require transfusion of washed or even frozen red blood cells to avoid future severe reactions.
Contaminated Blood
Rarely, blood is contaminated with gram-negative bacteria. Transfusion can lead to septicemia and shock from endotoxin. If this is suspected, the offending unit should be cultured and the patient treated with antibiotics as indicated.
Diseases Transmitted Through Transfusion
Despite the use of only volunteer blood donors and the routine screening of blood, transfusion-associated viral diseases remain a problem. All blood products (red blood cells, platelets, plasma, cryoprecipitate) can transmit viral diseases. All blood donors are screened with questionnaires designed to detect donors at high risk of transmitting diseases. All blood is now screened for hepatitis B surface antigen, antibody to hepatitis B core antigen, syphilis, p24 antigen and antibody to HIV, antibody to hepatitis C virus (HCV), antibody to human T cell lymphotropic/leukemia virus (HTLV), and antibody to West Nile virus.
With improved screening, the risk of posttransfusion hepatitis has steadily decreased. The risk of hepatitis B is 1:200,000 per unit and of HIV 1:250,000 per unit. The risk of seroconversion to HTLV is 1:70,000, but clinical sequelae when this occurs are rare. The major infectious risk of blood products is
P.547
hepatitis C, with a seroconversion rate of 1:3300 per unit transfused. Most of these cases are clinically silent, but there is a high incidence of chronic hepatitis.
Platelet Transfusion
Platelet transfusions are indicated in cases of thrombocytopenia due to decreased platelet production. They are not useful in immune thrombocytopenia, since transfused platelets will last no longer than the patient's endogenous platelets. The risk of spontaneous bleeding rises when the platelet count falls to less than 10,000/mcL, and the risk of life-threatening bleeding increases when the platelet count is less than 5000/mcL. Because of this, prophylactic platelet transfusions are often given at these very low levels. Platelet transfusions are also given prior to invasive procedures or surgery, and the goal should be to raise the platelet count to over 50,000/mcL.
Platelets are most commonly derived from donated blood units. One unit of platelets (derived from 1 unit of blood) usually contains 5 7 1010 platelets suspended in 35 mL of plasma. Ideally, 1 platelet unit will raise the recipient's platelet count by 10,000/mcL, and transfused platelets will last for 2 or 3 days. However, responses are often suboptimal, with poor platelet increments and short survival times. This may be due to sepsis, splenomegaly, or alloimmunization. Most alloantibodies causing platelet destruction are directed at HLA antigens. Patients requiring long periods of platelet transfusion support should be monitored to document adequate responses to transfusions so that the most appropriate product can be used. Patients may benefit from HLA-matched platelets derived from either volunteer donors or family members, with platelets obtained by plateletpheresis. Techniques of cross-matching platelets have been developed and appear to identify suitable platelet donors (nonreactive with the patient's serum) without the need for HLA typing. Such single-donor platelets usually contain the equivalent of six units of random platelets, or 30 50 1010 platelets suspended in 200 mL of plasma. Ideally, these platelet concentrates will raise the recipient's platelet count by 60,000/mcL. Leukocyte depletion of platelets has been shown to delay the onset of alloimmunization.
Granulocyte Transfusions
Granulocyte transfusions are seldom indicated and have largely been replaced by the use of myeloid growth factors (G-CSF and GM-CSF) that speed neutrophil recovery. However, they may be beneficial in patients with profound neutropenia (< 100/mcL) who have gram-negative sepsis or progressive soft tissue infection despite optimal antibiotic therapy. In these cases, it is clear that progressive infection is due to failure of host defenses. In such situations, daily granulocyte transfusions should be given and continued until the neutrophil count rises to above 500/mcL. Such granulocytes must be derived from ABO-matched donors. Although HLA matching is not necessary, it is preferred, since patients with alloantibodies to donor white blood cells will have severe reactions and no benefit.
The donor cells usually contain some immunocompetent lymphocytes capable of producing graft-versus-host disease in HLA-incompatible hosts whose immunocompetence may be impaired. Irradiation of the units of cells with 1500 cGy will destroy the lymphocytes without harm to the granulocytes or platelets.
Silliman CC et al: Transfusion-related acute lung injury. Blood 2005;105:2266.
Slichter SJ et al: Factors affecting posttransfusion platelet increments, platelet refractoriness, and platelet transfusion intervals in thrombocytopenic patients. Blood 2005;105:4106.
Stramer SL et al: West Nile virus among blood donors in the United States, 2003 and 2004. N Engl J Med 2005;353: 451.
Transfusion Of Plasma Components
Fresh-frozen plasma is available in units of approximately 200 mL. Fresh plasma contains normal levels of all coagulation factors (about 1 unit/mL). Fresh frozen plasma is used to correct coagulation factor deficiencies and to treat TTP. The risk of transmitting viral disease is comparable to that associated with transfusion of red blood cells.
Cryoprecipitate is made from fresh plasma. One unit has a volume of approximately 20 mL and contains approximately 250 mg of fibrinogen and between 80 and 100 units of factor VIII and vWF. Cryoprecipitate is used to supplement fibrinogen in cases of congenital deficiency of fibrinogen or DIC. One unit of cryoprecipitate will raise the fibrinogen level by about 8 mg/dL.
EAN: 2147483647
Pages: 49