7 - Eye
Editors: McPhee, Stephen J.; Papadakis, Maxine A.; Tierney, Lawrence M.
Title: Current Medical Diagnosis & Treatment, 46th Edition
Copyright 2007 McGraw-Hill
> Table of Contents > 10 - Heart
function show_scrollbar() {}
10
Heart
Thomas M. Bashore MD
Christopher B. Granger MD
Patrick Hranitzky MD
Symptoms & Signs
Common Symptoms
The most common symptoms of heart disease are chest pain, dyspnea, palpitations, syncope or presyncope, and fatigue. None is specific, and interpretation depends on the entire clinical picture and, in many cases, diagnostic testing.
Chest Pain or Discomfort
Chest pain and other forms of discomfort are common symptoms that can occur as a result of pulmonary, pleural, or musculoskeletal disease, esophageal or other gastrointestinal disorders, or anxiety states, as well as many cardiovascular diseases. Myocardial ischemia is a frequent cause of cardiac chest pain and the most important to identify to prevent complications, but it is often experienced more as a sensation of discomfort than actual pain, thereby increasing the potential for being ignored by the patient or misdiagnosed by the physician. This is usually described as dull, aching, or as a sensation of pressure, tightness, squeezing, or gas, rather than as sharp or spasmodic. Ischemic symptoms usually subside within 5 20 minutes but may last longer. Progressive symptoms or symptoms at rest may represent unstable angina due to coronary plaque rupture and thrombosis. Protracted episodes often represent myocardial infarction, although one-third of patients with acute myocardial infarction do not have chest pain. When present, the pain is commonly accompanied by a sense of anxiety or uneasiness. The location is usually retrosternal or left precordial. Because there are not the appropriate sensory nerves on the heart, the central nervous system (CNS) interpretation of pain location often results in pressure or heaviness being referred to the throat, lower jaw, shoulders, inner arms, upper abdomen, or back. Ischemic pain may be precipitated by exertion, cold temperature, meals, stress, or combinations of these factors and is usually relieved by rest, but many episodes do not conform to these patterns. It is not related to position or respiration and is usually not elicited by chest palpation. In myocardial infarction, a precipitating factor is frequently not apparent. One clue that the pain may be ischemic is other symptoms associated with the pain, such as shortness of breath, dizziness, a feeling of impending doom, and vagal symptoms, such as nausea and diaphoresis.
Hypertrophy of either ventricle or stenotic aortic valvular disease may also give rise to ischemic pain or pain with less typical features. Myocarditis, pulmonary hypertension, and mitral valve prolapse are also associated with chest pain atypical for angina pectoris. Pericarditis may produce pain that is greater supine than upright, and may increase with respiration, or swallowing. Pleuritic chest pain is not ischemic, and pain on palpation should signal a musculoskeletal etiology. Aortic dissection classically produces an abrupt onset of tearing pain of great intensity that often radiates to the back.
Boie ET: Initial evaluation of chest pain. Emerg Med Clin North Am 2005;23:937.
Canto JG et al: Prevalence, clinical characteristics, and mortality among patients with myocardial infarction presenting without chest pain. JAMA 2000;283:3223.
Kohn MA et al: Prevalence of acute myocardial infarction and other serious diagnoses in patients presenting to an urban emergency department with chest pain. J Emerg Med 2005;29:383.
Lee TH et al: Evaluation of the patient with acute chest pain. N Engl J Med 2000;342:1187.
Dyspnea
Dyspnea due to heart disease is generally precipitated or exacerbated by exertion and usually results from elevated left atrial (LA) and pulmonary venous pressures or from hypoxia, though some patients with purely right heart disease complain of dyspnea as well. The former are most commonly caused by left ventricular (LV) systolic dysfunction, LV diastolic dysfunction (due to hypertrophy, fibrosis, or pericardial disease), or valvular stenosis or regurgitation. Exertional dyspnea may be an anginal equivalent. The acute onset or worsening of LA hypertension may result in %pulmonary edema. Hypoxia may be a consequence of pulmonary edema, inherent lung disease, or shunting. Dyspnea should be quantified by the amount of activity that precipitates it. It is important to ask if commonly performed tasks can precipitate it, such as climbing stairs, housework, grocery shopping, mowing the lawn, vacuuming, etc. It is also a common symptom of primary and secondary pulmonary disease, and the etiologic distinction may be difficult. The diagnosis of congestive heart failure (CHF) may be aided by measurement
P.317
of B-type natriuretic peptide (BNP), though high levels of BNP may also result from right ventricular (RV) dysfunction and pulmonary embolism. Shortness of breath may also occur in sedentary or obese individuals and is associated with anxiety states, anemia, and many other illnesses.
Orthopnea is dyspnea that occurs in recumbency and results from an increase in central blood volume. It may also result from pulmonary disease and obesity. Paroxysmal nocturnal dyspnea is shortness of breath that occurs abruptly 30 minutes to 4 hours after going to bed and is relieved (after 10 or 20 minutes) by sitting up or standing up; this symptom is more specific for cardiac disease.
Palpitations, Dizziness, Syncope
Palpitations, or awareness of the heartbeat, may be a normal phenomenon or may reflect increased cardiac or stroke output in patients with many noncardiac conditions (eg, exercise, thyrotoxicosis, anemia, anxiety). It may also be due to cardiac abnormalities that increase stroke volume (regurgitant valvular disease, bradycardia) or may be a manifestation of cardiac dysrhythmias. Ventricular premature beats may be sensed as extra or skipped beats. Supraventricular or ventricular tachycardia may be felt as rapid, regular or irregular palpitations or fluttering ; many patients are asymptomatic, however.
If the abnormal rhythm is associated with a sufficient decline in arterial pressure or cardiac output, it may especially in the upright position impair cerebral blood flow, causing lightheadedness, blurring of vision, loss of consciousness (syncope), or other symptoms. However, dizziness in particular is nonspecific and is an uncommon symptom of cardiac disease or dysrhythmia.
Cardiogenic syncope most commonly results from bradyarrhythmias (sinus node arrest or exit block, atrioventricular [AV] conduction block), very rapid supraventricular rhythms or ventricular tachycardia or fibrillation. The absence of premonitory symptoms helps distinguish cardiogenic syncope from vasovagal faints, postural hypotension, or seizure, but is not a reliable screening tool. Although recovery is often immediate, some patients may exhibit seizure-like movements. Aortic stenosis and hypertrophic obstructive cardiomyopathy may also cause syncope, which is usually exertional or postexertional. Another form of syncope is termed neurocardiogenic syncope, commonly known as vasovagal syncope. In this syndrome, there is an inappropriate increase in vagal efferent activity, often resulting from a precedent increase in sympathetic cardiac stimulation. Syncope may follow presyncopal symptoms and may accompany a brief period of nausea and/or diaphoresis, or it may be abrupt in onset, mimicking arrhythmia-induced syncope. Autonomic dysfunction due to venous insufficiency or peripheral neuropathy can result in a positional fall in blood pressure (BP), and supine and upright BPs should be checked in all patients with syncope. Carotid sinus hypersensitivity may also result in syncope that is related to stimulation of the baroreceptors in the carotid sinus. Gentle carotid massage while monitoring the BP and cardiac rhythm usually can elicit the response.
Grubb BP: Clinical practice. Neurocardiogenic syncope. N Engl J Med 2005;352:1004.
Kapoor WN: Syncope. N Engl J Med 2000;343:1856.
Masotti G et al; Task Force on Syncope, European Society of Cardiology: Guidelines on management (diagnosis and treatment) of syncope update 2004. Europace 2004;6: 467.
Schnipper JL et al: Diagnostic evaluation and management of patients with syncope. Med Clin North Am 2001;85:423, xi.
Sloane PD et al: Dizziness: state of the science. Ann Intern Med 2001;134:823.
See also references in the section on syncope.
Functional Classification of Heart Disease
In the management of patients with heart disease, it is important to quantify and monitor the severity of symptoms. A commonly used classification system is that of the New York Heart Association, shown below. However, in following individual patients, it is better to document specific activities that produce symptoms, such as walking distance, stairs climbed, or activities of daily living.
Class I: No limitation of physical activity. Ordinary physical activity does not cause undue fatigue, dyspnea, or anginal pain.
Class II: Slight limitation of physical activity. Ordinary physical activity results in symptoms.
Class III: Marked limitation of physical activity. Comfortable at rest, but less than ordinary activity causes symptoms.
Class IV: Unable to engage in any physical activity without discomfort. Symptoms may be present even at rest.
Other classifications have been proposed, but these are universally accepted, and clinically can be applied to both heart failure and anginal symptoms.
Signs of Heart Disease
Although the cardiovascular examination centers on the heart, peripheral signs often provide important information.
Appearance
Although cardiac patients may appear healthy and comfortable at rest, many with acute myocardial infarction appear anxious and restless. Diaphoresis suggests hypotension or a hyperadrenergic state, such as during pericardial
P.318
tamponade, tachyarrhythmias, or myocardial infarction. Cold and clammy skin or pallor suggests low cardiac output and may be a sign of cardiogenic shock or anemia. Patients with severe chronic CHF or other long-standing low cardiac output states may appear cachectic.
Cyanosis may be central, due to arterial desaturation, or peripheral, reflecting impaired tissue delivery of adequately saturated blood in low-output states, polycythemia, or peripheral vasoconstriction. Clubbing may be present in chronic cyanotic states. Central cyanosis may be caused by pulmonary disease, left heart failure, or right-to-left intracardiac or intrapulmonary shunting; the latter will not be improved by increasing the inspired oxygen concentration. Edema may be present and its pitting nature and extent quantified. Note if presacral edema is present. Severe right heart failure may also present with ascites and scrotal edema.
Vital Signs
Although the normal resting heart rate usually ranges from 50 to 90 beats/min, both slower and more rapid rates may occur in normal individuals or may reflect noncardiac conditions such as anxiety or pain, medication effect, fever, thyroid disease, pulmonary disease, anemia, or hypovolemia. If symptoms or clinical suspicion warrants, an electrocardiogram (ECG) should be performed to diagnose arrhythmia, conduction disturbance, or other abnormalities. The range of normal BP is wide, but even in asymptomatic individuals systolic pressures below 90 mm Hg or above 140 mm Hg and diastolic pressures above 90 mm Hg warrant further clinical evaluation and follow-up. BP may vary between the upper extremities (often the left brachial is slightly lower than the right) and the BP measurement in the leg is usually higher than in the arm. Anxiety may increase the BP, and the patient should be asked if it has been checked in other settings. The ready availability of home BP monitoring or drugstore monitoring units should be considered before beginning antihypertensive therapy if the BP is borderline elevated. Tachypnea is also nonspecific, but pulmonary disease and heart failure should be considered when respiratory rates exceed 16/min under resting conditions. Cheyne-Stokes respiration, a form of periodic breathing, is not uncommon in severe heart failure.
Peripheral Pulses & Venous Pulsations
The quality of the pulses palpated is a reflection of the pulse pressure. Diminished peripheral pulses most commonly result from arteriosclerotic peripheral vascular disease and may be accompanied by localized bruits. Asymmetry of pulses should also arouse suspicion of coarctation of the aorta or aortic dissection, especially if a delay is noted between the brachial or radial pulse and the femoral pulse. Exaggerated upper extremity pulses may indicate aortic regurgitation, coarctation, patent ductus arteriosus (PDA), or other conditions that increase stroke volume. The carotid pulse is a valuable aid to assessment of LV ejection. It has a delayed upstroke in aortic stenosis and a bisferiens quality (two palpable peaks) in mixed aortic stenosis and regurgitation or hypertrophic obstructive cardiomyopathy. It may be difficult to feel in significant aortic stenosis or in low output states. Pulsus paradoxus (a decrease in systolic BP during inspiration) is a normal sign unless exaggerated to > 10 mm Hg. The most common cause of pulsus paradoxus is asthma and chronic obstructive pulmonary disease, though its presence may be a critical component to the diagnosis of pericardial tamponade. Pulsus alternans, in which the amplitude of the pulse alternates every other beat during sinus rhythm, occurs when cardiac contractility is very depressed. It is volume dependent and at times can be elicited by feeling the pulse on standing.
Jugular venous pulsations (JVP) provide insight into right atrial (RA) pressure and function. To identify the waveforms, it is important to palpate the opposite carotid pulse while simultaneously observing the JVP. During ventricular systole (a positive carotid pulse wave), the normal RA pressure falls due to both atrial diastole and the pulling of the tricuspid valve into the RV cavity (the x descent). In the JVP, a prominent waveform just prior to systole is the a wave. A prominent waveform just after systole is the v wave. A waveform during systole is the c-v wave. Prominent a waves imply poor RV compliance or atrial contraction against a closed tricuspid valve (due to AV dissociation associated with ventricular arrhythmias, pacing, or tricuspid stenosis). A prominent v wave implies rapid filling of the RA, such as in an atrial septal defect (ASD) or mild tricuspid regurgitation, and a c-v wave implies significant tricuspid regurgitation. The height of the JVP provides a measure of RA pressure with an elevated central venous pressure if it is visible above the angle of Louis with the patient upright. An increased central blood volume and reduced RV compliance can also be assumed if the JVP rise more than 1 cm and are sustained (> 200 seconds) by right upper quadrant abdominal pressure (hepatojugular reflux). Kussmaul's sign (failure of jugular venous pressure to decrease with inspiration) is commonly seen with RV infarction, postoperatively after cardiac surgery, with tricuspid regurgitation, and with constrictive pericarditis.
Abidov A et al: Prognostic significance of dyspnea in patients referred for cardiac stress testing. N Engl J Med 2005;353: 1889.
Constant J: Using internal jugular pulsations as a manometer for right atrial pressure measurements. Cardiology 2000;93:26.
Drazner MH et al: Prognostic importance of elevated jugular venous pressure and a third heart sound in patients with heart failure. N Engl J Med 2001;345:574.
Maisel A et al; Rapid Emergency Department Heart Failure Outpatient Trial investigators: Primary results of the Rapid Emergency Department Heart Failure Outpatient Trial (REDHOT). A multicenter study of B-type natriuretic peptide levels, emergency department decision making, and outcomes in patients presenting with shortness of breath. J Am Coll Cardiol 2004;44:1328.
P.319
Pulmonary Examination
Rales heard at the lung bases are a sign of CHF but may be caused by similarly localized pulmonary disease. Cardiac rales tend to occur late in inspiration and be fine in nature, while pulmonary rales tend to be more coarse and appear in early or mid inspiration. Rales are loudest at the bases in heart failure, and the examiner should note how far up from the diaphragm they are audible. Wheezing suggests obstructive pulmonary disease and only rarely occurs in left heart failure. Pleural effusions with bibasilar percussion dullness and reduced breath sounds are common in CHF and are more frequent or larger on the right. Egophony may be present due to pulmonary compression over a pleural effusion or to a pulmonary infiltration.
Precordial Pulsations
A parasternal lift usually indicates right ventricular hypertrophy (RVH), pulmonary hypertension (pulmonary artery [PA] systolic pressure > 50 mm Hg), or LA enlargement; PA pulsations may also be visible. The examiner should feel the LV apical impulse in the left lateral position and note if it is sustained or enlarged and whether an early impulse (A wave) precedes the main apical thrust. The A wave implies poor LV compliance and corresponds to a fourth heart sound. If the second heart sound is palpable along the left sternal border, it may imply an increased P2 and pulmonary hypertension.
Heart Sounds & Murmurs
Auscultation is helpful in the diagnosis of many heart diseases, and provides evidence for cardiac failure. Specific findings are discussed under individual diseases below.
The first heart sound (S1), the closing of the mitral valve and tricuspid valve, may be diminished with severe LV dysfunction or accentuated with mitral stenosis or short PR intervals. Separation of the components of S2 is due to the normally compliant lung, allowing for continuing forward cardiac output with systole compared to the stiffer arterial system. The pulmonary valve closes later than the aortic valve for that reason (splitting). Inspiration increases flow to the lung and reduces flow to the left heart, and splitting is increased. Splitting may be fixed in atrial septal defect, wide with right bundle branch block, and absent or reversed (paradoxic splitting) with aortic stenosis, LV failure, or left bundle branch block. With normal splitting, an accentuated P2 is an important sign of pulmonary hypertension. Third and fourth heart sounds (ventricular and atrial gallops, respectively) indicate ventricular volume overload or impaired compliance and may be heard over either ventricle. A right-sided gallop may increase with inspiration or may be confirmed if heard in the right subclavicular area (where a left-sided gallop does not usually radiate). A palpable A wave helps confirm an S4. An apical S3 is a normal finding in younger individuals and in high output states, such as pregnancy. Additional auscultatory findings include sharp, high-pitched sounds classified as clicks. These may be early systolic and represent ejection sounds (as with a bicuspid aortic valve or pulmonary stenosis) or may occur in mid or late systole, indicating myxomatous changes in the mitral valve.
Although many murmurs indicate valvular disease, a soft, short systolic murmur, usually localized along the left sternal border or toward the apex, may be innocent, reflecting pulmonary or aortic flow. Innocent murmurs often vary with inspiration, diminish in the upright position, and are most frequently heard in thin individuals. Systolic murmurs should be classified as holosystolic when they merge with S1 and persist through S2 or ejection when they begin after S1 and end before S2. Holosystolic murmurs tend to have a uniform intensity during systole, while ejection murmurs have a peak at some part of the systolic cycle. Holosystolic murmurs usually represent mitral regurgitation if maximal at the apex or in the axilla and tricuspid regurgitation or ventricular septal defect (VSD) if best heard at the sternal border. Short aortic ejection murmurs with a preserved A2 are common in older individuals, especially when hypertension has been present, and even if they are moderately loud they usually reflect thickening (sclerosis) of the valve rather than stenosis. Association of murmurs with palpable vibrations ( thrills ) is always clinically significant, as are diastolic murmurs. High-pitched diastolic murmurs imply flow from a high-pressure to low-pressure chamber (ie, pulmonic regurgitation in pulmonary hypertension, aortic regurgitation, or VSD) while low-pitched diastolic rumbles imply filling across an AV valve (ie, mitral stenosis). Further evaluation is warranted when the patient has symptoms of possible cardiac origin. An echocardiogram should be ordered when a murmur is heard that is of unclear etiology or significance.
Attenhofer Jost CG et al: Echocardiography in the evaluation of systolic murmurs of unknown cause. Am J Med 2000;108: 614.
Chun AA et al: Bedside diagnosis of coronary artery disease: a systematic review. Am J Med 2004;117:334.
Kobal SL et al: Comparison of effectiveness of hand-carried ultrasound to bedside cardiovascular physical examination. Am J Cardiol 2005;96:1002.
Richardson TR et al: Bedside cardiac examination: constancy in a sea of change. Curr Probl Cardiol 2000;25:783.
Edema
Subcutaneous fluid collections appear first in the lower extremities in ambulatory patients or in the sacral region of bedridden individuals. In heart disease, edema primarily results from elevated RA pressures or associated peripheral venous disease. Right heart failure most commonly results from left heart failure, pulmonary disease, or RV dysfunction and tricuspid
P.320
regurgitation, or constrictive pericarditis. Edema may also be due to nephrotic syndrome, low serum albumin, cirrhosis, premenstrual fluid retention, or drugs (especially vasodilators such as calcium channel blockers or salt-retaining medications such as nonsteroidal anti-inflammatory agents or thiazolidinedione diabetic agents), or it may be idiopathic. Ascites may predominate, especially in constrictive pericarditis or following aggressive diuretic usage (where peripheral edema is mobilized more readily than intra-abdominal fluid).
Cho S et al: Peripheral edema. Am J Med 2002;113:580.
O'Brien JG et al: Treatment of edema. Am Fam Physician 2005;71:2111.
Rasool A et al: Treatment of edematous disorders with diuretics. Am J Med Sci 2000;319:25.
Diagnostic Testing
The chest radiograph provides information about heart size (with cardiomegaly being a poor prognostic sign in chronic heart failure), the pulmonary circulation (with characteristic signs suggesting both pulmonary arterial or pulmonary venous hypertension), primary pulmonary disease, and aortic abnormalities. Individual chamber sizes can be estimated, and the presence of pleural effusions noted. The echocardiogram, though, provides much more reliable information about chamber size and hypertrophy, and the presence of pericardial effusions, valvular abnormalities, and congenital abnormalities and has replaced the radiograph for evaluation of structural cardiac disease. The ECG indicates cardiac rhythm, reveals conduction abnormalities, and provides evidence of ventricular hypertrophy, myocardial infarction, or ischemia. Nonspecific ST segment and T wave changes may reflect these processes but are also noted with electrolyte imbalance, drug effects, and many other conditions. Routine radiographs and ECGs are not recommended to screen for heart disease. Stress testing is useful in eliciting ischemia due to fixed coronary lesions, but its use in the typical asymptomatic patient is limited. For instance, it is useful to recall Bayes' theorem when contemplating stress testing patients as part of an executive physical. Bayes' theorem states that the results of a test depend on the risk of disease. If the patient is at low risk for coronary artery disease (CAD), for instance, one can expect a high number of false-positive stress results. If the patient is at high risk for CAD, one can expect a high number of false-negative results. Stress studies are most effective in those with an intermediate risk for CAD. The clinician's goal is to help define the CAD risk using the history, physical examination, and other available laboratory tests (see below). Stress testing may also be useful in non-CAD whenever symptoms seem disproportionate to anatomic defects. For instance, in valvular disease, if the patient is complaining of major symptoms but has only minor anatomic disease, a stress study may help define exercise capacity. Similarly, if there is significant disease by echocardiography, but few or no symptoms, the stress study may define an unrecognized disability. Blood chemistry tests play a major role in defining cardiac risk factors (ie, serum lipid levels, serum human C-reactive protein [hCRP] level, creatinine) and can help with determining etiology of symptoms (ie, BNP level).
Noninvasive Diagnostic Imaging for Noncoronary Heart Disease
The diagnostic procedures for CAD will be discussed in the following section on Coronary Heart Disease. Reviewed here is the use of noninvasive testing for noncoronary heart disease.
Echocardiography & Doppler Ultrasound Imaging
Echocardiography provides information regarding all four chamber sizes, regional and global systolic function, and chamber wall thickness. Excellent images of valve motion, intracardiac masses, abnormal or absent cardiac structures, and pericardial fluid can all be distinguished. %Pulsed Doppler ultrasound provides a semiquantitative or qualitative estimation of the severity of transvalvular gradients, RV systolic pressure, PA pressure, valvular regurgitation, and intracardiac shunts. The Doppler mitral inflow pattern can help confirm diastolic dysfunction and can help verify a restrictive cardiomyopathic picture or constrictive pericarditis. Color flow Doppler ultrasound provides a visual pattern of blood flow velocities superimposed over the anatomic two-dimensional (2-D) echocardiographic image. This allows for the demonstration of turbulence from stenotic or regurgitant valves, and for the visualization of intracardiac defects. Since some regurgitant flow occurs normally, especially when the AV valves close, the presence of minor amounts of regurgitant color flow should not be construed as pathology. Tissue Doppler methods are now being explored as a means of defining the extent of either mitral annular or ventricular wall motion independent of intracardiac flow velocity, and the relationship between the two may prove useful for defining diastolic pressure elevation, identifying abnormalities in ventricular contraction or relaxation, or evaluating pacemaker therapy.
Transesophageal echocardiography (TEE) with Doppler ultrasound is used to obtain echocardiographic data when surface sound transmission is poor, to derive information about posterior structures (especially the atria and AV valves), prosthetic heart valves, and intracardiac masses not seen on chest wall echocardiography (eg, vegetations in endocarditis or thrombi on pacemaker leads), and to monitor patients during surgery. It can confirm the location of the pulmonary veins and define septal defects or the presence of a
P.321
patent foramen ovale (PFO). It is superior to surface echocardiography in diagnosing LA appendage thrombi and regurgitant lesions associated with prosthetic valves. The absence of atrial thrombi identifies patients in atrial fibrillation at low risk for embolization, thus facilitating early cardioversion. It is also quite sensitive in detecting aortic dissection and severe atherosclerosis of the ascending aorta, which may be the source for transient ischemic attacks or embolic strokes.
Stress echocardiography can be used in valvular as well as ischemic heart disease. Echocardiograms may be performed during or immediately following exercise. Valvular gradient changes after exercise or during dobutamine or nitroprusside infusion can be evaluated. Transient segmental wall motion abnormalities during or immediately following exercise or pharmacologic stress suggest ischemia. Improvement in wall motion during low-dose dobutamine infusions is an indicator of myocardial viability.
Cheitlin MD et al: ACC/AHA/ASE 2003 guideline update for the clinical application of echocardiography summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASE Committee to Update the 1997 Guidelines for the Clinical Application of Echocardiography). J Am Coll Cardiol 2003;42:954.
Biagini E et al: The use of stress echocardiography for prognostication in coronary artery disease: an overview. Curr Opin Cardiol 2005;20:386.
Goldman ME et al: Echocardiography in search of a cardioembolic source. Curr Probl Cardiol 2002;27:342.
Gottdiener JS: Overview of stress echocardiography: uses, advantages, and limitations. Prog Cardiovasc Dis 2001;43:315.
Lerakis S et al: Part I: use of echocardiography in the evaluation of patients with suspected cardioembolic stroke. Am J Med Sci 2005;329:310.
Cardiac MRI & Multislice CT
Cardiac MRI continues to evolve rapidly. Currently available systems provide high-quality and high-resolution images of cardiac and adjacent vascular structures. MRI also provides excellent images that can be used to quantify cardiac function and structure. It is particularly useful for defining myocardial diseases such as sarcoidosis or amyloidosis. Flow measurements, valve orifice sizes, and shunt sizes can all be determined. With the use of gadolinium contrast agents, MRI can be used to assess myocardial perfusion during adenosine pharmacologic stress and viability. Contrast-enhanced images can provide accurate measurement of myocardial infarction size and location. MRI can also provide accurate 3-D reconstruction of the great vessels and abdominal aorta. The study can also be used to screen for renal artery stenosis in patients with hypertension. However, patients with metal pacemakers or defibrillators are not candidates for MRI.
Cardiac multislice CT (fast CT) is a new modality. At lower resolution, it has primarily been used to screen patients for CAD. The extent of calcium within the coronary vessels was found to correlate with the extent of atherosclerotic CAD. More recently, higher resolution imaging modes and faster acquisition have enabled noninvasive coronary angiography. At this stage, cardiac multislice CT is still undergoing investigation, but the predictive value of a negative study is high (around 95%), suggesting that it is an excellent test to confirm normal coronaries. Calcium in the arterial wall may limit its ability to define the severity of coronary disease, but the inherent resolution of fast CT allows it to better visualize coronaries than cardiac MRI. Multislice CT, with its impressive three-dimensional (3-D) reconstruction algorithms, also provides outstanding images in valvular, myocardial, and congenital heart disease. However, there is some concern regarding the radiation dose with multislice CT, which is not an issue with MRI. For most individuals, however, the effective dose equivalent is around 20 milliSieverts, about the same as a rest and exercise thallium study.
Becker CR et al: Past, present, and future perspective of cardiac computed tomography. J Magn Reson Imaging 2004;19: 676.
Berman DS et al: Roles of nuclear cardiology, cardiac computed tomography, and cardiac magnetic resonance: assessment of patients with suspected coronary artery disease. J Nucl Med 2006;47:74.
Constantine G et al: Role of MRI in clinical cardiology. Lancet 2004;363:2162.
Heatlie GJ et al: Cardiac magnetic resonance imaging. Postgrad Med J 2004;80:19.
Lima JA et al: Cardiovascular magnetic resonance imaging: current and emerging applications. J Am Coll Cardiol 2004; 44:1164.
Mahnken AH et al: Multislice spiral computed tomography of the heart: technique, current applications, and perspective. Cardiovasc Intervent Radiol 2005;28:388.
Raggi P et al: Computed tomography coronary calcium screening and myocardial perfusion imaging. J Nucl Cardiol 2005; 12:96.
Cardiac Catheterization & Angiography
Cardiac catheterization is evolving from a diagnostic procedure to a therapeutic one, as more and more types of cardiac lesions are approached with percutaneous techniques.
Right heart catheterization is convenient to perform in the laboratory, at the bedside, or in the operating room. It allows measurement of RA, RV, PA, and pulmonary capillary wedge pressures (PCWP; the latter an indicator of LA pressure), oxygen saturation, and cardiac output. These data may diagnose intracardiac shunts, physiologically significant pericardial disease, and right-sided valve lesions and can distinguish between cardiac and pulmonary disease. It is useful in pulmonary hypertension to document whether the elevated pressures are related to pulmonary disease or to left heart disease. In those with pulmonary vascular
P.322
disease, vasodilators can be administered to test for vasoactivity. Hemodynamic monitoring with a PA catheter may be very helpful in the assessment and treatment of shock, heart failure, complicated myocardial infarction, respiratory failure, and postoperative hemodynamic instability. However, this procedure is not without risk complications include pneumothorax, bleeding, arrhythmias, PA rupture, pulmonary emboli, infection, and patient immobility. Therefore, the role of this procedure remains unsettled, although it is still commonly used to monitor cardiac patients in surgery and in critical care settings. Bedside echocardiography can often fulfill this role for most situations.
Left heart catheterization is performed to assess the cardiac valves and LV function plus the presence and severity of CAD. Mitral stenosis and aortic stenosis are quantified by measuring the pressure gradients across the valves and, taking flow into account, the estimated valve areas. Mitral and aortic regurgitation are assessed semiquantitatively from contrast injections in the LV and aorta, respectively. The ejection fraction (EF) and regional wall motion are assessed by contrast left ventriculography. Stenotic valvular disease is well defined by echocardiography with Doppler ultrasound, but assessing the consequences of regurgitant valvular disease is more difficult and cardiac catheterization with hemodynamics is often more helpful.
Both coronary and noncoronary interventions are now common in the cardiac catheterization laboratory. Acute interventions include plain old balloon angioplasty (POBA) and stenting (now often with stents coated with agents to reduce restenosis). Coronary interventions are now done routinely in the acute as well as chronic setting. Other novel interventional procedures include balloon valvuloplasty (primarily for pulmonic or mitral valve stenosis); stenting of coarctation or branch PA stenosis; percutaneous occlusion of intracardiac defects, such as ASD, PFO or PDA; or alcohol ablation of the subaortic septum in hypertrophic cardiomyopathy.
Bernard GR et al: Pulmonary artery catheterization and clinical outcomes: National Heart, Lung, and Blood Institute and Food and Drug Administration Workshop Report. Consensus Statement. JAMA 2000;283:2568.
Harvey S et al; PAC-Man study collaboration: Assessment of the clinical effectiveness of pulmonary artery catheters in management of patients in intensive care (PAC-Man): a randomised controlled trial. Lancet 2005;366:472.
Ivanov R et al: The incidence of major morbidity in critically ill patients managed with pulmonary artery catheters: a meta-analysis. Crit Care Med 2000;28:615.
Shah MR et al: Impact of the pulmonary artery catheter in critically ill patients: meta-analysis of randomized clinical trials. JAMA 2005;294:1664.
Scanlon PJ et al: ACC/AHA guidelines for coronary angiography. A report of the American College of Cardiology/American Heart Association Task Force on practice guidelines (Committee on Coronary Angiography). Developed in collaboration with the Society for Cardiac Angiography and Interventions. J Am Coll Cardiol 1999;33:1756.
Congenital Heart Disease
Congenital lesions account for only about 2% of heart disease that presents in adulthood. As surgical and medical techniques have improved, more and more children are now reaching adulthood, and it is estimated that up to a million adults may now be surviving with congenital heart disease.
Bolger AP et al: Congenital heart disease: the original heart failure syndrome. Eur Heart J 2003;24:970.
Brickner ME et al: Congenital heart disease in adults. N Engl J Med 2000;342:256, 334.,
Corno AF: Surgery for congenital heart disease. Curr Opin Cardiol 2000;15:238.
Ellis CR et al: Clinical presentations of unoperated and operated adults with congenital heart disease. Curr Cardiol Rep 2005;7:291.
Krasuski RA et al: The emerging role of percutaneous intervention in adults with congenital heart disease. Rev Cardiovasc Med 2005;6:11.
Moodie DS: Diagnosis and management of congenital heart disease in the adult. Cardiol Rev 2001;9:276.
Perloff JK et al: Challenges posed by adults with repaired congenital heart disease. Circulation 2001;103:2637.
Pulmonary Stenosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
No symptoms in patients with mild or moderately severe lesions.
Severe cases may present with right-sided heart failure.
High-pitched systolic ejection murmur maximal in the second left interspace with radiation to the left shoulder.
P2 delayed and soft or absent. Ejection click often present and decreases with inspiration the only right heart auscultatory event that decreases with inspiration, all others increase.
Increased RV impulse.
Palpable thrill at second left intercostal space.
RVH on ECG; PA dilation on radiograph.
Echocardiography/Doppler is diagnostic.
Patients with peak pulmonic valve gradients > 50 mm Hg should undergo intervention regardless of symptoms.
General Considerations
Stenosis of the pulmonary valve or RV infundibulum increases the resistance to RV outflow, raises the RV pressure,
P.323
and limits pulmonary blood flow. Pulmonic stenosis is often associated with other cardiac lesions. Pulmonary blood flow preferentially goes to the left lung in valvular pulmonic stenosis. Most patients with valvular pulmonic stenosis have a domed valve, though some patients have a dysplastic valve. Patients with Noonan's syndrome have a high likelihood of a dysplastic pulmonary valve. The phenotype of Noonan's syndrome includes short stature, web neck, dental malocclusion, antimongoloid slanting of the eyes, mental retardation, and hypogonadism. Unlike those with a domed valve, patients with a dysplastic valve do not have a dilated main PA or commissural fusion. In the absence of associated shunts, arterial saturation is normal. Infundibular stenosis may be so severe that the RV is divided into a low-pressure and high-pressure chamber (double-chambered RV). Peripheral pulmonic stenosis can accompany valvular pulmonic stenosis and may be part of a variety of clinical syndromes, including the congenital rubella syndrome. Patients who have had the Ross procedure (transfer of the pulmonary valve to the aortic position with a homograft pulmonary valve placed in the pulmonary position) may experience postoperative pulmonic stenosis due to an immune response in the homograft.
Clinical Findings
A. Symptoms and Signs
Mild cases (RV-PA gradient < 30 mm Hg) are asymptomatic. Moderate pulmonic stenosis (gradients between 30 mm Hg and 50 mm Hg) to severe stenosis (gradients > 50 mm Hg) may cause symptoms of dyspnea on exertion, syncope, chest pain, and eventually RV failure.
There is often a palpable parasternal lift due to RVH and the pulmonary outflow tract may be palpable if it is enlarged. A loud, harsh systolic murmur and occasionally a prominent thrill are present in the left second and third interspaces parasternally. The murmur radiates toward the left shoulder and increases with inspiration. In mild to moderate pulmonic stenosis, a loud ejection click can be heard to precede the murmur; this sound decreases with inspiration as the increased RV filling from inspiration prematurely opens the valve during atrial systole. The valve excursion in systole, therefore, is less with inspiration, and the click diminishes in intensity. The second sound is obscured by the murmur in severe cases; the pulmonary component may be diminished, delayed, or absent. A right-sided S4 and a prominent a wave in the venous pulse are present when there is RV diastolic dysfunction or a c-v wave if there is tricuspid regurgitation present. Right-sided S4 gallops may be best heard in the right subclavicular area (where left-sided gallops would be distinctly uncommon). Pulmonic valve regurgitation is relatively uncommon in pulmonic stenosis and may be very difficult to hear, as the gradient between the reduced PA diastolic pressure and the elevated RV diastolic pressure may be quite small (low-pressure pulmonic regurgitation).
B. ECG and Chest Radiography
Right axis deviation or RVH is noted; peaked P waves provide evidence of RA overload. Heart size may be normal on radiographs, or there may be a prominent RV and RA or gross cardiac enlargement, depending on the severity. There is often poststenotic dilation of the main and left pulmonary arteries. Pulmonary vascularity is usually normal. A careful look at the chest radiograph may reveal greater vascular perfusion of the left than the right base (Chen's sign). In the adult, calcium may also be present in the main PA or pulmonic valve.
C. Diagnostic Studies
Echocardiography/Doppler is key to the diagnosis, can provide evidence for a doming valve versus a dysplastic valve, can determine the gradient across the valve, and can provide information regarding subvalvular obstruction. Echocardiography also helps define RV function and the presence or absence of tricuspid or pulmonic valvular regurgitation. Catheterization is usually unnecessary for the diagnosis, but is performed when percutaneous valvuloplasty is necessary. MRI and CT do not add additional information unless there is concern regarding associated cardiac lesions or peripheral pulmonary arterial stenosis.
Prognosis & Treatment
Patients with mild pulmonic stenosis (peak gradient < 30 mm Hg) may have a normal life span. Moderate stenosis may be asymptomatic in childhood and adolescence, but symptoms may appear as patients grow older. The degree of stenosis does worsen with time in many patients, so serial follow-up is important. Severe stenosis is associated with sudden death and can cause right heart failure in patients as early as in their 20s and 30s.
Class I indications for intervention include all symptomatic patients and all those with a resting gradient over 50 mm Hg, regardless of symptoms. The 2006 Class II guidelines from the American College of Cardiology/American Heart Association (ACC/AHA) suggest that intervention is reasonable in symptomatic patients when the peak gradient is > 30 mm Hg or in asymptomatic patients when the peak gradient is > 40 mm Hg. Percutaneous balloon valvuloplasty is highly successful in domed valve patients and is the treatment of choice. Surgical commissurotomy can also be done, or pulmonary valve replacement (with either a bioprosthetic valve or homograft) when pulmonic regurgitation is more severe or the valve is dysplastic.
Earing MG et al: Long-term follow-up of patients after surgical treatment for isolated pulmonary valve stenosis. Mayo Clin Proc 2005;80:871.
Khambadkone S et al: Percutaneous pulmonary valve implantation in humans: results in 59 consecutive patients. Circulation 2005;112:1189.
P.324
Latson LA: Critical pulmonary stenosis. J Interv Cardiol 2001; 14:345.
Coarctation of the Aorta
![]() Essentials of Diagnosis
Essentials of Diagnosis
Infants may have severe heart failure; children and adults are usually asymptomatic, presenting with hypertension.
At least 50% of patients with coarctation have an associated bicuspid aortic valve.
Some patients have a webbed neck (XO karyotype, Turner's syndrome).
Absent or weak femoral pulses. Delay of the palpable pulse between the femoral and brachial artery.
In some patients, the coarctation murmur can be heard in the back. In severe coarctation, the continuous murmurs of collaterals around the coarctation can be heard in the back.
Systolic pressure is higher in upper extremities than in lower extremities; diastolic pressures are similar.
ECG shows LV hypertrophy; chest radiograph shows rib notching; echocardiography/Doppler is diagnostic.
General Considerations
Coarctation of the aorta consists of localized narrowing of the aortic arch just distal to the origin of the left subclavian artery. Its development is thought to be related to accessory ductal material that contracts soon after birth. Therefore, it is not usually present in the fetus. Collateral circulation develops around the coarctation through the intercostal arteries and the branches of the subclavian arteries. Coarctation is one of the causes of secondary hypertension and should be considered in young patients with elevated BP. A bicuspid valve is seen in over 50% of the cases and there is an increased incidence of cerebral berry aneurysms. The renin-angiotensin system is reset and this contributes to the hypertension noted. Elevated BP may not return to normal after coarctation repair.
Clinical Findings
A. Symptoms and Signs
If cardiac failure does not occur in infancy, there are usually no symptoms until the hypertension produces LV failure or cerebral hemorrhage; the latter may also occur from the associated cerebral aneurysms. Strong arterial pulsations are seen in the neck and suprasternal notch. Hypertension is present in the arms, but the pressure is normal or low in the legs. This difference is exaggerated by exercise. Femoral pulsations are weak and are delayed in comparison with the brachial pulse. Patients may have severe coarctation, but with large collaterals may have relatively small gradients because of high flow through the collaterals to the aorta distal to the coarctation. Late systolic ejection murmurs at the base are often heard better posteriorly, especially over the spinous processes. There may be an associated aortic insufficiency or stenosis murmur due to the bicuspid aortic valve.
B. ECG and Chest Radiography
The ECG usually shows LV hypertrophy (LVH). Radiography shows scalloping of the ribs due to enlarged collateral intercostal arteries, dilation of the left subclavian artery and poststenotic aortic dilation, and LV enlargement. The coarctation region and the poststenotic dilation of the descending aorta may result in a 3 sign along aortic shadow on the PA chest radiograph (the notch in the 3 representing the area of coarctation).
C. Diagnostic Studies
Echocardiography/Doppler is usually confirmatory and may provide additional evidence for a bicuspid aortic valve. Both MRI and CT can also provide excellent images of the coarctation region to assess involvement of contiguous branch vessels. MRI and echocardiography/Doppler can provide estimates of the gradient across the lesion. Cardiac catheterization provides definitive gradient information and is necessary if percutaneous stenting is to be considered.
Prognosis & Treatment
Cardiac failure is common in infancy and in older untreated patients; it is uncommon in late childhood and young adulthood. Patients with a demonstrated gradient of > 20 mm Hg should be considered for intervention. Most untreated patients with the adult form of coarctation die before age 50 years from the complications of hypertension, rupture of the aorta, infective endarteritis, or cerebral hemorrhage. Aortic dissection also occurs with increased frequency in coarctation. Coarctation may be poorly tolerated in pregnancy because of the inability to support the placental flow.
Resection of the coarctation site has a surgical mortality rate of 1 4% and includes risk of spinal cord injury. Balloon angioplasty of the stenosis has been accomplished successfully, especially in coarctation restenosis after surgery, but the interventional procedure of choice is percutaneous stenting when feasible. Otherwise, surgical resection (usually with end-to-end anastomosis) should be performed. About 25% of surgically corrected patients continue to be hypertensive years after surgery because of the changes in the renin-angiotensin system described above, and they have all the complications associated with hypertension.
Hornung TS et al: Interventions for aortic coarctation. Cardiol Rev 2002;10:139.
P.325
Mahadevan V et al: Endovascular management of aortic coarctation. Int J Cardiol 2004;97(Suppl 1):75.
Oliver JM et al: Risk factors for aortic complications in adults with coarctation of the aorta. J Am Coll Cardiol 2004;44: 1641.
Ramnarine I: Role of surgery in the management of the adult patient with coarctation of the aorta. Postgrad Med J 2005; 81:243.
Toro-Salazar OH et al: Long-term follow-up of patients after coarctation of the aorta repair. Am J Cardiol 2002;89:541.
Vriend JW et al: Late complications in patients after repair of aortic coarctation: implications for management. Int J Cardiol 2005;101:399.
Atrial Septal Defect & Patent Foramen Ovale
![]() Essentials of Diagnosis
Essentials of Diagnosis
Often asymptomatic and discovered on routine physical examination.
RV lift; S2 widely split and fixed.
Grade I-III/VI systolic ejection murmur at pulmonary area.
ECG shows RV conduction delay; radiograph shows dilated pulmonary arteries and increased vascularity; echocardiography/Doppler diagnostic.
A PFO is present in 25% of the population but can lead to paradoxic emboli and cerebrovascular events. Suspicion should be highest in patients who had cryptogenic stroke before age 55 years.
General Considerations
Knowing how the atrial septum forms helps in understanding the different anatomic lesions that create a communication between the atria. Embryologically, the septum primum separates the two atria first, moving inferiorly toward the endocardial cushions. The ventricular septum forms by moving upward from the ventricles to the endocardial cushions at the same time. If the atrial septum does not make it all the way, the residual defect in the septum primum (ostium primum) results in the primum ASD. If the septum primum makes it all the way, a hole or holes (fenestrations) form in the middle of the septum (forming the ostium secundum). A second septum then moves down the right side of the first and normally covers the ostium secundum hole. If it does not cover the hole, a secundum ASD is present. The septum secundum normally completely covers the right side of the atrial septum except for an ovale hole in it (the foramen ovale). If the septae do not fuse, a patent path from the RA to the LA persists (the patent foramen ovale). The most common form of ASD (80% of cases) is persistence of the ostium secundum in the mid septum; less commonly, the ostium primum (which is low in the septum) persists. In many patients with an ostium primum defect, there are mitral or tricuspid valve clefts as part of the AV canal defect. A third form of ASD is the sinus venosus defect, a hole usually at of the upper part of the atrial septum due to failure of the embryonic superior vena cava (SVC) to merge with the atria properly. This latter lesion is often associated with anomalous drainage of the right upper pulmonary vein into the SVC. Rarer forms of ASD include a sinus venosus defect associated with failure of the inferior vena cava (IVC) to merge with the atria and a coronary sinus ASD that is basically an unroofed coronary sinus. In all cases, normally oxygenated blood from the higher-pressure LA passes into the RA, increasing RV output and pulmonary blood flow. In children, the degree of shunting across these defects may be quite large (3:1 or so). As the RV diastolic pressure rises from the chronic volume overload, the RA pressure may rise and the degree of left-to-right shunting may decrease. Eventually, the shunt may even be right-to-left and cyanosis appears.
The pulmonary pressures are modestly elevated in most patients with an ASD due to the high pulmonary blood flow, but severe pulmonary hypertension (Eisenmenger's physiology) is actually rare, occurring in only about 15% of the patients. Eventual RV failure may occur, and most shunts should be corrected unless they are quite small (< 1.5:1 right-to-left shunt). In adults, a large right-to-left shunt may have begun to reverse, so the absolute size at the time the patient is studied may underestimate what it was some years ago. In addition, in most patients the LV compliance normally declines more over time than the RV, and the natural history of small atrial septal shunts is to increase as the patient ages (unless RV failure ensues).
ASDs also predispose to atrial fibrillation due to RA enlargement, and paradoxic right-to-left emboli do occur. Interestingly, paradoxic emboli may be more common in patients with a PFO than a true ASD, as the eustachian valve in the RA directs flow from the inferior vena cava (IVC) toward the septum, and the usual significant left-to-right flow from an ASD is often not present with a simple PFO. An aneurysm of the atrial septum is when there is redundancy of the septum primum through the foramen ovale; it may include the septum secundum. When present with a PFO, there is more shunting right to left as the atrial septum swings back and forth. This latter anatomic issue may explain why more right to left shunting occurs in patients with an atrial septal aneurysm and PFO than in those with a PFO alone.
Clinical Findings
A. Symptoms and Signs
Patients with small or moderate ASDs and with a PFO are asymptomatic unless a complication occurs. With large shunts, exertional dyspnea or cardiac failure may develop, most commonly in the fourth decade of life or later. Prominent RV and PA pulsations
P.326
are readily visible and palpable. A moderately loud systolic ejection murmur can be heard in the second and third interspaces parasternally as a result of increased PA flow. S2 is widely split and does not vary with breathing due to the fact that the left-to-right shunt decreases as the RA pressure increases with inspiration.
B. ECG and Chest Radiography
Right axis deviation or RVH may be present depending on the size of the RV volume overload. Incomplete or complete right bundle branch block is present in nearly all cases of ASD, and superior axis deviation is noted in the AV canal defect, where complete heart block is often seen as well. With sinus venosus defects, the P axis is leftward of +15 due to abnormal atrial activation with loss of the upper RA tissue from around the sinus node. The chest radiograph shows large pulmonary arteries, increased pulmonary vascularity, an enlarged RA and RV, and a small aortic knob with all pre-tricuspid cardiac left-to-right shunts.
C. Diagnostic Studies
Echocardiography demonstrates evidence of RA and RV volume overload. The atrial defect is usually observed, though sinus venosus defects may be elusive. Many patients with a PFO also have a redundant atrial septum (atrial septal aneurysm) that promotes right-to-left shunting. Echocardiography with agitated saline bubble contrast can demonstrate a right-to-left shunt and both pulsed and colorflow Doppler flow studies can demonstrate shunting in either direction. A TEE is helpful when transthoracic echocardiography quality is not optimal, and it improves the sensitivity for small shunts and provides a better assessment of PFO anatomy. Radionuclide flow studies quantify left-to-right shunting by observing the bolus of contrast within the lung fields and demonstrating early recirculation. Both CT and MRI can also elucidate the atrial septal anatomy as well, and allow for observation of associated lesions. Cardiac catheterization is often helpful, especially if there are associated anomalous pulmonary veins. The size and location of the shunt can be determined and the pulmonary pressure and pulmonary vascular resistance (PVR) measured. Cardiac catheterization is required if percutaneous closure is to be contemplated.
Prognosis & Treatment
Patients with small atrial shunts may live a normal life span. Large shunts usually cause disability by age 40 years. Because left-to-right shunts tend to increase with age-related changes in LV compliance, most clinicians believe that closure of all shunts over 1.5:1 should be accomplished. Increased PVR and hypertension secondary to pulmonary vascular disease rarely occur in childhood or young adult life in secundum defects but are more common in primum defects. Significant pulmonary hypertension rarely develops in older patients. After age 40 years, cardiac arrhythmias (especially atrial fibrillation) and heart failure may occur due to the chronic right heart volume overload. Paradoxical systemic arterial embolization becomes more of a concern as RV compliance is lost and the left-to-right shunt begins to reverse.
PFOs are not associated with significant shunting, and therefore the patients are asymptomatic and the heart size is normal. However, PFOs are responsible for most paradoxical emboli and are one of the most frequent causes of cryptogenic strokes in patients under age 55 years. That is likely because there is often right-to-left or bidirectional shunting, and the IVC blood is directed toward the foramen ovale by the eustachian valve.
Small ASDs do not require intervention. The easiest way to decide if a shunt is small is by echocardiography. If the shunt is not creating an RV volume overload, then one can assume the shunt needs no further evaluation. For larger deficits (those with an RV volume overload), surgery can be done at very low risk. Surgery involves anything from simple stitching of the foramen closed to patching of the hole with Dacron or a pericardial patch. Anomalous pulmonary venous connections are baffled to the LA through the sinus venosus defect when such anomalous veins are present. For ostium secundum ASDs, percutaneous closure by use of a variety of devices is now preferred over surgery. The percutaneous closure devices often resemble double umbrellas that lock the septum between the Dacron umbrellas when opened.
Patients with a PFO may have symptoms related to stroke or transient ischemic attack (especially if the age is under 55) or have hypoxemia (especially upon standing so called platypnea orthodeoxia). There are data that suggest that migraine headaches may be more common in patients with a PFO, suggesting some unknown substance normally metabolized in the lung is entering the systemic circulation through the PFO. For patients with cryptogenic stroke or transient ischemic attack, it is uncertain whether closure of the PFO, either by open surgical or percutaneous techniques, has any advantage over anticoagulation with either warfarin or aspirin. Although there are no data yet suggesting that PFO closure is better than medical therapy, ongoing randomized trials should help settle this issue.
Azarbal B et al: Association of interatrial shunts and migraine headaches: impact of transcatheter closure. J Am Coll Cardiol 2005;45:489.
Diener HC et al: Patent foramen ovale: paradoxical connection to migraine and stroke. Curr Opin Neurol 2005;18:299.
Hara H et al: Patent foramen ovale: current pathology, pathophysiology, and clinical status. J Am Coll Cardiol 2005;46:1768.
Martin F et al: Percutaneous transcatheter closure of patent foramen ovale in patients with paradoxical embolism. Circulation 2002;106:1121.
Mas JL et al: Recurrent cerebrovascular events associated with patent foramen ovale, atrial septal aneurysm, or both. N Engl J Med 2001;345:1740.
P.327
Messe SR et al: Practice parameter: recurrent stroke with patent foramen ovale and atrial septal aneurysm: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2004;62:1042.
Wahl A et al: Transcatheter treatment of atrial septal aneurysm associated with patent foramen ovale for prevention of recurrent paradoxical embolism in high-risk patients. J Am Coll Cardiol 2005;45:377.
Ventricular Septal Defect
![]() Essentials of Diagnosis
Essentials of Diagnosis
A restrictive VSD is small and makes a louder murmur than an unrestricted one.
Adults may remain asymptomatic if the defect is small.
Larger defects may result in pulmonary hypertension (Eisenmenger physiology) if not repaired.
A grade II-VI/VI pansystolic murmur maximal at the left sternal border is heard; an associated thrill is common.
ECG may show LVH, RVH, or both; radiograph shows increased pulmonary vascularity, and increased PA and LA size.
Echocardiography/Doppler is diagnostic. The higher the gradient across the septum, the smaller the left-to-right shunt.
General Considerations
De novo VSDs are uncommon in adults. Congenital VSDs occur in various parts of the ventricular septum, but in adults most are in the membranous septum. Others occur high where both ventricles may be committed to the defect or in the muscular septum. Membranous and muscular septal defects may spontaneously close in childhood as the septum grows and hypertrophies. A left-to-right shunt is present unless there is associated RV hypertension. The smaller the defect, the greater the gradient from the LV to the RV and the louder the murmur. The presentation in adults is dependent on the size of the shunt and whether there is associated pulmonic or subpulmonic stenosis that has protected the lung from the systemic pressure and volume. Unprotected lungs with large shunts invariably lead to pulmonary vascular disease and severe pulmonary hypertension (Eisenmenger physiology).
Clinical Findings
A. Symptoms and Signs
The clinical features depend on the size of the defect and the presence or absence of increased pulmonary vascular resistance. Small shunts are associated with loud, harsh holosystolic murmurs in the left third and fourth interspaces along the sternum. Larger shunts may create RV volume and pressure overload. If pulmonary hypertension occurs, high-pressure pulmonic regurgitation may result. A systolic thrill is common. Right heart failure may gradually become evident late in the course, and the shunt will begin to reverse as RV and LV systolic pressures equalize with the advent of pulmonary hypertension. Cyanosis from right-to-left shunting may then occur.
B. ECG and Chest Radiography
The ECG may be normal or may show right, left, or biventricular hypertrophy, depending on the size of the defect and the pulmonary vascular resistance. With large shunts, the RV, the LV the LA, and the pulmonary arteries are enlarged, and pulmonary vascularity is increased on chest radiographs. If pulmonary vascular disease (pulmonary hypertension) evolves, an enlarged PA with pruning of the distal pulmonary vascular bed is seen. In rare cases of a very high VSD, an aortic cusp may prolapse into the VSD and reduce the VSD shunt but result in acute aortic regurgitation.
C. Diagnostic Studies
Echocardiography can demonstrate the size of the overloaded chambers (RV, PA, LA, and LV) and can readily define the defect anatomy. Doppler ultrasound can qualitatively assess the magnitude of shunting by noting the gradient from LV to RV and, if some tricuspid regurgitation is present, the RV systolic pressure can be estimated. Colorflow Doppler helps delineate the shunt defect and the presence of valvular regurgitation. MRI and cardiac CT can often visualize the defect and describe any other anatomic abnormalities. MRI can provide quantitative shunt data as well. Radionuclide flow studies are sometimes used to quantify the relative size of the left-to-right shunt. Cardiac catheterization is usually reserved for those with at least moderate shunting to determine the PVR and the degree of pulmonary hypertension. A PVR of > 7.0 absolute units or a PVR/SVR ratio of > 0.7 usually implies inoperability.
Prognosis & Treatment
Patients with the typical murmur as the only abnormality have a normal life expectancy except for the threat of infective endocarditis. Endocarditis is more typical of smaller shunts due to the high velocity of the jet lesion. Antibiotic prophylaxis is recommended. With large shunts, CHF may develop early in life, and survival beyond age 40 years is unusual.
Small shunts (pulmonary-to-systemic flow ratio < 1.5) in asymptomatic patients do not require surgery or other intervention. Defects causing large shunts should be repaired to prevent pulmonary hypertension or late heart failure. The presence of RV infundibular stenosis distal or pulmonary valve stenosis may protect the pulmonary circuit such that some patients even with a large VSD may still be operable as adults.
Surgical repair of a VSD is generally a low risk procedure unless there is significant Eisenmenger physiology
P.328
as noted above. Currently, several new percutaneous closure devices are under review for nonsurgical closure of VSDs; devices for muscular VSDs are now approved and those for membranous VSDs are being implanted with promising results.
Ammash NM et al: Ventricular septal defects in adults. Ann Intern Med 2001;135:812.
Fu YC et al: Transcatheter closure of perimembranous ventricular septal defects using the new Amplatzer membranous VSD occluder: results of the U.S. phase I trial. J Am Coll Cardiol 2006;47:319.
Gabriel HM et al: Long-term outcome of patients with ventricular septal defect considered not to require surgical closure during childhood. J Am Coll Cardiol 2002;39:1066.
Knauth AL et al: Transcatheter device closure of congenital and postoperative residual ventricular septal defects. Circulation 2004;110:501.
Tetralogy of Fallot
![]() Essentials of Diagnosis
Essentials of Diagnosis
Four features are characteristic: VSD, RVH, RV outflow obstruction from infundibular stenosis, and an overriding aorta (< 50%). A right-sided aortic arch is common (seen in 25%).
Most adult patients with tetralogy of Fallot have been operated upon, usually with an RV ouflow patch and VSD closure.
Physical examination may be deceptive after classic tetralogy repair, with wide-open pulmonic regurgitation often present if a transannular patch was used.
Echocardiography/Doppler may underestimate significant pulmonic regurgitation. Be wary if the RV is enlarged.
Arrhythmias are common and periodic Holter monitoring is recommended.
If the QRS width is > 180 msec, then the patient is subject to serious arrhythmias and sudden death.
General Considerations
Patients with tetralogy of Fallot have a VSD, RV infundibular stenosis, RVH, and a dilated aorta (in about 50% of patients it overrides the septum). There may or may not be pulmonary valve stenosis as well, usually due to a bicuspid pulmonary valve. The aorta can be quite enlarged and aortic regurgitation may occur. If more than 50% of the aorta overrides into the RV outflow tract, the situation is not unlike a double outlet RV. Two vascular abnormalities are common: a right-sided aortic arch and anomalous left anterior descending coronary artery from the right cusp. The latter is important in that surgical correction must avoid cutting this vessel and producing an anterior myocardial infarction. A right-sided aortic arch occurs in 25% of patients and has no functional significance.
Most adult patients have undergone prior surgery. If significant RV outflow obstruction is present in infancy, a Blalock-Taussig (or similar) shunt is often the initial surgical procedure to improve pulmonary blood flow. This procedure enables blood to reach the underperfused lung either by directly attaching one of the subclavian arteries to the PA (classic Blalock shunt) or by creating a conduit between the two (modified Blalock shunt). Other types of systemic to pulmonary shunts no longer in use include a window between the right PA and the aorta (Waterston-Cooley shunt) or a window between the left PA and the descending aorta (Potts shunt). In the adult, there may be a reduced upper extremity pulse on the side used for the Blalock procedure. Total repair of the tetralogy of Fallot generally includes a VSD patch and usually a larger RV outflow tract patch, as well as a take-down of the Blalock shunt. Often the RV outflow tract patch extends through the pulmonary valve into the PA (transannular patch), and the patient is left with wide-open pulmonic regurgitation. Over the years, the volume overload from the severe pulmonary regurgitation becomes the major hemodynamic problem. Patients with tetralogy of Fallot should be monitored to ensure the RV volume does not increase. If it does, then pulmonary valve replacement should be done to correct the pulmonary insufficiency. Ventricular arrhythmias can also originate from the edge of the patch, and tend to correlate with the size of the RV.
Clinical Findings
Most patients are relatively asymptomatic unless right heart failure occurs or arrhythmias become an issue. Patients can be active and generally require no specific therapy except endocarditis prophylaxis. However, low-pressure pulmonic regurgitation is difficult to diagnose except at cardiac catheterization due to the fact that the RV diastolic pressures tend to be high and the pulmonary arterial diastolic pressure is low. This means there is little gradient between the PA and the RVin diastole, so that there may be little murmur or evidence for turbulence on colorflow Doppler. If the RV begins to enlarge, it must be assumed that this is due to pulmonic regurgitation until proven otherwise. Patients often do well into their 40s and 50s.
A. Symptoms and Signs
Physical examination should include checking both arms for any loss of pulse from a prior shunt procedure in infancy. The JVP may reveal an increased a wave from poor RV compliance or a c-v wave due to tricuspid regurgitation. The right-sided arch has no consequence. The precordium may be active, often with a persistent pulmonary outflow murmur. P2 may or may not be present. A right-sided gallop may be heard. A residual
P.329
VSD or aortic regurgitation may also be present. At times, the insertion site of a prior Blalock or other shunt may create a stenotic area in the PA and a continuous murmur occurs as a result.
B. ECG and Chest Radiography
The ECG reveals RVH and right axis deviation; in repaired tetralogy, there is often a right bundle branch block pattern. The chest radiograph shows a classic boot-shaped heart with prominence of the RV and a concavity in the RV outflow tract. This may be less impressive following repair. The aorta may be enlarged and right-sided. Importantly, the width of the QRS should be examined yearly. There are data that persons at greatest risk for sudden death are those with a QRS width of greater than 180 msec. The width of the QRS corresponds to the RV size, and in many patients, the QRS width actually decreases following repair of the pulmonary insufficiency.
C. Diagnostic Studies
Echocardiography/Doppler usually establishes the diagnosis by noting the unrestricted (large) VSD, the RV infundibular stenosis, and the enlarged aorta. In patients who have had tetralogy of Fallot repaired, echocardiography/Doppler also provides data regarding the amount of pulmonic regurgitation, RV and LV function, and the presence of aortic regurgitation.
Cardiac MRI and CT are evolving to have more major diagnostic roles, since they can quantitate both the pulmonary insufficiency and the RV volumes. In addition, cardiac MRI and CT can identify whether there is either a native pulmonary arterial branch stenosis or a stenosis at the distal site of a prior Blalock or other shunt. Cardiac catheterization is sometimes required to document the degree of pulmonic regurgitation because noninvasive studies depend on velocity gradients. Pulmonary angiography demonstrates the degree of pulmonic regurgitation, and RV angiography helps assess any postoperative outflow tract aneurysm.
Prognosis & Treatment
A few patients with just the right amount of pulmonic stenosis enter adulthood without having had surgery. However, most patients have had surgical repair of tetralogy of Fallot, including VSD closure, resection of infundibular muscle, and insertion of an outflow tract patch. Many have a transannular patch resulting in pulmonic regurgitation. If an anomalous coronary is present, then an extracardiac conduit around it from the RV to the PA may be necessary. By 20-year follow-up, reoperation is needed in about 10 15%, not only for severe pulmonic regurgitation but also for residual infundibular stenosis. Usually the pulmonary valve is replaced with a pulmonary homograft, though a porcine bioprosthetic valve is also suitable. Cryoablation of tissue giving rise to arrhythmias is sometimes performed at the time of reoperation. Branch pulmonary stenosis may be percutaneously opened by stenting. All patients require endocarditis prophylaxis.
Atik FA et al: Long-term results of correction of tetralogy of Fallot in adulthood. Eur J Cardiothorac Surg 2004;25:250.
Therrien J et al: Pulmonary valve replacement in adults late after repair of tetralogy of Fallot: are we operating too late? J Am Coll Cardiol 2000;36:1670.
Warnes CA: The adult with congenital heart disease: born to be bad? J Am Coll Cardiol 2005;46:1.
Patent Ductus Arteriosus
![]() Essentials of Diagnosis
Essentials of Diagnosis
Rare in adults.
Adults with small or moderate size PDA are usually asymptomatic, at least until middle age.
Widened pulse pressure; loud S2.
Continuous murmur over left pulmonary area; thrill common.
Echocardiography/Doppler is helpful, but the lesion is best visualized by MRI, CT, or contrast angiography.
General Considerations
The embryonic ductus arteriosus allows shunting of blood from the PA to the aorta in utero. The ductus arteriosus normally closes immediately after birth so that pulmonary blood flows to the pulmonary arteries. Failure to close normally results in a persistent shunt connecting the left PA and aorta, usually near the origin of the left subclavian artery. Prior to birth, the ductus is kept patent by the effect of circulating prostaglandins; in the neonate, a patent ductus can often be closed by administration of a prostaglandin inhibitor such as indomethacin. The effect of the persistent left-to-right shunt on the pulmonary circuit is dependent on the size of the ductus. If large enough, pulmonary hypertension (Eisenmenger physiology) may occur. A small ductus may be well tolerated until adulthood.
Clinical Findings
A. Symptoms and Signs
There are no symptoms unless LV failure or pulmonary hypertension develops. The heart is of normal size or slightly enlarged, with a hyperdynamic apical impulse. The pulse pressure is wide, and diastolic pressure is low. A continuous rough machinery murmur, accentuated in late systole at the time of S2, is heard best in the left first and second interspaces at the left sternal border. Thrills are common. If pulmonary hypertension is present, the shunt may reverse and the lower body receives desaturated blood, while the upper body receives saturated blood. Thus, the hands appear normal while the toes are cyanotic and clubbed.
P.330
B. ECG and Chest Radiography
A normal tracing or LVH is found, depending on the magnitude of shunting. On chest radiographs, the heart is normal in size and contour, or there may be LV and LA enlargement. The PA, aorta, and LA are prominent because they all are in the shunt pathway.
C. Diagnostic Studies
Echocardiography/Doppler can determine LV, RV, and atrial dimensions. Colorflow Doppler allows visualization of the high velocity shunt jet into the proximal left PA. Cardiac MRI and CT can demonstrate the abnormality and assess the size of the pulmonary arteries. Cardiac catheterization can establish the shunt size and direction, and define the size of the ductus. It can also help determine whether pulmonary hypertension has occurred. If percutaneous closure is feasible, catheterization can be therapeutic.
Prognosis & Treatment
Large shunts cause a high mortality rate from cardiac failure early in life. Smaller shunts are compatible with long survival, CHF being the most common complication. Infective endocarditis or endarteritis may also occur, and antibiotic prophylaxis is recommended.
Surgical ligation of the patent ductus can be accomplished with excellent results in patients with few complications, even in very young children. If the lumen size of the ductus is small enough (< 4 mm), percutaneous approaches using either coils or occluder devices can be used with excellent results. Duct closure is usually feasible unless pulmonary hypertension and right-to-left shunting is present.
Arora R: Transcatheter closure of patent ductus arteriosus. Expert Rev Cardiovasc Ther 2005;3:865.
Bilkis AA et al: The Amplatzer duct occluder: experience in 209 patients. J Am Coll Cardiol 2001;37:258.
Pass RH et al: Multicenter USA Amplatzer patent ductus arteriosus occlusion device trial: initial and one-year results. J Am Coll Cardiol 2004;44:513.
Valvular Heart Disease
Although most cases of valvular disease in the United States were at one time due to rheumatic heart disease (still true in developing countries), other causes are now much more common. In the elderly, degenerative aortic valvular disease appears to be due to the same process that produces atherosclerosis, and studies have suggested that about 25% of adults over age 65 have some thickening of their aortic valve (aortic sclerosis) while 2 3% have frank aortic stenosis. Aortic sclerosis alone is a marker for future cardiovascular events and death. Calcium deposition may also occur in the mitral annulus creating enough dysfunction of the valve that either stenosis or regurgitation (or both) results. Endocarditis remains a serious problem and its incidence has risen over the last 10 years. Mitral valve prolapse is still frequently seen and may be associated with the hyperadrenergic syndrome in younger patients. Valvular regurgitation may be due to LV dysfunction (mitral regurgitation) or RV dysfunction (tricuspid regurgitation).
The typical findings of each native lesion are described in Table 10-1. Table 10-2 shows how to use bedside maneuvers to distinguish the various murmurs.
Echocardiography yields key information about valve morphology, LV mass and function, and atrial and ventricular chamber size. Doppler ultrasound provides quantitative measurements of transvalvular gradients and RV systolic pressure (a surrogate for peak PA pressure when there is no pulmonic stenosis) and gives more qualitative estimates of the presence and severity of valvular regurgitation. TEE often provides improved image quality and valve morphology (particularly prosthetic valves), vegetations, thrombi, and eccentric regurgitant jets are more easily identified with TEE. Cardiac MRI and CT generally add only confirmatory information, though abnormalities of the great vessels are best seen by these modalities; the procedures are useful in tracking the size of ascending aortic aneurysms in patients with bicuspid aortic valve and in quantitating RV function.
Bach DS et al: Perioperative assessment and management of patients with valvular heart disease undergoing noncardiac surgery. Minerva Cardioangiol 2004;52:255.
Bonow RO et al: Guidelines for the management of patients with valvular heart disease. Circulation 1998;98:1949.
Boon NA: The medical management of valvar heart disease. Heart 2002;87:395.
Botkin NF et al: Asymptomatic valvular disease: who benefits from surgery? Curr Cardiol Rep 2005;7:87.
Elkayam U et al: Valvular heart disease and pregnancy part I: native valves. Am Coll Cardiol 2005;46:223.
Elkayam U et al: Valvular heart disease and pregnancy: part II: prosthetic valves. J Am Coll Cardiol 2005;46:403.
Rahimtoola SH: The year in valvular heart disease. J Am Coll Cardiol 2006;47:427.
Rahimtoola SH: Valvular heart disease/cardiac surgery. J Am Coll Cardiol 2005;45(11 Suppl B):20B.
Sachdev M et al: Effect of fenfluramine-derivative diet pills on cardiac valves: a meta-analysis of observational studies. Am Heart J 2002;44:1065.
Seiler C: Management and follow up of prosthetic heart valves. Heart 2004;90:818.
Mitral Stenosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Exertional dyspnea, orthopnea, and paroxysmal nocturnal dyspnea when the stenosis becomes severe.
Symptoms often precipitated by onset of atrial fibrillation or pregnancy.
Two syndromes occur; one with moderate mitral stenosis and pulmonary edema, and one with severe mitral stenosis, pulmonary hypertension, and low cardiac output.
Prominent mitral first sound, opening snap (usually), and apical diastolic rumble.
ECG shows LA abnormality and, commonly, atrial fibrillation. Echocardiography/Doppler confirms diagnosis and quantitates severity.
Surgery indicated for symptoms or evidence of pulmonary hypertension. Most symptomatic patients have a valve area less than 1.5 cm2.
P.331
P.332
P.333
P.334
Table 10-1. Differential diagnosis of valvular heart disease. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 10-2. Effect of various interventions on systolic murmurs. | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
General Considerations
Patients with mitral stenosis are usually presumed to have underlying rheumatic heart disease, though a history of rheumatic fever is usually noted in only about one-third. Rheumatic mitral stenosis results in thickening of the leaflets, fusion of the mitral commissures, retraction, thickening and fusion of the chordae, and calcium deposition in the valve. Mitral stenosis can also occur due to congenital disease due to chordal fusion or papillary muscle malposition. The papillary muscles may be abnormally close together, sometimes so close they merge into a single papillary muscle (the parachute mitral valve). In these patients, the chordae and/or valvular tissue may also be fused. In other patients, mitral annular calcification may build up enough to produce a mitral gradient, most often in the elderly or patients with end-stage renal disease. Calcium in the mitral annulus virtually invades the mitral leaflet from the annulus inward. Mitral valve obstruction may also develop in patients who have had mitral valve repair with a ring that is too small, or in patients who have had a surgical valve replacement.
Clinical Findings
A. Symptoms and Signs
A characteristic finding of mitral stenosis is a localized mid-diastolic murmur low in pitch whose duration increases with the severity of the stenosis and the heart rate (Table 10-1). Because the valve is stiff and the LA pressure is high relative to the LV diastolic pressure, the valve opens in early diastole with a snap. The sound is sharp and widely distributed over the chest. The interval between the opening snap and aortic closure sound is long when the LA pressure is low but shortens as the LA pressure rises and approaches the aortic diastolic pressure. The gradient across the mitral valve produces a low-pitched rumble, best heard at the apex with the patient in the left lateral position. Brief exercise, such as sit ups, increase the heart rate and cardiac output resulting in increased flow across the mitral valve and increased audibility of the mitral rumble. Mitral regurgitation may accompany mitral stenosis. Generally, if a regurgitant murmur is audible, there is too much mitral regurgitation to allow either percutaneous valvuloplasty or surgical commissurotomy and valve replacement is the only alternative.
Two clinical syndromes occur with mitral stenosis. In mild to moderate stenosis, LA pressure and cardiac output may be essentially normal and the patient is either asymptomatic or symptomatic only with extreme exertion. The measured valve area is usually between 1.8 cm2 and 1.3 cm2 in those cases. In severe mitral stenosis (valve area < 1.0 cm2), severe pulmonary hypertension develops due to a secondary stenosis of the pulmonary vasculature. In this condition, pulmonary edema is uncommon, but symptoms of low cardiac output and right heart failure predominate.
Paroxysmal or chronic atrial fibrillation develops in 50 80% of patients. Any increase in the heart rate reduces diastolic time and increases the mitral gradient. A sudden increase in heart rate may precipitate pulmonary edema. Therefore, heart rate control is important to maintain, with slow heart rates preferred.
P.335
B. Diagnostic Studies
Echocardiography is the most valuable technique for assessing mitral stenosis. A scoring system is used to help define which patients are eligible for valvuloplasty. One to four points are assigned to each of four observed parameters, with one being the least involvement and four the greatest: mitral leaflet thickening, mitral leaflet mobility, submitral scarring, and commissural calcium. Patients with a total valve score of 8 or less respond better to balloon valvuloplasty techniques than patients with scores less than 8. LA size can also be determined by echocardiography: increased size denotes an increased likelihood of atrial fibrillation and thrombus formation. The effective mitral valve area can be determined by planimetering the smallest mitral orifice or by using the continuous-wave Doppler gradient. Some determination of the pulmonary pressure can also be quantitated by measuring the peak RV pressure from the tricuspid velocity jet signal.
Because echocardiography and careful symptom evaluation provide most of the needed information, cardiac catheterization is used primarily to detect associated valve, coronary, or myocardial disease usually after the decision to intervene has been made.
Treatment & Prognosis
Mitral stenosis may be present for a lifetime with few or no symptoms, or it may become severe in a few years. In most cases, there is a long asymptomatic phase, followed by subtle limitation of activity. Pregnancy and its associated increase in cardiac output and the increased transmitral pressure gradient that results often precipitate symptoms. Toward the end of pregnancy, the cardiac output is also maintained by an increase in heart rate, further increasing the mitral gradient. The onset of atrial fibrillation often precipitates more severe symptoms, which usually improve with control of the ventricular rate or restoration of sinus rhythm. Conversion to and subsequent maintenance of sinus rhythm are most commonly successful when the duration of atrial fibrillation is brief (< 6 12 months) and the LA is not severely dilated (diameter < 4.5 cm). Once atrial fibrillation occurs, the patient should receive warfarin anticoagulation therapy even if sinus rhythm is restored, since atrial fibrillation often recurs even with antiarrhythmic therapy and 20 30% of these patients will have systemic embolization if untreated. Systemic embolization in the presence of only mild to moderate disease is not an indication for surgery but should be treated with warfarin anticoagulation.
Indications for intervention focus on symptoms such as an episode of pulmonary edema, a decline in exercise capacity, or evidence for pulmonary hypertension.
Open mitral commissurotomy is now rarely performed and has given way to percutaneous balloon valvuloplasty. Ten-year follow-up data comparing surgery to balloon valvuloplasty suggest no real difference in outcome between the two modalities. Replacement of the valve is indicated when combined stenosis and regurgitation are present or when the mitral valve echo score is > 8. Percutaneous mitral valvuloplasty has a very low mortality rate (< 0.5%) and low morbidity rate (3 5%). Operative mortality rates are also low: 1 3% in most institutions. Repeat valvuloplasty can be done if the morphology of the valve is suitable.
Mechanical mitral prosthetic valves are more prone to thrombosis than aortic valves. Bioprosthetic valves degenerate after about 10 12 years and percutaneous balloon valvuloplasty procedures cannot be done on bioprosthetic valves should stenosis occur. Younger patients and those with end-stage renal disease do least well with bioprosthetic heart valves. Endocarditis prophylaxis is always indicated. Percutaneous balloon valvuloplasty can safely be done during pregnancy if symptoms warrant.
Arora R et al: Percutaneous transvenous mitral commissurotomy: immediate and long-term follow-up results. Catheter Cardiovasc Interv 2002;55:450.
Fawzy ME et al: Immediate and long-term results of mitral balloon valvotomy for restenosis following previous surgical or balloon mitral commissurotomy. Am J Cardiol 2005;96:971.
Guerios EE et al: Mitral stenosis and percutaneous mitral valvuloplasty (part 1). J Invasive Cardiol 2005;17:382.
Guerios EE et al: Mitral stenosis and percutaneous mitral valvuloplasty (part 2). J Invasive Cardiol 2005;17:440.
Krasuski RA et al. Comparison of results of percutaneous balloon mitral commissurotomy in patients aged 65 years with those in patients aged < 65 years. Am J Cardiol 2001;88:994.
Iung B et al: The long-term outcome of balloon valvuloplasty for mitral stenosis. Curr Cardiol Rep 2002;4:118.
Rahimtoola SH: Current evaluation and management of patients with mitral stenosis. Circulation 2002;106;1183.
Mitral Regurgitation (Mitral Insufficiency)
![]() Essentials of Diagnosis
Essentials of Diagnosis
Variable causes determine clinical presentation.
May be asymptomatic for many years (or for life) or may cause left-sided heart failure.
Pansystolic murmur at the apex, radiating into the axilla; associated with S3 when regurgitant volume is great.
ECG shows LA abnormality or atrial fibrillation and LVH; radiograph shows LA and LV enlargement.
Echocardiographic findings can help decide when to operate.
For primary mitral regurgitation, surgery is indicated for symptoms or when LV EF is < 60% or the echocardiographic LV end-systolic diameter is > 4.5 cm.
P.336
General Considerations
The components of the mitral valve apparatus include the myocardium below the papillary muscles, the papillary muscles themselves, the chordae, the leaflets, and the mitral annulus. Chordae from both the anterior and posterior leaflets attach to both papillary muscles. When ventricular contraction occurs, the papillary muscles contract first and pull the leaflets toward each other. As the LV pressure rises, the leaflets touch and the rising pressure in the LV pushes the leaflets together (so-called keystone effect). The chordae continue to rein in the mitral valve as systole commences and the annulus contracts. All of these components keep the mitral valve from leaking. Failure of any of these components results in mitral regurgitation. Thus, if the papillary muscles are displaced (as in dilated cardiomyopathy), the chordae are too long, the leaflets are too baggy (as in mitral prolapse), or the annulus does not contract (as in annular calcification or cardiomyopathy), then mitral regurgitation will result. Mitral regurgitation places a volume load on the heart (increases preload), but reduces afterload. The result is an enlarged LV with an increased EF. Over time, the stress of the volume overload weakens the LV; when this occurs, there is a drop in EF and a rise in end-systolic volume.
Clinical Findings
A. Symptoms and Signs
During LV systole, the mitral leaflets do not close normally, and blood is ejected into the LA as well as through the aortic valve. In acute mitral regurgitation, LA pressure rises abruptly, leading to pulmonary edema if severe. When chronic, the LA enlarges progressively and the increased volume can be handled without a major rise in the LA pressure; the pressure in pulmonary veins and capillaries may rise only transiently during exertion. Exertional dyspnea and fatigue progress gradually over many years.
Mitral regurgitation leads to LA enlargement and may cause subsequent atrial fibrillation. Systemic embolization is relatively unusual compared with other conditions causing atrial fibrillation. Mitral regurgitation may predispose to infective endocarditis.
Clinically, mitral regurgitation is characterized by a pansystolic murmur maximal at the apex, radiating to the axilla and occasionally to the base; a hyperdynamic LV impulse and a brisk carotid upstroke; and a prominent third heart sound due to the increased volume returning to the LV in early diastole. LA enlargement is at times considerable in chronic mitral regurgitation; the degree of LV enlargement usually reflects the severity of regurgitation. Calcification of the mitral valve is less common than in pure mitral stenosis. Hemodynamically, LV volume overload may ultimately lead to LV failure and reduced cardiac output, but for many years the LV end-diastolic pressure and the cardiac output may be normal at rest.
Nonrheumatic mitral regurgitation may develop abruptly, such as with papillary muscle dysfunction following myocardial infarction, valve perforation in infective endocarditis, or ruptured chordae tendineae in mitral valve prolapse.
Mitral valve prolapse ( floppy or myxomatous mitral valve) is usually asymptomatic but may be associated with nonspecific chest pain, dyspnea, fatigue, or palpitations. Most patients are female, many are thin, and some have skeletal deformities such as pectus excavatum or scoliosis. A hyperadrenergic syndrome has been described, especially in young females, that may be responsible for some of the noncardiac symptoms observed. This syndrome attenuates with age. Some patients have findings of a systemic collagen abnormality (Marfan or Ehler-Danlos syndrome). In these conditions, a dilated aortic root and aortic regurgitation may coexist with the mitral valve prolapse.
On auscultation, there are characteristic mid-systolic clicks that emanate from the chordae or redundant valve tissue and that may be multiple. If leaflets fail to come together properly, the clicks will be followed by a late systolic murmur. As the mitral regurgitation worsens, the murmur is heard more and more throughout systole. The smaller the LV chamber, the greater the degree of prolapse, and thus auscultatory findings are often accentuated in the standing position. The diagnosis is primarily clinical but can be confirmed echocardiographically.
The significance of mitral valve prolapse is in dispute because of the frequency with which it is diagnosed in healthy young women (up to 10%), but in occasional patients this lesion is not benign. Patients who have only a mid-systolic click usually have no sequelae, but significant mitral regurgitation may develop, occasionally due to rupture of chordae tendineae (flail leaflet), in patients with a late or pansystolic murmur. The need for valve repair or replacement is most common in men and increases with aging, so that approximately 2% of patients with clinically significant regurgitation over age 60 years will require surgery. Mitral valve repair is favored over valve replacement, and its efficacy has led many to recommend intervention earlier and earlier in the course of the disease process. Mitral repair may include shortening of chordae, chordae transfers, wedge resection of redundant valve tissue, and mitral annular ring to reduce the annular size. Stitching of the leaflets together to create a double orifice mitral valve is also used at times (Alfieri procedure). Mitral repair or replacement can be done through a right minithoracotomy. Robotic devices are also being used to repair these valves. A variety of new percutaneous methods for repair of the regurgitant mitral valve are being investigated.
P.337
These methods include devices to create a double orifice mitral valve by clipping the anterior and posterior leaflets together, devices that are inserted in the coronary sinus to cinch the annulus and make it smaller, and devices that can be inserted onto the annulus from the ventricular side that can pull the annulus inward and reduce annular circumference. Most of these percutaneous methods are in phase 1 or phase 2 trials.
Infective endocarditis may occur, primarily in patients with murmurs; such patients should have antibiotic prophylaxis prior to dental work and surgical procedures, though the data supporting the use of antibiotic prophylaxis are actually quite poor. New guidelines regarding prophylaxis for endocarditis are due out by the end of 2006. -Adrenergic blocking agents are often effective for supraventricular arrhythmias and may be useful if there are symptoms of the hyperadrenergic syndrome. Sudden death is rare in mitral prolapse, but when symptomatic ventricular tachycardia is present, aggressive management with an implantable cardioverter-defibrillator is usually indicated. An association between mitral prolapse and embolic cerebrovascular events has also been reported but has not been confirmed in subsequent studies. Echocardiographic evidence of marked thickening or redundancy of the valve is associated with a higher incidence of most complications.
Papillary muscle dysfunction or infarction following acute myocardial infarction is less common. When mitral regurgitation is due to papillary dysfunction, it may subside as the infarction heals or LV dilation diminishes. The cause of the regurgitation in most situations is displacement of the papillary muscles and an enlarged mitral annulus rather than true papillary muscle ischemia. In acute infarction, rupture of the papillary muscle may occur with catastrophic results. Transient but sometimes severe mitral regurgitation may occur during episodes of myocardial ischemia and contribute to flash pulmonary edema. Patients with dilated cardiomyopathies of any origin may have secondary mitral regurgitation due to papillary muscle displacement or dilation of the mitral annulus. In patients with ischemic cardiomyopathy, ventricular reconstructive surgery to restore the mitral apparatus anatomy is under investigation (eg, the Dor procedure). If mitral valve replacement is performed, preservation of the chordae to the native valve helps prevent further ventricular dilation following surgery. Several groups have reported good results with mitral valve repair in patients with LV EFs greater than 30% and secondary mitral insufficiency.
B. Diagnostic Studies
Echocardiography is useful in demonstrating the underlying pathologic process (rheumatic, prolapse, flail leaflet, cardiomyopathy), and Doppler techniques provide qualitative and semiquantitative estimates of the severity of mitral regurgitation. It should be noted that a minor degree of retrograde flow from the closing of the mitral valve occurs almost uniformly and echocardiography/Doppler detects this clinically insignificant regurgitation in many normal individuals. Echocardiographic information concerning LV size and function, LA size, PA pressure, and RV function can be invaluable in planning treatment as well as in recognizing associated lesions. TEE may help reveal the cause of regurgitation and is especially useful in patients who have had mitral valve replacement, in endocarditis, and in identifying candidates for valvular repair. Echocardiographic dimensions and measures of systolic function are critical in deciding the timing of surgery. In the past, exercise radionuclide angiography for measurements of exercise EF or determination of myocardial stress-EF relationships was recommended, but this is now not often done. Cardiac MRI is occasionally useful, for example, if specific myocardial causes are being sought (such as amyloid or myocarditis) or if myocardial viability is needed prior to deciding whether to add coronary artery bypass grafting to mitral repair in patients with chronic ischemic mitral regurgitation.
Cardiac catheterization provides a further assessment of regurgitation and its hemodynamic impact along with LV function, resting cardiac output, and PA pressure. Coronary angiography is often indicated to determine the presence of CAD prior to valve surgery in patients with risk factors or those older than age 45 years. In the near future, it may be that multidetector CT methods will be adequate to screen patients with valvular heart disease for asymptomatic CAD.
Treatment & Prognosis
Acute mitral regurgitation due to endocarditis, myocardial infarction, and ruptured chordae tendineae often requires emergency surgery. Some patients can be stabilized with vasodilators or intra-aortic balloon counterpulsation, which reduce the amount of regurgitant flow by lowering systemic vascular resistance. Patients with chronic lesions may remain asymptomatic for many years. Surgery is necessary when symptoms develop. However, because progressive and irreversible deterioration of LV function may occur prior to the onset of symptoms, early operation is indicated even in asymptomatic patients with a reduced EF (< 60%) or marked LV dilation (end-systolic dimension > 4.5 cm on echocardiography). There is controversy regarding the role of afterload reduction in mitral regurgitation, since the lesion inherently results in a reduction in afterload. A heightened sympathetic state has led some to suggest -blockade as well. Most clinicians who monitor patients with chronic mitral regurgitation do prescribe medications, eg, angiotensin-converting enzyme (ACE) inhibitors, to reduce afterload, extrapolating data from studies of patients with cardiomyopathy or aortic regurgitation. In mitral regurgitation, studies suggest that such afterload reduction reduces chamber sizes and the amount of regurgitation, but there has yet to be prospective studies that show a definite long-term benefit.
Badhwar V et al: Mitral valve surgery: when is it appropriate? Congest Heart Fail 2002;8:210.
P.338
Block PC: Percutaneous mitral valve repair for mitral regurgitation. J Interv Cardiol 2003;16:93.
Feldman T et al: Percutaneous mitral valve repair using the edge-to-edge technique: six-month results of the EVEREST Phase I Clinical Trial. J Am Coll Cardiol 2005;46: 2134.
Freed LA et al: Mitral valve prolapse in the general population: the benign nature of echocardiographic features in the Framingham Heart Study. J Am Coll Cardiol 2002;40:1298.
Hayek E et al: Mitral valve prolapse. Lancet 2005;365:507.
Mohty D et al: The long-term outcome of mitral valve repair for mitral valve prolapse. Curr Cardiol Rep 2002;4:104.
Otto CM: Timing of surgery in mitral regurgitation. Heart 2003; 89:100.
Webb JG et al: Percutaneous transvenous mitral annuloplasty: initial human experience with device implantation in the coronary sinus. Circulation 2006;113:851.
Aortic Stenosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
In adults with a bicuspid aortic valve, usually asymptomatic until middle or old age.
Growing evidence that degenerative or calcific aortic stenosis is related to the same process as atherosclerosis.
Delayed and diminished carotid pulses.
Soft, absent, or paradoxically split S2.
Harsh systolic murmur, sometimes with thrill along left sternal border, often radiating to the neck; may be louder at apex in older patients.
ECG usually shows LVH; calcified valve on radiography or fluoroscopy; echocardiography/Doppler is diagnostic.
Surgery indicated for symptoms. Surgical risk is typically low even in the very elderly.
Symptoms likely once the peak echo gradient is > 64 mm Hg.
Surgery often considered for asymptomatic patients when severe aortic stenosis is documented by echocardiography/Doppler.
General Considerations
There are two common clinical scenarios in which aortic stenosis is prevalent. The first is due to a congenitally abnormal valve, usually bicuspid rather than tricuspid. Symptoms at times occur in young or adolescent individuals if severe, but more often symptoms emerge at age 50 65 years when calcification and degeneration of the valve becomes manifest. A dilated ascending aorta, primarily due to an intrinsic defect in the aortic media, may accompany the bicuspid valve. Coarctation of the aorta is also seen in a small number of patients with aortic stenosis. A second group develops what has traditionally been called degenerative or calcific aortic stenosis, which is thought to be related to calcium deposition due to processes similar to what occurs in atherosclerotic vascular disease. Approximately 25% of patients over age 65 years and 35% of those over age 70 years have echocardiographic evidence of aortic sclerosis. About 10 20% of these will progress to hemodynamically significant aortic stenosis over a period of 10 15 years. Thus, aortic stenosis has become the most common surgical valve lesion in developed countries, and many patients are elderly. The risk factors for aortic stenosis in the elderly are similar to those for atherosclerosis, including hypertension, hypercholesterolemia, and smoking. LV outflow tract obstruction may also be caused by other rare congenital lesions, such as supravalvular aortic stenosis (seen in Williams' syndrome) or by subvalvular obstruction such as a subvalvular membrane. Hypertrophic obstructive cardiomyopathy may also coexist with valvular aortic stenosis.
Clinical Findings
A. Symptoms and Signs
Aortic stenosis produces a progressive afterload increase on the LV. To reduce wall stress, the ventricle hypertrophies by laying sarcomeres on top of each other, and increasing the wall thickness. At times, severe LVH ensues. Eventually, the hypertrophy may lead to myocardial dysfunction. Since the EF measurement is afterload dependent, at times it is difficult to sort out whether the low EF is due to increased afterload or to myocardial dysfunction.
Slightly narrowed, thickened, or roughened valves (aortic sclerosis) or aortic dilation may produce the typical ejection murmur of aortic stenosis. In mild or moderate cases where the valve is still pliable, an ejection click may precede the murmur. The characteristic systolic ejection murmur is heard at the aortic area and is usually transmitted to the neck and apex. In some cases, only the high-pitched components of the murmur are heard at the apex, and the murmur may sound like mitral regurgitation (so-called Gallaverdin phenomenon). In severe cases, a palpable LV heave or thrill, a weak to absent aortic second sound, or reversed splitting of the second sound is present (see Table 10-1). When the valve area is less than 0.8 1 cm2 (normal, 3 4 cm2), ventricular systole becomes prolonged and the typical carotid pulse pattern of delayed upstroke and low amplitude is present. This may be an unreliable finding, however, in older patients with extensive arteriosclerotic vascular disease. LVH increases progressively, with resulting elevation in ventricular end-diastolic pressure, and consequently LA and PCWPs. Cardiac output is maintained until the stenosis is severe (with a valve area < 0.8 cm2). LV failure,
P.339
angina pectoris, or syncope may be presenting symptoms and signs of aortic stenosis; importantly, all occur with exertion.
Symptoms of failure may be sudden in onset or may progress gradually. Angina pectoris frequently occurs in aortic stenosis due to underperfusion of the endocardium. Of patients with calcific aortic stenosis and angina, 50% have significant associated CAD, whereas CAD is noted at only half this rate in the absence of angina. Syncope is typically exertional and a late finding. Very rarely, it is a presenting symptom in otherwise asymptomatic patients. Syncope occurs with exertion due to stimulation of reflex baroreceptors. With exertion, the LV pressures rise, stimulating these LV baroreceptors to peripherally vasodilate. This results in an increase in stroke volume in an attempt to further increase cardiac output, which increases the LV systolic pressure again due to the obstructed aortic valve; a cycle of vasodilation and stimulation of the baroreceptors thus occurs and eventually the stenotic valve will not allow an adequate increase in cardiac output and systemic BP falls. Less commonly, syncope may be due to arrhythmias (usually ventricular tachycardia but sometimes sinus bradycardia as calcific invasion of the conduction system from the aortic valve may occur).
B. Diagnostic Studies
The clinical assessment of the severity of aortic stenosis may be difficult, especially when there is reduced cardiac output or significant associated aortic regurgitation. The ECG reveals LVH or suggestive repolarization changes in most patients, but can be normal in up to 10%. The chest radiograph may show a normal or enlarged cardiac silhouette, calcification of the aortic valve, and dilation and calcification of the ascending aorta. The echocardiogram provides useful data about aortic valve calcification and opening and LV thickness and function, while Doppler can provide a good estimate of the aortic valve gradient. These data can usually reliably exclude or diagnose severe stenosis. The maximal Doppler gradient reflects the peak instantaneous gradient across the aortic valve and this differs from the catheterization peak-to-peak gradient. The mean gradients from Doppler and catheterization are more often in agreement. The peak Doppler gradient is derived by multiplying the maximal flow velocity through the valve orifice squared (m/s) times 4; thus, a 4 m/s maximum velocity gradient translates into a 64 mm Hg peak Doppler gradient. Valve area estimation by echocardiography is less reliable. With improvement in the reliability of echocardiography/Doppler, cardiac catheterization mostly provides confirmatory data, an assessment of the hemodynamic consequence of the aortic stenosis, and a look at the coronary arteries. In younger patients, and in patients with high aortic gradients the aortic valve need not be crossed at catheterization. If it is crossed, the valve gradient can be measured at catheterization and an estimated valve area calculated; a valve area below 1.0 cm2 indicates significant stenosis in the newest ACC/AHA guidelines. Aortic regurgitation can be semiquantified by aortic root angiography. In patients with a low EF and both low output and a low valve gradient, the valve area calculations may erroneously indicate significant aortic stenosis, and it may be unclear if an increased afterload is responsible for the low EF or if there is an associated cardiomyopathy. To sort this out, the patient should be studied at baseline and then during an intervention that increases cardiac output (eg, dobutamine or nitroprusside infusion). If the valve area calculation increases, the flow-limiting problem is not the valve, but rather the cardiomyopathy, and surgery is not warranted. If the valve area remains unchanged at the higher outputs, then the valve is considered flow limiting and surgery is indicated.
Prognosis & Treatment
Following the onset of heart failure, angina, or syncope, the prognosis without surgery is poor (50% 3-year mortality rate). Medical treatment may stabilize patients in heart failure, but surgery is indicated for all symptomatic patients with evidence of significant aortic stenosis, as described above. Valve replacement is usually not indicated in asymptomatic individuals, though a Class II indication is to operate once the peak valve gradient by Doppler exceeds 64 mm Hg or the mean exceeds 40 mm Hg because of the high likelihood of symptoms developing over the next 2 years.
The surgical mortality rate for valve replacement is remarkably low even in the elderly, and ranges from 2% to 5%. This low risk is due to the dramatic hemodynamic improvement that occurs with relief of the increased afterload. Mortality rates are substantially higher when there is an associated ischemic cardiomyopathy. Severe coronary lesions are usually bypassed at the same time and bypass adds only a small amount to the overall mortality and morbidity.
The interventional options in patients with aortic stenosis are variable and dependent on the patient's lifestyle and age. In the young and adolescent patient, percutaneous valvuloplasty still has a role. Balloon valvuloplasty is less effective and is associated with early restenosis in the elderly, and thus is rarely used. Data suggest aortic balloon valvuloplasty has an advantage only in those with preserved LV function, and such patients are usually excellent candidates for surgical aortic valve replacement (AVR). There is continuing interest in using the Ross procedure in younger patients. The Ross procedure is performed by moving the patient's own pulmonary valve to the aortic position and replacing the pulmonary valve with a homograft (or rarely a bioprosthetic valve). However, dilation of the pulmonary valve autograft and consequent aortic regurgitation, plus early stenosis of the pulmonary homograft in the pulmonary position, has reduced the enthusiasm for this approach in some institutions. Middle-aged adults generally can take the anticoagulation necessary for the use of mechanical AVR, so most undergo AVR with a bileaflet mechanical valve. If the aortic root is severely
P.340
dilated as well (> 5.5 cm), then the valve may be housed in a Dacron sheath (Bentall procedure) and the root replaced as well. Alternatively, a human homograft root and valve replacement may be used. In the elderly, bioprosthetic (either porcine or bovine pericardial) valves with a life expectancy of about 10 15 years are routinely used to avoid need for anticoagulation. Recent data favor the bovine over the porcine pericardial valve. If the aortic anulus is small, a bioprosthetic valve with a short sheath can be sewn to the aortic wall (the stentless AVR) rather than sewing the prosthetic annulus to the aortic annulus.
Anticoagulation is required with the use of mechanical valves, and the international normalized ratio (INR) should be maintained between 2.0 and 2.5. Mechanical aortic valves are less subject to thrombosis than mechanical mitral valves.
Recently, there has been interest in developing a percutaneous approach to AVR. Both a retrograde approach (from the aorta) and an antegrade approach (from the ventricles by way of a transseptal catheter across the atrial septum) are being investigated. The devices being tested use either a stent with a trileaflet bovine pericardial valve constructed in it, or a stent with a large valve from a cow's jugular vein mounted inside. Preliminary data are encouraging.
Alborino D et al: Value of exercise testing to evaluate the indication for surgery in asymptomatic patients with valvular aortic stenosis. J Heart Valve Dis 2002;11:204.
Carabello B: Clinical practice. Aortic stenosis. N Engl J Med 2002; 346:677.
Cosmi JE et al: The risk of the development of aortic stenosis in patients with benign aortic valve thickening. Arch Intern Med 2002;162:2345.
Cowell SJ et al; Scottish Aortic Stenosis and Lipid Lowering Trial, Impact on Regression (SALTIRE) Investigators: A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med 2005;352:2389.
Palta S et al: New insights into the progression of aortic stenosis: implications for secondary prevention. Circulation 2000; 101:2497.
Park MH: Timely intervention in asymptomatic aortic stenosis. Emerging clinical parameters may help predict outcomes. Postgrad Med 2001;110:28.
Pereira JJ et al: Survival after aortic valve replacement for severe aortic stenosis with low transvalvular gradients and severe left ventricular dysfunction. J Am Coll Cardiol 2002;39: 1356.
Touati GD et al: Management of patients with asymptomatic moderate aortic stenosis undergoing coronary artery bypass grafting. J Heart Valve Dis 2002;11:210.
Aortic Regurgitation (Chronic Regurgitation)
![]() Essentials of Diagnosis
Essentials of Diagnosis
Usually asymptomatic until middle age; presents with left-sided failure or chest pain.
Wide pulse pressure with associated peripheral signs.
Hyperactive, enlarged LV.
Diastolic murmur along left sternal border.
ECG shows LVH; radiograph shows LV dilation. Echocardiography/Doppler confirms diagnosis and estimates severity.
Afterload reduction proven to be of benefit if the LV is dilated (LV end-diastolic dimension > 5.0 cm).
Surgery indicated for symptoms or EF < 55% or LV end-systolic dimension by echocardiography > 5.0 cm.
General Considerations
Rheumatic aortic regurgitation has become much less common than in the preantibiotic era, and nonrheumatic causes now predominate. These include congenitally bicuspid valves, infective endocarditis, and hypertension. Many patients have aortic regurgitation secondary to aortic root diseases such as cystic medial necrosis, Marfan syndrome, or aortic dissection. Rarely, inflammatory diseases, such as ankylosing spondylitis or Reiter's syndrome, may be causative.
Chronic aortic regurgitation presents both an increased preload and an increased afterload to the LV. The response to these effects is to hypertrophy by laying sarcomeres end to end, increasing the LV chamber size greater than the wall thickness (eccentric hypertrophy). The amount of hypertrophy is substantial and greater than that seen in aortic stenosis or mitral regurgitation.
Clinical Findings
A. Symptoms and Signs
The clinical presentation is determined by the rapidity with which regurgitation develops. In chronic regurgitation, the only sign for many years may be a soft aortic diastolic murmur. As the valve deformity increases, the severity of the aortic regurgitation increases, diastolic BP falls, and the LV progressively enlarges. Most patients remain asymptomatic even at this point, and an often prolonged plateau phase, characterized by stable LV dilation, occurs. LV failure is a late event and may be sudden in onset. Exertional dyspnea and fatigue are the most frequent symptoms, but paroxysmal nocturnal dyspnea and pulmonary edema may also occur. Angina pectoris or atypical chest pain may occasionally be present. Associated CAD and presyncope or syncope are less common than in aortic stenosis.
Hemodynamically, because of compensatory LV dilation, patients eject a large stroke volume, which is adequate to maintain forward cardiac output until late in the course of the disease. LV diastolic pressure remains normal also but may rise when heart failure occurs.
P.341
Abnormal LV systolic function, as manifested by reduced EF and increasing end-systolic LV volume, is a sign that surgical intervention is warranted.
The major physical findings in chronic aortic regurgitation relate to the high stroke volume being ejected into the systemic vascular system with rapid runoff as the regurgitation takes place. This results in a wide arterial pulse pressure. The pulse has a rapid rise and fall (water-hammer pulse or Corrigan's pulse), with an elevated systolic and low diastolic pressure. The large stroke volume is also responsible for characteristic findings such as Quincke's pulses (subungual capillary pulsations), Duroziez's sign (to and fro murmur over a partially compressed peripheral artery, commonly the femoral), and Musset's sign (head bob with each pulse). In younger patients, the increased stroke volume may summate with the reflected wave from the periphery and create an even higher systolic pressure in the extremity compared with the central aorta. Since the peripheral bed is much larger in the leg than the arm, the BP in the leg may be over 40 mm Hg higher than in the arm (Hill's sign). The apical impulse is prominent, laterally displaced, usually hyperdynamic, and may be sustained. A systolic murmur is usually present and may be quite soft and localized; the aortic diastolic murmur is usually high-pitched and decrescendo. A mid or late diastolic low-pitched mitral murmur (Austin Flint murmur) may be heard in advanced aortic regurgitation, owing to obstruction of mitral flow produced by partial closure of the mitral valve by the regurgitant jet and the rapidly rising LV diastolic pressure.
When aortic regurgitation develops acutely (as in aortic dissection or infective endocarditis), LV failure, manifested primarily as pulmonary edema, may develop rapidly, and surgery is urgently required. Patients with acute aortic regurgitation do not have the dilated LV of chronic aortic regurgitation and the extra volume is handled poorly. For the same reason, the diastolic murmur is shorter and may be minimal in intensity, and the pulse pressure may not be widened, making clinical diagnosis difficult. The mitral valve may close prematurely before systole has been initiated (pre-closure) due to the rapid rise in the LV diastolic pressure and the first heart sound is thus diminished or inaudible.
B. Diagnostic Studies
The ECG usually shows moderate to severe LVH. Radiographs show cardiomegaly with LV prominence and sometimes dilated aorta.
Echocardiography demonstrates the major diagnostic features, including whether the lesion involves the proximal aortic root and what valvular disease is present. Serial assessments of LV size and function are critical in determining the timing for valve replacement. Color Doppler techniques can qualitatively estimate the severity of regurgitation, though some mild regurgitation due to aortic valve closure is not uncommon and should not be overinterpreted. Cardiac MRI and CT have a role in estimating aortic root size, particularly when there is concern for an ascending aneurysm. MRI can provide a regurgitant fraction to help confirm severity. Scintigraphic studies are infrequently used but can quantify LV function and functional reserve during exercise. Exercise increases the heart rate and reduces the diastolic time, resulting in less aortic regurgitation per beat; this complicates interpretation of the exercise EF. Cardiac catheterization may be unnecessary in younger patients, particularly those with acute aortic regurgitation, but can help define hemodynamics, aortic root abnormalities, and associated CAD preoperatively in older patients.
Treatment & Prognosis
Aortic regurgitation that appears or worsens during or after an episode of infective endocarditis or aortic dissection may lead to acute severe LV failure or subacute progression over weeks or months. The former usually presents as pulmonary edema; surgical replacement of the valve is indicated even during active infection. These patients may be transiently improved or stabilized by vasodilators.
Chronic regurgitation has a long natural history, but the prognosis without surgery becomes poor when symptoms occur. Since aortic regurgitation places both a volume and afterload increase on the LV, vasodilators, such as hydralazine, nifedipine, and ACE inhibitors, can reduce the severity of regurgitation, and prophylactic treatment, which may postpone or avoid surgery in asymptomatic patients with severe regurgitation and dilated LVs, is indicated. Most clinicians prescribe ACE inhibitors whenever the LV diastolic size is increased > 5.0 cm as shown on echocardiogram. -Blocker therapy may slow the rate of aortic dilation in Marfan syndrome by reducing the dP/dt, though the slower heart rates that result may theoretically increase the diastolic time and the amount of regurgitation per beat. Patients with aortic regurgitation need to be monitored serially by echocardiography. Surgery is indicated once symptoms emerge or for any evidence of LV dysfunction. LV dysfunction in this situation can be defined by echocardiography if the EF is < 55% or if the LV end-systolic dimension is > 5.0 cm, even in the asymptomatic patient. In addition, aortic root diameters of > 5.0 cm in Marfan or > 5.5 cm in non-Marfan patients are indications for surgery to avoid rapid expansion. Although the operative mortality rate is higher when LV function is severely impaired, valve replacement or repair is still indicated, since LV function often improves and the long-term prognosis is thereby enhanced even in this situation. The issues with AVR covered in the above section concerning aortic stenosis pertain here. Currently, however, there are no percutaneous approaches to aortic regurgitation. The choice of prosthetic valve for AVR depends on the patient's age and compatibility with warfarin anticoagulation.
The operative mortality rate is usually in the 3 5% range. Aortic regurgitation due to aortic root disease requires
P.342
repair or replacement of the root. Though valve-sparing operations have improved recently, most patients with root replacement undergo valve replacement at the same time. Root replacement procedures include the Ross procedure (moving the pulmonary valve to the aortic position and replacing the pulmonary valve with a homograft or, less commonly, bioprosthetic valve), the direct homograft for the aortic root and valve, and the Bentall procedure (the use of a Dacron sheath with a mechanical or bioprosthetic valve sewn in place). Root replacement in association with valve replacement requires reanastomosis of the coronary arteries, and thus the procedure is more complex than valve replacement alone. Following surgery, LV size usually decreases and LV function generally improves even when the baseline EF is depressed. For that reason, a surgical approach is often recommended. However, the improvement in EF is generally much less, and the surgical risk is higher than in aortic stenosis patients with a similar EF.
Chaliki HP et al: Outcomes after aortic valve replacement in patients with severe aortic regurgitation and markedly reduced left ventricular function. Circulation 2002;106:2687.
Enriquez-Sarano M et al: Clinical practice. Aortic regurgitation. N Engl J Med 2004;351:1539.
Evangelista A et al: Long-term vasodilator therapy in patients with severe aortic regurgitation. N Engl J Med 2005;353: 1342.
Ha JW et al: Is prophylactic aortic valve replacement indicated during mitral valve surgery for mild to moderate aortic valve disease? Ann Thorac Surg 2002;74:1115.
Hicks GL Jr et al: Update on indications for surgery in aortic insufficiency. Curr Opin Cardiol 2002;17:172.
Scognamiglio R et al: Long-term survival and functional results after aortic valve replacement in asymptomatic patients with chronic severe aortic regurgitation and left ventricular dysfunction. J Am Coll Cardiol 2005;45:1025.
Tricuspid Stenosis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Female predominance.
History of rheumatic heart disease. Carcinoid disease more common in the United States.
Elevated JVP with prominent a wave.
Right heart failure after tricuspid surgery or in rheumatic disease or carcinoid syndrome.
Echocardiography/Doppler key to diagnosis.
Mean valve gradient > 5 mm Hg by echocardiography indicates severe tricuspid stenosis.
General Considerations
Tricuspid stenosis is usually rheumatic in origin, though in the United States, tricuspid stenosis is more commonly due to tricuspid valve repair or replacement or to carcinoid syndrome than to rheumatic fever. Tricuspid regurgitation frequently accompanies the lesion. It should be suspected when right heart failure appears in the course of mitral valve disease without significant pulmonary hypertension, or in the postoperative period after tricuspid valve repair or replacement.
Clinical Findings
Tricuspid stenosis is characterized by right heart failure and hepatomegaly, ascites, and dependent edema. A giant a wave is seen in the JVP, which is elevated. The typical diastolic rumble along the lower left sternal border mimics mitral stenosis, with the rumble increasing upon inspiration. In sinus rhythm, a presystolic liver pulsation may be found.
Diagnostic Studies
In the absence of atrial fibrillation, the ECG reveals RA enlargement. The chest radiograph may show marked cardiomegaly with a normal PA size. A dilated SVC and azygous vein may be evident.
The normal valve area of the tricuspid valve is 10 cm2, so significant stenosis must be present to produce a gradient. Hemodynamically, a mean diastolic pressure gradient of > 5 mm Hg is considered significant, though even a 2 mm Hg gradient can be considered abnormal. This can be demonstrated by echocardiography or at cardiac catheterization. The echocardiogram reveals constricted motion and often thickening of the leaflets. At catheterization, the RA pressures demonstrate prominent a waves and with a slow y descent because of the slow RV filling. If there is associated tricuspid regurgitation a c-v wave will be observed.
Treatment & Prognosis
Tricuspid stenosis may be progressive, eventually causing severe right heart failure. Initial therapy is directed at reducing the fluid congestion, with diuretics the mainstay. When there is considerable bowel edema, torsemide may have an advantage over other loop diuretics, such as furosemide. Aldactone may also help if there is ascites. Neither surgical nor percutaneous valvuloplasty is effective for residual tricuspid regurgitation after tricuspid valve procedures, and tricuspid valve replacement is clearly the preferred surgical approach. Opening the commissure between the anterior and posterior leaflets is particularly prone to producing tricuspid regurgitation. Mechanical tricuspid valve replacement is rarely done because the low flow predisposes to thrombosis and because the mechanical valve cannot be crossed should the need arise for right heart catheterization or pacemaker implantation. Therefore, bioprosthetic valves are almost always used. Often tricuspid valve replacement is done in conjunction with mitral valve replacement for mitral stenosis.
P.343
Garcia-Pinilla JM et al: Reversible tricuspid stenosis secondary to massive ascites in hepatic cirrhosis. Ann Intern Med 2004; 140:233.
Staicu I et al: Tricuspid stenosis: a rare cause of heart failure in the United States. Congest Heart Fail 2002;8:281.
Thatipelli MR et al: Isolated tricuspid stenosis and heart failure: a focus on carcinoid heart disease. Congest Heart Fail 2003; 9:294.
Tricuspid Regurgitation
![]() Essentials of Diagnosis
Essentials of Diagnosis
Frequently occurs in patients with pulmonary or cardiac disease, especially if pulmonary hypertension is present (functional tricuspid regurgitation).
Systolic c-v wave in jugular venous pulsations.
Holosystolic murmur along left sternal border, which increases with inspiration.
Echocardiography useful in determining cause (low- or high-pressure tricuspid regurgitation).
General Considerations
The tricuspid valve apparatus differs in many ways from the mitral valve apparatus. Besides having three leaflets rather than two, the tricuspid valve has many chordae that attach to the RV endocardium rather than to discrete papillary muscles, and chordal attachments to the RV septum. The result of this anatomic feature is that tricuspid valvular incompetence often occurs whenever there is RV dilation from any cause. As tricuspid regurgitation increases, the RV size increases further, and this in turn worsens the tricuspid regurgitation. The causes of tricuspid regurgitation thus relate to anatomic issues with either the valve itself or to the RV geometry. An enlarged, dilated RV may be present if there is pulmonary hypertension for any reason, in severe pulmonic regurgitation, or in cardiomyopathy. The RV may be injured from myocardial infarction or may be inherently dilated due to infiltrative diseases (RV dysplasia or sarcoidosis). Usually the RV dilation is secondary to left heart failure. Inherent abnormalities of the tricuspid valve include Ebstein's anomaly (displacement of the septal and posterior leaflets into the RV), tricuspid valve prolapse, carcinoid plaque formation, collagen disease inflammation, tricuspid endocarditis, or RV pacemaker catheter injury.
Clinical Findings
The symptoms and signs of tricuspid regurgitation are identical to those resulting from RV failure due to any cause. As a generality, the diagnosis can be made by careful inspection of the JVP. The JVP waveform should decline during ventricular systole (the x descent). The timing of this decline can be observed by palpating the opposite carotid artery. As tricuspid regurgitation worsens, more and more of this valley in the JVP is filled with the regurgitant wave until all of the x descent is obliterated and a positive systolic waveform will be noted in the JVP. An associated tricuspid regurgitation murmur may or may not be present and can be distinguished from mitral regurgitation by the left parasternal location and increase with inspiration. An S3 may accompany the murmur. Cyanosis may be present if the increased RA pressure stretches the atrial septum and opens a PFO or there is a true ASD (eg, in about 50% of patients with Ebstein's anomaly).
The ECG is usually nonspecific, though atrial fibrillation is not uncommon. The chest radiograph may reveal evidence for an enlarged RA or dilated azygous vein and pleural effusion. The echocardiogram helps assess severity of tricuspid regurgitation, PA pressure, and RV size and function. A paradoxically moving interventricular septum may be present. Catheterization confirms the presence of the regurgitant wave in the RA and elevated RA pressures. If the PA or RV systolic pressure is < 40 mm Hg, primary tricuspid regurgitation should be suspected.
Treatment & Prognosis
Minor tricuspid regurgitation is well tolerated. Severe tricuspid regurgitation results in hepatomegaly, edema, and ascites. In patients in the intensive care unit, tricuspid regurgitation may result in erroneous thermodilution cardiac output measurements because the recirculation of the saline bolus back to the RA reduces the temperature signal to noise required to perform the measurement. When present, bowel edema may reduce the effectiveness of oral furosemide, and intravenous diuretics should initially be used. Torsemide is better absorbed in this situation. Aldosterone antagonists have a role as well, particularly if ascites is present. At times, the efficacy of loop diuretics can be enhanced by adding a thiazide diuretic.
Definitive treatment usually requires elimination of the cause of the tricuspid regurgitation. If the problem is left heart disease (cardiomyopathy, aortic or mitral valve disease), then its treatment may lower pulmonary pressures, reduce RV size, and resolve the tricuspid regurgitation. Other causes for pulmonary hypertension, such as hypoxemia or pneumonia, may also need to be addressed and treated appropriately. Treatment for primary and secondary causes of pulmonary hypertension will generally reduce the tricuspid regurgitation. If surgery is contemplated for other reasons, especially mitral valve disease, then tricuspid annuloplasty is generally performed at the same time. Annuloplasty without insertion of a prosthetic ring (DeVega annuloplasty) may be effective to reduce the annular dilation. The valve can occasionally be repaired in tricuspid valve endocarditis. In heroin addicts with tricuspid regurgitation due to endocarditis, the tricuspid valve may be temporarily removed to aid in cure of the endocarditis, though it must eventually be replaced (usually by 6 months). If there is an inherent defect in the
P.344
tricuspid apparatus that cannot be repaired, then replacement of the tricuspid valve is warranted. Generally, a bioprosthetic valve, and not a mechanical valve, is used. Anticoagulation is not required unless there is associated atrial fibrillation.
Behm CZ et al: Clinical correlates and mortality of hemodynamically significant tricuspid regurgitation. J Heart Valve Dis 2004;13:784.
Filsoufi F et al: Long-term outcomes of tricuspid valve replacement in the current era. Ann Thorac Surg 2005;80:845.
Hung MJ et al: Reversible left ventricular function after tricuspid valve replacement for a patient with congenital isolated severe tricuspid regurgitation. Echocardiography 2002;19: 517.
Koelling TM et al: Prognostic significance of mitral regurgitation and tricuspid regurgitation in patients with left ventricular systolic dysfunction. Am Heart J 2002;144:524.
Nath J et al: Impact of tricuspid regurgitation on long-term survival. J Am Coll Cardiol 2004;43:405.
Pulmonic Regurgitation
![]() Essentials of Diagnosis
Essentials of Diagnosis
Most cases are due to pulmonary hypertension.
Loud diastolic (Graham-Steell) murmur in high-pressure pulmonic regurgitation.
Soft or no murmur in low-pressure pulmonic regurgitation.
Echocardiogram is definitive in high-pressure but may be less helpful in low-pressure pulmonic regurgitation.
Low-pressure pulmonic regurgitation is well tolerated.
General Considerations
Pulmonary valve regurgitation can be divided into high-pressure causes (due to pulmonary hypertension) and low-pressure causes (usually due to a dilated pulmonary annulus [idiopathic or traumatic] or to plaque from carcinoid disease or following surgical repair). It may be iatrogenic, eg, frequently occurring after repair of tetralogy of Fallot. Because the RV tolerates a volume load more than a pressure load, it tends to tolerate pulmonic regurgitation well.
Clinical Findings
On examination, a hyperdynamic RV can usually be palpated. If the PA is enlarged, it may be palpated along the left sternal border. P2 will be palpable in pulmonary hypertension and both systolic and diastolic thrills are occasionally noted. On auscultation, the second heart sound may sound widely split due to prolonged RV systole. Systolic clicks may be noted as well as a right-sided gallop. In high-pressure pulmonic regurgitation, the pulmonary diastolic (Graham-Steell) murmur is readily audible. It is often due to a dilated pulmonary annulus. The murmur increases with inspiration and diminishes with the Valsalva maneuver. It is often confused with the murmur of aortic regurgitation. In low-pressure pulmonic regurgitation, the PA diastolic pressure may be only a few mm Hg higher than the RV diastolic pressure and there is little diastolic gradient to produce a murmur or characteristic echocardiography/Doppler findings. At times, only contrast angiography of the main PA will show the free flowing pulmonic regurgitation in this situation.
The ECG is generally of little value. The chest radiograph may show only the enlarged RV and PA. Echocardiography may demonstrate evidence of RV volume overload (paradoxic septal motion), and Doppler can determine peak systolic RV pressure and reveal any associated tricuspid regurgitation. The size of the main PA can be determined and colorflow Doppler can demonstrate the pulmonic regurgitation, particularly in the high-pressure situation. Cardiac MRI and CT can be useful for assessing the size of the PA, for imaging the jet lesion, for excluding other causes of pulmonary hypertension (eg, thromboembolic disease, peripheral PA stenosis), and for evaluating RV function.
Treatment & Prognosis
Pulmonic regurgitation rarely needs specific therapy other than treatment of the primary cause. In low-pressure pulmonic regurgitation due to surgical patch repair of tetralogy of Fallot, pulmonary valve replacement may be indicated if RV enlargement or dysfunction is present. In carcinoid heart disease, pulmonary valve replacement with a porcine bioprosthesis may be undertaken, though the plaque from this disorder eventually covers the prosthetic valve and this tends to limit the lifespan of these valves. In high-pressure pulmonic regurgitation, treatment to control the cause of the pulmonary hypertension is key.
Backer CL: Severe pulmonary valvar insufficiency should be aggressively treated. Cardiol Young 2005;15(Suppl 1):64.
Bouzas B et al: Pulmonary regurgitation: not a benign lesion. Eur Heart J 2005;26:433.
Gaynor JW: Severe pulmonary insufficiency should be conservatively treated. Cardiol Young 2005;15(Suppl 1):68.
Choice & Management of Prosthetic Valves & Percutaneous Approaches
In general, surgical results for valve replacement have improved over the past few decades. There is clearly interest, particularly among patients, for less invasive surgical procedures. This interest has led to mini-incisional
P.345
approaches to aortic, mitral, and tricuspid valve replacement and repair. Percutaneous valvuloplasty has replaced surgical commissurotomy for mitral stenosis in most cases. However, percutaneous valvuloplasty is effective in aortic stenosis only in children and adolescents; it has a very limited role in adults. Neither surgical nor percutaneous tricuspid valvuloplasty is very effective.
Repair of the mitral valve is successful in appropriate patients and has lowered the threshold for intervention in mitral regurgitation. Some experts recommend that all mitral prolapse patients with mitral regurgitation undergo mitral valve repair even when other indications are not present. However, to recommend this option, the surgeon performing the procedure must have extensive experience with excellent results. When tricuspid regurgitation is present and mitral valve repair or replacement is planned, tricuspid repair is now commonplace. Aortic valve sparing procedures are also improving and may obviate the need for AVR, especially in patients undergoing root replacement. Early experience, though, with aortic valve repair is mixed. Direct valvular repair and removal of vegetations are also viable options in some patients with endocarditis.
The choice of prosthetic valve depends on a variety of considerations, including the expected survival of the patient versus the durability of the valve and the safety of warfarin for the patient. Bioprosthetic valves usually have a life expectancy of 10 15 years, but less in young patients and those on dialysis. The lifespan of bovine pericardial valves is somewhat longer than porcine valves in some series. Bioprosthetic valves, homografts, and the Ross procedure do not require anticoagulation with warfarin. Mechanical valves have a much longer lifespan, but all require use of warfarin (and frequently aspirin as an adjunct). The long-term risk of warfarin depends on patient compliance and whether there is coexisting disease that may predispose to bleeding.
Mechanical mitral valve prostheses pose a greater risk for thrombosis than mechanical aortic valves. For that reason, the INR should be kept between 2.5 and 3.5 for mechanical mitral prosthetic valves but can be kept between 2.0 and 2.5 for mechanical aortic prosthetic valves. Enteric-coated aspirin 81 mg once daily is given to patients with both types of mechanical valves, but appears to be more important for mitral valve prostheses.
Warfarin causes fetal skeletal abnormalities in about 2% of women who become pregnant while taking warfarin, so every effort is made to defer valve replacement in women until after childbearing age. However, if a woman with a mechanical valve becomes pregnant while taking warfarin, the risk of stopping warfarin is higher for the mother than the risk of continuing warfarin for the fetus. The risk of warfarin to the fetal skeleton is greatest during the first trimester, so if pregnancy is planned in a woman with a mechanical valve, unfractionated heparin is often used temporarily during the first trimester. After the first trimester, warfarin use is safe again until 2 weeks before planned delivery, when the patient should be switched back to unfractionated heparin. Low-molecular-weight heparin has not been shown to be effective and should not be substituted for unfractionated heparin in the pregnant patient.
When patients with mechanical valves must undergo noncardiac surgery, the risk of thrombosis from stopping warfarin versus the risk of excessive bleeding from continuing it must be weighed. In general, for bileaflet aortic mechanical valves, warfarin can be stopped 3 4 days ahead of the surgical procedure and resumed the night of the procedure without bridging by use of unfractionated or low-molecular-weight heparin. In patients with a bileaflet mitral valve, warfarin may be stopped 3 4 days ahead of time and bridging low-molecular-weight heparin used until the warfarin is restarted and the INR is > 1.9. The highest risk for thromboembolic events occurs in patients with a ball-in-cage mechanical valve in any position, and in patients with a bileaflet mitral valve prosthesis and concurrent atrial fibrillation. In this latter group, bridging with either unfractionated or low-molecular-weight heparin is warranted once the INR has dropped below 2.0 before surgery; heparin is continued until warfarin is restarted and the INR is once again > 1.9.
Block PC et al: Percutaneous approaches to valvular heart disease. Curr Cardiol Rep 2005;7:108.
Bloomfield P: Choice of heart valve prosthesis. Heart 2002;87: 583.
Puvimanasinghe JP et al: Comparison of outcomes after aortic valve replacement with a mechanical valve or a bioprosthesis using microsimulation. Heart 2004;90:1172.
Salem DN et al: Antithrombotic therapy in valvular heart disease native and prosthetic: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126(3 Suppl):457S.
Thamilarasan M: Choosing the most appropriate valve operation and prosthesis. Cleve Clin J Med 2002;69:688.
Thistlethwaite PA et al: Tricuspid valvular disease in the patient with chronic pulmonary thromboembolic disease. Curr Opin Cardiol 2003;18:111.
Coronary Heart Disease (Atherosclerotic Cad; Ischemic Heart Disease)
Coronary heart disease, or atherosclerotic CAD, is the number one killer in the United States and worldwide. Every minute, an American dies of coronary heart disease. About 40% of people who experience a coronary attack will die of it in the same year. Coronary heart disease is responsible for more than one in five deaths
P.346
and nearly 700,000 deaths per year in the United States. More than 10 times the number of women die each year from cardiovascular disease than from breast cancer. Coronary heart disease afflicts over 13 million Americans and the prevalence rises steadily with age; thus, the aging of the U.S. population promises to increase the overall burden of coronary heart disease.
Risk Factors for CAD
Epidemiologic studies have identified a number of important risk factors for CAD. Most patients with coronary heart disease have some identifiable risk factor. These include a positive family history (the younger the onset in a first-degree relative, the greater the risk), male gender, blood lipid abnormalities, diabetes mellitus, hypertension, physical inactivity and obesity, and cigarette smoking. A recent large international epidemiologic study of myocardial infarction shows that most of the population-attributable risk is explained by eight factors: abnormal lipids, smoking, hypertension, diabetes mellitus, abdominal obesity, psychosocial factors, consumption of too few fruits and vegetables and too much alcohol, and lack of regular physical activity. Smoking remains the number one preventable cause of cardiovascular disease worldwide. Although smoking rates have declined in the United States in recent decades, 21% of women and 25% of men smoke. According to the World Health Organization, 1 year after quitting, the risk of coronary heart disease decreases by 50%. Various interventions, including physician counseling, formal smoking cessation programs, nicotine replacement therapy, and bupropion, have been shown to increase the likelihood of successful cessation (see Chapter 1).
Overwhelming evidence indicates that hypercholesterolemia and other lipid abnormalities provide an important modifiable risk factor for coronary heart disease. Risk increases progressively with higher levels of low-density lipoprotein (LDL) cholesterol and declines with higher levels of high-density lipoprotein (HDL) cholesterol. Composite risk scores, such as the Framingham score (see Table 28-2), provide estimates of 10-year probability of development of coronary heart disease that can guide primary prevention strategies.
The metabolic syndrome is defined as a constellation of three or more of the following: abdominal obesity, triglycerides 150 mg/dL, HDL cholesterol < 40 mg/dL for men and < 50 mg/dL for women, fasting glucose 110 mg/dL, and hypertension. This syndrome, recognized as a major contributor to coronary heart disease risk, is increasing in prevalence at an alarming rate. Related to the metabolic syndrome, the epidemic of obesity in the United States is likewise a major factor contributing to coronary heart disease risk. In 2001, over 20% of the population was obese (body mass index [BMI] > 30 kg/m2), a 74% increase compared to 1991. Particularly alarming is the rapidly increasing incidence of obesity in adolescents in the United States. Increasing physical activity is an important goal to help combat obesity and its consequences. Although the American Heart Association continues to promote a diet based largely on low saturated fat, more information is needed on the health consequences of all diets, especially given the lack of protection from a low-fat diet in the largest randomized study ever done, the Women's Health Initiative trial. Surprisingly, low carbohydrate diets, even when high in saturated fat, may improve the cholesterol profile in overweight men, at least temporarily. Fish, rich in omega-3 fatty acids, may help protect against vascular disease and it is recommended that it be eaten three times a week by patients at risk.
It is now clear that markers of inflammation are strong risk factors for CAD. High sensitivity CRP is the best-characterized inflammatory marker and is available for clinical use, but others include interleukin-6, CD-40 ligand, myelopyroxidase, and placental growth factor. Although CRP levels > 10 mcg/mL are often found in systemic inflammation, levels < 1, 1 3, and > 3 mcg/mL, respectively, identify patients at low, intermediate, and high risk for future cardiovascular events. The prognostic value of CRP levels is independent and additive to lipid levels. Use of CRP may be helpful in determining which patients at intermediate risk according to the Framingham 10-year risk of coronary heart disease calculation (score of 10 20%) are at high enough risk to warrant more intensive primary prevention, including use of statins to lower LDL cholesterol. CRP levels are often elevated in patients who have other conditions associated with accelerated atherosclerosis, such as diabetes, the metabolic syndrome, and obesity. In patients presenting with acute coronary syndromes, these elevations identify a group that is at high risk for early recurrent events.
Pathophysiology
Knowledge concerning the pathophysiology of atherosclerosis has accumulated rapidly. Abnormal lipid metabolism or excessive intake of cholesterol and saturated fats especially when superimposed on a genetic predisposition is important in early stages of the atherosclerotic process. The initial step is the fatty streak, or subendothelial accumulation of lipids and lipid-laden monocytes (macrophages). LDLs are the major atherogenic lipid. HDLs, in contrast, are protective by virtue of their role in reverse cholesterol transport, removing cholesterol from the vascular wall. The pathogenetic role of other lipids, including triglycerides, is less clear. LDLs undergo in situ oxidation, which makes them more difficult to mobilize as well as locally cytotoxic.
Macrophages migrate into the subendothelial space and take up lipids, giving them the appearance of foam cells. As the plaque progresses, smooth muscle cells also migrate into the lesion. At this stage, the lesion may be hemodynamically insignificant, but endothelial function is abnormal and its ability to limit the entry of lipoproteins into the vessel wall is impaired. If the plaque remains stable, a fibrous cap forms, the lesion
P.347
becomes calcified, remodeling of the vessel wall occurs, and ultimately the vessel lumen may become narrowed, although extensive atherosclerosis may be present even before this occurs.
Although many atherosclerotic plaques remain stable or progress only gradually, others may rupture, often related to the inflammatory process and metalloproteinase activity. The rupture causes turbulent flow, extrusion of lipids and fatty gruel, and exposure of tissue factor that result in a cascade of events culminating in intravascular thrombosis. The outcome of these events is determined in large part by whether the vessel becomes occluded, which depends on the lesion anatomy as well as the balance of pro- and antithrombotic and pro- and antifibrinolytic forces. The result may be partial or complete vessel occlusion (causing the symptoms of unstable angina or myocardial infarction), or the plaque may become restabilized, often with more severe stenosis. Transient occlusion and/or embolization of platelet and thrombin debris, which may result in elevation in serum troponin, predispose to clinical events and portend a worse prognosis.
Several features are associated with enhanced plaque vulnerability, including a higher lipid content, a higher concentration of macrophages, especially in the plaque shoulder, and a very thin fibrous cap. Lesions with these characteristics are often relatively early lesions that can be responsible for acute myocardial infarction or sudden death as the first manifestation of coronary disease. Such lesions may occur in up to 50% of cases. This abrupt progression explains why most infarctions do not occur at the site of preexisting critical stenosis. Conversely, the relatively greater reduction in clinical events than in lesion severity in lipid-lowering treatment trials is probably explained by the stabilization of these early nonfibrotic lesions.
American Heart Association: Heart Disease and Stroke Statistics 2006 Update. Dallas, TX: American Heart Association, 2006. (http://www.aha.org)
Centers for Disease Control and Prevention. Overweight and Obesity: Home (http://www.cdc.gov/nccdphp/dnpa/obesity; accessed March 2006)
Haffner SM: Insulin resistance, inflammation, and the prediabetic state. Am J Cardiol 2003;92:18J.
Howard BV et al: Low-fat dietary pattern and risk of cardiovascular disease: the Women's Health Initiative Randomized Dietary Modification Trial. JAMA 2006;295:655.
Libby P: Current concepts of the pathogenesis of the acute coronary syndromes. Circulation 2001;104:365.
Libby P et al: Inflammation and atherosclerosis. Circulation 2002;105:1135.
Nissen SE et al: Intravascular ultrasound: novel pathophysiological insights and current clinical applications. Circulation 2001;103:604.
Pearson TA et al; Centers for Disease Control and Prevention; American Heart Association: Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003;107:499.
Resnick HE et al: Diabetes and cardiovascular disease. Annu Rev Med 2002;53:245.
Ridker PM: Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation 2003; 107:363.
Yusuf S et al; INTERHEART Study Investigators: Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004;364:937.
Primary & Secondary Prevention of Coronary Heart Disease
Although many risk factors for CAD are not modifiable, it is now clear that interventions such as smoking cessation, treatment of dyslipidemia, and lowering of BP can both prevent coronary disease and delay its progression and complications after it is manifest. Treatment of lipid abnormalities delays the progression of atherosclerosis and in some cases may produce regression. Even in the absence of regression, fewer new lesions develop, endothelial function may be restored, and coronary event rates are markedly reduced in patients with clinical evidence of vascular disease.
A series of clinical trials has demonstrated the efficacy of hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) in preventing death, coronary events, and strokes. Beneficial results have been found in patients who have already experienced coronary events (secondary prevention), in those at particularly high risk for events (diabetics and patients with peripheral artery disease), and those with elevated LDL cholesterol without multiple risk factors. Benefits occurred regardless of age, race, or the presence of hypertension. There is now clear evidence that treatment with statins can prevent coronary events and stroke in patients without clinically manifest atherosclerosis (primary prevention) and LDL levels as low as 130 mg/dL. It is also clear that for patients with vascular disease, statins provide benefit for those with normal cholesterol levels, and that more aggressive LDL lowering is associated with greater benefits. The Heart Protection Study demonstrated that simvastatin 40 mg a day reduces vascular events by more than 20% in patients with prior myocardial infarction, stroke, peripheral vascular disease, or diabetes with total cholesterol levels as low as 135 mg/dL. The treatment benefit was similar regardless of baseline LDL cholesterol, with equal benefit above or below 100 mg/dL. This result suggests that all patients at significant risk for vascular events should receive a statin regardless of their cholesterol levels. The PROVE-IT trial showed that vascular events were reduced with more aggressive lipid lowering (atorvastatin 80 mg/d compared to pravastatin 40 mg/d following an acute coronary syndrome), providing more evidence of lower is better for patients with vascular disease. The TNT (Treating to New Targets) trial likewise found greater benefit with more aggressive LDL lowering (atorvastatin 80 mg versus 10 mg) in a population of patients with coronary heart disease and LDL cholesterol < 130 mg/dL.
P.348
The IDEAL (Incremental Decrease in End Points Through Aggressive Lipid Lowering) trial provided only modest support for very aggressive lipid lowering, showing a nonstatistically significant reduction in major coronary events with 80 mg/d of atorvastatin compared with 20 mg/d of simvastatin in patients with prior myocardial infarction. Although true regression of plaque is uncommon even with intensive lipid therapy (as in the REVERSAL and ASTEROID trials), progression can be prevented at least in the short run in most patients.
Treatment of abnormally low HDL levels or elevations of lipoprotein(a) and small, dense LDL particles is more difficult, but oral niacin in high dosages (2 3 g/d or more) may be effective. A trial in postinfarction patients has demonstrated that an increase in HDL levels with gemfibrozil (600 mg twice daily) in patients with relatively low LDL levels prolongs reinfarction-free survival, although the larger FIELD trial failed to show prevention of nonfatal myocardial infarction or coronary heart disease death with fenofibrate for patients with type 2 diabetes mellitus. The value of reducing elevated triglyceride levels is less clear, but since elevated triglycerides are often associated with other lipid abnormalities, treatment of high-risk patients with niacin, gemfibrozil, or fenofibrate for levels above 400 mg/dL is appropriate.
Because LDL oxidation appears to play a role in the atherogenicity of lipid molecules that have passed into the vessel wall, antioxidant therapy has been advocated as a preventive measure. Thus far, however, there are few data to support this popular concept, and many large, well-controlled studies have failed to demonstrate a benefit with vitamin E therapy. In fact, the Heart Protection Study and the Heart Outcomes Prevention Evaluation (HOPE) trial found that vitamin E may even be harmful by increasing the likelihood of heart failure and other trials have suggested that vitamin E may hinder the effectiveness of statin therapy.
Elevated plasma homocysteine levels are associated with an increased risk of vascular events. Although homocysteine levels can be reduced with dietary supplements of folic acid (1 mg/d) in combination with vitamin B6 and vitamin B12, two randomized clinical trials have shown that they are of little or no value in preventing vascular events.
Antiplatelet therapy is another very effective preventive measure. Aspirin (325 mg every other day) in males over the age of 50 years reduces the incidence of myocardial infarction. A similar approach (100 mg every other day), however, did not prevent myocardial infarction in women age 45 years or older, although stroke did appear to be reduced. Thus, the role of aspirin in primary prevention, including the dose, remains controversial. A prudent approach would be to administer 81 325 mg daily to men with multiple coronary risk factors or concomitant diabetes starting at age 45 50 years if no contraindication is present. While clopidogrel was found to be effective at preventing vascular events for 9 12 months after acute coronary syndromes, it was not found to be effective at preventing vascular events in the CHARISMA trial. This large trial included patients with clinically evident stable atherothrombosis or with multiple risk factors; all were treated with aspirin and observed for a median of 28 months.
The previously mentioned GISSI Prevention Trial found a significant reduction in mortality with administration of omega-3 fatty acid (1 g daily) which has an antiplatelet effect in addition to other purported mechanisms in postinfarction patients.
The effect of hormone replacement therapy in postmenopausal women has now been clarified, and it is clear that neither combined estrogen-progesterone nor estrogen alone therapy is protective (in fact both cause harm). Control of BP has been shown to prevent infarctions. Individuals who exercise for at least 30 minutes a week are at lower risk for subsequent coronary events, and 30 minutes of exercise five times a week reduces the risk of developing diabetes in half among patients at risk.
The HOPE and the EUROPA trials demonstrated that ACE inhibitors (ramipril 10 mg/d and perindopril 8 mg/d, respectively) reduce fatal and nonfatal vascular events (cardiovascular deaths, nonfatal myocardial infarctions, and nonfatal strokes) by 20 25% in patients at high risk, including diabetics with additional risk factors or patients with clinical coronary, cerebral, or peripheral arterial atherosclerotic disease. The PEACE trial did not show benefit of the ACE inhibitor trandolapril in a lower-risk population of patients with coronary heart disease, most of whom had been treated aggressively with medical and revascularization therapies. Therefore, while low-risk patients may not derive substantial benefits from ACE inhibitors, higher-risk patients with vascular disease, even in the absence of heart failure or LV dysfunction, should be treated with an ACE inhibitor.
Bhatt DL et al: Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006;354:1706.
Bonaa KH et al; NORVIT Trial Investigators: Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med 2006;354:1578.
Braunwald E et al; PEACE Trial Investigators: Angiotensin-converting-enzyme inhibition in stable coronary artery disease. N Engl J Med 2004;351:2058.
Cannon CP et al: Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495.
Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high-risk patients. BMJ 2002;324:71.
Fox KM et al: Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet 2003;362:782.
P.349
Grundy SM et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association: Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004;110:227.
Hu FB et al: Optimal diets for prevention of coronary heart disease. JAMA 2002;288:2569.
Keech A et al; FIELD study investigators: Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 2005;366:1849.
Khot UN et al: Prevalence of conventional risk factors in patients with coronary heart disease. JAMA 2003;290:898.
Larosa JC et al: Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005;352: 1423.
Lauer MS: Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med 2002;346:1468.
Lonn E et al; Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators: Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med 2006;354:1567.
MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002;360:7.
Nelson HD et al: Postmenopausal hormone replacement therapy: scientific review. JAMA 2002;288:872.
Nissen SE et al: Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004;291:1071.
Pearson TA et al: AHA Guidelines for Primary Prevention of Cardiovascular Disease and Stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee. Circulation 2002; 106:388.
Pedersen TR et al; Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) Study Group: High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005;294:2437.
Ridker PM et al: A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005;352:1293.
Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002;106:3143.
van den Hoogen PC et al: The relation between blood pressure and mortality due to coronary heart disease among men in different parts of the world. Seven Countries Study Research Group. N Engl J Med 2000;342:1.
Women's Health Initiative Investigators: Risks and benefits of estrogen plus progestin in healthy post-menopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA 2002;288:321.
Pathophysiology of Chronic Ischemia & Acute Coronary Syndromes
Chronic ischemia, including stable angina, is classically caused by supply and demand mismatch, where significant fixed coronary stenosis and/or excess myocardial demand result in ischemia. Precipitants include exercise, eating, cold weather, and emotional stress.
The acute coronary syndromes of unstable angina and myocardial infarction are generally caused by a combination of plaque disruption, platelet and thrombin-mediated coronary thrombosis, coronary spasm, and microvascular dysfunction. Of interest is the predilection for these episodes to occur in the early morning or shortly after arising. Antithrombotic therapy is directed toward inhibition of platelet activity (aspirin, clopidogrel, IIb/IIIa receptor antagonists), inhibition of coagulation (unfractionated or low-molecular-weight heparin), and fibrinolysis for ST-segment elevation myocardial infarction.
Some episodes of myocardial ischemia are symptomatic, causing angina pectoris; others are completely silent. Many silent episodes are brought on by emotional and mental stress. In patients with diagnosed coronary disease, as evidenced by prior myocardial infarction or angina, silent ischemic episodes have the same prognostic import as symptomatic ones. The prognosis for patients with only silent ischemia is not well established, nor is the potential benefit of preventing silent ischemia.
Faxon DP; American Heart Association: Atherosclerotic Vascular Disease Conference: Writing Group III: pathophysiology. Circulation 2004;109:2617.
Servoss SJ et al: Triggers of acute coronary syndromes. Prog Cardiovasc Dis 2002;44:369.
Myocardial Hibernation & Stunning
Areas of myocardium that are persistently underperfused but still viable may develop sustained contractile dysfunction. This phenomenon, which is termed myocardial hibernation, appears to represent an adaptive response but may lead to LV failure. It is important to recognize this phenomenon, since this form of dysfunction is reversible following coronary revascularization. Hibernating myocardium can be identified by radionuclide testing, positron emission tomography (PET), contrast-enhanced MRI, or its retained response to inotropic stimulation with dobutamine. A related phenomenon, termed myocardial stunning, is the occurrence of persistent contractile dysfunction following prolonged or repetitive episodes of myocardial ischemia.
Angina Pectoris
![]() Essentials of Diagnosis
Essentials of Diagnosis
Precordial chest pain, usually precipitated by stress or exertion, relieved rapidly by rest or nitrates.
ECG or scintigraphic evidence of ischemia during pain or stress testing.
Angiographic demonstration of significant obstruction of major coronary vessels.
P.350
General Considerations
Angina pectoris is usually due to atherosclerotic heart disease. Coronary vasospasm may occur at the site of a lesion or, less frequently, in apparently normal vessels. Other unusual causes of coronary artery obstruction such as congenital anomalies, emboli, arteritis, or dissection may cause ischemia or infarction. Angina may also occur in the absence of coronary artery obstruction as a result of severe myocardial hypertrophy, severe aortic stenosis or regurgitation, or in response to increased metabolic demands, as in hyperthyroidism, marked anemia, or paroxysmal tachycardias with rapid ventricular rates. Rarely, angina occurs with angiographically normal coronary arteries and without other identifiable causes. This presentation has been labeled syndrome X and is most likely due to inadequate flow reserve in the resistance vessels (microvasculature). Although treatment is often not very successful in relieving symptoms, the prognosis of syndrome X is good.
Clinical Findings
A. History
The diagnosis of angina pectoris depends principally upon the history, which should specifically include the following information: circumstances that precipitate and relieve angina, characteristics of the discomfort, location and radiation, duration of attacks, and effect of nitroglycerin.
1. Circumstances that precipitate and relieve angina
Angina occurs most commonly during activity and is relieved by resting. Patients may prefer to remain upright rather than lie down, as increased preload in recumbency increases myocardial work. The amount of activity required to produce angina may be relatively consistent under comparable physical and emotional circumstances or may vary from day to day. The threshold for angina is usually less after meals, during excitement, or on exposure to cold. It is often lower in the morning or after strong emotion; the latter can provoke attacks in the absence of exertion. In addition, discomfort may occur during sexual activity, at rest, or at night as a result of coronary spasm.
2. Characteristics of the discomfort
Patients often do not refer to angina as pain but as a sensation of tightness, squeezing, burning, pressing, choking, aching, bursting, gas, indigestion, or an ill-characterized discomfort. It is often characterized by clenching a fist over the mid chest. The distress of angina is rarely sharply localized and is not spasmodic.
3. Location and radiation
The distribution of the distress may vary widely in different patients but is usually the same for each patient unless unstable angina or myocardial infarction supervenes. In most cases, the discomfort is felt behind or slightly to the left of the mid sternum. When it begins farther to the left or, uncommonly, on the right, it characteristically moves centrally substernally. Although angina may radiate to any dermatome from C8 to T4, it radiates most often to the left shoulder and upper arm, frequently moving down the inner volar aspect of the arm to the elbow, forearm, wrist, or fourth and fifth fingers. Radiation to the right shoulder and distally is less common, but the characteristics are the same. Occasionally, angina may be felt initially in the lower jaw, the back of the neck, the interscapular area, high in the left back, or in the volar aspect of the wrist.
4. Duration of attacks
Angina is of short duration and subsides completely without residual discomfort. If the attack is precipitated by exertion and the patient promptly stops to rest, it usually lasts less than 3 minutes. Attacks following a heavy meal or brought on by anger often last 15 20 minutes. Attacks lasting more than 30 minutes are unusual and suggest the development of unstable angina, myocardial infarction, or an alternative diagnosis.
5. Effect of nitroglycerin
The diagnosis of angina pectoris is strongly supported if sublingual nitroglycerin promptly and invariably shortens an attack and if prophylactic nitrates permit greater exertion or prevent angina entirely.
B. Signs
Examination during a spontaneous or induced attack frequently reveals a significant elevation in systolic and diastolic BP, although hypotension may also occur, and may reflect more severe ischemia or inferior ischemia (especially with bradycardia) due to a Bezold-Jarisch reflex. Occasionally, a gallop rhythm and an apical systolic murmur due to transient mitral regurgitation from papillary muscle dysfunction are present during pain only. Supraventricular or ventricular arrhythmias may be present, either as the precipitating factor or as a result of ischemia.
It is important to detect signs of diseases that may contribute to or accompany atherosclerotic heart disease, eg, diabetes mellitus (retinopathy or neuropathy), xanthelasma, tendinous xanthomas, hypertension, thyrotoxicosis, myxedema, or peripheral vascular disease. Aortic stenosis or regurgitation, hypertrophic cardiomyopathy, and mitral valve prolapse should be sought, since they may produce angina or other forms of chest pain.
Differential Diagnosis
Angina can usually be diagnosed from a proper history. When atypical features are present such as prolonged duration (hours or days) or darting, knifelike pains at the apex or over the precordium ischemia is less likely.
Anterior chest wall syndrome is characterized by sharply localized tenderness of intercostal muscles. Inflammation of the chondrocostal junctions, which
P.351
may be warm, swollen, and red, may result in diffuse chest pain that is also reproduced by local pressure (Tietze's syndrome). Intercostal neuritis (due to herpes zoster, diabetes mellitus, etc) also mimics angina.
Cervical or thoracic spine disease involving the dorsal roots produces sudden sharp, severe chest pain suggesting angina in location and radiation but related to specific movements of the neck or spine, recumbency, and straining or lifting. Pain due to cervical or thoracic disk disease involves the outer or dorsal aspect of the arm and the thumb and index fingers rather than the ring and little fingers.
Peptic ulcer, chronic cholecystitis, esophageal spasm, and functional gastrointestinal disease may produce pain suggestive of angina pectoris. Reflux esophagitis is characterized by lower chest and upper abdominal pain after heavy meals, occurring in recumbency or upon bending over, and awakening patients several hours after eating. The pain is relieved by antacids, sucralfate, H2-receptor antagonists, or proton pump inhibitors. The picture may be especially confusing because ischemic pain may also be associated with upper gastrointestinal symptoms, and esophageal motility disorders may be improved by nitrates and calcium channel blockers. Assessment of esophageal motility may be helpful.
Degenerative and inflammatory lesions of the left shoulder and thoracic outlet syndromes may cause chest pain due to nerve irritation or muscular compression; the symptoms are usually precipitated by movement of the arm and shoulder and are associated with paresthesias.
Spontaneous pneumothorax may cause chest pain as well as dyspnea and may create confusion with angina as well as myocardial infarction. The same is true of pneumonia and pulmonary embolism. Dissection of the thoracic aorta can cause severe chest pain that is commonly felt in the back; it is sudden in onset, reaches maximum intensity immediately, and may be associated with changes in pulses. Other cardiac disorders such as mitral valve prolapse, hypertrophic cardiomyopathy, myocarditis, pericarditis, aortic valve disease, or RVH may cause atypical chest pain or even myocardial ischemia. Noninvasive testing and, in many cases, cardiac catheterization may be required to establish the diagnosis.
Evaluation of Patients with Angina Pectoris
A. Laboratory Findings
Serum lipid levels should be determined in all patients with suspected angina. Anemia and diabetes may also be investigated if clinically appropriate.
B. ECG
The resting ECG is normal in about 25% of patients with angina. In the remainder, abnormalities include old myocardial infarction, nonspecific ST-T changes, AV or intraventricular conduction defects, and changes of LVH. During anginal episodes, the characteristic ECG change is horizontal or downsloping ST-segment depression that reverses after the ischemia disappears. T wave flattening or inversion may also occur. Less frequently, ST-segment elevation is observed; this finding suggests severe (transmural) ischemia and often occurs with coronary spasm.
C. Exercise ECG
Exercise testing is the most useful noninvasive procedure for evaluating the patient with angina. Ischemia that is not present at rest is detected by precipitation of typical chest pain or ST-segment depression (or, rarely, elevation). Exercise testing is often combined with imaging studies (nuclear, echocardiography, or MRI [see below]), but in patients without baseline ST segment abnormalities or in whom anatomic localization is not necessary, the exercise ECG remains the recommended initial procedure because of considerations of cost and convenience.
Exercise testing can be done on a motorized treadmill or with a bicycle ergometer. A variety of exercise protocols are utilized, the most common being the Bruce protocol, which increases the treadmill speed and elevation every 3 minutes until limited by symptoms. At least two ECG leads should be monitored continuously.
1. Precautions and risks
The usually quoted risk of exercise testing is one infarction or death per 1000 tests, but individuals who continue to have pain at rest or minimal activity are at higher risk and should not be tested. Many of the traditional exclusions, such as recent myocardial infarction or CHF, are no longer used if the patient is stable and ambulatory, but aortic stenosis remains a contraindication. Although most tests are carried to a symptom-limited end point (except submaximal testing early postinfarction), the test should be terminated when hypotension, significant ventricular or supraventricular arrhythmias, more than mild to moderate angina, or more than 3- to 4-mm ST-segment depression occurs.
2. Indications
Exercise testing is used (1) to confirm the diagnosis of angina; (2) to determine the severity of limitation of activity due to angina; (3) to assess prognosis in patients with known coronary disease, including those recovering from myocardial infarction, by detecting groups at high or low risk; (4) to evaluate responses to therapy; and (5) less successfully, to screen asymptomatic populations for silent coronary disease. The latter application is controversial. Because false-positive tests often exceed true positives, leading to much patient anxiety and self-imposed or mandated disability, exercise testing of asymptomatic individuals should be done only for those at high risk (usually a strong family history of premature coronary disease or hyperlipidemia), those whose occupations place them or others at special risk
P.352
(eg, airline pilots), and older individuals commencing strenuous activity.
3. Interpretation
The usual ECG criterion for a positive test is 1 mm (0.1 mV) horizontal or downsloping ST-segment depression (beyond baseline) measured 80 milliseconds after the J point. By this criterion, 60 80% of patients with anatomically significant coronary disease will have a positive test, but 10 30% of those without significant disease will also be positive. False positives are uncommon when a 2-mm depression is present. Additional information is inferred from the time of onset and duration of the ECG changes, their magnitude and configuration, BP and heart rate changes, the duration of exercise, and the presence of associated symptoms. In general, patients exhibiting more severe ST-segment depression (> 2 mm) at low workloads (< 6 minutes on the Bruce protocol) or heart rates (< 70% of age-predicted maximum) especially when the duration of exercise and rise in BP are limited or when hypotension occurs during the test have more severe disease and a poorer prognosis. Depending on symptom status, age, and other factors, such patients should be referred for coronary arteriography and possible revascularization. On the other hand, less impressive positive tests in asymptomatic patients are often false positives. Therefore, exercise testing results that do not conform to the clinical picture should be confirmed by stress scintigraphy or echocardiography.
D. Scintigraphic Assessment of Ischemia
Two nuclear medicine studies provide additional information about the presence, location, and extent of CAD.
1. Myocardial perfusion scintigraphy
This test provides images in which radionuclide uptake is proportionate to blood flow at the time of injection. Thallium-201, technetium-99m sestamibi, and tetrafosmin are most frequently used. Areas of diminished uptake reflect relative hypoperfusion (compared with other myocardial regions). If the radiotracer is injected during exercise or dipyridamole- or adenosine-induced coronary vasodilation, scintigraphic defects indicate a zone of hypoperfusion that may represent either ischemia or scar. If the myocardium is viable, as relative blood flow equalizes over time or during a scintigram performed under resting conditions, these defects tend to fill in or reverse, indicating reversible ischemia. Defects observed when the radiotracer is injected at rest or still present 3 4 hours after an injection during exercise or pharmacologic vasodilation (intravenous adenosine or dipyridamole) usually indicate myocardial infarction (old or recent) but may be present with severe ischemia. Occasionally, other conditions, including infiltrative diseases (sarcoidosis, amyloidosis), left bundle branch block, and dilated cardiomyopathy, may produce resting or persistent perfusion defects.
In experienced laboratories, stress perfusion scintigraphy is positive in 75 90% of patients with anatomically significant coronary disease and in 20 30% of those without it. False-positive tests may occur as a result of diaphragmatic attenuation or, in women, attenuation through breast tissue. Tomographic imaging (single-photon emission computed tomography, SPECT) can reduce the severity of artifacts. Gated imaging allows for analysis of ventricular size, EF, and regional wall motion.
Myocardial scintigraphy is indicated (1) when the resting ECG makes an exercise ECG difficult to interpret (left bundle branch block, baseline ST-T changes, low voltage, etc); (2) for confirmation of the results of the exercise ECG when they are contrary to the clinical impression (eg, a positive test in an asymptomatic patient); (3) to localize the region of ischemia; (4) to distinguish ischemic from infarcted myocardium; (5) to assess the completeness of vascularization following bypass surgery or coronary angioplasty; or (6) as a prognostic indicator in patients with known coronary disease.
2. Radionuclide angiography
This procedure images the LV and measures its EF and wall motion. In coronary disease, resting abnormalities usually represent infarction, and those that occur only with exercise usually indicate stress-induced ischemia. Normal subjects usually exhibit an increase in EF with exercise or no change; patients with coronary disease may exhibit a decrease. Exercise radionuclide angiography has approximately the same sensitivity as thallium-201 scintigraphy, but it is less specific in older individuals and those with other forms of heart disease. The indications are similar to those for thallium-201 scintigraphy.
3. Positron emission tomography
PET scanning uses positron-emitting agents to demonstrate either perfusion or metabolism of myocardium. PET can accurately distinguish transiently dysfunctional ( stunned ) myocardium from scar by showing persistent glycolytic metabolism with the tracer fluorodeoxyglucose (FDG) in regions with reduced blood flow. A nearby cyclotron is required to produce this tracer. The newer SPECT camera can provide acceptable images without the more expensive PET technology.
E. Echocardiography
Echocardiography can image the LV and reveal segmental wall motion abnormalities, which may indicate ischemia or prior infarction. It is a convenient technique for assessing LV function, which is an important indicator of prognosis and determinant of therapy. Echocardiograms performed during supine exercise or immediately following upright exercise may demonstrate exercise-induced segmental wall motion abnormalities as an indicator of ischemia. This technique requires considerable expertise; however, in experienced laboratories, the test accuracy is comparable to that obtained with scintigraphy though a higher proportion of tests is technically inadequate. Pharmacologic stress with high-dose (20 40 mcg/kg/min) dobutamine can be used as an alternative to exercise. Echocardiography contrast agents allow for perfusion
P.353
imaging and may improve the diagnostic accuracy of this form of testing.
F. CT and MRI Scanning
Many new imaging techniques have been developed, but their application in cardiovascular disease remains to be determined. CT scan can image the heart and, with contrast medium and thin slice technology, the coronary arteries with increasing resolution, but with a need for relatively large radiation exposure and contrast load. An important application of CT is the evaluation of pericardial disease. Ultrafast or electron beam CT (EBCT) involves a specially designed instrument with high temporal resolution. Its availability is limited, but it provides excellent assessments of cardiac structure and function. EBCT is increasingly being used to detect and quantify coronary artery calcification, but proper application of this highly sensitive test is uncertain. False-negative studies may occur in patients under 50 years of age, and positive studies in older patients do not necessarily provide a quantitative assessment of the severity of coronary arteriosclerosis. Thus, although this test can stratify patients into lower and higher risk groups, the appropriate management of individual patients with asymptomatic coronary artery calcification beyond aggressive risk factors modification is unclear.
Cardiac MRI provides high-resolution images of the heart and great vessels without radiation exposure or use of iodinated contrast media. It provides excellent anatomic definition, permitting assessment of pericardial disease, neoplastic disease of the heart, myocardial thickness, chamber size, and many congenital heart defects. It is an excellent noninvasive test for nonemergently evaluating dissection of the aorta. Rapid acquisition sequences can produce excellent cine-mode images demonstrating LV function and wall motion, and it is thus a useful alternative when the echocardiogram is suboptimal. Perfusion imaging can be done with gadolinium first pass perfusion using dobutamine or adenosine to produce pharmacologic stress. Recent advances have been made in imaging the proximal coronary arteries, but this application remains investigational.
G. Ambulatory ECG Monitoring
With current ambulatory ECG recorders and with trained technicians, episodes of ischemic ST-segment depression can be monitored. In patients with CAD, these episodes usually signify ischemia, even when asymptomatic ( silent ). In many, silent episodes are more frequent than symptomatic ones. In most cases, they occur in patients with other evidence of ischemia, and they respond to the same treatments, so that the role of ambulatory monitoring is unclear, as is the benefit of abolishing all such episodes in patients who are otherwise being managed properly.
H. Coronary Angiography
Selective coronary arteriography is the definitive diagnostic procedure for CAD. It can be performed with low mortality (about 0.1%) and morbidity (1 5%), but the cost is high, and with currently available noninvasive techniques it is usually not indicated solely for diagnosis.
Coronary arteriography should be performed in the following groups:
Patients being considered for coronary artery revascularization because of limiting stable angina who have not improved on an adequate medical regimen.
Patients in whom coronary revascularization is being considered because the clinical presentation (unstable angina, postinfarction angina, etc) or noninvasive testing suggests high-risk disease (see Indications for Revascularization).
Patients with aortic valve disease who also have angina pectoris, to determine whether the angina is due to accompanying coronary disease. Coronary angiography is also performed in asymptomatic older patients undergoing valve surgery so that concomitant bypass may be done if the anatomy is propitious.
Patients who have had coronary revascularization with subsequent recurrence of symptoms, to determine whether bypass grafts or native vessels are occluded.
Patients with cardiac failure in whom a surgically correctable lesion, such as LV aneurysm, mitral regurgitation, or reversible ischemic dysfunction, is suspected.
Patients surviving sudden death or with symptomatic or life-threatening arrhythmias in whom CAD may be a correctable cause.
Patients with chest pain of uncertain cause or cardiomyopathy of unknown cause.
Coronary arteriography visualizes the location and severity of stenoses. Narrowing greater than 50% of the luminal diameter is considered clinically significant, although most lesions producing ischemia are associated with narrowing in excess of 70%. This information has important prognostic value, since mortality rates are progressively higher in patients with one-, two-, and three-vessel disease and those with left main coronary artery obstruction (ranging from 1% per year to 25% per year). In those with strongly positive exercise ECGs or scintigraphic studies, three-vessel or left main disease may be present in 75 95% depending on the criteria used. Coronary arteriography also shows whether the obstructions are amenable to bypass surgery or percutaneous transluminal coronary angioplasty.
Coronary angiography may underestimate the degree of atherosclerosis because it images only the lumen of the vessel. If there is concentric plaque with arterial enlargement (remodeling), then the lumen may appear relatively normal. Intravascular ultrasound (IVUS) uses a small ultrasound transducer that can be positioned within the artery and image beneath the endothelial surface. This technique is useful when the angiogram is equivocal as well as for assessing the results of angioplasty or stenting.
P.354
I. LV Angiography
LV angiography is usually performed at the same time as coronary arteriography. Global and regional LV function are visualized, as well as mitral regurgitation if present. LV function is a major determinant of prognosis in coronary heart disease.
Acampa W et al: Nuclear medicine procedures in cardiovascular diseases. An evidence based approach. Q J Nucl Med 2002; 46:323.
ACC/AHA 2002 guideline update for exercise testing: summary article. Circulation 2002;106:1833.
ACC/AHA 2002 guideline update for the management of patients with chronic stable angina summary article. Circulation 2003;107:149.
Botoman VA: Noncardiac chest pain. J Clin Gastroenterol 2002; 34:6.
Gottdiener JS: Overview of stress echocardiography: uses, advantages, and limitations. Prog Cardiovasc Dis 2001;43:315.
Kim WY et al: Coronary magnetic resonance angiography for the detection of coronary stenoses. N Engl J Med 2001;345: 1863.
Lee TH et al: Clinical practice. Noninvasive tests in patients with stable coronary artery disease. N Engl J Med 2001;344:1840.
O'Rourke RA et al: American College of Cardiology/American Heart Association Expert Consensus Document on electron-beam computed tomography for the diagnosis and prognosis of coronary artery disease. J Am Coll Cardiol 2000;36:326.
Paetsch I et al: Comparison of dobutamine stress magnetic resonance, adenosine stress magnetic resonance, and adenosine stress magnetic resonance perfusion. Circulation 2004;110: 835.
Scanlon PJ et al: ACC/AHA guidelines for coronary angiography: executive summary and recommendations. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Coronary Angiography) developed in collaboration with the Society for Cardiac Angiography and Interventions. Circulation 1999;99:2345.
Williams SV et al: Guidelines for the management of patients with chronic stable angina: diagnosis and risk stratification. Ann Intern Med 2001;135:530.
Coronary Vasospasm & Angina with Normal Coronary Arteriograms
Although most symptoms of myocardial ischemia result from fixed stenosis of the coronary arteries or intraplaque hemorrhage or thrombosis at the site of lesions, some ischemic events may be precipitated or exacerbated by coronary vasoconstriction.
Spasm of the large coronary arteries with resulting decreased coronary blood flow may occur spontaneously or may be induced by exposure to cold, emotional stress, or vasoconstricting medications, such as ergot derivative drugs. Spasm may occur both in normal and in stenosed coronary arteries and may be silent or result in angina pectoris. Even myocardial infarction may occur as a result of spasm in the absence of visible obstructive coronary heart disease, although most instances of such coronary spasm occur in the presence of coronary stenosis.
Cocaine can induce myocardial ischemia and infarction by causing coronary artery vasoconstriction or by increasing myocardial energy requirements.
Prinzmetal's (variant) angina is a clinical syndrome in which chest pain occurs without the usual precipitating factors and is associated with ST-segment elevation rather than depression. It often affects women under 50 years of age. It characteristically occurs in the early morning, awakening patients from sleep, tends to involve the right coronary artery, and is apt to be associated with arrhythmias or conduction defects. There may be no fixed stenoses. Ischemia usually results from coronary vasoconstriction and may be diagnosed by challenge with ergonovine (a vasoconstrictor), although such provocation entails risk.
Patients with this pattern of pain or any chest pain syndrome associated with ST-segment elevation should undergo coronary arteriography to determine whether fixed stenotic lesions are present. If they are, aggressive medical therapy or revascularization is indicated, since this may represent an unstable phase of the disease. If significant lesions are not seen and spasm is suspected, avoidance of precipitants such as cigarette smoking and cocaine is the top priority. Episodes of coronary spasm generally respond well to nitrates, and both nitrates and calcium channel blockers (including long-acting nifedipine, diltiazem, or amlopidine) are effective prophylactically. By allowing unopposed 1-mediated vasoconstriction, -blockers have exacerbated coronary vasospasm, but they may have a role in management of patients in whom spasm is associated with fixed stenoses.
There is a growing consensus that myocardial ischemia may also occur in patients with normal coronary arteries as a result of disease of the coronary microcirculation or abnormal vascular reactivity. This has been termed syndrome X.
Al Suwaidi J et al: Pathophysiology, diagnosis, and current management strategies for chest pain in patients with normal findings on angiography. Mayo Clin Proc 2001;76:813.
Frishman WH et al: Cardiovascular manifestations of substance abuse part 1: cocaine. Heart Dis 2003;5:187.
Lange RA et al: Cardiovascular complications of cocaine use. N Engl J Med 2001;345:351.
Treatment
A. Treatment of Anginal Episodes
Sublingual nitroglycerin is the drug of choice; it acts in about 1 2 minutes. Nitrates decrease arteriolar and venous tone, reduce preload and afterload, and lower the oxygen demand of the heart. Nitrates may also improve myocardial blood flow by dilating collateral channels and, in the presence of increased vasomotor tone, coronary stenoses. As soon as the attack begins, one fresh tablet is placed under the tongue. This may be repeated
P.355
at 3- to 5-minute intervals. The dosage (0.3, 0.4, or 0.6 mg) and the number of tablets to be used before seeking further medical attention must be individualized. Nitroglycerin buccal spray is also available as a metered (0.4 mg) delivery system. It has the advantage of being more convenient for patients who have difficulty handling the pills and of being more stable. Nitroglycerin can also be used prophylactically before activities likely to precipitate angina. Pain not responding to three tablets or lasting more than 20 minutes may represent evolving infarction, and the patient should be instructed to seek immediate medical attention.
B. Prevention of Further Attacks
1. Aggravating factors
Angina may be aggravated by hypertension, LV failure, arrhythmia (usually tachycardias), strenuous activity, cold temperatures, and emotional states. These factors should be identified and treated when possible.
2. Nitroglycerin
Nitroglycerin, 0.3 0.6 mg sublingually or 0.4 0.8 mg translingually by spray, should be taken 5 minutes before any activity likely to precipitate angina. Sublingual isosorbide dinitrate (2.5 10 mg) is only slightly longer-acting than sublingual nitroglycerin.
3. Long-acting nitrates
A number of longer-acting nitrate preparations are available. These include isosorbide dinitrate, 10 40 mg orally three times daily; isosorbide mononitrate, 10 40 mg orally twice daily or 60 120 mg once daily in a sustained-release preparation; oral sustained-release nitroglycerin preparations, 6.25 12.5 mg two to four times daily; nitroglycerin ointment, 6.25 25 mg applied two to four times daily; and transdermal nitroglycerin patches that deliver nitroglycerin at a predetermined rate (usually 5 20 mg/24 h). The main limitation to long-term nitrate therapy is tolerance, which occurs to some degree in most patients. The degree of tolerance can be limited by using a regimen that includes a minimum 8- to 10-hour period per day without nitrates. Isosorbide dinitrate can be given three times daily, with the last dose after dinner, or longer-acting isosorbide mononitrate once daily. Transdermal nitrate preparations should be removed overnight in most patients.
Nitrate therapy is often limited by headache. Other side effects include nausea, light-headedness, and hypotension.
4. -Blockers
-Blockers prevent angina by reducing myocardial oxygen requirements during exertion and stress. This is accomplished by reducing the heart rate, myocardial contractility, and, to a lesser extent, BP. The -blockers are the only antianginal agents that have been demonstrated to prolong life in patients with coronary disease (post-myocardial infarction). They are at least as effective at relieving angina as alternative agents in studies employing exercise testing, ambulatory monitoring, and symptom assessment. As a result, they should be considered for first-line therapy in most patients with chronic angina.
-Blockers with intrinsic sympathomimetic activity, such as pindolol, are less desirable because they may exacerbate angina in some individuals and have not been effective in secondary prevention trials. The pharmacology and side effects of the -blockers are discussed in Chapter 11 (see Table 11-7). The dosages of all these drugs when given for angina are similar. The major contraindications are severe bronchospastic disease, bradyarrhythmias, and decompensated heart failure.
5. Calcium channel blocking agents
Verapamil, diltiazem, and the dihydropyridine group of calcium blockers are chemically and pharmacologically heterogeneous agents that prevent angina by reducing myocardial oxygen requirements and by inducing coronary artery vasodilation. Myocardial oxygen demand is decreased by reducing BP, LV wall stress and, in the case of verapamil and diltiazem, resting or exercise heart rate. Though these agents are all potent coronary vasodilators, it is unclear whether they improve myocardial blood flow in most patients with stable exertional angina. In those with coronary vasospasm, the calcium channel blockers may be the agents of choice.
Most calcium channel blockers have negative inotropic, chronotropic, and dromotropic properties in vitro, but the reflex sympathetic response may obscure these effects in vivo (except in the presence of -blockade or severely depressed LV function). Unlike the -blockers, calcium channel blockers have not been shown to reduce mortality postinfarction and in some cases have increased ischemia and mortality rates. This appears to be the case with some dihydropyridines and with diltiazem and verapamil in patients with clinical heart failure or moderate to severe LV dysfunction. Meta-analyses have suggested that short-acting nifedipine in moderate to high doses causes an increase in mortality. It is uncertain whether these findings are relevant to longer-acting dihydropyridines. Nevertheless, considering the uncertainties and the lack of demonstrated favorable effect on outcomes, calcium channel blockers should be considered third-line anti-ischemic drugs in the postinfarction patient. Similarly, with the exception of amlodipine, which in the PRAISE trial proved safe in patients with heart failure, these agents should be avoided in patients with CHF or low EFs.
The pharmacologic effects and side effects of the calcium channel blockers are discussed in Chapter 11 and summarized in Table 11-9. Although all have been shown to be efficacious for angina, not all preparations and agents are approved for this indication. By and large, diltiazem and verapamil are preferable as first-line agents because they produce less reflex tachycardia and because the former, at least, may cause fewer side effects. Nifedipine, nicardipine, and amlodipine are also approved agents for angina. Isradipine, felodipine, and nisoldipine are not approved for angina but probably are as effective as the other dihydropyridines.
6. Alternative and combination therapies
Patients who do not respond to one class of antianginal medication often respond to another. It may, therefore, be worthwhile to use an alternative agent before progressing
P.356
to combinations. If the patient remains symptomatic, a -blocker and a long-acting nitrate or a -blocker and a calcium channel blocker (other than verapamil, where the risk of AV block or heart failure is higher) are the most appropriate combinations. A few patients will have a further response to a regimen including all three agents.
7. Ranolazine
Ranolazine, an agent that involves selective inhibition of the late sodium current, has been approved by the US Food and Drug Administration to prevent angina, the first such new drug in more than 10 years. The approval was based on two trials showing that ranolazine reduced angina compared with -blockers or calcium channel blockers.
The usual dose is 500 mg orally twice a day. Because it can cause QT prolongation, it is contraindicated in patients with existing QT prolongation, concurrent use of QT prolonging drugs such as class I or III antiarrhythmics (eg, quinidine, dofetilide, sotalol), and potent and moderate CYP450 3A inhibitors. An ECG is recommended both at baseline and during treatment. It is also contraindicated in patients with significant liver and renal disease. Ranolazine is not to be used for treatment of acute anginal episodes.
8. Platelet-inhibiting agents
Coronary thrombosis is responsible for most episodes of myocardial infarction and many unstable ischemic syndromes. Several studies have demonstrated the benefit of antiplatelet drugs following unstable angina and infarction. Therefore, unless contraindicated, small doses of aspirin (81 325 mg daily) should be prescribed for patients with angina. Clopidogrel is an antiplatelet agent that acts by blocking the ADP receptor and resulting platelet aggregation. Unlike its older congener ticlopidine, clopidogrel does not cause agranulocytosis but may rarely induce thrombotic thrombocytopenic purpura. It can reduce cardiac events in patients with acute coronary syndromes and is an appropriate alternative in aspirin-intolerant patients.
9. Risk reduction
As discussed above, patients with coronary disease should undergo aggressive risk factor modification. This approach, with a particular focus on lowering LDL cholesterol, treating hypertension, stopping smoking, and exercise and weight loss (especially for patients with metabolic syndrome or at risk for diabetes), may markedly improve outcome.
10. Revascularization
The indications for coronary artery revascularization and the choice of procedure are discussed below.
11. Mechanical extracorporeal counterpulsation
Extracorporeal counterpulsation (ECP) entails repetitive inflation of a high-pressure chamber surrounding the lower half of the body during the diastolic phase of the cardiac cycle for daily 1-hour sessions over a period of 7 weeks. Randomized trials have shown that ECP reduces angina, improves exercise tolerance, and can reduce symptoms of heart failure. However, the response has been variable, often time limited, and not shown to be associated with improved myocardial perfusion so a placebo effect may be responsible.
Table 10-3. Duke treadmill score: calculation and interpretation. | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Prognosis
The prognosis of angina pectoris has improved with development of therapies aimed at secondary prevention. Mortality rates vary depending on the number of vessels diseased, the severity of obstruction, the status of LV function, and the presence of complex arrhythmias. The outlook in individual patients is unpredictable, and nearly half of the deaths are sudden. Therefore, risk stratification is often attempted. Patients with accelerating symptoms have a poorer outlook. Among stable patients, those whose exercise tolerance is severely limited by ischemia (less than 6 minutes on the Bruce treadmill protocol) and those with extensive ischemia by exercise ECG or scintigraphy have more severe anatomic disease and a poorer prognosis. The Duke Treadmill Score, based on a standard Bruce protocol exercise treadmill test, provides an estimate of risk of death at 1 year. The score uses time on the treadmill, amount of ST-segment depression, and presence of angina (Table 10-3).
Chaitman BR et al; Combination Assessment of Ranolazine In Stable Angina (CARISA) Investigators: Effects of ranolazine with atenolol, amlodipine, or diltiazem on exercise tolerance and angina frequency in patients with severe chronic angina: a randomized controlled trial. JAMA 2004;291:309.
Gibbons RJ et al: ACC/AHA 2002 guideline update for the management of patients with chronic stable angina summary article: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines (Committee on the Management of Patients With Chronic Stable Angina). J Am Coll Cardiol 2003;41:159.
U.S. Food and Drug Administration: FDA Approves New Treatment for Chest Pain. http://www.fda.gov/bbs/topics/news/2006/NEW01306.html
Revascularization Procedures for Patients with Angina Pectoris
Indications
There is general agreement that otherwise healthy patients in the following groups should undergo revascularization:
P.357
(1) Patients with unacceptable symptoms despite medical therapy to its tolerable limits. (2) Patients with left main coronary artery stenosis greater than 50% with or without symptoms. (3) Patients with three-vessel disease with LV dysfunction (EF < 50% or previous transmural infarction). (4) Patients with unstable angina who after symptom control by medical therapy continue to exhibit ischemia on exercise testing or monitoring. (5) Post-myocardial infarction patients with continuing angina or severe ischemia on noninvasive testing. (See sections on Acute Coronary Syndromes and Myocardial Infarction.)
In addition, many cardiologists believe that patients with less severe symptoms should be revascularized if they have two-vessel disease associated with underlying LV dysfunction, anatomically critical lesions (> 90% proximal stenoses, especially of the proximal left anterior descending artery), or physiologic evidence of severe ischemia (early positive exercise tests, large exercise-induced thallium scintigraphic defects, or frequent episodes of ischemia on ambulatory monitoring). This trend toward aggressive intervention has accelerated as a result of the growing use of coronary angioplasty and stenting. Although such patients are at increased risk, it has not been proved that their prognosis is better after coronary revascularization by either surgery or angioplasty.
Type of Procedure
A. Coronary Artery Bypass Grafting
Coronary artery bypass grafting (CABG) can be accomplished with a very low mortality rate (1 3%) in otherwise healthy patients with preserved cardiac function. However, the mortality rate of this procedure rises to 4 8% in older individuals and in patients who have had a prior CABG. Increasingly, younger individuals with focal lesions of one or several vessels are undergoing coronary angioplasty as the initial revascularization procedure.
Grafts using one or both internal mammary arteries (usually to the left anterior descending artery or its branches) provide the best long-term results in terms of patency and flow. Segments of the saphenous vein (or, less optimally, other veins) or the radial artery interposed between the aorta and the coronary arteries distal to the obstructions are also used. One to five distal anastomoses are commonly performed. After successful surgery, symptoms generally abate. The need for antianginal medications diminishes, and LV function may improve.
Minimally invasive surgical techniques utilize different approaches to the heart than standard sternotomy and cardiopulmonary bypass. The surgical approach may involve a limited sternotomy, lateral thoracotomy (MIDCAB), or thoracoscopy (port-access). These approaches may be used in conjunction with standard cardiopulmonary bypass, with peripheral cardiopulmonary bypass, or with operating on the beating heart utilizing a mechanical coronary stabilizer. Avoiding bypass may decrease the risk of cerebral complications. These techniques allow earlier postoperative mobilization and discharge. They are more technically demanding, usually not suitable for more than two grafts, and do not have established durability.
The operative mortality rate is increased in patients with poor LV function (LV EF < 35%) or those requiring additional procedures (valve replacement or ventricular aneurysmectomy). Patients over 70 years of age, patients undergoing repeat procedures, or those with important noncardiac disease (especially renal insufficiency and diabetes) or poor general health also have higher operative mortality and morbidity rates, and full recovery is slow. Thus, CABG should be reserved for more severely symptomatic patients in this group. Early (1 6 months) graft patency rates average 85 90% (higher for internal mammary grafts), and subsequent graft closure rates are about 4% annually. Early graft failure is common in vessels with poor distal flow, while late closure is more frequent in patients who continue smoking and those with untreated hyperlipidemia. Antiplatelet therapy with aspirin improves graft patency rates. Smoking cessation and vigorous treatment of blood lipid abnormalities are necessary, with a goal for LDL cholesterol of 100 mg/dL and of HDL cholesterol 45 mg/dL. Repeat revascularization (see below) is often necessitated by progressive native vessel disease and graft occlusions. Reoperation is technically demanding and less often fully successful than the initial operation.
B. Percutaneous Coronary Intervention Including Stenting
Coronary artery stenoses can be effectively dilated by inflation of a balloon under high pressure. This procedure is performed in the cardiac catheterization laboratory under local anesthesia either at the same time as diagnostic coronary arteriography or at a later time. The mechanism of dilation involves both rupture of the atheromatous plaque and remodeling of the vessel.
This procedure was at one time reserved for proximal single-vessel disease, but now it is widely used in multivessel disease with multiple lesions, though only rarely in left main disease. Percutaneous transluminal coronary angioplasty (PTCA) is possible but often less successful in bypass graft stenoses. Bypass graft patients with multivessel disease have lower mortality rates and fewer nonfatal myocardial infarctions with surgery than with percutaneous interventions. Optimal lesions for PTCA are relatively proximal, noneccentric, free of significant calcification or plaque dissection, and removed from the origin of large branches. With improved catheter systems, experienced operators are able to successfully dilate 90% of lesions attempted. The major early complication is intimal dissection with vessel occlusion. This can usually be treated by repeat PTCA or by deployment of an intracoronary stent. The use of platelet glycoprotein IIb/IIIa inhibitors (abciximab, eptifibatide, tirofiban) has substantially reduced the rate of acute vessel closure, and placement of intracoronary stents has
P.358
markedly improved initial and long-term angiographic results, especially with complex and long lesions. Although the early experience with stents was complicated by an unacceptable rate of acute thrombosis, this problem has largely been prevented by aggressive antithrombotic therapy (long-term aspirin plus clopidogrel for between 30 days and 1 year, with acute use of platelet glycoprotein IIb/IIIa inhibitors in high-risk patients). Restenosis rates have fallen with the use of stents. Stents are now used in the majority of patients undergoing percutaneous revascularization.
The major limitation with PTCA has been restenosis, which occurs in the first 6 months in 30 40% of vessels dilated, though it can often be treated successfully by repeat PTCA. Factors associated with higher restenosis rates include diabetes, small luminal diameter, longer and more complex lesions, and lesions at coronary ostia or in the left anterior descending coronary artery. The use of stents has reduced the restenosis rate by 50%. Drug-eluting stents that elute antiproliferative agents such as sirolimus or paclitaxel have substantially reduced restenosis to rates of < 10%. These devices have had a significant impact on clinical practice, leading to increased use of percutaneous interventions in patients with multiple and less favorable lesions. However, because prevention of tissue proliferation around the stent may leave exposed stent and predispose to thrombosis, drug-eluting stents are associated with a small but poorly defined incidence of late stent thrombosis. Full stent deployment as well as use of clopidogrel for at least 3 (for sirolimus) to 6 (for paclitaxel) months are important to prevent stent thrombosis.
Currently, in-stent restenosis is often treated with either brachytherapy or restenting with drug-eluting stents.
The number of PTCA and stenting procedures now exceeds the number of CABG operations, but the justification for many of the procedures performed in patients with stable angina is unclear. Several studies have shown PTCA to be superior to medical therapy for symptom relief but not in preventing infarction or death. In patients with no or only mild symptoms, aggressive lipid-lowering and antianginal therapy may be preferable to PTCA.
Several studies of PTCA versus CABG in patients with multivessel disease have been reported. The consistent finding has been comparable mortality and infarction rates over follow-up periods of 1 3 years but a high rate (approximately 40%) of repeat procedures following PTCA. As a result, the choice of revascularization procedure is often a matter of patient preference. However, it should be noted that less than 20% of patients with multivessel disease met the entry criteria, so these results cannot be generalized to all multivessel disease patients. Outcomes with percutaneous revascularization in diabetics have been inferior to those with CABG. However, these trials preceded the widespread use of stenting.
C. Experimental Approaches
Several experimental approaches have been studied in patients with refractory angina who are not candidates for percutaneous or surgical revascularization procedures. Laser transmyocardial revascularization has been used either from the epicardial surface of the LV during surgery or from the ventricular cavity by catheter-based techniques. Several studies reporting an improvement in symptoms have not shown objective evidence of improvement in perfusion, raising the possibility of a placebo effect, and clinical outcomes are either not improved or worsened. Other studies have evaluated intracoronary injections of growth factors, such as vascular endothelial growth factor (VEGF) or fibroblast growth factors, or cell therapy using endothelial progenitor cells, but thus far there is inconclusive evidence of benefit.
Summary of Results of Treatment
Several randomized trials have shown that over follow-up periods of several years, the mortality and infarction rates with percutaneous revascularization and CABG are generally comparable. An exception may be diabetic patients, who have had better outcomes with CABG. Recovery after PTCA is obviously faster, but the intermediate-term success rate of CABG is higher both because of the high restenosis rate with PTCA and, less importantly, with stenting. The increasing popularity of PTCA and stenting primarily reflects the lower cost and shorter hospitalization, the perception that CABG is best done only once and can be reserved for later, and the preference of patients for less invasive treatment. These arguments make PTCA the procedure of choice for revascularization of single-vessel disease. The situation is less clear with multivessel disease. It should also be noted that the excellent outcome of patients treated medically has made it difficult to show an advantage with either revascularization approach except in patients who remain symptom limited or have left main lesions or three-vessel disease and LV dysfunction. The availability of drug-eluting stents is shifting the balance toward percutaneous revascularization.
Arora RR et al: The multicenter study of enhanced external counterpulsation (MUST-EECP): effect of EECP on exercise-induced myocardial ischemia and anginal episodes. J Am Coll Cardiol 1999;33:1833.
Eagle KA et al: ACC/AHA Guidelines for Coronary Artery Bypass Graft Surgery: a Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1991 Guidelines for Coronary Artery Bypass Graft Surgery). American College of Cardiology/American Heart Association. J Am Coll Cardiol 1999;34:1262.
Kim MC: Refractory angina pectoris. J Am Coll Cardiol 2002; 39:923.
Lowe HC: Coronary in-stent restenosis: current status and future strategies. J Am Coll Cardiol 2002;39:183.
Moses JW et al; SIRIUS Investigators: Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N Engl J Med 2003;349:1315.
O'Shea JC et al: Platelet glycoprotein IIb/IIIa integrin blockade with eptifibatide in coronary stent intervention. JAMA 2001;285:2468.
Schofield PM: Indications for percutaneous and surgical revascularisation: how far does the evidence base guide us? Heart 2003;89:565.
P.359
Steinhubl SR et al: Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention. A randomized controlled trial. JAMA 2002;288:2411.
Stone GW et al; TAXUS-IV Investigators: One-year clinical results with the slow-release, polymer-based, paclitaxel-eluting TAXUS stent: the TAXUS-IV trial. Circulation 2004;109:1942.
Trial of invasive versus medical therapy in elderly patients with chronic symptomatic coronary-artery disease (TIME): a randomised trial. Lancet 2001;358:951.
Acute Coronary Syndromes
Acute coronary syndromes comprise the spectrum of unstable cardiac ischemia from unstable angina to acute myocardial infarction. Rather than the traditional nomenclature of unstable angina, non-Q wave and Q wave myocardial infarction, acute coronary syndromes are now classified based on the presenting ECG as either ST elevation or non-ST elevation. This allows for immediate classification and guides determination of whether patients should be considered for acute reperfusion therapy. The evolution of cardiac markers then allows determination of whether myocardial infarction has occurred. Acute coronary syndromes represent a dynamic state in which patients frequently shift from one category to another, as new ST elevation can develop after presentation and cardiac markers can become abnormal with recurrent ischemic episodes.
Diagnosis
Many patients with acute coronary syndromes will exhibit ECG changes during pain either ST-segment elevation, ST-segment depression, or T wave flattening or inversion. They may exhibit signs of LV dysfunction during pain and for a time thereafter.
Chest pain is one of the most frequent reasons for emergency department visits. Algorithms have been developed to aid in determining the likelihood that a patient has an acute coronary syndrome, and for those patients that do have an acute coronary syndrome, the risk of death or death and ischemic events.
Many hospitals have developed chest pain observation units to provide a systematic approach toward serial risk stratification to improve the triage process. In many cases those who have not experienced new chest pain and have no ECG changes or cardiac enzyme elevations undergo treadmill exercise tests or imaging procedures to exclude ischemia at the end of an 8- to 24-hour period and are discharged directly from the emergency department if these tests are negative.
Treatment
Table 10-4 provides a summary of the ACC/AHA Guideline recommendations for selected medical treatments.
A. General Measures
Treatment of acute coronary syndromes without ST elevation should be multifaceted and vigorous. Patients who are at high risk should be hospitalized, maintained at bed rest or at very limited activity, monitored, and given supplemental oxygen. Sedation with a benzodiazepine agent may help if anxiety is present.
B. Anticoagulation, Antiplatelet, and Thrombolytic Therapy
Coronary thrombosis plays a prominent role in the pathophysiology of unstable angina and its progression to myocardial infarction, and antithrombotic therapy plays an important role in treatment. Patients should receive a combination of antiplatelet and anticoagulant agents. Aspirin, 81 325 mg daily, and heparin (low-molecular-weight or unfractionated) should be commenced on presentation. Several trials have shown that low-molecular-weight heparin (and specifically enoxaparin 1 mg/kg subcutaneously every 12 hours) is somewhat more effective than unfractionated heparin in preventing recurrent ischemic events in the setting of acute coronary syndromes. However, the SYNERGY trial showed that unfractionated heparin and enoxaparin had similar rates of death or (re)infarction in the setting of frequent early coronary intervention. Fondaparinux, a specific factor Xa inhibitor given in a dose of 2.5 mg subcutaneously once a day, was found in the OASIS-6 trial to be equally effective as enoxaparin among 20,000 patients at preventing early death, myocardial infarction, and refractory ischemia, and resulted in a 50% reduction in major bleeding. This reduction in major bleeding appeared to translate into a reduction in mortality (and in death and/or myocardial infarction) at 30 days. While catheter-related thrombosis was more common during coronary intervention procedures with fondaparinux, it appears that this can be controlled by adding unfractionated heparin during the procedure. This trial not only established fondaparinux as an excellent treatment for acute coronary syndromes but also highlighted the importance of bleeding and its prevention.
The ACUITY trial showed that the direct thrombin inhibitor bivalirudin appears to be a reasonable alternative to heparin or enoxaparin plus a glycoprotein IIb/IIIa antagonist for many patients with acute coronary syndromes who are undergoing early coronary intervention.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial demonstrated a 20% reduction in the composite end point of cardiovascular death, myocardial infarction, and stroke with the addition of clopidogrel (300 mg loading dose, 75 mg/d for 9 12 months) in patients with non-ST-segment elevation acute coronary syndromes. When treated with clopidogrel, the optimal aspirin dose appears to be 81 mg/d (versus 160 mg/d or 325 mg/d) based on similar thrombotic event rates and lower rates of bleeding.
Table 10-4. Summary of the current ACC/AHA guideline recommendations for medical management of acute coronary syndromes (ACS) and acute myocardial infarction (AMI).1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Small molecule inhibitors of the platelet glycoprotein IIb/IIIa receptor are useful adjuncts in high-risk patients (usually defined by fluctuating ST-segment depression or positive biomarkers) with acute coronary syndromes, particularly when they are undergoing PTCA or stenting.
P.360
Tirofiban, 0.4 mcg/kg/min for 30 minutes, followed by 0.1 mcg/kg/min, and eptifibatide, 180 mcg/kg bolus followed by a continuous infusion of 0.1 mcg/kg/min, have both have been shown to be effective when added to heparin. Downward dose adjustments are required in patients with reduced renal function. For example, if the estimated creatinine clearance is below 50 mL/min, the eptifibatide infusion should be cut in half to 1 mcg/kg/min.
Fibrinolytic therapy should be avoided in patients without ST-segment elevation since they generally have a patent culprit artery, and since the risk of such therapy appears to outweigh the benefit.
C. Nitroglycerin
The nitrates are first-line anti-ischemic therapy for acute coronary syndromes. Nonparenteral therapy with sublingual or oral agents or nitroglycerin ointment is usually sufficient. If pain persists or recurs, intravenous nitroglycerin should be started. The usual initial dosage is 10 mcg/min. The dosage should be titrated upward by 10 20 mcg/min (to a maximum of 200 mcg/min) until angina disappears or mean arterial pressure drops by 10%. Careful usually continuous BP monitoring is required when intravenous nitroglycerin is used. Avoid hypotension (systolic BP < 100 mm Hg). Tolerance to continuous nitrate infusion is common.
D. -Blockers
These agents are also a part of the initial treatment of unstable angina unless otherwise contraindicated. If the patient has no physical findings of heart failure, these agents can usually be started without measurements of
P.361
LV function. Patients with evidence of large or multiple old infarctions are an exception. The pharmacology of these agents is discussed in Chapter 11 and summarized in Table 11-7. Use of agents with intrinsic sympathomimetic activity should be avoided in this setting. The goal of acute treatment is to reduce the heart rate below 60 70 beats/min. Oral medication is adequate in most patients, but intravenous treatment with metoprolol, given as three 5 mg doses 5 minutes apart, achieves a more rapid effect. Oral therapy should be aggressively titrated upward as BP permits.
E. Calcium Channel Blockers
Calcium channel blockers have not been shown to favorably affect outcome in unstable angina, and they should be used primarily as third-line therapy in patients with continuing symptoms on nitrates and -blockers or those who are not candidates for these drugs. In the presence of nitrates and without accompanying -blockers, diltiazem or verapamil is preferred, since nifedipine and the other dihydropyridines are more likely to cause reflex tachycardia or hypotension. The initial dosage should be low, but upward titration should proceed rapidly (see Table 11-9).
F. Statins
The PROVE-IT trial provides evidence for starting a statin in the days immediately following an acute coronary syndrome. In this trial, more intensive therapy with atorvastatin 80 mg a day, regardless of total or LDL cholesterol level, improved outcome compared to pravastatin 40 mg a day, with the curves of death or major cardiovascular event separating as early as 3 months after starting therapy.
G. Intra-aortic Balloon Counterpulsation
Intra-aortic balloon counterpulsation (IABC) can both reduce myocardial energy requirements (systolic unloading) and improve diastolic coronary blood flow. This approach is sometimes used to stabilize patients prior to angiography or revascularization, but with modern techniques it is rarely necessary.
Prognosis & Indications for Revascularization
Risk stratification is important for determining intensity of care. Several therapies, including glycoprotein IIb/IIIa receptor antagonists, low-molecular-weight heparin, and early invasive catheterization, have been shown to have the greatest benefit in higher-risk patients. As outlined in the ACC/AHA guidelines, patients with any high-risk feature (Table 10-5) warrant an early invasive strategy with catheterization and revascularization. For patients without these high-risk features, either an invasive or noninvasive approach, using exercise (or pharmacologic stress for patients unable to exercise) stress testing to identify patients who have residual ischemia and/or high risk, can be used.
Table 10-5. Indications for catheterization and percutaneous coronary intervention. | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Two risk-stratification tools are available that can be used at the bedside, the TIMI Risk Score and the GRACE Risk Score. The TIMI Risk Score includes nine variables: age 65, three or more cardiac risk factors, prior coronary stenosis 50%, ST-segment deviation, two anginal events in prior 24 hours, acetylsalicylic acid in prior 7 days, and elevated cardiac markers. The GRACE risk score, which applies to patients with or without ST elevation, includes Killip class, BP, ST-segment deviation, cardiac arrest at presentation, serum creatinine, elevated creatine kinase (CK)-MB or troponin, and heart rate. The TIMI
P.362
Risk Score is available for PDA download at http://www.timi.org.
ACC/AHA 2002 guideline update for the management of patients with unstable angina and non-ST-segment elevation myocardial infarction summary article. J Am Coll Cardiol 2002;40:1366.
Boden WE et al: Optimizing management of non-ST segment elevation acute coronary syndromes. J Am Coll Cardiol 2003; 41(Suppl):1S.
Boersma E et al: Platelet glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: a meta-analysis of all major randomised clinical trials. Lancet 2002;359:189.
Cannon CP et al: Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med 2001;344:1879.
de Feyter PJ et al: Bypass surgery versus stenting for the treatment of multivessel disease in patients with unstable angina compared with stable angina. Circulation 2002;105:2367.
Eagle KA et al; GRACE Investigators: A validated prediction model for all forms of acute coronary syndrome: estimating the risk of 6-month postdischarge death in an international registry. JAMA 2004;291:2727.
Fox KA et al: Interventional versus conservative treatment for patients with unstable angina or non-ST-elevation myocardial infarction: the British Heart Foundation RITA 3 randomised trial. Lancet 2002;360:743.
Granger CB et al; Global Registry of Acute Coronary Events Investigators: Predictors of hospital mortality in the global registry of acute coronary events. Arch Intern Med 2003; 163:2345.
Hamm CW et al: Acute coronary syndrome without ST elevation: implementation of new guidelines. Lancet 2001;358: 1533.
Levine GN et al: Antithrombotic therapy in patients with acute coronary syndromes. Arch Intern Med 2001;161:937.
Mahoney EM et al: Cost and cost-effectiveness of an early invasive vs conservative strategy for the treatment of unstable angina and non-ST-segment elevation myocardial infarction. JAMA 2002;288:1851.
Schofield PM: Indications for percutaneous and surgical revascularisation: how far does the evidence base guide us? Heart 2003;89:565.
Stone GW: ACUITY Trial. Presented at Scientific Sessions of American College of Cardiology Late Breaking Clinical Trial. March 2006, Atlanta, GA. http://www.medscape.com/viewarticle/529634
Udelson JE et al: Myocardial perfusion imaging for evaluation and triage of patients with suspected acute cardiac ischemia: a randomized controlled trial. JAMA 2002;288:2693.
Wong GC et al: Use of low-molecular-weight heparins in the management of acute coronary artery syndromes and percutaneous coronary intervention. JAMA 2003;289:331.
Yusuf S et al: Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001;345:494.
Yusuf S et al; Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators: Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006;354:1464.
Acute Myocardial Infarction
![]() Essentials of Diagnosis
Essentials of Diagnosis
Sudden but not instantaneous development of prolonged (> 30 minutes) anterior chest discomfort (sometimes felt as gas or pressure) that may produce arrhythmias, hypotension, shock, or cardiac failure.
Sometimes painless, masquerading as acute CHF, syncope, stroke, or shock.
ECG: ST-segment elevation or depression, evolving Q waves, symmetric inversion of T waves.
Elevation of cardiac markers (CK-MB, troponin T, or troponin I).
Appearance of segmental wall motion abnormality by imaging techniques.
General Considerations
Myocardial infarction results from prolonged myocardial ischemia, precipitated in most cases by an occlusive coronary thrombus at the site of a preexisting (though not necessarily severe) atherosclerotic plaque. More rarely, infarction may result from prolonged vasospasm, inadequate myocardial blood flow (eg, hypotension), or excessive metabolic demand. Very rarely, myocardial infarction may be caused by embolic occlusion, vasculitis, aortic root or coronary artery dissection, or aortitis. Cocaine is a cause of infarction, which should be considered in young individuals without risk factors.
A condition that may mimic ST elevation acute myocardial infarction is stress cardiomyopathy (also referred to as Tako-Tsubo cardiomyopathy or apical ballooning syndrome). This is a reversible acute cardiomyopathy involving the cardiac apex, without angiographic evidence of coronary obstruction. It is often associated with emotional stress in older women and is generally associated with small increases in cardiac biomarkers.
The location and extent of infarction depend on the anatomic distribution of the occluded vessel, the presence of additional stenotic lesions, and the adequacy of collateral circulation. Thrombosis in the anterior descending branch of the left coronary artery results in infarction of the anterior LV and interventricular septum. Occlusion of the left circumflex artery produces anterolateral or posterolateral infarction. Right coronary thrombosis leads to infarction of the posteroinferior portion of the LV and generally involves the RV myocardium if the obstruction is proximal. The arteries supplying the AV node and the sinus node more commonly arise from the right coronary artery; thus, AV block at the nodal level and sinus node dysfunction occur more frequently during inferior or right-sided infarctions. Individual variation in coronary anatomy and the presence
P.363
of collateral vessels can make the prediction of coronary anatomy from infarct location imperfect.
Infarctions are often classified as Q wave versus non-Q wave infarction. This classification is generally related to whether there was ST-segment elevation on the presenting ECG, and whether there is transmural or subendocardial involvement. However, Q waves may be transient, and the relationships among Q waves, ST elevation, and extent of infarction are often misleading. Patients presenting without ST elevation are more likely to be older, have multivessel disease, and more vascular disease in general, and thus this population is at higher risk for adverse outcomes following hospital discharge.
The size and anatomic location of an infarction influence the acute course, the early complications, and the long-term prognosis. Hemodynamic stability is related to both noninfarct zone LV function and extent of acute necrosis. Patients with LV dysfunction or clinical heart failure are an important population because of their poor prognosis and the availability of several medical treatments able to improve their survival. The complications of acute infarction are discussed below.
Boersma E et al: Acute myocardial infarction. Lancet 2003;361: 847.
Myocardial infarction redefined a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol 2000;36:959.
Sharkey SW et al: Acute and reversible cardiomyopathy provoked by stress in women from the United States. Circulation 2005;111:472.
Clinical Findings
A. Symptoms
1. Premonitory pain
Many patients give a history of alteration in the pattern of angina preceding the time of onset of symptoms of myocardial infarction, classically the onset of angina with minimal exertion or at rest.
2. Pain of infarction
Unlike anginal episodes, most infarctions occur at rest, and more commonly in the early morning. The pain is similar to angina in location and radiation but it may be more severe, and it builds up rapidly or in waves to maximum intensity over a few minutes or longer. Nitroglycerin has little effect; even opioids may not relieve the pain.
3. Associated symptoms
Patients may break out in a cold sweat, feel weak and apprehensive, and move about, seeking a position of comfort. They prefer not to lie quietly. Light-headedness, syncope, dyspnea, orthopnea, cough, wheezing, nausea and vomiting, or abdominal bloating may be present singly or in any combination.
4. Painless infarction
One-third of patients with acute myocardial infarction present without chest pain, and these patients tend to be undertreated and have poor outcomes. Older patients, women, and patients with diabetes mellitus are more likely to present without classic chest pain. As many as 25% of infarctions are detected on routine ECG without any recallable acute episode.
5. Sudden death and early arrhythmias
Of all deaths from myocardial infarction, about 50% occur before the patients arrive at the hospital, with death presumably caused by ventricular fibrillation.
B. Signs
1. General
Patients may appear anxious and sometimes are sweating profusely. The heart rate may range from marked bradycardia (most commonly in inferior infarction) to tachycardia resulting from increased sympathetic nervous system activity, low cardiac output, or arrhythmia. The BP may be high, especially in former hypertensives, or low in patients with shock. Respiratory distress usually indicates heart failure. Fever, usually low grade, may appear after 12 hours and persist for several days.
2. Chest
Listening for rales on lung examination as evidence for pulmonary edema is a very important part of the physical examination. The Killip classification is a common way to classify heart failure in patients with acute myocardial infarction and has powerful prognostic value. Killip Class I is absence of rales and S3, Class II is rales that do not clear with coughing over one-third or less of the lung fields or presence of an S3, Class III is rales that do not clear with coughing over more than one-third of the lung fields, and Class IV is cardiogenic shock (rales, hypotension, and signs of hypoperfusion).
3. Heart
The cardiac examination may be unimpressive or very abnormal. An abnormally located ventricular impulse often represents the dyskinetic infarcted region. Jugular venous distention reflects RA hypertension, which may indicate RV infarction or elevated LV filling pressures. The absence of elevated central venous pressure, however, does not indicate normal LA or LV diastolic pressures. Soft heart sounds may indicate LV dysfunction. Atrial gallops (S4) are the rule, whereas ventricular gallops (S3) are less common and indicate significant LV dysfunction. Mitral regurgitation murmurs are not uncommon and usually indicate papillary muscle dysfunction or, rarely, rupture. Pericardial friction rubs are uncommon in the first 24 hours but may appear later.
4. Extremities
Edema is usually not present. Cyanosis and cold temperature indicate low output. The peripheral pulses should be noted, since later shock or emboli may alter the examination.
C. Laboratory Findings
The most valuable laboratory tests are cardiac-specific markers of myocardial damage, including quantitative determinations of CK-MB, troponin I, and
P.364
troponin T. Troponins are more sensitive and specific but stay elevated for days and, therefore, are generally not useful for evaluating suspected early reinfarction. Each of these tests may become positive as early as 4 6 hours after the onset of a myocardial infarction and should be abnormal by 8 12 hours. Circulating levels of troponins may remain elevated for 5 7 days or longer.
D. ECG
Most patients with acute infarction have ECG changes, and a normal tracing is uncommon. The extent of the ECG abnormalities, especially the sum of the total amount of ST-segment deviation, is a good indicator of extent of acute infarction and risk of subsequent adverse events. The classic evolution of changes is from peaked ( hyperacute ) T waves, to ST-segment elevation, to Q wave development, to T wave inversion. This may occur over a few hours to several days. The evolution of new Q waves (> 30 milliseconds in duration and 25% of the R wave amplitude) is diagnostic, but Q waves do not occur in 30 50% of acute infarctions (non-Q wave infarctions).
E. Chest Radiography
The chest radiograph may demonstrate signs of CHF, but these changes often lag behind the clinical findings. Signs of aortic dissection, including mediastinal widening, should be sought as a possible alternative diagnosis.
F. Echocardiography
Echocardiography provides convenient bedside assessment of LV global and regional function. This can help with the diagnosis and management of infarction; echocardiography has been used successfully to make judgments about admission and management of patients with suspected infarction, since normal wall motion makes an infarction unlikely. Doppler echocardiography is probably the most convenient procedure for diagnosing postinfarction mitral regurgitation or VSD.
G. Scintigraphic Studies
Technetium-99m pyrophosphate scintigraphy can be used to diagnose acute myocardial infarction. When injected at least 18 hours postinfarction, the radiotracer complexes with calcium in necrotic myocardium to provide a hot spot image of the infarction. This test is insensitive to small infarctions, and false-positive studies occur, so its use is limited to patients in whom the diagnosis by ECG and enzymes is not possible principally those who present several days after the event or have intraoperative infarctions. Radiolabeled antimyosin antibody fragments are more sensitive and specific imaging agents, but scintigraphy must be performed 24 and 48 hours postinjection, so this test has limited clinical usefulness in the diagnosis of acute myocardial infarction.
Scintigraphy with thallium-201 or the newer technetium-based perfusion tracers will demonstrate cold spots in regions of diminished perfusion, which usually represent infarction when the radiotracer is administered at rest, but abnormalities do not distinguish recent from old damage.
Radionuclide angiography demonstrates akinesis or dyskinesis in areas of infarction and also measures EF, which can be valuable. RV dysfunction may indicate infarction of this chamber.
MRI with gadolinium contrast enhancement has emerged as one of the most sensitive tests to detect and quantitate extent of infarction.
H. Hemodynamic Measurements
These can be helpful in managing the patient with suspected cardiogenic shock. Their use is described below and in Table 10-6. Use of PA catheters, however, has generally not been associated with better outcomes and should be limited to patients with severe hemodynamic compromise.
Table 10-6. Hemodynamic subsets in acute myocardial infarction. | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
Ammann P et al: Characteristics and prognosis of myocardial infarction in patients with normal coronary arteries. Chest 2000;117:333.
Cohen MG et al: Pulmonary artery catheterization in acute coronary syndromes: insights from the GUSTO IIb and GUSTO III trials. Am J Med 2005;18:482.
Mahrholdt H et al: Relationship of contractile function to transmural extent of infarction in patients with chronic coronary artery disease. J Am Coll Cardiol 2003;42:505.
Myocardial infarction redefined a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol 2000;36:959.
Zimetbaum PJ et al: Current concepts. Use of the electrocardiogram in acute myocardial infarction. N Engl J Med 2003; 348:933.
Treatment
Table 10-4 provides a summary of the ACC/AHA Guideline recommendations for selected medical treatments.
A. Aspirin and Clopidogrel
All patients with definite or suspected myocardial infarction should receive aspirin at a dose of 162 mg or 325 mg at once regardless of whether thrombolytic therapy is being considered or the patient has been taking aspirin. Chewable aspirin provides more rapid blood levels. Patients with a definite aspirin allergy should be treated with clopidogrel; a 300 mg (or 600 mg) loading dose will result in faster onset of action than the standard 75 mg dose.
Clopidogrel, in addition to aspirin, has also been shown to provide important benefits in patients with acute ST elevation myocardial infarction. In the CLARITY trial, a loading dose of 300 mg of clopidogrel given with thrombolytic therapy, followed by 75 mg a day, led to substantial improvement in coronary
P.365
patency on catheterization 3.5 days following thrombolysis. Moreover, there was no increase in serious bleeding in this population of patients up to 75 years of age. The COMMIT/CCS-2 trial randomized over 45,000 patients in China with acute myocardial infarction to clopidogrel 75 mg a day or placebo, and found a small but statistically significant reduction in early death, myocardial reinfarction, and stroke, with no excess in major bleeding. For patients who have received thrombolytic therapy but will undergo angiography in the first day or two, the early benefits of clopidogrel need to be weighed against the need to delay bypass surgery for approximately 5 days for those patients found to require surgical revascularization.
B. Thrombolytic Therapy
Thrombolytic therapy reduces mortality and limits infarct size in patients with acute myocardial infarction associated with ST-segment elevation (defined as 0.1 mV in two inferior or lateral leads or two contiguous precordial leads), or with left bundle branch block. The greatest benefit occurs if treatment is initiated within the first 3 hours, when up to a 50% reduction in mortality rate can be achieved. The magnitude of benefit declines rapidly thereafter, but a 10% relative mortality reduction can be achieved up to 12 hours after the onset of chest pain. The survival benefit is greatest in patients with large usually anterior infarctions. Patients without ST-segment elevation (previously labeled non-Q wave infarctions) do not benefit, and may derive harm, from thrombolysis.
Major bleeding complications occur in 0.5 5% of patients, the most serious of which is intracranial hemorrhage. The major risk factors for intracranial bleeding are age over 65 years, hypertension at presentation, low body weight (< 70 kg), and the use of clot-specific thrombolytic agents (alteplase, reteplase, tenecteplase). Although patients over age 75 years have a much higher mortality rate with acute myocardial infarction and therefore may derive greater benefit, the risk of severe bleeding is also higher, particularly among patients with risk factors from bleeding or intracranial hemorrhage, such as severe hypertension or recent stroke. Patients presenting more than 12 hours after the onset of chest pain may also derive a small benefit, particularly if pain and ST-segment elevation persist, but rarely does this benefit outweigh the attendant risk.
Therefore, the current recommendation is to treat patients with ST-segment elevation infarction who seek medical attention within 6 12 hours of the onset of symptoms with reperfusion therapy, either primary percutaneous coronary intervention (PCI) or thrombolytic therapy. Contraindications include previous hemorrhagic stroke, other strokes or cerebrovascular events within 1 year, known intracranial neoplasm, active internal bleeding (excluding menstruation), or suspected aortic dissection. Relative contraindications are BP > 180/110 mm Hg at presentation, other intracerebral pathology not listed above as a contraindication, known bleeding diathesis, trauma within 2 4 weeks, major surgery within 3 weeks, prolonged (> 10 minutes) or traumatic cardiopulmonary resuscitation, recent (within 2 4 weeks) internal bleeding, noncompressible vascular punctures, active diabetic retinopathy, pregnancy, active peptic ulcer disease, a history of severe hypertension, current use of anticoagulants (INR > 2.0 3.0), and prior allergic reaction or exposure to streptokinase or anistreplase within 2 years.
The following thrombolytic agents are available for acute myocardial infarction and are characterized in Table 10-7.
Streptokinase is not commonly used for treatment of acute myocardial infarction since it is less effective at opening occluded arteries and less effective at reducing mortality. It is non-fibrin-specific, causes depletion of circulating fibrinogen, and has a tendency to induce hypotension, particularly if infused rapidly.
P.366
This can be managed by slowing or interrupting the infusion and administering fluids. There is controversy as to whether adjunctive heparin is beneficial in patients given streptokinase, unlike its administration with the more clot-specific agents. Allergic reactions, including anaphylaxis, occur in 1 2% of patients, and this agent should generally not be administered to patients with prior exposure.
Table 10-7. Thrombolytic therapy for acute myocardial infarction. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Alteplase (recombinant tissue plasminogen activator; t-PA) is a naturally occurring plasminogen activator that is modestly fibrin specific, resulting in about a 50% reduction in circulating fibrinogen. In the first GUSTO trial, which compared t-PA (with unfractionated heparin) with streptokinase, the 30-day mortality rate with t-PA was one absolute percentage point lower (one additional life saved per 100 patients treated), though there was also a small increase in the rate of intracranial hemorrhage. An angiographic substudy confirmed a higher 90-minute patency rate and a higher rate of normal (TIMI grade 3) flow in patients.
Reteplase is a recombinant deletion mutant of t-PA that is slightly less fibrin specific. In comparative trials, it appears to have efficacy similar to that of alteplase, but it has a longer duration of action and can be administered as two boluses 30 minutes apart.
Tenecteplase (TNK-t-PA) is a genetically engineered substitution mutant of native t-PA that has reduced plasma clearance, increased fibrin sensitivity, and increased resistance to plasminogen activator inhibitor-1. It can be given as a single weight-adjusted bolus. In a large comparative trial, this agent was equivalent to t-PA with regard to efficacy and resulted in significantly less noncerebral bleeding.
1. Selection of a thrombolytic agent
In the United States, most patients are treated with alteplase, reteplase, or tenecteplase. The differences in efficacy between them are small compared with the potential benefit of treating a greater proportion of appropriate candidates in a more prompt manner. The principal objective should be to administer a thrombolytic agent within 30 minutes of presentation or even during transport. The ability to administer tenecteplase as a single bolus is an attractive feature that may facilitate earlier treatment. The combination of a reduced-dose thrombolytic given with a platelet glycoprotein IIb/IIIa antagonist has been investigated in several trials, with no evidence of reduction in mortality but a modest increase in bleeding complications.
2. Postthrombolytic management
After completion of the thrombolytic infusion, aspirin should be
P.367
continued. Anticoagulation with intravenous heparin (initial dose of 60 units/kg bolus to a maximum of 4000 units, followed by an infusion of 12 units/kg/min to a maximum of 1000 units, then adjusted to maintain an activated partial thromboplastin time [aPTT] of 50 75 seconds beginning with an aPTT drawn 3 hours after thrombolytic) is continued for at least 24 hours after alteplase, reteplase, or tenecteplase.
With tenecteplase, enoxaparin, a 30 mg intravenous bolus, followed by 1 mg/kg every 12 hours, resulted in a lower incidence of the composite of death, myocardial infarction, refractory ischemia, and disabling stroke in the ASSENT-3 trial, but bleeding rates were increased in the elderly. In the larger EXTRACT trial, enoxaparin reduced death and myocardial infarction at day 30 (compared with unfractionated heparin), at the expense of a modest increase in bleeding. In patients younger than age 75, enoxaparin was given as a 30 mg intravenous bolus and 1 mg/kg every 12 hours; in patients age 75 years and older, it was given with no bolus and 0.75 mg/kg intravenously every 12 hours. Another antithrombotic option is fondaparinux, given at a dose of 2.5 mg subcutaneously once a day. In the OASIS-6 trial, it resulted in significant reductions in death and reinfarction when compared with control (unfractionated heparin when indicated, otherwise placebo). Similar to the findings of the OASIS-5 trial, fondaparinux tended to result in less bleeding, despite its longer duration compared with heparin and despite its comparison to placebo in about half of the enrolled patients. There was no benefit of fondaparinux among patients undergoing primary PCI.
For all patients with acute myocardial infarction treated with intensive antithrombotic therapy, prophylactic treatment with antacids and an H2-blocker is advisable.
Myocardial reperfusion can be recognized clinically by the early cessation of pain and the resolution of ST-segment elevation. Although at least 50 70% resolution of ST-segment elevation by 90 minutes may occur without coronary reperfusion, ST resolution is a strong predictor of better outcome. Even with anticoagulation, 10 20% of reperfused vessels will reocclude during hospitalization, although reocclusion and reinfarction appear to be reduced following intervention. Reinfarction, indicated by recurrence of pain and ST-segment elevation, can be treated by readministration of a thrombolytic agent or immediate angiography and PCI. The role of catheterization and intervention after thrombolysis is controversial. For patients who do not reperfuse based on lack of at least 50% resolution of ST elevation, rescue angioplasty has been shown to reduce the composite of death, reinfarction, stroke, or severe heart failure. Many cardiologists advocate routine catheterization and revascularization following acute myocardial infarction, in otherwise suitable candidates, although the data to support this are inconclusive. Patients with recurrent ischemic pain prior to discharge should undergo catheterization and, if indicated, revascularization. Asymptomatic, clinically stable patients should undergo predischarge evaluation to determine whether residual jeopardized myocardium is present. This can be accomplished by submaximal exercise or pharmacologic stress scintigraphy. Those with significantly positive tests or a low threshold for symptomatic ischemia should undergo angiography and revascularization where feasible.
C. Primary Percutaneous Coronary Intervention for ST Segment Elevation Myocardial Infarction
Immediate coronary angiography and primary PCI (including stenting) of the infarct-related artery have been shown to be superior to thrombolysis when done by experienced operators in high-volume centers with rapid time from first medical contact to intervention ( door-to-balloon ). U.S. and European guidelines call for first medical contact or door-to-balloon times of < 90 minutes. Several trials have shown that if efficient transfer systems are in place, transfer of patients with acute myocardial infarction from hospitals without to hospitals with primary PCI capability can improve outcome compared with thrombolytic therapy at the presenting hospital, although this requires sophisticated systems to ensure rapid identification, transfer, and PCI. Primary PCI is the approach of choice in patients with absolute and many relative contraindications to thrombolytic therapy. The results of this approach in specialized centers are excellent, exceeding those obtainable by thrombolytic therapy even in good candidates, but this experience may not be generalizable to centers and operators with less experience or expertise. Stenting in conjunction with the platelet glycoprotein IIb/IIIa antagonist abciximab is now widely used in patients with acute myocardial infarction. In the subgroup of patients with cardiogenic shock, early catheterization and percutaneous or surgical revascularization are the preferred management. Because an acute interventional approach carries a lower risk of hemorrhagic complications, it may also be the preferred strategy in many older patients (see Tables 10-4 and 10-5 for factors to consider in choosing thrombolytic therapy or primary PCI).
In part because patients in the United States who are transferred for primary PCI tend to have long delays from first hospital arrival to balloon inflation, there has been interest in developing facilitated PCI whereby a combination of medications (full or reduced dose fibrinolytic agents with or without glycoprotein IIb/IIIa inhibitors) is given to establish patency followed by immediate PCI. While this approach has been shown to establish patency at the time of catheterization laboratory arrival in a substantial portion of patients, it has not yet been shown to improve outcome. In fact, in the ASSENT-4 PCI trial, patients with a delay in median time to PCI balloon inflation did better if they did not receive full-dose tenecteplase on the way to the catheterization laboratory. Thus, for
P.368
the time being, patients should be treated either with fibrinolytic agents (and immediate rescue PCI for reperfusion failure) or with primary PCI, if it can be done in the rapid timecourse outlined in the ACC/AHA guidelines.
D. General Measures
Cardiac care unit monitoring should be instituted as soon as possible. Patients without complications can be transferred to a telemetry unit after 24 48 hours. Activity should initially be limited to bed rest but can be advanced within 24 hours. Progressive ambulation should be started after 24 72 hours if tolerated. For patients without complications, discharge by day 4 appears to be appropriate. Low-flow oxygen therapy (2 4 L/min) should be given if oxygen saturation is reduced.
E. Analgesia
An initial attempt should be made to relieve pain with sublingual nitroglycerin. However, if no response occurs after two or three tablets, intravenous opioids provide the most rapid and effective analgesia and may also reduce pulmonary congestion. Morphine sulfate, 4 8 mg, or meperidine, 50 75 mg, should be given. Subsequent small doses can be given every 15 minutes until pain abates.
F. -Adrenergic Blocking Agents
Although trials have shown modest short-term benefit from intravenous -blockers given immediately after acute myocardial infarction, it has not been clear that this provides a major advantage over simply beginning an oral -blocker. The Chinese COMMIT/CCS-2 trial involving 45,000 patients found no overall benefit to intravenous followed by oral metoprolol; the aggressive dosing (three 5 mg intravenous boluses followed by 200 mg/d orally) appeared to prevent reinfarction at the cost of increasing shock in patients presenting with heart failure. Thus, -blockade should be avoided in patients with decompensated heart failure, decompensated asthma, or high degrees of AV block. The CAPRICORN trial showed the benefits of carvedilol following the acute phase of large myocardial infarction with contemporary care.
G. Nitrates
Nitroglycerin is the agent of choice for continued or recurrent ischemic pain and is useful in lowering BP or relieving pulmonary congestion. However, routine nitrate administration is not recommended, since no improvement in outcome has been observed in the ISIS-4 or GISSI-3 trials, in which a total of over 70,000 patients were randomized to nitrate treatment or placebo. Nitrates should be avoided in patients who received phosphodiesterase inhibitors (sildenafil, vardenafil, and tadalafil) in the prior 24 hours.
H. ACE Inhibitors
A series of trials (SAVE, AIRE, SMILE, TRACE, GISSI-III, and ISIS-4) have shown both short- and long-term improvement in survival with ACE inhibitor therapy. The benefits are greatest in patients with low EFs, large infarctions, or clinical evidence of heart failure. Because substantial amounts of the survival benefit occur on the first day, ACE inhibitor treatment should be commenced early in patients without hypotension, especially patients with large or anterior myocardial infarction.
I. Angiotensin Receptor Blockers
Although there has been inconsistency in the effects of different angiotensin receptor blockers (ARBs) on mortality for patients post-myocardial infarction with heart failure and/or LV dysfunction, the VALIANT trial showed that valsartan 160 mg twice a day is equivalent to captopril in reducing mortality. Thus, valsartan should be used for all patients with ACE inhibitor intolerance, and is a reasonable, albeit more expensive, alternative to captopril. The combination of captopril and valsartan (at reduced dose) was no better than either agent alone and resulted in more side effects.
J. Aldosterone Antagonists
The RALES trial showed that spironolactone can reduce the mortality rate of patients with advanced heart failure, and the EPHESUS trial showed a 15% relative risk reduction in mortality with eplerenone for patients post-myocardial infarction with LV dysfunction and heart failure. Patients must be monitored carefully for development of hyperkalemia.
K. Antiarrhythmic Prophylaxis
The incidence of ventricular fibrillation in hospitalized patients is approximately 5%, with 80% of episodes occurring in the first 12 24 hours. Prophylactic lidocaine infusions (1 2 mg/min) prevent most episodes, but this therapy has not reduced the mortality rate and it increases the risk of asystole, so this approach is no longer recommended except in patients with sustained ventricular tachycardia.
L. Calcium Channel Blockers
There are no studies to support the routine use of calcium channel blockers in most patients with acute myocardial infarction and indeed, they have the potential to exacerbate ischemia and cause death from reflex tachycardia or myocardial depression. Long-acting calcium channel blockers should generally be reserved for management of hypertension or ischemia as second- or third-line drugs after -blockers and nitrates.
M. Long-Term Antithrombotic Therapy
Discharge on aspirin, since it is highly effective, inexpensive, and well tolerated, is a key quality indicator of myocardial infarction care. In the WARIS-II trial, long-term anticoagulation with warfarin post-myocardial infarction was associated with a reduction in the composite of death, reinfarction, and stroke. However, whether the results of this trial are transferable to the United States where anticoagulation services may
P.369
be less organized and effective than in Norwegian hospitals is unknown. In the CURE trial, clopidogrel for 3 12 months for non-ST elevation acute coronary syndromes resulted in a similar 20% relative risk reduction in cardiovascular death, myocardial infarction, and stroke.
Antman EM et al; ExTRACT-TIMI 25 Investigators: Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006;354:1477.
Assessment of the Safety and Efficacy of a New Thrombolytic Regimen (ASSENT-3) Investigators: Efficacy and safety of tenecteplase in combination with enoxaparin, abciximab, or unfractionated heparin: the ASSENT-3 randomised trial in acute myocardial infarction. Lancet 2001;358:605.
Assessment of the Safety and Efficacy of a New Treatment Strategy with Percutaneous Coronary Intervention (ASSENT-4 PCI) investigators. Primary versus tenecteplase-facilitated percutaneous coronary intervention in patients with ST-segment elevation acute myocardial infarction (ASSENT-4 PCI): randomised trial. Lancet 2006;367: 569.
Chen Z: Results of the COMMIT/CCS-2 Trial. American College of Cardiology, Orlando, FL, March 2005. Results online at http://www.ctsu.ox.ac.uk/~ccs2/live/content/presentations/slideshow/ (accessed March 28, 2005).
Collins R: Results of the COMMIT/CCS-2 Trial. American College of Cardiology, Orlando, FL, March 2005. Results online at http://www.ctsu.ox.ac.uk/~ccs2/live/content/presentations/slideshow/ (accessed March 28, 2005).
Hurlen M et al: Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002;347:969.
Keeley EC et al: Primary coronary intervention for acute myocardial infarction. JAMA 2004;291:736.
Lincoff AM et al: Mortality at 1 year with combination platelet glycoprotein IIb/IIIa inhibition and reduced-dose fibrinolytic therapy vs conventional fibrinolytic therapy for acute myocardial infarction: GUSTO V randomized trial. JAMA 2002;288:2130.
Pfeffer MA et al: Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003;349:1893.
Rapaport E: ACC/AHA American College of Cardiology/American Heart Association. Guidelines for the acute coronary syndromes. Curr Cardiol Rep 2001;3:289.
Ryan TJ et al: 1999 update: ACC/AHA guidelines for the management of acute myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Acute Myocardial Infarction). J Am Coll Cardiol 1999;34:890.
Sabatine MS et al; CLARITY-TIMI 28 Investigators: Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med 2005;352:1179.
Stone GW et al: Comparison of angioplasty with stenting, with or without abciximab, in acute myocardial infarction. N Engl J Med 2002;346:957.
Yusuf S et al; OASIS-6 Trial Group: Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: the OASIS-6 randomized trial. JAMA 2006;295:1519.
Complications
A variety of complications can occur after myocardial infarction even when treatment is initiated promptly.
A. Postinfarction Ischemia
In recent clinical trials of thrombolysis, recurrent ischemia occurred in about one-third of patients, was more common following non-ST elevation than ST elevation myocardial infarction, and had important short- and long-term prognostic implications. Vigorous medical therapy should be instituted, including nitrates and -blockers as well as aspirin, heparin, and consideration of platelet glycoprotein IIb/IIIa antagonists. Most patients with postinfarction angina and all who are refractory to medical therapy should undergo early catheterization and revascularization by PTCA or CABG.
B. Arrhythmias
Abnormalities of rhythm and conduction are common.
1. Sinus bradycardia
This is most common in inferior infarctions or may be precipitated by medications. Observation or withdrawal of the offending agent is usually sufficient. If accompanied by signs of low cardiac output, atropine, 0.5 1 mg intravenously, is usually effective. Temporary pacing is rarely required.
2. Supraventricular tachyarrhythmias
Sinus tachycardia is common and may reflect either increased adrenergic stimulation or hemodynamic compromise due to hypovolemia or pump failure. In the latter, -blockade is contraindicated. Supraventricular premature beats are common and may be premonitory for atrial fibrillation. Electrolyte abnormalities and hypoxia should be corrected and causative agents (especially aminophylline) stopped. Atrial fibrillation should be rapidly controlled or converted to sinus rhythm. Intravenous -blockers such as metoprolol (2.5 5 mg/h) or short-acting esmolol (50 200 mcg/kg/min) are the agents of choice if cardiac function is adequate. Intravenous diltiazem (5 15 mg/h) may be used if -blockers are contraindicated or ineffective. Digoxin (0.5 mg as initial dose, then 0.25 mg every 90 120 minutes [up to 1 1.25 mg] for a loading dose, followed by 0.25 mg daily if renal function is normal) is preferable if heart failure is present with atrial fibrillation, but the onset of action is delayed. Electrical cardioversion (commencing with 100 J) may be necessary if atrial fibrillation is complicated by hypotension, heart failure, or ischemia, but the arrhythmia often recurs. Amiodarone (150 mg intravenous bolus and then 15 30 mg/h intravenously, or rapid oral loading with 400 mg three times daily) may be helpful to restore or maintain sinus rhythm.
3. Ventricular arrhythmias
Ventricular arrhythmias are most common in the first few hours after infarction. Ventricular premature beats may be premonitory for ventricular tachycardia or fibrillation but generally should not be treated in the absence of frequent
P.370
nonsustained ventricular tachycardia (usually more than six consecutive beats). Lidocaine is recommended as a prophylactic measure. Toxicity (tremor, anxiety, confusion, seizures) is common, especially in older patients and those with hypotension, heart failure, or liver disease.
Sustained ventricular tachycardia should be treated with a 1 mg/kg bolus of lidocaine if the patient is stable or by electrical cardioversion (100 200 J) if not. If the arrhythmia cannot be suppressed with lidocaine, procainamide (100 mg boluses over 1 2 minutes every 5 minutes to a cumulative dose of 750 1000 mg) or intravenous amiodarone (150 mg over 10 minutes, which may be repeated as needed, followed by 360 mg over 6 hours and then 540 mg over 18 hours) should be initiated, followed by an infusion of 20 80 mg/kg/min. Ventricular fibrillation is treated electrically (300 400 J). Unresponsive ventricular fibrillation should be treated with additional amiodarone and repeat cardioversion while cardiopulmonary resuscitation (CPR) is administered.
Accelerated idioventricular rhythm is a regular, wide-complex rhythm at a rate of 70 100/min. It may occur with or without reperfusion and does not require specific therapy.
4. Conduction disturbances
All degrees of AV block may occur in the course of acute myocardial infarction. Block at the level of the AV node is more common than infranodal block and occurs in approximately 20% of inferior myocardial infarctions. First-degree block is the most common and requires no treatment. Second-degree block is usually of the Mobitz type I form (Wenckebach), is often transient, and requires treatment only if associated with a heart rate slow enough to cause symptoms. Complete AV block occurs in up to 5% of acute inferior infarctions, usually is preceded by Mobitz I second-degree block, and generally resolves spontaneously, though it may persist for hours to several weeks. The escape rhythm originates in the distal AV node or AV junction and hence has a narrow QRS complex and is reliable, albeit often slow (30 50 beats/min). Treatment is often necessary because of resulting hypotension and low cardiac output. Intravenous atropine (1 mg) usually restores AV conduction temporarily, but if the escape complex is wide or if repeated atropine treatments are needed, temporary ventricular pacing is indicated. The prognosis for these patients is only slightly worse than for patients in whom AV block did not develop.
In anterior infarctions, the site of block is distal, below the AV node, and usually a result of extensive damage of the His-Purkinje system and bundle branches. New first-degree block (prolongation of the PR interval) is unusual in anterior infarction; Mobitz type II AV block or complete heart block may be preceded by intraventricular conduction defects or may occur abruptly. The escape rhythm, if present, is an unreliable wide-complex idioventricular rhythm. Urgent ventricular pacing is mandatory, but even with successful pacing, morbidity and mortality are high because of the extensive myocardial damage. New conduction abnormalities such as right or left bundle branch block or fascicular blocks may presage progression, often sudden, to second- or third-degree AV block. Temporary ventricular pacing is recommended for new-onset alternating bilateral bundle branch block, bifascicular block, or bundle branch block with worsening first-degree AV block. Patients with anterior infarction who progress to second- or third-degree block even transiently should be considered for insertion of a prophylactic permanent ventricular pacemaker before discharge.
C. Myocardial Dysfunction
The severity of cardiac dysfunction is proportionate to the extent of myocardial necrosis but is exacerbated by preexisting dysfunction and ongoing ischemia. Patients who have normal BP, no signs of heart failure, and normal urinary output have a good prognosis. Those with hypotension or evidence of more than mild heart failure should have bedside right heart catheterization and continuous measurements of arterial pressure. These measurements permit the accurate assessment of cardiac function, facilitate the correct choice of therapy, and provide important prognostic information. Table 10-6 categorizes patients based on these hemodynamic findings.
1. Acute LV failure
Basilar rales are common in acute myocardial infarction, but dyspnea, more diffuse rales, and arterial hypoxemia usually indicate LV failure. Because both the physical examination and chest radiograph correlate poorly with hemodynamic measurements and because the central venous pressure does not correlate with the PCWP, right heart catheterization may be useful in cases of suspected cardiogenic shock. General measures include supplemental oxygen to increase arterial saturation to above 95% and elevation of the trunk. Diuretics are usually the initial therapy unless RV infarction is present. Intravenous furosemide (10 40 mg) or bumetanide (0.5 1 mg) is preferred because of the reliably rapid onset and short duration of action of these drugs. Higher dosages can be given if an inadequate response occurs. Morphine sulfate (4 mg intravenously followed by increments of 2 mg) is valuable in acute pulmonary edema.
Diuretics are usually effective; however, because most patients with acute infarction are not volume overloaded, the hemodynamic response may be limited and may be associated with hypotension. Vasodilators will reduce PCWP and improve cardiac output by a combination of venodilation (increasing venous capacitance) and arteriolar dilation (reducing afterload and LV wall stress). In mild heart failure, sublingual isosorbide dinitrate (2.5 10 mg every 2 hours) or nitroglycerin ointment (6.25 25 mg every 4 hours) may be adequate to lower PCWP. In more severe failure, especially if cardiac output is reduced, sodium nitroprusside
P.371
is the preferred agent. It should be initiated only with arterial pressure monitoring; the initial dosage should be low (0.25 mcg/kg/min) to avoid excessive hypotension, but the dosage can be increased by increments of 0.5 mcg/kg/min every 5 10 minutes up to 5 10 mcg/kg/min until the desired hemodynamic response is obtained. Excessive hypotension (mean BP < 65 75 mm Hg) or tachycardia (> 10/min increase) should be avoided.
Intravenous nitroglycerin (starting at 10 mcg/min) also may be effective but may lower PCWP with less hypotension. Oral or transdermal vasodilator therapy with nitrates or ACE inhibitors is often necessary after the initial 24 48 hours (see below).
Inotropic agents should be avoided if possible, because they often increase heart rate and myocardial oxygen requirements and worsen clinical outcomes. Dobutamine has the best hemodynamic profile, increasing cardiac output and modestly lowering PCWP, usually without excessive tachycardia, hypotension, or arrhythmias. The initial dosage is 2.5 mcg/kg/min, and it may be increased by similar increments up to 15 20 mcg/kg/min at intervals of 5 10 minutes. Dopamine is more useful in the presence of hypotension (see below), since it produces peripheral vasoconstriction, but it has a less beneficial effect on PCWP. Amrinone is a positive inotrope and vasodilator that produces hemodynamic effects similar to those of dobutamine but with a greater decrease in PCWP. However, its longer duration of action makes it less useful in unstable situations. Milrinone is a more potent and newer congener of amrinone with fewer side effects. It should be commenced in a loading dose of 50 mcg/kg over 10 minutes, followed by an infusion of 0.375 0.75 mcg/kg/min. Digoxin has not been helpful in acute infarction except to control the ventricular response in atrial fibrillation, but it may be beneficial if chronic heart failure persists.
2. Hypotension and shock
Patients with hypotension (systolic BP < 100 mm Hg, individualized depending on prior BP) and signs of diminished perfusion (low urinary output, confusion, cold extremities) that does not respond to fluid resuscitation should be considered for hemodynamic monitoring with a PA catheter. Up to 20% will have findings indicative of intravascular hypovolemia (due to diaphoresis, vomiting, decreased venous tone, medications such as diuretics, nitrates, morphine, -blockers, calcium channel blockers, and thrombolytic agents and lack of oral intake). These should be treated with successive boluses of 100 mL of normal saline until PCWP reaches 15 18 mm Hg to determine whether cardiac output and BP respond. Pericardial tamponade due to hemorrhagic pericarditis (especially after thrombolytic therapy or cardiopulmonary resuscitation) or ventricular rupture should be considered and excluded by echocardiography if clinically indicated. RV infarction, characterized by a normal PCWP but elevated RA pressure, can produce hypotension. This is discussed below.
Most patients with cardiogenic shock will have moderate to severe LV systolic dysfunction, with a mean EF of 30% in the SHOCK trial. If hypotension is only modest (systolic pressure > 90 mm Hg) and the PCWP is elevated, diuretics and an initial trial of nitroprusside (see above for dosing) are indicated. If the BP falls, inotropic support will need to be added or substituted. Such patients may also be treated with IABC. This device unloads the LV during systole and increases diastolic coronary artery filling pressure. It often facilitates the use of vasodilators in patients who previously did not tolerate them.
Dopamine is the most appropriate pressor for cardiogenic hypotension. It should be initiated at a rate of 2 4 mcg/kg/min and increased at 5-minute intervals to the appropriate hemodynamic end point. At low dosages (< 5 mcg/kg/min), it improves renal blood flow; at intermediate dosages (2.5 10 mcg/kg/min), it stimulates myocardial contractility; at higher dosages (> 8 mcg/kg/min), it is a potent 1-adrenergic agonist. In general, BP and cardiac index rise, but PCWP does not fall. Dopamine may be combined with nitroprusside or dobutamine (see above for dosing), or the latter may be used in its place if hypotension is not severe. Milrinone has hemodynamic effects similar to those of dobutamine, but the longer duration of action precludes rapid dosage adjustment. Norepinephrine (0.1 0.5 mcg/kg/min) is the usual pressor of last resort, since isoproterenol and epinephrine produce less vasoconstriction and do not increase coronary perfusion pressure (aortic diastolic pressure), but both tend to worsen the balance between myocardial oxygen delivery and utilization.
Patients with cardiogenic shock not due to hypovolemia have a poor prognosis, with 30-day mortality rates of 50 80%. If they do not respond rapidly, IABC should be instituted. Surgically implanted ventricular assist devices may be used in extreme cases. Emergent cardiac catheterization and coronary angiography followed by percutaneous or surgical revascularization offer the best chance of survival, particularly in patients under 75 years of age.
D. RV Infarction
RV infarction is present in one-third of patients with inferior wall infarction but is clinically significant in less than 50% of these. It presents as hypotension with relatively preserved LV function and should be considered whenever patients with inferior infarction exhibit low BP, raised venous pressure, and clear lungs. Hypotension is often exacerbated by medications that decrease intravascular volume or produce venodilation, such as diuretics, nitrates, and narcotics. RA pressure and jugular venous pulsations are high, while PCWP is normal or low and the lungs are clear. The diagnosis is suggested by ST-segment elevation in right-sided anterior chest leads, particularly RV4. The diagnosis can be confirmed by echocardiography or hemodynamic
P.372
measurements. Treatment consists of fluid loading to improve LV filling, and inotropic agents if necessary.
E. Mechanical Defects
Partial or complete rupture of a papillary muscle or of the interventricular septum occurs in less than 1% of acute myocardial infarctions and carries a poor prognosis. These complications occur in both anterior and inferior infarctions, usually 3 7 days after the acute event. They are detected by the appearance of a new systolic murmur and clinical deterioration, often with pulmonary edema. The two lesions are distinguished by the location of the murmur (apical versus parasternal) and by Doppler echocardiography. Hemodynamic monitoring is essential for appropriate management and demonstrates an increase in oxygen saturation between the RA and PA in VSD and, often, a large v wave with mitral regurgitation. Treatment by nitroprusside and, preferably, IABC reduces the regurgitation or shunt, but surgical correction is mandatory. In patients remaining hemodynamically unstable or requiring continuous parenteral pharmacologic treatment or counterpulsation, early surgery is recommended, though mortality rates are high (15% to nearly 100%, depending on residual ventricular function and clinical status). Patients who are stabilized medically can have delayed surgery with lower risks (10 25%), although this may be due to the death of sicker patients, some of whom may have been saved by earlier surgery.
F. Myocardial Rupture
Complete rupture of the LV free wall occurs in less than 1% of patients and usually results in immediate death. It occurs 2 7 days postinfarction, usually involves the anterior wall, and is more frequent in older women. Incomplete or gradual rupture may be sealed off by the pericardium, creating a pseudoaneurysm. This may be recognized by echocardiography, radionuclide angiography, or LV angiography, often as an incidental finding. It demonstrates a narrow-neck connection to the LV. Early surgical repair is indicated, since delayed rupture is common.
G. LV Aneurysm
An LV aneurysm, a sharply delineated area of scar that bulges paradoxically during systole, develops in 10 20% of patients surviving an acute infarction. This usually follows anterior Q wave infarctions. Aneurysms are recognized by persistent ST-segment elevation (beyond 4 8 weeks), and a wide neck from the LV can be demonstrated by echocardiography, scintigraphy, or contrast angiography. They rarely rupture but may be associated with arterial emboli, ventricular arrhythmias, and CHF. Surgical resection may be performed for these indications if other measures fail. The best results (mortality rates of 10 20%) are obtained when the residual myocardium contracts well and when significant coronary lesions supplying adjacent regions are bypassed.
H. Pericarditis
The pericardium is involved in approximately 50% of infarctions, but pericarditis is often not clinically significant. Twenty percent of patients with Q wave infarctions will have an audible friction rub if examined repetitively. Pericardial pain occurs in approximately the same proportion after 2 7 days and is recognized by its variation with respiration and position (improved by sitting). Often, no treatment is required, but aspirin (650 mg every 4 6 hours) will usually relieve the pain. Indomethacin and corticosteroids can cause impaired infarct healing and predispose to myocardial rupture, and therefore should generally be avoided in the early post-myocardial infarction period. Likewise, anticoagulation should be used cautiously, since hemorrhagic pericarditis may result.
One week to 12 weeks after infarction, Dressler's syndrome (post-myocardial infarction syndrome) occurs in less than 5% of patients. This is an autoimmune phenomenon and presents as pericarditis with associated fever, leukocytosis and, occasionally, pericardial or pleural effusion. It may recur over months. Treatment is the same as for other forms of pericarditis. A short course of nonsteroidal agents or corticosteroids may help relieve symptoms.
I. Mural Thrombus
Mural thrombi are common in large anterior infarctions but not in infarctions at other locations. Arterial emboli occur in approximately 2% of patients with known infarction, usually within 6 weeks. Anticoagulation with heparin followed by short-term (3-month) warfarin therapy prevents most emboli and should be considered in all patients with large anterior infarctions. Mural thrombi can be detected by echocardiography or CT scan (MRI has yielded frequent false-positive results) but with only moderate reliability, and only a small percentage (up to 25%) embolize, so these procedures should not be relied upon for determining the need for anticoagulation.
Brady WJ et al: Diagnosis and management of bradycardia and atrioventricular block associated with acute coronary ischemia. Emerg Med Clin North Am 2001;19:371.
Crenshaw BS et al: Risk factors, angiographic patterns, and outcomes in patients with ventricular septal defect complicating acute myocardial infarction. Circulation 2000;101:27.
Goldstein JA: Pathophysiology and management of right heart ischemia. J Am Coll Cardiol 2002;40:841.
Hochman JS et al: Early revascularization in acute myocardial infarction complicated by cardiogenic shock. SHOCK Investigators. Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock. N Engl J Med 1999;341:625.
Mangrum JM: Tachyarrhythmias associated with acute myocardial infarction. Emerg Med Clin North Am 2001;19:385.
P.373
Menon V et al: Management of cardiogenic shock complicating acute myocardial infarction. Heart 2002;88:531.
Pfisterer M: Right ventricular involvement in myocardial infarction and cardiogenic shock. Lancet 2003;362:392.
Prieto A et al: Nonarrhythmic complications of acute myocardial infarction. Emerg Med Clin North Am 2001;19:397.
Rathore SS et al: Acute myocardial infarction complicated by heart block in the elderly: prevalence and outcomes. Am Heart J 2001;141:47.
Sayer JW et al: Prognostic implications of ventricular fibrillation in acute myocardial infarction: new strategies required for mortality reduction. Heart 2000;84:258.
Postinfarction Management
After the first 24 hours, the focus of patient management is to prevent recurrent ischemia, improve infarct healing and prevent remodeling, and prevent recurrent vascular events. Patients with hemodynamic compromise, who are at high risk for death, need careful monitoring and management of volume status.
A. Risk Stratification
Just as for non-ST-segment elevation acute coronary syndrome, risk stratification is important for management of ST-segment elevation acute myocardial infarction. GRACE and TIMI risk scores can be helpful tools. Patients with recurrent ischemia (spontaneous or provoked), hemodynamic instability, impaired LV function, heart failure, or serious ventricular arrhythmias should undergo cardiac catheterization (Table 10-5). ACE inhibitor (or ARB) therapy is indicated in patients with clinical heart failure or LV EF < 40%.
For patients not undergoing cardiac catheterization, submaximal exercise (or pharmacologic stress testing for patients unable to exercise) before discharge or a maximal test after 3 6 weeks (the latter being more sensitive for ischemia) helps patients and physicians plan the return to normal activity. Imaging in conjunction with stress testing adds additional sensitivity for ischemia and provides localizing information. Both exercise and pharmacologic stress imaging have successfully predicted subsequent outcome. One of these tests should be used prior to discharge in patients who have received thrombolytic therapy as a means of selecting appropriate candidates for coronary angiography.
B. Secondary Prevention
Postinfarction management should begin with identification and modification of risk factors. Treatment of hyperlipidemia and smoking cessation both prevent recurrent infarction and death. LDL cholesterol levels should be lowered below 100 mg/dL, and probably to a goal of 70 mg/dL, with a statin commencing prior to discharge. BP control and cardiac rehabilitation or exercise are also recommended.
-Blockers improve survival rates, primarily by reducing the incidence of sudden death in high-risk subsets of patients, though their value may be less in patients without complications with small infarctions and normal exercise tests. While a variety of -blockers have been shown to be beneficial, for patients with LV dysfunction managed with contemporary treatment, carvedilol has been shown to reduce mortality. -Blockers with intrinsic sympathomimetic activity have not proved beneficial in postinfarction patients.
Antiplatelet agents are beneficial; aspirin (81 325 mg daily) is recommended, and adding clopidogrel (75 mg daily) has been shown to provide additional benefit short term after ST-segment elevation myocardial infarction and for up to 1 year after non-ST elevation acute coronary syndromes. Warfarin anticoagulation for 3 months reduces the incidence of arterial emboli after large anterior infarctions, and according to the results of at least one study it improves long-term prognosis. An advantage to combining low-dose aspirin and warfarin has not been demonstrated, except perhaps in patients with atrial fibrillation.
Calcium channel blockers have not been shown to improve prognoses overall and should not be prescribed purely for secondary prevention. Antiarrhythmic therapy other than with -blockers has not been shown to be effective except in patients with symptomatic arrhythmias. Amiodarone has been studied in several trials of postinfarct patients with either LV dysfunction or frequent ventricular ectopy. Although survival was not improved, amiodarone was not harmful unlike other agents in this setting. Therefore, it is the agent of choice for individuals with symptomatic postinfarction supraventricular arrhythmias. While implantable defibrillators improve survival for patients with postinfarction LV dysfunction and heart failure, the DINAMIT trial found no benefit to implantable defibrillators implanted in the 40 days following acute myocardial infarction.
Cardiac rehabilitation programs and exercise training can be of considerable psychological benefit and appear to improve prognosis.
C. ACE Inhibitors and Angiotensin Receptor Blockers in Patients with LV Dysfunction
Patients who sustain substantial myocardial damage often experience subsequent progressive LV dilation and dysfunction, leading to clinical heart failure and reduced long-term survival. In patients with EFs less than 40%, long-term ACE inhibitor (or ARB) therapy prevents LV dilation and the onset of heart failure and prolongs survival. The HOPE trial also demonstrated a reduction of approximately 20% in mortality rates and the occurrence of nonfatal myocardial infarction and stroke with ramipril treatment of postinfarction patients without confirmed LV systolic dysfunction. Therefore, ACE inhibitor therapy should be strongly considered in this broader group of patients and especially in diabetics and patients with even mild systolic hypertension, in whom the greatest benefit was observed.
P.374
D. Revascularization
Because of the increasing use of thrombolytic therapy and accumulating experience with PTCA, the indications for revascularization are rapidly evolving. Postinfarction patients who appear likely to benefit from early revascularization if the anatomy is appropriate are (1) those who have undergone thrombolytic therapy and have residual symptoms or laboratory evidence of ischemia; (2) patients with LV dysfunction (EF < 30 40%) and evidence of ischemia; (3) patients with non-Q wave infarction and evidence of more than mild ischemia; and (4) patients with markedly positive exercise tests and multivessel disease. The value of revascularization in the following groups is less clear: (1) patients treated with thrombolytic agents, with little evidence of reperfusion or residual ischemia; (2) patients with LV dysfunction but no detectable ischemia; and (3) patients with preserved LV function who have mild ischemia and are not symptom limited. Patients who survive infarctions without complications, have preserved LV function (EF > 50%), and have no exercise-induced ischemia have an excellent prognosis and do not require invasive evaluation.
Ades PA: Cardiac rehabilitation and secondary prevention of coronary heart disease. N Engl J Med 2001;345:892.
Dargie HJ et al: Effect of carvedilol on outcome after myocardial infarction in patients with left ventricular dysfunction: the CAPRICORN randomized trial. Lancet 2001;357:1385.
Hohnloser SH et al; DINAMIT Investigators: Prophylactic use of an implantable cardioverter-defibrillator after acute myocardial infarction. N Engl J Med 2004;351:2481.
Hurlen M et al: Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002;347:969.
JAMA patient page: Heart attack. JAMA 1998;280:1462.
Michaels AD et al: Risk stratification after acute myocardial infarction in the reperfusion era. Prog Cardiovasc Dis 2000; 42:273.
Sutton MGS et al: Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 2000; 101:2981.
See also references for section on Primary and Secondary Prevention of Ischemic Heart Disease.
Disturbances of Rate & Rhythm
Abnormalities of cardiac rhythm and conduction can be lethal (sudden cardiac death), symptomatic (syncope, near syncope, dizziness, fatigue, or palpitations), or asymptomatic. They are dangerous to the extent that they reduce cardiac output, so that perfusion of the brain or myocardium is impaired, or tend to deteriorate into more serious arrhythmias with the same consequences. Stable supraventricular tachycardia is generally well tolerated in patients without underlying heart disease but may lead to myocardial ischemia or CHF in patients with coronary disease, valvular abnormalities, and systolic or diastolic myocardial dysfunction. Ventricular tachycardia, if prolonged (lasting more than 10 30 seconds), often results in hemodynamic compromise and is more likely to deteriorate into ventricular fibrillation.
Whether slow heart rates produce symptoms at rest or on exertion depends on whether cerebral perfusion can be maintained, which is generally a function of whether the patient is upright or supine and whether LV function is adequate to maintain stroke volume. If the heart rate abruptly slows, as with the onset of complete heart block or sinus arrest, syncope or convulsions may result.
Arrhythmias are detected either because they present with symptoms or because they are detected during the course of monitoring. Arrhythmias causing sudden death, syncope, or near syncope require further evaluation and treatment unless they are related to conditions that are unlikely to recur (eg, electrolyte abnormalities or acute myocardial infarction). In contrast, there is controversy over when and how to evaluate and treat rhythm disturbances that are not symptomatic but are possible markers for more serious abnormalities (eg, nonsustained ventricular tachycardia). This uncertainty reflects two issues: (1) the difficulty of reliably stratifying patients into high-risk and low-risk groups; and (2) the lack of treatments that are both effective and safe. Thus, screening patients for these so-called premonitory abnormalities is often not productive.
A number of procedures are used to evaluate patients with symptoms who are believed to be at risk for life-threatening arrhythmias, including in-hospital and ambulatory ECG monitoring, event recorders (instruments that can be used for prolonged periods to record or transmit rhythm tracings when infrequent episodes occur), exercise testing, intracardiac electrophysiologic studies (to assess sinus node function, AV conduction, and inducibility of arrhythmias), signal-averaged ECGs, and tests of autonomic nervous system function (especially tilt-table testing). These are discussed below and in the subsequent sections on individual rhythm disturbance and symptomatic presentation. In general, these techniques are more successful in diagnosing symptomatic arrhythmias than in predicting the outcome of asymptomatic ones.
Mechanisms of Arrhythmias
Susceptibility to arrhythmias results from genetic abnormalities (most often affecting ion channels) and acquired structural heart disease. Susceptibility may be increased by electrolyte abnormalities, hormonal imbalances (thyrotoxicosis, hypercatecholaminergic states), hypoxia, drug effects (such as QT interval prolongation or changes in automaticity, conduction, and refractoriness), and myocardial
P.375
ischemia. Ongoing research has given us important information on the genetic basis of arrhythmias and knowledge in this arena is expanding rapidly. Most arrhythmias can be classified as (1) disorders of impulse formation or automaticity, (2) abnormalities of impulse conduction, (3) reentry, and (4) triggered activity.
Altered automaticity is the mechanism for sinus node arrest, many premature beats, and automatic rhythms as well as an initiating factor in reentry arrhythmias.
Abnormalities of impulse conduction can occur at the sinus or AV node, in the intraventricular conduction system, and within the atria or ventricles. These are responsible for sinoatrial exit block, for AV block at the node or below, and for establishing reentry circuits.
Reentry is the underlying mechanism for many arrhythmias, including premature beats, most paroxysmal supraventricular tachycardias, atrial flutter, and infarct-related ventricular tachycardia. For reentry to occur, there must be an area of unidirectional block with an appropriate delay to allow repeat depolarization at the site of origin. Reentry is confirmed if the arrhythmia can be terminated by interruption of the circuit by a spontaneous or induced premature beat.
Triggered activity occurs when afterdepolarizations (abnormal electrical activity persisting after repolarization) reach the threshold level required to trigger a new depolarization. Triggered activity can be divided into two different categories: pause-dependent and catecholamine-dependent. Pause-dependent triggered activity is caused by early afterdepolarizations in phase 3 of the action potential. These ventricular tachycardias are usually polymorphic.
Catecholamine-dependent triggered activity is caused by late afterdepolarizations in phase 4 of the action potential. They may be seen in patients with digitalis toxicity, cardiac ischemia, or congenitally prolonged QT intervals. Increased sympathetic tone also plays a part in these arrhythmias. RV outflow tract ventricular tachycardia is attributed to this mechanism.
Eckardt L et al: Arrhythmias in heart failure: current concepts of mechanisms and therapy. J Cardiovasc Electrophysiol 2000; 11:106.
Napolitano C et al: Genetics of ventricular tachycardia. Curr Opin Cardiol 2002;17:222.
Wall TS et al: Ventricular tachycardia in structurally normal hearts. Curr Cardiol Rep 2002;4:388.
Wilde AA et al: Genetics of cardiac arrhythmias. Heart 2005; 91:1352.
Techniques for Evaluating Rhythm Disturbances
Ecg Monitoring
The ideal way of establishing a causal relationship between a symptom and a rhythm disturbance is to demonstrate the presence of the rhythm during the symptom. Unfortunately, this is not always easy because symptoms are usually sporadic.
Patients with aborted sudden death and recent or recurrent syncope are often monitored in the hospital. Those with less ominous symptoms may be monitored as outpatients. When episodes are infrequent, use of an event recorder (either implantable or external) is preferable to 24-hour continuous monitoring. Exercise testing may be helpful when the symptoms are associated with exertion or stress. If symptomatic bradyarrhythmias or supraventricular tachyarrhythmias are detected, therapy can usually be initiated without additional diagnostic studies. Further electrophysiologic studies may be useful in evaluating ventricular tachyarrhythmias.
Caution is required before attributing a patient's symptom to rhythm or conduction abnormalities observed during monitoring without concomitant symptoms. In many cases, the symptoms are due to a different arrhythmia or to noncardiac causes. For instance, dizziness or syncope in older patients may be unrelated to concomitantly observed bradycardia, sinus node abnormalities, and ventricular ectopy. Ambulatory monitoring is frequently used to quantify ventricular ectopy and detect asymptomatic ventricular tachycardia in post-myocardial infarction or heart failure patients.
Abbott AV: Diagnostic approach to palpitations. Am Fam Physician 2005;71:743.
Kadish AH et al: ACC/AHA clinical competence statement on electrocardiography and ambulatory electrocardiography: a report of the ACC/AHA/ACP-ASIM task force on clinical competence (ACC/AHA Committee to develop a clinical competence statement on electrocardiography and ambulatory electrocardiography) endorsed by the International Society for Holter and Noninvasive Electrocardiology. Circulation 2001;104:3169.
Sarasin FP et al: Usefulness of 24-h Holter monitoring in patients with unexplained syncope and a high likelihood of arrhythmias. Int J Cardiol 2005;101:203.
Heart Rate Variability
Although it has long been appreciated that there are periodical fluctuations in heart rate even under basal conditions, considerable recent interest has been focused on measurements of heart rate variability. These measurements can be made under controlled conditions in the ECG laboratory or from recordings obtained during ambulatory monitoring. Greater fluctuations in heart rate correspond to greater parasympathetic activity, and several studies have indicated that greater heart rate variability is associated with a better prognosis and fewer life-threatening arrhythmias in a variety of cardiac conditions More recently, analyses have used frequency transformation of RR cycle length variability to provide indices of the relative balance between parasympathetic and sympathetic activity, with the greater contribution of the parasympathetic system being considered to confer a better prognosis. In studies of postinfarction patients and patients with symptomatic arrhythmias, these indices have had some prognostic value. More recent studies of heart rate variability in patients with CHF have
P.376
shown that decreases in heart rate variability are associated with worse outcomes.
De Jong MJ: Heart rate variability analysis in the assessment of autonomic function in heart failure. J Cardiovasc Nurs 2005;20:186.
Jouven X et al: Heart-rate profile during exercise as a predictor of sudden death. N Engl J Med 2005;352:1951.
Pumprla J et al: Functional assessment of heart rate variability: physiological basis and practical applications. Int J Cardiol 2002;84:1.
Sugiura H et al: Heart rate variability is a useful parameter for evaluation of anticholinergic effect associated with inducibility of atrial fibrillation. Pacing Clin Electrophysiol 2005;28:1208.
Signal-Averaged ECG
Another technique is the signal-averaged ECG. Most commonly, an orthogonal three-lead system is used to record 300 consecutive beats during basal conditions. Using appropriate electrical filtering and computer averaging of the signal, very low frequency signals called late potentials can be identified in the period following the QRS complex. Abnormal late potentials are considered markers for potential ventricular arrhythmias. Adequate data are not yet available to define the role of this technique with confidence, but it may be useful in detecting groups of patients at increased risk for arrhythmic events after myocardial infarction. Approximately one-third of post-myocardial infarction patients will have abnormal late potentials, and these individuals are at higher risk for arrhythmic events, though the positive predictive value of this finding is relatively low (10 15%). More importantly, the absence of late potentials identifies a group of patients at low risk for arrhythmic events, so post-myocardial infarction patients found to have frequent ventricular ectopy or nonsustained ventricular tachycardia in the absence of late potentials may not require further investigation or treatment. The prognostic value of late potentials in patients with chronic ischemic heart disease who are more than 6 12 months removed from myocardial infarction and in patients with other forms of heart disease is not yet known.
Gomes JA et al: Prediction of long-term outcomes by signal-averaged electrocardiography in patients with unsustained ventricular tachycardia, coronary artery disease and left ventricular dysfunction. Circulation 2001;104:436.
Iravanian S et al: Role of electrophysiologic studies, signal-averaged electrocardiography, heart rate variability, T-wave alternans, and loop recorders for risk stratification of ventricular arrhythmias. Am J Geriatr Cardiol 2005;14:16.
Electrophysiologic Testing
Electrophysiologic testing using intracardiac ECG recordings and programmed atrial or ventricular (or both) stimulation is useful in the diagnosis and management of complex arrhythmias. The primary indications for electrophysiologic testing are (1) evaluation of recurrent syncope of possible cardiac origin, when the ambulatory ECG has not provided the diagnosis; (2) differentiation of supraventricular from ventricular arrhythmias; (3) evaluation of therapy in patients with accessory atrioventricular pathways; and (4) evaluation of patients for catheter ablation procedures or antitachycardia devices.
Autonomic Testing (Tilt-Table Testing)
In many patients with recurrent syncope or near syncope, arrhythmias are not the cause. This is particularly true when the patient has no evidence of associated heart disease by history, examination, standard ECG, or noninvasive testing. Syncope may be neurocardiogenic in origin, mediated by excessive vagal stimulation or an imbalance between sympathetic and parasympathetic autonomic activity. With assumption of upright posture, there is venous pooling in the lower limbs. However, instead of the normal response, which consists of an increase in heart rate and vasoconstriction, a sympathetically mediated increase in myocardial contractility activates mechanoreceptors that trigger reflex bradycardia and vasodilation. Autonomic testing can be an important component of the evaluation in these individuals and, if performed, should usually precede invasive electrophysiologic procedures. Carotid sinus massage in patients who do not have carotid bruits or a history of cerebral vascular disease can precipitate sinus node arrest or AV block in patients with carotid sinus hypersensitivity. Head-up tilt-table testing can identify patients whose syncope may be on a vasovagal basis. Although different testing protocols are used, passive tilting to at least 70 degrees for 10 40 minutes in conjunction with isoproterenol infusion, if necessary is typical. Syncope due to bradycardia, hypotension, or both will occur in approximately one-third of patients with recurrent syncope. Some recent studies have suggested that, at least with some of the more extreme protocols, false-positive responses may occur.
Fitzpatrick AP et al: Tilt methodology in reflex syncope: emerging evidence. J Am Coll Cardiol 2000;36:179.
Sheldon R: Tilt testing for syncope: a reappraisal. Curr Opin Cardiol 2005;20:38
Antiarrhythmic Drugs (Table 10-8)
Antiarrhythmic drugs have variable efficacy and produce frequent side effects. They are often divided into classes based upon their electropharmacologic actions. Some have multiple actions. The most frequently used classification scheme is the Vaughn-Williams classification, which consists of four classes.
Class I agents block membrane sodium channels. Three subclasses are further defined by the effect of agents on the Purkinje fiber action potential. Class Ia drugs (ie, quinidine, procainamide, disopyramide,
P.377
P.378
P.379
moricizine) slow the rate of rise of the action potential (Vmax) and prolong its duration, thus slowing conduction and increasing refractoriness (moderate depression of phase 0 upstroke of the action potential). Class Ib agents (ie, lidocaine, mexiletine, tocainide, phenytoin) shorten action potential duration; they do not affect conduction or refractoriness (minimal depression of phase 0 upstroke of the action potential). Class Ic agents (ie, flecainide, propafenone) prolong Vmax and slow repolarization, thus slowing conduction and prolonging refractoriness, but more so than class Ia drugs (maximal depression of phase 0 upstroke of the action potential).
Class II agents are the -blockers, which decrease automaticity, prolong AV conduction, and prolong refractoriness.
Class III agents (ie, amiodarone, sotalol, dofetilide, azimilide, ibutilide) block potassium channels and prolong repolarization, widening the QRS and prolonging the QT interval. They decrease automaticity and conduction and prolong refractoriness.
Class IV agents are the calcium channel blockers, which decrease automaticity and AV conduction.
Table 10-8. Antiarrhythmic drugs. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Although the in vitro electrophysiologic effects of most of these agents have been defined, their use remains largely empirical. All can exacerbate arrhythmias (proarrhythmic effect), and most depress LV function.
The risk of antiarrhythmic agents has been highlighted by the Coronary Arrhythmia Suppression Trial (CAST), in which two class Ic agents (flecainide, encainide) and a class Ia agent (moricizine) increased mortality rates in patients with asymptomatic ventricular ectopy after myocardial infarction. A similar result has been reported in the Mortality in the Survival With Oral D-sotalol (SWORD) study with D-sotalol class III agent without the -blocking activity of D,L-sotalol, the currently marketed formulation. Therefore, these agents (and perhaps any antiarrhythmic drug) should not be used except for life-threatening ventricular arrhythmias and symptomatic supraventricular tachyarrhythmias and patients receiving these agents should be monitored regularly.
The use of antiarrhythmic agents for specific arrhythmias is discussed below.
Cordina J et al: Pharmacological cardioversion for atrial fibrillation and flutter. Cochrane Database Syst Rev 2005;(2): CD003713.
Khan MH: Oral class III antiarrhythmics: what is new? Curr Opin Cardiol 2004;19:47.
Kowey PR et al: Classification and pharmacology of antiarrhythmic drugs. Am Heart J 2000;140:12.
Roden DM: Antiarrhythmic drugs: from mechanisms to clinical practice. Heart 2000;84:339.
Radiofrequency Ablation for Cardiac Arrhythmias
Catheter ablation techniques have become first-line therapy for treatment of many arrhythmias. This growing trend reflects the increasing ability to localize the origin or conduction pathways of many arrhythmias and safely deliver lesions to destroy the arrhythmia focus, the improved technology for delivering radiofrequency energy, and the growing dissatisfaction with the efficacy and safety of pharmacologic therapy. Ablation has become the primary modality of therapy for many symptomatic supraventricular arrhythmias, including AV nodal reentry tachycardia, reentry tachycardias involving accessory pathways, paroxysmal atrial tachycardia, inappropriate sinus tachycardia, atrial flutter and automatic junctional tachycardia. Ablation of atrial fibrillation is more complex
P.380
and involves electrical isolation of the pulmonary veins, which are often the site of initiation of atrial fibrillation, or placing linear lesions within the atria to prevent spread of the rhythm. This technique has become mainstream and is considered a reasonable second-line therapy for certain patients with symptomatic atrial fibrillation. Catheter ablation of ventricular arrhythmias has proved more difficult, but experienced centers have had reasonable success with all types of ventricular tachycardias including: bundle-branch reentry, tachycardia originating in the ventricular outflow tract, tachycardias originating in the left side of the interventricular septum (also called fascicular ventricular tachycardia), and even ventricular tachycardias occurring in patients with CAD and dilated cardiomyopathy.
These procedures are generally safe, though there is a low incidence of perforation of the atria or RV that results in pericardial tamponade and sufficient damage to the AV node to require permanent cardiac pacing in less than 5% of patients. In addition, some procedures involve transseptal or retrograde LV catheterization, with the attendant potential complications of aortic perforation, damage to the heart valves, or left-sided emboli. A potentially lethal complication during the ablation of atrial fibrillation is the development of an atrio-esophageal fistula resulting from ablation lesions placed on the posterior wall of the LA just overlying the esophagus.
Oral H et al: Catheter ablation for paroxysmal atrial fibrillation: segmental pulmonary vein ostial ablation versus left atrial ablation. Circulation 2003;108:2355.
Pappone C et al: Atrio-esophageal fistula as a complication of percutaneous transcatheter ablation of atrial fibrillation. Circulation 2004;109:2724.
Saad EB et al: Ablation of atrial fibrillation. Curr Cardiol Rep 2002;4:379.
Wu RC et al: Catheter ablation of atrial flutter and macroreentrant atrial tachycardia. Curr Opin Cardiol 2002;17:58.
See also references in sections devoted to specific rhythm disturbances.
Supraventricular Arrhythmias
1. Sinus Arrhythmia, Bradycardia, & Tachycardia
Sinus arrhythmia is a cyclic increase in normal heart rate with inspiration and decrease with expiration. It results from reflex changes in vagal influence on the normal pacemaker and disappears with breath holding or increase of heart rate due to any cause. It has no clinical significance. It is common in both the young and the elderly.
Sinus bradycardia is a heart rate slower than 50 beats/min due to increased vagal influence on the normal pacemaker or organic disease of the sinus node. The rate usually increases during exercise or administration of atropine. In healthy individuals, and especially in patients who are in excellent physical condition, sinus bradycardia to a rate of 50 beats/min or even lower is a normal finding. However, severe sinus bradycardia may be an indication of sinus node pathology (see below), especially in elderly patients and individuals with heart disease. It may cause weakness, confusion, or syncope if cerebral perfusion is impaired. Atrial, junctional and ventricular ectopic rhythms are more apt to occur with slow sinus rates. Pacing may be required if symptoms correlate with the bradycardia.
Sinus tachycardia is defined as a heart rate faster than 100 beats/min that is caused by rapid impulse formation from the normal pacemaker; it occurs with fever, exercise, emotion, pain, anemia, heart failure, shock, thyrotoxicosis, or in response to many drugs. Alcohol and alcohol withdrawal are common causes of sinus tachycardia and other supraventricular arrhythmias. The onset and termination are usually gradual, in contrast to paroxysmal supraventricular tachycardia due to reentry. The rate infrequently exceeds 160 beats/min but may reach 180 beats/min in young persons. The rhythm is basically regular, but serial 1-minute counts of the heart rate indicate that it varies five or more beats per minute with changes in position, with breath holding, or with sedation. Rare individuals have persistent or episodic inappropriate sinus tachycardia that may be very symptomatic or may lead to LV contractile dysfunction. Radiofrequency modification of the sinus node has mitigated this problem.
Still AM et al: Prevalence, characteristics and natural course of inappropriate sinus tachycardia. Europace 2005;7:104.
Yusuf S et al: Deciphering the sinus tachycardias. Clin Cardiol 2005;28:267.
2. Atrial Premature Beats (Atrial Extrasystoles)
Atrial premature beats occur when an ectopic focus in the atria fires before the next sinus node impulse or a reentry circuit is established. The contour of the P wave usually differs from the patient's normal complex, unless the ectopic focus is near the sinus node. The subsequent RR cycle length is usually unchanged or only slightly prolonged. Such premature beats occur frequently in normal hearts and are never a sufficient basis for a diagnosis of heart disease. Speeding of the heart rate by any means usually abolishes most premature beats. Early atrial premature beats may cause aberrant QRS complexes (wide and bizarre) or may be nonconducted to the ventricles because the latter are still refractory.
3. Differentiation of Aberrantly Conducted Supraventricular Beats from Ventricular Beats
This distinction can be very difficult in patients with a wide QRS complex; it is important because of the differing prognostic and therapeutic implications of each
P.381
type. Findings favoring a ventricular origin include (1) AV dissociation; (2) a QRS duration exceeding 0.14 second; (3) capture or fusion beats (infrequent); (4) left axis deviation with right bundle branch block morphology; (5) monophasic (R) or biphasic (qR, QR, or RS) complexes in V1; and (6) a qR or QS complex in V6. Supraventricular origin is favored by (1) a triphasic QRS complex, especially if there was initial negativity in leads I and V6; (2) ventricular rates exceeding 170 beats/min; (3) QRS duration longer than 0.12 second but not longer than 0.14 second; and (4) the presence of preexcitation syndrome.
The relationship of the P waves to the tachycardia complex is helpful. A 1:1 relationship usually means a supraventricular origin, except in the case of ventricular tachycardia with retrograde P waves.
4. Paroxysmal Supraventricular Tachycardia
This is the most common paroxysmal tachycardia and often occurs in patients without structural heart disease. Episodes begin and end abruptly and may last a few seconds to several hours or longer. The heart rate may be 140 240 beats/min (usually 160 220 beats/min) and is perfectly regular (despite exercise or change in position). The P wave usually differs in contour from sinus beats. Patients may be asymptomatic except for awareness of rapid heart action, but some experience mild chest pain or shortness of breath, especially when episodes are prolonged, even in the absence of associated cardiac abnormalities. Paroxysmal supraventricular tachycardia may result from digitalis toxicity and then is commonly associated with AV block.
The most common mechanism for paroxysmal supraventricular tachycardia is reentry, which may be initiated or terminated by a fortuitously timed atrial or ventricular premature beat. The reentry circuit most commonly involves dual pathways (a slow and a fast pathway) within the AV node. This is referred to as AV nodal reentry tachycardia (AVNRT). Less commonly, reentry is due to an accessory pathway between the atria and ventricles (AVRT). Approximately one-third of patients with supraventricular tachycardia have aberrant pathways to the ventricles. The pathophysiology and management of arrhythmias due to accessory pathways differ in important ways and are discussed separately below.
Treatment of the Acute Attack
In the absence of heart disease, serious effects are rare, and most attacks break spontaneously. Particular effort should be made to terminate the attack quickly if cardiac failure, syncope, or anginal pain develops or if there is underlying cardiac or (particularly) coronary disease. Because reentry is the most common mechanism for paroxysmal atrial tachycardia, effective therapy requires that conduction be interrupted at some point in the reentry circuit and the majority of these circuits involve the AV node.
A. Mechanical Measures
A variety of methods have been used to interrupt attacks, and patients may learn to perform these themselves. These include Valsalva's maneuver, stretching the arms and body, lowering the head between the knees, coughing, splashing cold water on the face, and breath holding. Carotid sinus massage is often performed by physicians but should be avoided if the patient has carotid bruits or a history of transient cerebral ischemic attacks. Firm but gentle pressure and massage are applied first over the right carotid sinus for 10 20 seconds and, if unsuccessful, then over the left carotid sinus. Pressure should not be exerted on both sides at the same time! Continuous ECG or auscultatory monitoring of the heart rate is essential so that pressure can be relieved as soon as the rhythm is broken or if excessive bradycardia occurs. Carotid sinus pressure will interrupt up to half of the attacks, especially if the patient has received a digitalis glycoside or other agent (such as adenosine or a calcium channel blocker) that delays AV conduction. These maneuvers stimulate the vagus nerve, delay AV conduction, and block the reentry mechanism, terminating the arrhythmia.
B. Drug Therapy
If mechanical measures fail, two rapidly acting intravenous agents will terminate more than 90% of episodes. Intravenous adenosine has a very brief duration of action and minimal negative inotropic activity. Initially, a 6 mg bolus is administered. If no response is observed after 1 2 minutes, a second 12 mg bolus should be given, followed by a third if necessary. Because the half-life of adenosine is less than 10 seconds, the drug must be given rapidly (in 1 2 seconds from a peripheral intravenous line); use half the dose if given through a central line. Adenosine causes block of electrical conduction through the AV node. Adenosine is very well tolerated, but nearly 20% of patients will experience transient flushing, and some patients experience severe chest discomfort. Caution must be taken when adenosine is given to elderly patients because the resulting pause can be prolonged. Adenosine must also be used with caution in patients with reactive airways disease because it can promote bronchospasm.
Calcium channel blockers also rapidly induce AV block and break most episodes of reentry supraventricular tachycardia. Intravenous verapamil may be given as a 2.5 mg bolus, followed by additional doses of 2.5 5 mg every 1 3 minutes up to a total of 20 mg if blood pressure and rhythm are stable. If the rhythm recurs, further doses can be given. Oral verapamil, 80 120 mg every 4 6 hours, can be used as well in stable patients who are tolerating the rhythm without difficulty, but avoid it if there is any concern that the arrhythmia may be ventricular in origin. Intravenous
P.382
diltiazem (0.25 mg/kg over 2 minutes, followed by a second bolus of 0.35 mg/kg if necessary and then an infusion of 5 15 mg/h) may cause less hypotension and myocardial depression.
Esmolol, a short-acting -blocker, may also be effective; the initial dose is 500 mcg/kg intravenously over 1 minute followed by an infusion of 25 200 mcg/min. Metoprolol is also effective and can be given in 5 mg boluses every 5 minutes and repeated up to two times. Digoxin is effective, but it often requires several hours to safely administer an adequate dose. An initial dose of 0.5 0.75 mg intravenously over 20 minutes, followed by 0.25-mg or 0.125-mg increments every 2 4 hours up to a total of 1 1.25 mg, is used. If the tachycardia is believed to be mediated by an accessory pathway, intravenous procainamide may terminate supraventricular tachycardia by prolonging refractoriness in the accessory pathway; however, because it facilitates AVconduction and an initial increase in rate may occur, it is usually not given until after digoxin, verapamil, or a -blocker has been administered. Although intravenous amiodarone is safe, it is usually not required and often ineffective for treatment of these arrhythmias.
C. Cardioversion
If the patient is hemodynamically unstable or if adenosine and verapamil are contraindicated or ineffective, synchronized electrical cardioversion (beginning at 100 J) is almost universally successful. If digitalis toxicity is present or strongly suspected, as in the case of paroxysmal tachycardia with block, electrical cardioversion should be avoided.
Prevention of Attacks
A. Radiofrequency Ablation
Because of concerns about the safety and the intolerability of antiarrhythmic medications, radiofrequency ablation is the preferred approach to patients with recurrent symptomatic reentry supraventricular tachycardia, whether it is due to dual pathways within the AV node or to accessory pathways.
B. Drugs
AV nodal blocking agents are the drugs of choice as first-line medical therapy. Non-dihydropyridine calcium channel blockers, such as diltiazem and verapamil, or -blockers are typically used first. For patients with heart failure, digoxin can be effective as initial treatment. Patients who do not respond to agents that increase refractoriness of the AV node may be treated with antiarrhythmics. The class Ic agents (flecainide, propafenone) can be used in patients without underlying structural heart disease. In patients with evidence of structural heart disease, class III agents, such as sotalol or amiodarone, are probably a better choice because of the lower incidence of ventricular proarrhythmia during long-term therapy.
Blomstrom-Lundqvist C et al; European Society of Cardiology Committee, NASPE-Heart Rhythm Society: ACC/AHA/ESC guidelines for the management of patients with supraventricular arrhythmias executive summary. A report of the American college of cardiology/American heart association task force on practice guidelines and the European society of cardiology committee for practice guidelines (writing committee to develop guidelines for the management of patients with supraventricular arrhythmias) developed in collaboration with NASPE-Heart Rhythm Society. J Am Coll Cardiol 2003;42:1493.
Chauhan VS et al: Supraventricular tachycardia. Med Clin North Am 2001;85:193.
Hebbar AK et al: Management of common arrhythmias: Part I. Supraventricular arrhythmias. Am Fam Physician 2002;65:2479.
5. Supraventricular Tachycardias Due to Accessory AV Pathways (Preexcitation Syndromes)
Pathophysiology & Clinical Findings
Accessory pathways between the atria and the ventricle that avoid the conduction delay of the AV node predispose to reentry tachycardias, such as AVRT, atrial flutter, and atrial fibrillation. These may be wholly or partly within the node (Mahaim fibers), yielding a short PR interval and normal QRS morphology (Lown-Ganong-Levine syndrome). More commonly, they make direct connections between the atria and ventricle through Kent bundles (Wolff-Parkinson-White syndrome). This produces a short PR interval but an early delta wave at the onset of the wide, slurred QRS complex owing to early ventricular depolarization of the region adjacent to the pathway. Although the morphology and polarity of the delta wave can suggest the location of the bypass tract, mapping by intracardiac recordings is required for precise anatomic localization.
Accessory pathways occur in 0.1 0.3% of the population and facilitate reentry arrhythmias owing to the disparity in refractory periods of the AV node and accessory pathway. Whether the tachycardia is associated with a narrow or wide QRS complex is determined by whether antegrade conduction is through the node (narrow) or the bypass tract (wide). Many patients with Wolff-Parkinson-White syndrome never conduct in an antegrade direction through the bypass tract, which is therefore concealed. Orthodromic tachycardia is a reentrant rhythm that conducts antegrade down the AV node and retrograde up the accessory pathway, resulting in a narrow QRS complex unless an underlying bundle branch block or interventricular conduction delay is present. Antidromic tachycardia conducts down the accessory pathway and retrograde through the AV node, resulting in a wide QRS complex. Because accessory pathways are less refractory than specialized conduction tissue, tachycardias proceeding in this direction have the potential to be more rapid. Up to 30% of patients with Wolff-Parkinson-White syndrome will develop atrial fibrillation or flutter with antegrade conduction down the accessory pathway and a rapid ventricular response.
P.383
If this conduction is very rapid, it can potentially degenerate to ventricular fibrillation
Treatment
Some patients have a delta wave found incidentally on ECG. In the absence of palpitations, light-headedness, or syncope, these patients do not require specific therapy. They should be advised to report the onset of any of these symptoms. Patients found incidentally to have delta waves who have jobs that could potentially put others at risk (ie, pilot, bus driver, etc) may need to undergo electrophysiologic testing and prophylactic catheter ablation to ensure that they are not at risk for sudden death.
A. Radiofrequency Ablation
As with AVNRT, radiofrequency ablation has become the procedure of choice in patients with accessory pathways and recurrent symptoms. Patients with preexcitation syndromes who have episodes of atrial fibrillation or flutter should be tested by induction of atrial fibrillation in the electrophysiologic laboratory, noting duration of the RR cycle; if it is less than 220 ms, a short refractory period is present. These individuals are at highest risk for sudden death, and prophylactic ablation is indicated. Success rates for ablation of accessory pathways with radiofrequency catheters exceed 90% in appropriate patients.
B. Pharmacologic Therapy
Narrow-complex reentry rhythms involving a bypass tract can be managed as discussed for AVNRT. Atrial fibrillation and flutter must be managed differently, since agents such as digoxin, calcium channel blockers, and even -blockers may decrease the refractoriness of the accessory pathway or increase that of the AV node, leading to sometimes faster ventricular rates. Therefore, these agents should be avoided. The class Ia, class Ic, and class III antiarrhythmic agents will increase the refractoriness of the bypass tract and are the drugs of choice for wide-complex tachycardias. If hemodynamic compromise is present, electrical cardioversion is warranted.
Long-term therapy often involves a combination of agents that increases refractoriness in the bypass tract (class Ia or Ic agents) and in the AV node (verapamil, digoxin, and -blockers), provided that atrial fibrillation or flutter with short RR cycle lengths is not present (see above). The class III agents sotalol and amiodarone are effective in refractory cases. Patients who are difficult to manage should undergo electrophysiologic evaluation.
Antz M et al: Risk of sudden death after successful accessory atrioventricular pathway ablation in resuscitated patients with Wolff-Parkinson-White syndrome. J Cardiovasc Electrophysiol 2002;13:231.
Hamada T et al: Mechanisms for atrial fibrillation in patients with Wolff-Parkinson-White syndrome. J Cardiovasc Electrophysiol 2002;13:223.
Jezior MR et al: Exercise testing in Wolff-Parkinson-White syndrome: case report with ECG and literature review. Chest 2005;127:1454.
Keating L et al: Electrocardiographic features of Wolff-Parkinson-White syndrome. Emerg Med J 2003;20:491.
Pappone C et al: A randomized study of prophylactic catheter ablation in asymptomatic patients with the Wolff-Parkinson-White syndrome. N Engl J Med 2003;349:1803.
6. Atrial Fibrillation
Atrial fibrillation is the most common chronic arrhythmia, with an incidence and prevalence that rise with age, so that it affects nearly 10% of individuals over age 80 years. It occurs in rheumatic and other forms of valvular heart disease, dilated cardiomyopathy, ASD, hypertension, and coronary heart disease as well as in patients with no apparent cardiac disease; it may be the initial presenting sign in thyrotoxicosis, and this condition should be excluded with the initial episode. Atrial fibrillation often appears paroxysmally before becoming the established rhythm. Pericarditis, chest trauma, thoracic or cardiac surgery, or pulmonary disease (as well as medications such as theophylline and -adrenergic agonists) may cause attacks in patients with normal hearts. Acute alcohol excess and alcohol withdrawal and, in predisposed individuals, even consumption of small amounts of alcohol may precipitate atrial fibrillation. This latter presentation, which is often termed holiday heart, is usually transient and self-limited. Short-term rate control usually suffices as treatment.
Atrial fibrillation is the only common arrhythmia in which the ventricular rate is rapid and the rhythm very irregular. Because of the varying stroke volumes resulting from varying periods of diastolic filling, not all ventricular beats produce a palpable peripheral pulse. The difference between the apical rate and the pulse rate is the pulse deficit ; this deficit is greater when the ventricular rate is high.
Atrial fibrillation itself is rarely life-threatening; however, it can have serious consequences if the ventricular rate is sufficiently rapid to precipitate hypotension, myocardial ischemia, or tachycardia-induced myocardial dysfunction. Although many patients particularly older or inactive individuals have relatively few symptoms if the rate is controlled, some patients are aware of the irregular rhythm and may experience it as very uncomfortable. Most patients will complain of fatigue whether they experience other symptoms or not. Perhaps the most serious consequence of atrial fibrillation is the propensity for thrombus formation due to stasis in the atria (particularly the atrial appendages) and consequent embolization, most devastatingly to the cerebral circulation. Overall, the rate of stroke is approximately five events per 100 patient-years of follow-up. However, patients with significant obstructive valvular disease, chronic heart failure or LV dysfunction, diabetes, hypertension, or age over 75 years and those with a history of prior embolic events are at substantially higher risk (up to nearly 20 events per 100 patient-years in patients with multiple risk factors) (Table 10-9). A simple score derived from the Framingham study to estimate risk of stroke is available
P.384
for PDA download at http://www.statcoder.com/a-fib_stroke.htm. Patients with one or more risk factors for stroke should be treated with warfarin. Patients with none of these factors may be treated with aspirin if conditions are present that increase the risk of warfarin. While there was hope that clopidogrel and aspirin might be reasonable alternatives for warfarin for some patients, the ACTIVE trial was stopped early because of substantially lower rates of stroke with warfarin compared with clopidogrel. Anticoagulation clinics with systematic management of warfarin dosing and adjustment have been shown to result in better maintanance of target anticoagulation. Patients below the age of 60 65 years without any of these stroke risk factors ( lone atrial fibrillation ) may be treated with aspirin or no antithrombotic therapy.
Table 10-9. Risk factors for ischemic stroke and systemic embolism in patients with nonvalvular atrial fibrillation. | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
Newly Diagnosed Atrial Fibrillation
A. Initial Management
The approach to the initial management of atrial fibrillation depends on the clinical presentation. If, as is often the case particularly in older individuals the patient has no symptoms, hemodynamic instability, or evidence of important precipitating conditions (such as silent myocardial infarction or ischemia, decompensated heart failure, pulmonary embolism, or hemodynamically significant valvular disease), hospitalization is usually not necessary. In most of these cases, atrial fibrillation is an unrecognized chronic or paroxysmal condition and should be managed accordingly (see below). For new onset atrial fibrillation, thyroid function tests and assessment for occult valvular or myocardial disease should be performed.
In contrast, if the patient is hemodynamically unstable usually as a result of a rapid ventricular rate or associated cardiac or noncardiac conditions hospitalization and immediate treatment of atrial fibrillation are required. Urgent cardioversion is usually indicated in patients with shock or severe hypotension, pulmonary edema, or ongoing myocardial infarction or ischemia. Although if atrial fibrillation has been present for more than 48 hours, there is a potential risk of thromboembolism; however, the need for immediate rate control in these very unstable patients outweighs that risk. Electrical cardioversion is usually preferred in unstable patients. An initial shock with 100 200 J is administered in synchrony with the R wave. If sinus rhythm is not restored, an additional attempt with 360 J is indicated. If this fails, cardioversion may be successful after loading with intravenous ibutilide (1 mg over 10 minutes, repeated in 10 minutes if necessary) or intravenous procainamide (500 1000 mg administered at a rate of 20 mg/min with careful monitoring of blood pressure).
In less unstable patients or those at particularly high risk for embolism (underlying mitral stenosis, a history of prior emboli, or severe heart failure), a strategy of rate control is appropriate. A rate control strategy is appropriate both when the conditions that precipitated atrial fibrillation are likely to persist (such as following cardiac or noncardiac surgery, respiratory failure, or pericarditis) and when these conditions might resolve spontaneously over a period of hours to days (such as alcohol-induced atrial fibrillation, electrolyte or fluid imbalances, excessive exposure to theophylline or sympathomimetic agents, or some of the same previously cited conditions). The choice of agent is guided by the hemodynamic status of the patient, associated conditions, and the urgency of achieving rate control. Although both hypotension and heart failure may improve when the ventricular rate is slowed, calcium channel blockers and -blockers may themselves precipitate hemodynamic deterioration. Digoxin is less risky, but even when used aggressively (0.5 mg intravenously over 30 minutes, followed by increments of 0.25 mg every 1 2 hours to a total dose of 1 1.5 mg over 24 hours in patients not previously receiving this agent), rate control is rather slow and may be inadequate, particularly in patients with sympathetic activation. In the setting of myocardial infarction or ischemia, -blockers are the preferred agent. The most frequently used agents are either metoprolol (administered as a 5 mg intravenous bolus, repeated twice at intervals of 5 minutes and then given as needed by repeat boluses or orally at total daily doses of 50 400 mg) or, in very unstable patients, esmolol (0.5 mg/kg intravenously, repeated if necessary, followed by a titrated infusion of 0.05 0.2 mg/kg/min). If hypertension is present or -blockers are contraindicated, calcium channel blockers are immediately effective. Diltiazem (20 mg bolus, repeated after 15 minutes if necessary, followed by a maintenance infusion of 5 15 mg/h) is the preferred calcium blocker if hypotension
P.385
or LV dysfunction is present. Otherwise, verapamil (5 10 mg intravenously over 2 3 minutes, repeated after 30 minutes if necessary) may be used. Amiodarone, even when administered intravenously, has a relatively slow onset but is often a useful adjunct when rate control with the previously cited agents is incomplete or contraindicated or when cardioversion is planned in the near future. However, amiodarone should not be used in this setting if long-term therapy is planned with other antiarrhythmic agents.
If rate control proves unsuccessful or early cardioversion is considered necessary and the duration of atrial fibrillation exceeds 2 3 days or is unknown, a strategy of transesophageal echocardiography-guided cardioversion should be considered. By this approach, the presence of atrial thrombus is excluded and electrical cardioversion can be attempted while the patient remains under sedation. If thrombus is present, the cardioversion is delayed until after a 3- to 4-week period of therapeutic anticoagulation. In any case, because atrial contractile activity may not recover for several weeks after restoration of sinus rhythm in patients who have been in atrial fibrillation for more than several days, cardioversion is usually followed by anticoagulation for at least 1 month unless it is contraindicated.
B. Subsequent Management
Up to two-thirds of patients experiencing a first episode of atrial fibrillation will spontaneously revert to sinus rhythm within 24 hours. If atrial fibrillation persists or has been present for more than a week, spontaneous conversion is unlikely. In most cases early cardioversion is not required, so management consists of rate control and anticoagulation whether or not the patient has been admitted to hospital. Rate control is usually relatively easy to achieve with -blockers, rate-slowing calcium blockers, and digoxin, used as single agents or more often in combination. Good rate control should consist of a ventricular rate between 50 and 100 beats/min with usual daily activities and a ventricular rate not exceeding 120 beats/min except with moderate to strenuous activity. In older patients, who often have diminished AV nodal function and relatively limited activity, this can often be achieved with a single agent. Most younger or more active individuals require a combination of two agents. Choice of the initial medication is best based on the presence of accompanying conditions: Hypertensive patients should be given -blockers or calcium blockers; coronary patients should usually receive a -blocker; and patients with heart failure should be given a -blocker with consideration of adding digoxin. Adequacy of rate control should be evaluated by recording the apical pulse rate both at rest and with an appropriate level of activity (such as after brisk walking around the corridor or climbing stairs).
For patients with atrial fibrillation, even when it is paroxysmal or occurs rarely, anticoagulation with warfarin to an INR target of 2.0 3.0 should be established and maintained indefinitely, at least for patients with at least one risk factor for stroke (Table 10-9). An exception is the patient with lone atrial fibrillation (eg, no evidence of associated heart disease, hypertension, atherosclerotic vascular disease, or diabetes) who is under age 60 years. Cardioversion, if planned, should be performed after at least 3 weeks of anticoagulation at a therapeutic level.
C. Rate Control or Elective Cardioversion
Two recent large randomized controlled trials (the 4060-patient Atrial Fibrillation Follow-up Investigation of Rhythm Management, or AFFIRM trial; and the Rate Control Versus Electrical Cardioversion for Persistent Atrial Fibrillation, or RACE trial) compared strategies of rate control and rhythm control. In both, a strategy of rate control and long-term anticoagulation was associated with no higher rates of death or stroke both, if anything, favored rate control and only a modestly increased risk of hemorrhagic events than a strategy of restoring sinus rhythm and maintaining it with antiarrhythmic drug therapy. Of note is that exercise tolerance and quality of life were not significantly better in the rhythm control group. Nonetheless, the decision as to whether to attempt to restore sinus rhythm following the initial episode remains controversial. Elective cardioversion following an appropriate period of anticoagulation is generally recommended for the initial episode in patients in whom atrial fibrillation is thought to be of recent onset and when there is an identifiable precipitating factor. Similarly, cardioversion is appropriate in patients who remain symptomatic from the rhythm despite aggressive efforts to achieve rate control. However, it should be noted that even in patients for whom this is the initial episode of atrial fibrillation, the recurrence rate is sufficiently high that longer-term anticoagulation is generally appropriate until persistence of sinus rhythm can be confirmed for at least 6 months.
In cases in which elective cardioversion is required, it may be accomplished electrically (as described above) or pharmacologically. Intravenous ibutilide may also be used as described above in a setting in which the patient can undergo continuous ECG monitoring for at least 3 hours following administration. In patients in whom a decision has been made to continue antiarrhythmic therapy to maintain sinus rhythm (see next paragraph), cardioversion can be attempted with an agent that is being considered for long-term use. For instance, after therapeutic anticoagulation has been established, amiodarone can be initiated on an outpatient basis (300 400 mg twice daily for 2 weeks, followed by 200 mg twice daily for at least a 2 4 weeks and then a maintenance dose of 200 mg daily). Because amiodarone increases the prothrombin time in patients taking warfarin and digoxin levels, careful monitoring of anticoagulation and drug levels is required. Other agents that may be used for both cardioversion and maintenance therapy include propafenone (150 300 mg twice daily; should be avoided in patients with structural heart disease), flecainide (50 150 mg twice daily; should be avoided in patients with structural heart disease and should be used
P.386
in conjunction with an AV nodal blocking drug if there is a history of atrial flutter), and dofetilide (0.5 mg twice daily; downward dose adjustment is required with renal dysfunction and dosing must be initiated in hospital because of risk of torsades de pointes). Sotalol (80 160 mg twice daily; should be initiated in hospital in patients with structural heart disease because of risk of torsades de pointes) is not very effective for converting atrial fibrillation but can be used to maintain sinus rhythm following cardioversion.
Unfortunately, sinus rhythm will persist in only 25% of patients who have had a sustained (lasting more than several days) or recurrent episode of atrial fibrillation. However, if the patient is treated long-term with an antiarrhythmic agent, sinus rhythm will persist in approximately 50%. The most commonly used medications are amiodarone, sotalol, propafenone, flecainide, and dofetilide, but the latter four agents are associated with a clear risk of proarrhythmia, and amiodarone frequently causes other adverse effects. Therefore, it may be prudent to determine whether atrial fibrillation recurs during a period of 6 months without antiarrhythmic drugs during which anticoagulation is maintained. If it does recur, the decision as to whether to restore sinus rhythm and initiate long-term antiarrhythmic therapy can be based on how well the patient tolerates atrial fibrillation. In such a patient, long-term anticoagulation is probably indicated in any case, because of the high rate of recurrence and the likely occurrence of asymptomatic paroxysmal episodes.
Paroxysmal & Refractory Atrial Fibrillation
A. Recurrent Paroxysmal Atrial Fibrillation
It is now well established that patients with recurrent paroxysmal atrial fibrillation are at similar stroke risk as those who are in atrial fibrillation chronically. Although these episodes may be apparent to the patient, many are not recognized and may be totally asymptomatic. Thus, ambulatory ECG monitoring or event recorders are indicated in those in whom paroxysmal atrial fibrillation is suspected. Antiarrhythmic agents are usually not successful in preventing all paroxysmal atrial fibrillation episodes. However, dofetilide has been shown to be as effective as amiodarone in maintaining sinus rhythm in certain patients and does not have as many untoward effects. Long-term anticoagulation is indicated except in those who are under 60 65 years of age and have no additional stroke risk factors (see above).
B. Refractory Atrial Fibrillation
Because of trial results indicating that important adverse clinical outcomes (death, stroke, hemorrhage, heart failure) are no more common with rate control than rhythm control, atrial fibrillation should generally be considered refractory if it causes persistent symptoms or limits activity. This is much more likely in younger individuals and those who are very active or engage in strenuous exercise. Even in such individuals, two-drug or three-drug combinations of a -blocker, rate-slowing calcium blocker, and digoxin usually can prevent excessive ventricular rates, though in some cases they are associated with excessive bradycardia during sedentary periods.
If no drug works, radiofrequency AV node ablation and permanent pacing ensure rate control and may facilitate a more physiologic rate response to activity, but this is used only as a last resort. There is growing experience with focal ablation of foci in and around the pulmonary veins that initiate atrial fibrillation, following which sinus rhythm may be restored or maintained. This therapy has become more widely accepted and is a reasonable second-line therapy for individuals with symptomatic atrial fibrillation that is refractory to pharmacologic therapy. This procedure is routinely performed in the electrophysiology laboratory using a catheter-based approach and can also be performed thorascopically in the operating room by experienced surgeons.
Aasbo JD et al: Amiodarone prophylaxis reduces major cardiovascular morbidity and length of stay after cardiac surgery: a meta-analysis. Ann Intern Med 2005;143:327.
Corley SD et al; AFFIRM Investigators: Relationships between sinus rhythm, treatment, and survival in the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) Study. Circulation 2004;109:1509.
Daoud EG: Management of atrial fibrillation in the post-cardiac surgery setting. Cardiol Clin 2004;22:159.
de Denus S et al: Rate vs rhythm control in patients with atrial fibrillation: a meta-analysis. Arch Intern Med 2005;165:258.
Fang MC et al: Anticoagulation for atrial fibrillation. Cardiol Clin 2004;22:47.
Finta B et al: Catheter ablation therapy for atrial fibrillation. Cardiol Clin 2004;22:127.
Gage BF et al: Selecting patients with atrial fibrillation for anticoagulation: stroke risk stratification in patients taking aspirin. Circulation 2004;110:2287.
Gillinov AM et al: Advances in the surgical treatment of atrial fibrillation. Cardiol Clin 2004;22:147.
Joglar JA et al: Electrical cardioversion of atrial fibrillation. Cardiol Clin 2004;22:101.
McKeown PP et al; American College of Chest Physicians: Executive summary: American College of Chest Physicians guidelines for the prevention and management of postoperative atrial fibrillation after cardiac surgery. Chest 2005;128(2 Suppl):1S.
McNamara RL et al: Management of atrial fibrillation: review of the evidence for the role of pharmacologic therapy, electrical cardioversion, and echocardiography. Ann Intern Med 2003;139:1018.
Rockson SG et al: Comparing the guidelines: anticoagulation therapy to optimize stroke prevention in patients with atrial fibrillation. J Am Coll Cardiol 2004;43:929.
Singh BN et al; Sotalol Amiodarone Atrial Fibrillation Efficacy Trial (SAFE-T) Investigators: Amiodarone versus sotalol for atrial fibrillation. N Engl J Med 2005;352:1861.
Tamariz LJ et al: Pharmacological rate control of atrial fibrillation. Cardiol Clin 2004;22:35.
P.387
VerNooy RA et al: Antiarrhythmic drug therapy of atrial fibrillation. Cardiol Clin 2004;22:21.
Wang TJ et al: A risk score for predicting stroke or death for individuals with new-onset atrial fibrillation in the community: the Framingham Heart Study. JAMA 2003;290:1049.
Wazni OM et al: Radiofrequency ablation vs antiarrhythmic drugs as first-line treatment of symptomatic atrial fibrillation: a randomized trial. JAMA 2005;293:2634.
Wijffels MC et al: Rate versus rhythm control in atrial fibrillation. Cardiol Clin 2004;22:63.
7. Atrial Flutter
Atrial flutter is less common than fibrillation. It occurs most often in patients with chronic obstructive pulmonary disease (COPD) but may be seen also in those with rheumatic or coronary heart disease, CHF, ASD, or surgically repaired congenital heart disease. Ectopic impulse formation occurs at atrial rates of 250 350 beats/min, with transmission of every second, third, or fourth impulse through the AV node to the ventricles. Ventricular rate control is accomplished using the same agents used in atrial fibrillation, but it is much more difficult with atrial flutter than with atrial fibrillation. Conversion of atrial flutter to sinus rhythm with class I antiarrhythmic agents is also difficult to achieve, and administration of these drugs has been associated with slowing of the atrial flutter rate to the point at which 1:1 AV conduction can occur at rates in excess of 200 beats/min, with subsequent hemodynamic collapse. The intravenous class III antiarrhythmic agent ibutilide has been significantly more successful in converting atrial flutter. About 50 70% of patients return to sinus rhythm within 60 90 minutes following the infusion of 1 2 mg of this agent. Electrical cardioversion is also very effective for atrial flutter, with approximately 90% of patients converting following shocks of as little as 25 50 J.
The persistence of atrial contractile function in this arrhythmia provides some protection against thrombus formation, though the risk of systemic embolization remains increased. Precardioversion anticoagulation is not necessary for atrial flutter of less than 48 hours duration except in the setting of mitral valve disease. However, anticoagulation is prudent in chronic atrial flutter, particularly since transient periods of atrial fibrillation are common in these patients.
Chronic atrial flutter is often a difficult management problem, as rate control is difficult. If pharmacologic therapy is chosen, amiodarone and dofetilide are the antiarrhythmics of choice. Dofetilide is often given in conjunction with an AV nodal blocker.
Atrial flutter can follow a typical or atypical reentry circuit around the atrium. The anatomy of the typical circuit has been well defined and allows for catheter ablation within the atrium to interrupt the circuit and eliminate atrial flutter. Catheter ablation has become the preferred treatment for recurrent typical atrial flutter.
Ghali WA et al: Atrial flutter and the risk of thromboembolism: a systematic review and meta-analysis. Am J Med 2005;118: 101.
Lee KW et al: Atrial flutter: a review of its history, mechanisms, clinical features, and current therapy. Curr Probl Cardiol 2005;30:121.
Knight BP et al; American Heart Association Council on Clinical Cardiology (Subcommittee on Electrocardiography and Arrhythmias); Quality of Care and Outcomes Research Interdisciplinary Working Group; Heart Rhythm Society; AHA Writing Group: Role of permanent pacing to prevent atrial fibrillation: science advisory from the American Heart Association Council on Clinical Cardiology (Subcommittee on Electrocardiography and Arrhythmias) and the Quality of Care and Outcomes Research Interdisciplinary Working Group, in collaboration with the Heart Rhythm Society. Circulation 2005;111: 240.
Sun JL et al: Clinical comparison of ibutilide and propafenone for converting atrial flutter. Cardiovasc Drugs Ther 2005;19:57.
Wu RC et al: Catheter ablation of atrial flutter and macroreentrant atrial tachycardia. Curr Opin Cardiol 2002;17:58.
8. Multifocal (Chaotic) Atrial Tachycardia
This is a rhythm characterized by varying P wave morphology (by definition, three or more foci) and markedly irregular PP intervals. The rate is usually between 100 and 140 beats/min, and AV block is unusual. Most patients have severe associated COPD. Treatment of the underlying condition is the most effective approach; verapamil, 240 480 mg daily in divided doses, is also of value in some patients, but this particular arrhythmia is very difficult to manage.
Spodick DH: Multifocal atrial arrhythmia. Am J Geriatr Cardiol 2005;14:162.
9. AV Junctional Rhythm
The atrial-nodal junction or the nodal-His bundle junctions may assume pacemaker activity for the heart, usually at a rate of 40 60 beats/min. This may occur in patients with myocarditis, CAD, and digitalis toxicity as well as in individuals with normal hearts. The rate responds normally to exercise, and the diagnosis is often an incidental finding on ECG monitoring, but it can be suspected if the jugular venous pulse shows cannon a waves. Junctional rhythm is often an escape rhythm because of depressed sinus node function with sinoatrial block or delayed conduction in the AV node. Nonparoxysmal junctional tachycardia results from increased automaticity of the junctional tissues in digitalis toxicity or ischemia and is associated with a narrow QRS complex and a rate usually less than 120 130 beats/min. It is usually considered benign when it occurs in acute myocardial infarction, but the ischemia that induces it may also cause ventricular tachycardia and ventricular fibrillation.
P.388
Ventricular Arrhythmias
1. Ventricular Premature Beats (Ventricular Extrasystoles)
Ventricular premature beats are characterized by wide QRS complexes that differ in morphology from the patient's normal beats. They are usually not preceded by a P wave, although retrograde ventriculoatrial conduction may occur. Unless the latter is present, there is a fully compensatory pause (ie, without change in the PP interval). Bigeminy and trigeminy are arrhythmias in which every second or third beat is premature; these patterns confirm a reentry mechanism for the ectopic beat. Exercise generally abolishes premature beats in normal hearts, and the rhythm becomes regular. The patient may or may not sense the irregular beat, usually as a skipped beat. Ambulatory ECG monitoring or monitoring during graded exercise may reveal more frequent and complex ventricular premature beats than occur in a single routine ECG. An increased frequency of ventricular premature beats during exercise is associated with a higher risk of cardiovascular mortality, though there is no evidence that specific therapy has a role.
Sudden death occurs more frequently (presumably as a result of ventricular fibrillation) when ventricular premature beats occur in the presence of organic heart disease but not in individuals with no known cardiac disease. If no associated cardiac disease is present and if the ectopic beats are asymptomatic, no therapy is indicated. If they are frequent, electrolyte abnormalities (especially hypokalemia or hyperkalemia and hypomagnesemia), hyperthyroidism, and occult heart disease should be excluded. Pharmacologic treatment is indicated only for patients who are symptomatic. Because of concerns about worsening arrhythmia and sudden death with most antiarrhythmic agents, -blockers are the agents of first choice. If the underlying condition is mitral prolapse, hypertrophic cardiomyopathy, LVH, or coronary disease or if the QT interval is prolonged -blocker therapy is appropriate. The class I and III agents (see Table 10-8) are all effective in reducing ventricular premature beats but often cause side effects and may exacerbate serious arrhythmias in 5 20% of patients. Therefore, every attempt should be made to avoid using class I or III antiarrhythmic agents in patients without symptoms.
Conti CR: Ventricular arrhythmias: a general cardiologist's assessment of therapies in 2005. Clin Cardiol 2005;28:314.
Morshedi-Meibodi A et al: Clinical correlates and prognostic significance of exercise-induced ventricular premature beats in the community: the Framingham Heart Study. Circulation 2004;109:2417.
O'Neill JO et al: Severe frequent ventricular ectopy after exercise as a predictor of death in patients with heart failure. J Am Coll Cardiol 2004;44:820.
2. Ventricular Tachycardia
Ventricular tachycardia is defined as three or more consecutive ventricular premature beats. The usual rate is 160 240 beats/min and is moderately regular but less so than atrial tachycardia. The distinction from aberrant conduction of supraventricular tachycardia may be difficult. The usual mechanism is reentry, but abnormally triggered rhythms occur. Ventricular tachycardia is either nonsustained (lasting less than 30 seconds) or sustained. It may be asymptomatic or associated with syncope or milder symptoms of impaired cerebral perfusion.
Ventricular tachycardia is a frequent complication of acute myocardial infarction and dilated cardiomyopathy but may occur in chronic coronary disease, hypertrophic cardiomyopathy, mitral valve prolapse, myocarditis, and in most other forms of myocardial disease. Torsades de pointes, a form of ventricular tachycardia in which QRS morphology twists around the baseline, may occur spontaneously in the setting of hypokalemia or hypomagnesemia or after any drug that prolongs the QT interval; it has a particularly poor prognosis. In nonacute settings, most patients with ventricular tachycardia have known or easily detectable cardiac disease, and the finding of ventricular tachycardia is an unfavorable prognostic sign.
Treatment
A. Acute Ventricular Tachycardia
The treatment of acute ventricular tachycardia is determined by the degree of hemodynamic compromise and the duration of the arrhythmia. The management of ventricular tachycardia in acute infarction has been discussed. In other patients, if ventricular tachycardia causes hypotension, heart failure, or myocardial ischemia, synchronized DC cardioversion with 100 360 J should be performed immediately. If the patient is tolerating the rhythm, lidocaine, 1 mg/kg as an intravenous bolus injection, or amiodarone 150 mg as a slow intravenous bolus over 10 minutes, followed by a slow infusion of 1 mg/min for 6 hours and then a maintenance infusion of 0.5 mg/min for an additional 18 42 hours can be used. If the ventricular tachycardia recurs, supplemental amiodarone infusions of 150 mg over 10 minutes can be given. If the patient is stable, intravenous procainamide, 20 mg/min intravenously (up to 1000 mg), followed by an infusion of 20 80 mcg/kg/min could also be tried. Empiric magnesium replacement (1 g intravenously) may help. Ventricular tachycardia can also be terminated by ventricular overdrive pacing, and this approach is useful when the rhythm is recurrent.
B. Chronic Recurrent Ventricular Tachycardia
1. Sustained ventricular tachycardia
Patients with symptomatic or sustained ventricular tachycardia in the absence of a reversible precipitating cause (acute
P.389
myocardial infarction or ischemia, electrolyte imbalance, drug toxicity, etc) are at high risk for recurrence. In those with significant LV dysfunction, subsequent sudden death is common. Several trials, including the Antiarrhythmic Drug Versus Implantable Defibrillator (AVID) and the Canadian Implantable Defibrillator trials, strongly suggest that these patients should be managed with implantable cardioverter-defibrillator devices (ICDs). In those with preserved LV function, the mortality rate is lower and the etiology is often different than in those with depressed ventricular function. Treatment with amiodarone, optimally in combination with a -blocker, may be adequate. Sotalol may be an alternative, though there is less supporting evidence. However, many times if ventricular tachycardia occurs in a patient with preserved ventricular function, it is either an outflow tract tachycardia or a fascicular ventricular tachycardia, and these arrhythmias will often respond to AV nodal blockers and can be effectively treated with catheter ablation. The role of electrophysiologic studies in this group is less clear than was previously thought, but they may help identify patients who are candidates for radiofrequency ablation of a ventricular tachycardia focus. This is particularly the case for arrhythmias that originate in the RV outflow tract (appearing as left bundle branch block with inferior axis on the surface ECG), the posterior fascicle (right bundle branch block, superior axis morphology), or sustained bundle branch reentry. Catheter ablation can be used as a palliative therapy for those patients with recurrent tachycardia who receive ICD shocks despite antiarrhythmic therapy.
2. Nonsustained ventricular tachycardia (NSVT)
NSVT is defined as runs of three or more ventricular beats lasting less than 30 seconds. These may be symptomatic (usually experienced as light-headedness) or asymptomatic. In individuals without heart disease, NSVT is not clearly associated with a poor prognosis. However, in patients with structural heart disease, particularly when they have reduced EFs, there is an increased risk of subsequent symptomatic ventricular tachycardia or sudden death. -Blockers reduce these risks in patients who have coronary disease with significant LV systolic dysfunction (EFs < 35 40%), but if sustained ventricular tachycardia has been induced during electrophysiologic testing, an implantable defibrillator may be indicated. In patients with chronic heart failure and reduced EFs whether due to coronary disease or primary cardiomyopathy and regardless of the presence of asymptomatic ventricular arrhythmias -blockers reduce the incidence of sudden death by 40 50% and should be routine therapy (see section on Heart Failure).
Although there are no definitive data with amiodarone in this group, trends from a number of studies suggest that it may be beneficial. Other antiarrhythmic agents should generally be avoided because their proarrhythmic risk appears to outweigh any benefit, even in patients with inducible arrhythmias that are successfully suppressed in the electrophysiology laboratory.
Brodsky MA et al: Prognostic value of baseline electrophysiology studies in patients with sustained ventricular tachyarrhythmia: the Antiarrhythmics Versus Implantable Defibrillators (AVID) trial. Am Heart J 2002;144:478.
Ermis C et al: Comparison of ventricular arrhythmia frequency in patients with ischemic cardiomyopathy versus nonischemic cardiomyopathy treated with implantable cardioverter defibrillators. Am J Cardiol 2005;96:233.
Kadish A et al; Defibrillators in Non-Ischemic Cardiomyopathy Treatment Evaluation (DEFINITE) Investigators: Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N Engl J Med 2004;350: 2151.
Katritsis DG et al: Nonsustained ventricular tachycardia: where do we stand? Eur Heart J 2004;25:1093.
Klein RC et al: Analysis of implantable cardioverter defibrillator therapy in the Antiarrhythmics Versus Implantable Defibrillators (AVID) Trial. J Cardiovasc Electrophysiol 2003;14: 940.
Sarkozy A et al: Advances in the acute pharmacologic management of cardiac arrhythmias. Curr Cardiol Rep 2003;5:387.
Wall TS et al: Ventricular tachycardia in structurally normal hearts. Curr Cardiol Rep 2002;4:388.
Wietholt D et al: Prevention of sustained ventricular tachyarrhythmias in patients with implantable cardioverter-defibrillators the PREVENT study. J Interv Card Electrophysiol 2003;9:383.
Zimetbaum PJ et al: Electrocardiographic predictors of arrhythmic death and total mortality in the multicenter unsustained tachycardia trial. Circulation 2004;110:766.
3. Ventricular Fibrillation & Sudden Death
Sudden cardiac death is defined as unexpected nontraumatic death in clinically well or stable patients who die within 1 hour after onset of symptoms. The causative rhythm in most cases is ventricular fibrillation, which is usually preceded by ventricular tachycardia except in the setting of acute ischemia or infarction. Complete heart block and sinus node arrest may also cause sudden death. A disproportionate number of sudden deaths occur in the early morning hours. Over 75% of victims of sudden cardiac death have severe CAD. Many have old infarctions. Sudden death may be the initial manifestation of coronary disease in up to 20% of patients and accounts for approximately 50% of deaths from coronary disease. When ventricular fibrillation occurs in the initial 24 hours after infarction, long-term management is no different from that of other patients with acute infarction. Other conditions that predispose to sudden death include severe LVH, hypertrophic cardiomyopathy, congestive cardiomyopathy, aortic stenosis, pulmonary stenosis, primary pulmonary hypertension, cyanotic congenital heart disease, atrial myxoma, mitral valve prolapse, hypoxia, electrolyte abnormalities, prolonged QT interval syndrome, the Brugada syndrome
P.390
and conduction system disease. Late potentials (after the QRS complex) on a signal-averaged surface ECG in patients with prior myocardial infarction may identify a group of patients at risk for ventricular arrhythmias and sudden death.
Unless ventricular fibrillation occurred shortly after myocardial infarction, is associated with ischemia, or is seen with an unusual correctable process (such as an electrolyte abnormality, drug toxicity, or aortic stenosis), surviving patients require evaluation and intervention since recurrences are frequent. Exercise testing or coronary arteriography should be performed to exclude coronary disease as the underlying cause, since revascularization may prevent recurrence. Conduction disturbances should be managed as described in the next section. If prodromal supraventricular arrhythmias or ventricular arrhythmias, such as sustained or nonsustained ventricular tachycardia, are found by ambulatory ECG monitoring, their elimination by pharmacologic therapy or ablation may prevent further episodes. There is growing consensus that if myocardial infarction or ischemia, other precipitating causes of ventricular fibrillation, or bradyarrhythmias and conduction disturbances are not found to be the cause of the sudden death episode, an implantable defibrillator is the treatment of choice for appropriate patients. In addition, evidence from the MADIT II study and Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) suggest that patients with severe LV dysfunction whether due to an ischemic cause such as a remote myocardial infarction or a nonischemic cause of advanced heart failure have a reduced risk of death with the prophylactic implantation of a implantable cardioverter-defibrillator. However, there is also evidence that implanting prophylactic ICDs in patients early after myocardial infarction is associated with a trend toward worse outcomes.
ACC/AHA/NASPE 2002 guideline update for implantation of cardiac pacemakers and antiarrhythmia devices: summary article. J Am Coll Cardiol 2002;40:1703.
Bardy GH et al; Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) Investigators. Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med 2005;352:225. Erratum in: N Engl J Med 2005;352: 2146.
Bokhari F et al: Long-term comparison of the implantable cardioverter defibrillator versus amiodarone: eleven-year follow-up of a subset of patients in the Canadian Implantable Defibrillator Study (CIDS). Circulation 2004;110:112.
Brodine WN et al; MADIT-II Research Group: Effects of beta-blockers on implantable cardioverter defibrillator therapy and survival in the patients with ischemic cardiomyopathy (from the Multicenter Automatic Defibrillator Implantation Trial-II). Am J Cardiol 2005;96:691.
Dorian P et al: Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med 2002; 346:884.
Eckardt L et al: Long-term prognosis of individuals with right precordial ST-segment-elevation Brugada syndrome. Circulation 2005;111:257.
Hohnloser SH et al; DINAMIT Investigators: Prophylactic use of an implantable cardioverter-defibrillator after acute myocardial infarction. N Engl J Med 2004;351:2481.
Kadish A et al; Defibrillators in Non-Ischemic Cardiomyopathy Treatment Evaluation (DEFINITE) Investigators: Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N Engl J Med 2004;350: 2151.
Klein RC et al: Analysis of implantable cardioverter defibrillator therapy in the Antiarrhythmics Versus Implantable Defibrillators (AVID) Trial. J Cardiovasc Electrophysiol 2003;14: 940.
Moss AJ: Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med 2002;346:877.
Sanders GD et al: Cost-effectiveness of implantable cardioverter-defibrillators. N Engl J Med 2005;353:1471.
Wilde AA et al: Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 2002;106:2514.
4. Accelerated Idioventricular Rhythm
Accelerated idioventricular rhythm is a regular wide complex rhythm with a rate of 60 120 beats/min, usually with a gradual onset. Because the rate is often similar to the sinus rate, fusion beats and alternating rhythms are common. Two mechanisms have been invoked: (1) an escape rhythm due to suppression of higher pacemakers resulting from sinoatrial and AV block or from depressed sinus node function; and (2) slow ventricular tachycardia due to increased automaticity or, less frequently, reentry. It occurs commonly in acute infarction and following reperfusion after thrombolytic drugs. The incidence of associated ventricular fibrillation is much less than that of ventricular tachycardia with a rapid rate, and treatment is not indicated unless there is hemodynamic compromise or more serious arrhythmias. This rhythm also is common in digitalis toxicity.
Accelerated idioventricular rhythm must be distinguished from the idioventricular or junctional rhythm with rates less than 40 45 beats/min that occurs in the presence of complete AV block. AV dissociation where ventricular rate exceeds sinus but not AV block occurs in most cases of accelerated idioventricular rhythm.
5. Long QT Syndrome
Congenital long QT syndrome is an uncommon disease that is characterized by recurrent syncope, a long QT interval (usually 0.5 0.7 second), documented ventricular arrhythmias, and sudden death. It may occur in the presence (Jervell syndrome, Lange-Nielsen syndrome) or absence (Romano-Ward syndrome) of congenital deafness. Inheritance may be autosomal recessive or autosomal dominant (Romano-Ward). Specific genetic mutations affecting membrane potassium and sodium channels have been identified and help delineate the mechanisms of susceptibility to arrhythmia.
Because this is a primary electrical disorder, usually with no evidence of structural heart disease or LV dysfunction, the long-term prognosis is excellent if arrhythmia is controlled. Long-term treatment with -blockers,
P.391
permanent pacing, or left cervicothoracic sympathectomy is frequently effective. ICD implantation is recommended for patients in whom recurrent syncope, sustained ventricular arrhythmias, or sudden cardiac death occurs despite drug therapy. The ICD should be considered as primary therapy in certain patients, such as those in whom aborted sudden cardiac death is the initial presentation of the long-QT syndrome, when there is a strong family history of sudden cardiac death, or when compliance or intolerance to drugs is a concern.
Acquired long QT interval secondary to use of antiarrhythmic agents or antidepressant drugs, electrolyte abnormalities, myocardial ischemia, or significant bradycardia may result in ventricular tachycardia (particularly torsades de pointes, ie, twisting about the baseline into varying QRS morphology). Notably, many drugs that are in some settings effective for the treatment of ventricular arrhythmias prolong the QT interval. Prudence dictates that drug therapy that prolongs the QT interval beyond 500 ms be discontinued.
The management of torsades de pointes differs from that of other forms of ventricular tachycardia. Class I, Ic, or III antiarrhythmics, which prolong the QT interval, should be avoided or withdrawn immediately if being used. Intravenous -blockers may be effective, especially in the congenital form; intravenous magnesium should be given acutely. An effective approach is temporary ventricular or atrial pacing, which can both break and prevent the rhythm.
Chiang CE: Congenital and acquired long QT syndrome. Current concepts and management. Cardiol Rev 2004;12:222.
Goldenberg I et al: Sudden cardiac death without structural heart disease: update on the long QT and Brugada syndromes. Curr Cardiol Rep 2005;7:349.
Monnig G et al: Implantable cardioverter-defibrillator therapy in patients with congenital long-QT syndrome: a long-term follow-up. Heart Rhythm 2005;2:497.
Passman R et al: Polymorphic ventricular tachycardia, long Q-T syndrome, and torsades de pointes. Med Clin North Am 2001;85:321.
Priori SG et al: Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA 2004;292:1341.
Wehrens XH et al: Novel insights in the congenital long QT syndrome. Ann Intern Med 2002;137:981.
Welde AA: Is there a role for implantable cardioverter defibrillators in long QT syndrome? J Cardiovasc Electrophysiol 2002;13(1 Suppl):S110.
Bradycardias & Conduction Disturbances
Abnormalities of conduction can occur between the sinus node and atrium, within the AV node, and in the intraventricular conduction pathways.
Sick Sinus Syndrome
This imprecise diagnosis is applied to patients with sinus arrest, sinoatrial exit block (recognized by a pause equal to a multiple of the underlying PP interval or progressive shortening of the PP interval prior to a pause), or persistent sinus bradycardia. These rhythms are often caused or exacerbated by drug therapy (digitalis, calcium channel blockers, -blockers, sympatholytic agents, antiarrhythmics), and agents that may be responsible should be withdrawn prior to making the diagnosis. Another presentation is of recurrent supraventricular tachycardias (paroxysmal reentry tachycardias, atrial flutter, and atrial fibrillation), associated with bradyarrhythmias ( tachy-brady syndrome ). The long pauses that often follow the termination of tachycardia cause the associated symptoms.
Sick sinus syndrome occurs most commonly in elderly patients. The pathologic changes are usually nonspecific, characterized by patchy fibrosis of the sinus node and cardiac conduction system. Sick sinus syndrome may be caused by other conditions, including sarcoidosis, amyloidosis, Chagas' disease, and various cardiomyopathies. Coronary disease is an uncommon cause.
Most patients with ECG evidence of sick sinus syndrome are asymptomatic, but rare individuals may experience syncope, dizziness, confusion, palpitations, heart failure, or angina. Because these symptoms are either nonspecific or are due to other causes, it is essential that they be demonstrated to coincide temporally with arrhythmias. This may require prolonged ambulatory monitoring or the use of an event recorder. Pharmacologic therapy for sick sinus syndrome has been difficult, but recent studies have indicated that oral theophylline may be effective, especially when sinus bradycardia is the major manifestation. Most symptomatic patients will require permanent pacing. Dual-chamber pacing is preferred because ventricular pacing is associated with a higher incidence of subsequent atrial fibrillation, and subsequent atrioventricular block occurs at a rate of 2% per year. Treatment of associated tachyarrhythmias is often difficult without first instituting pacing, since digoxin and other antiarrhythmic agents may exacerbate the bradycardia. Unfortunately, symptomatic relief following pacing has not been consistent, largely because of inadequate documentation of the etiologic role of bradyarrhythmias in producing the symptom. Furthermore, many of these patients may have associated ventricular arrhythmias that may require treatment; however, carefully selected patients may become asymptomatic with permanent pacing alone.
Adan V et al: Diagnosis and treatment of sick sinus syndrome. Am Fam Physician 2003;67:1725.
Brignole M: Sick sinus syndrome. Clin Geriatr Med 2002;18: 211.
Dretzke J et al: Dual chamber versus single chamber ventricular pacemakers for sick sinus syndrome and atrioventricular block. Cochrane Database Syst Rev 2004;(2):CD003710.
P.392
Lamas GA et al: Quality of life and clinical outcomes in elderly patients treated with ventricular pacing as compared with dual-chamber pacing. Pacemaker Selection in the Elderly Investigators. N Engl J Med 1998;338:1097.
AV Block
AV block is categorized as first-degree (PR interval > 0.21 second with all atrial impulses conducted), second-degree (intermittent blocked beats), or third-degree (complete heart block, in which no supraventricular impulses are conducted to the ventricles).
Second-degree block is subclassified. In Mobitz type I (Wenckebach) AV block, the AV conduction time (PR interval) progressively lengthens, with the RR interval shortening, before the blocked beat; this phenomenon is almost always due to abnormal conduction within the AV node. In Mobitz type II AV block, there are intermittently nonconducted atrial beats not preceded by lengthening AV conduction. It is usually due to block within the His bundle system. The classification as Mobitz type I or Mobitz type II is only partially reliable because patients may appear to have both types on the surface ECG, and the site of origin of the 2:1 AV block cannot be predicted from the ECG. The width of the QRS complexes assists in determining whether the block is nodal or infranodal. When they are narrow, the block is usually nodal; when they are wide, the block is usually infranodal. Electrophysiologic studies may be necessary for accurate localization. Management of AV block in acute myocardial infarction has already been discussed. This section deals with patients in the nonischemic setting.
First-degree and Mobitz type I block may occur in normal individuals with heightened vagal tone. They may also occur as a drug effect (especially digitalis, calcium channel blockers, -blockers, or other sympatholytic agents), often superimposed on organic disease. These disturbances also occur transiently or chronically due to ischemia, infarction, inflammatory processes, fibrosis, calcification, or infiltration. The prognosis is usually good, since reliable alternative pacemakers arise from the AV junction below the level of block if higher degrees of block occur.
Mobitz type II block is almost always due to organic disease involving the infranodal conduction system. In the event of progression to complete heart block, alternative pacemakers are not reliable. Thus, prophylactic ventricular pacing is required.
Complete (third-degree) heart block is a more advanced form of block often due to a lesion distal to the His bundle and associated with bilateral bundle branch block. The QRS is wide and the ventricular rate is slower, usually less than 50 beats/min. Transmission of atrial impulses through the AV node is completely blocked, and a ventricular pacemaker maintains a slow, regular ventricular rate, usually less than 45 beats/min. Exercise does not increase the rate. The first heart sound varies in intensity; wide pulse pressure, a changing systolic blood pressure level, and cannon venous pulsations in the neck are also present. Patients may be asymptomatic or may complain of weakness or dyspnea if the rate is less than 35 beats/min; symptoms may occur at higher rates if the left ventricle cannot increase its stroke output. During periods of transition from partial to complete heart block, some patients have ventricular asystole that lasts several seconds to minutes. Syncope occurs abruptly.
Patients with episodic or chronic infranodal complete heart block require permanent pacing, and temporary pacing is indicated if implantation of a permanent pacemaker is delayed.
Barold SS: Atrioventricular block revisited. Compr Ther 2002; 28:74.
Bourke JP: Atrioventricular block and problems with atrioventricular conduction. Clin Geriatr Med 2002;18:229.
Toff WD et al; United Kingdom Pacing and Cardiovascular Events Trial Investigators: Single-chamber versus dual-chamber pacing for high-grade atrioventricular block. N Engl J Med 2005;353:145.
AV Dissociation
When a ventricular pacemaker is firing at a rate faster than or close to the sinus rate (accelerated idioventricular rhythm, ventricular premature beats, or ventricular tachycardia), atrial impulses arriving at the AV node when it is refractory may not be conducted. This phenomenon is AV dissociation but does not necessarily indicate AV block. No treatment is required aside from management of the causative arrhythmia.
Intraventricular Conduction Defects
Intraventricular conduction defects, including bundle branch block, are common in individuals with otherwise normal hearts and in many disease processes, including ischemic heart disease, inflammatory disease, infiltrative disease, cardiomyopathy, and postcardiotomy. Below the AV node and bundle of His, the conduction system trifurcates into a right bundle and anterior and posterior fascicles of the left bundle. Conduction block in each of these fascicles can be recognized on the surface ECG. Although such conduction abnormalities are often seen in normal hearts, they are more commonly due to organic heart disease either an isolated process of fibrosis and calcification or more generalized myocardial disease. Bifascicular block is present when two of these right bundle, left anterior and posterior hemibundle are involved. Trifascicular block is defined as right bundle branch block with alternating left hemiblock, alternating right and left bundle branch block, or bifascicular block with documented prolonged infranodal conduction (long His-ventricular interval).
The prognosis of intraventricular block is generally that of the underlying myocardial process. Patients with
P.393
no apparent heart disease have an overall survival rate similar to that of matched controls. However, left bundle branch block but not right is associated with a higher risk of development of overt cardiac disease and cardiac mortality. Even in bifascicular block, the incidence of occult complete heart block or progression to it is low, and pacing is not usually warranted. In patients with symptoms (eg, syncope) consistent with heart block and intraventricular block, pacing should be reserved for those with documented concomitant complete heart block on monitoring or those with a very prolonged HV interval (> 90 ms) with no other cause for symptoms. Even in the latter group, prophylactic pacing has not improved the prognosis significantly, probably because of the high incidence of ventricular arrhythmias in the same population.
Permanent Pacing
The indications for permanent pacing have been discussed: symptomatic bradyarrhythmias, asymptomatic Mobitz II AV block, or complete heart block. The versatility of pacemaker generator units has increased markedly, and dual-chamber multiple programmable units are being implanted with increasing frequency. A standardized nomenclature for pacemaker generators is used, usually consisting of four letters. The first letter refers to the chamber that is stimulated (A = atrium, V = ventricle, D = dual, for both). The second letter refers to the chamber in which sensing occurs (also A, V, or D). The third letter refers to the sensory mode (I = inhibition by a sensed impulse, T = triggering by a sensed impulse, D = dual modes of response). The fourth letter refers to the programmability or rate modulation capacity (usually P for programming for two functions, M for programming more than two, and R for rate modulation).
A pacemaker that senses and paces in both chambers is the most physiologic approach to pacing patients who remain in sinus rhythm. AV synchrony is particularly important in patients in whom atrial contraction produces a substantial increment in stroke volume and in those in whom sensing the atrial rate to provide rate-responsive ventricular pacing is useful. Dual-chamber pacing is most useful for individuals with LV systolic or perhaps more importantly diastolic dysfunction and for physically active individuals. In patients with single-chamber pacemakers, the lack of an atrial kick may lead to the so-called pacemaker syndrome, in which the patient experiences signs of low cardiac output while upright.
Pulse generators are also available that can increase their rate in response to motion or respiratory rate when the atrial rate is not an indication of the optimal heart rate. These are most useful in active individuals. Follow-up after pacemaker implantation, usually by telephonic monitoring, is essential. All pulse generators and lead systems have an early failure rate that is now below 5% and an expected battery life varying from 4 years to 10 years.
ACC/AHA/NASPE 2002 guideline update for implantation of cardiac pacemakers and antiarrhythmia devices: summary article. J Am Coll Cardiol 2002;40:1703.
Bryce M et al: Evolving indications for permanent pacemakers. Ann Intern Med 2001;134:1130.
Faddis MN et al: Pacing interventions for falls and syncope in the elderly. Clin Geriatr Med 2002;18:279.
Gregoratos G: Indications and recommendations for pacemaker therapy. Am Fam Physician 2005;71:1563.
JAMA patient page: Heart pacemakers. JAMA 2001;286:878.
Trohman RG et al: Cardiac pacing: the state of the art. Lancet 2004;364:1701.
Vlietstra RE et al: Choice of pacemakers in patients aged 75 years and older: ventricular pacing mode vs. dual-chamber pacing mode. Am J Geriatr Cardiol 2005;14:35.
Wong GC et al: Single chamber ventricular compared with dual chamber pacing: a review. Can J Cardiol 2002;18: 301.
Evaluation of Syncope
Syncope, defined as a transient loss of consciousness and postural tone due to inadequate cerebral blood flow with prompt recovery without resuscitative measures, is a common clinical problem, especially in the elderly. Thirty percent of the adult population will experience at least one episode, and syncope accounts for approximately 3% of emergency department visits. Causes include cardiac abnormalities (either disturbances of rhythm or hemodynamics), vascular disorders, or neurologic processes. A specific cause is identified in about 50% of cases during the initial evaluation. The prognosis is relatively benign except when accompanying cardiac disease is present. Syncope is more likely to occur in patients with known heart disease, older men, and young women (who are prone to vasovagal episodes). Syncope is characteristically abrupt in onset, often resulting in injury, transient (lasting for seconds to a few minutes), and followed by prompt recovery of full consciousness.
Vasomotor syncope may be due to excessive vagal tone or impaired reflex control of the peripheral circulation. The most frequent type of vasodepressor syncope is vasovagal hypotension or the common faint, which is often initiated by a stressful, painful, or claustrophobic experience, especially in young women. Premonitory symptoms, such as nausea, diaphoresis, tachycardia, and pallor, are usual. Episodes can be aborted by lying down or removing the inciting stimulus. Enhanced vagal tone with resulting hypotension is the cause of syncope in carotid sinus hypersensitivity and postmicturition syncope; vagal-induced sinus bradycardia, sinus arrest, and AV block are common accompaniments and may themselves be the cause of syncope. Carotid sinus massage under carefully monitored conditions or tilt-table testing may be diagnostic (see above under Autonomic Testing). Treatment consists largely of counseling patients to avoid predisposing situations. Paradoxically, -blockers have been used in patients with altered autonomic function uncovered by head-up tilt testing but they have provided only minimal benefit. Permanent pacing has little benefit
P.394
except in patients with documented severe pauses and bradycardiac responses.
Volume expanders, such as fludrocortisone, or vasoconstrictors, such as midodrine, have also been tried but with minimal benefit. Selective serotonin reuptake inhibitors have shown some benefit in select patients.
Orthostatic (postural) hypotension is another common cause of vasomotor syncope, especially in the elderly, in diabetics or other patients with autonomic neuropathy, in patients with blood loss or hypovolemia, and in patients taking vasodilators, diuretics, and adrenergic-blocking drugs. In addition, a syndrome of chronic idiopathic orthostatic hypotension exists primarily in older men. In most of these conditions, the normal vasoconstrictive response to assuming upright posture, which compensates for the abrupt decrease in venous return, is impaired. A greater than normal decline (20 mm Hg) in blood pressure immediately upon arising from the supine to the standing position is observed, with or without tachycardia depending on the status of autonomic (baroreceptor) function. Studying patients with a tilt table can establish the diagnosis with more certainty. Autonomic function can be assessed by observing blood pressure and heart rate responses to Valsalva's maneuver and by tilt testing. In older patients, vasoconstrictor abnormalities and autonomic insufficiency are perhaps the most common causes of syncope. Thus, tilt testing should be done before proceeding to invasive studies unless clinical and ambulatory ECG evaluation suggests a cardiac abnormality.
Cardiogenic syncope can occur on a mechanical or arrhythmic basis. Mechanical problems that can cause syncope include aortic stenosis (where syncope may occur from autonomic reflex abnormalities or ventricular tachycardia), pulmonary stenosis, hypertrophic obstructive cardiomyopathy, congenital lesions associated with pulmonary hypertension or right-to-left shunting, and LA myxoma obstructing the mitral valve. Episodes are commonly exertional or postexertional. More commonly, cardiac syncope is due to disorders of automaticity (sick sinus syndrome), conduction disorders (AV block), or tachyarrhythmias (especially ventricular tachycardia and supraventricular tachycardia with rapid ventricular rate).
The evaluation for syncope depends on findings from the history and physical examination (especially orthostatic blood pressure evaluation, examination of carotid and other arteries, cardiac examination, and, if appropriate, carotid sinus massage). The resting ECG may reveal arrhythmias, evidence of accessory pathways, prolonged QT interval, and other signs of heart disease (such as infarction or hypertrophy). If the history is consistent with syncope, ambulatory ECG monitoring is essential. This may need to be repeated several times, since yields increase with longer periods of monitoring, at least up to 3 days. Event recorder and transtelephone ECG monitoring may be helpful in patients with intermittent presyncopal episodes. Electrophysiologic studies to assess sinus node function and AV conduction and to induce supraventricular or ventricular tachycardia are indicated in patients with recurrent episodes and nondiagnostic ambulatory ECGs. They reveal an arrhythmic cause in 20 50% of patients, depending on the study criteria, and are most often diagnostic when the patient has had multiple episodes and has identifiable cardiac abnormalities.
Faddis MN et al: Pacing interventions for falls and syncope in the elderly. Clin Geriatr Med 2002;18:279.
Goldschlager N: Etiologic considerations in the patient with syncope and an apparently normal heart. Arch Intern Med 2003;163:151.
Grubb BP: Neurocardiogenic syncope. N Engl J Med 2005;352: 1004.
Kapoor WN: Current evaluation and management of syncope. Circulation 2002;106:1606.
Kenny RA: Syncope in the elderly: diagnosis, evaluation, and treatment. J Cardiovasc Electrophysiol 2003;14(9 Suppl): S74.
Weimer LH et al: Syncope and orthostatic intolerance. Med Clin North Am 2003;87:835.
Recommendations for Resumption of Driving
An important management problem in patients who have experienced syncope, symptomatic ventricular tachycardia, or aborted sudden death is to provide recommendations concerning automobile driving. According to a survey published in 1991, only eight states had specific laws dealing with this issue, whereas 42 had laws restricting driving in patients with seizure disorders. Patients with syncope or aborted sudden death thought to have been due to temporary factors (acute myocardial infarction, bradyarrhythmias subsequently treated with permanent pacing, drug effect, electrolyte imbalance) should be strongly advised after recovery not to drive for at least 1 month. Other patients with symptomatic ventricular tachycardia or aborted sudden death, whether treated pharmacologically, with antitachycardia devices, or with ablation therapy, should not drive for at least 6 months. Longer restrictions are warranted in these patients if spontaneous arrhythmias persist. The physician should comply with local regulations and consult local authorities concerning individual cases.
Akiyama T et al: Resumption of driving after life-threatening ventricular tachyarrhythmia. N Engl J Med 2001;345:391.
Baessler C et al; DAVID Investigators: Time to resumption of driving after implantation of an automatic defibrillator (from the Dual chamber and VVI Implantable Defibrillator [DAVID] trial). Am J Cardiol 2005;95:665.
Congestive heart Failure
![]() Essentials of Diagnosis
Essentials of Diagnosis
LV failure: Exertional dyspnea, cough, fatigue, orthopnea, paroxysmal nocturnal dyspnea,
P.395
cardiac enlargement, rales, gallop rhythm, and pulmonary venous congestion.RV failure: Elevated venous pressure, hepatomegaly, dependent edema; usually due to LV failure.
Assessment of LV function is a crucial part of diagnosis and management.
Epidemiology
Heart failure is a common syndrome that is increasing in incidence and prevalence. Approximately 5 million patients in the United States have heart failure, and there are nearly 500,000 new cases each year. It is primarily a disease of aging, with over 75% of existing and new cases occurring in individuals over 65 years of age. The prevalence of heart failure rises from < 1% in individuals below 60 years to nearly 10% in those over 80 years of age.
Pathophysiology
Systolic function of the heart is governed by four major determinants: the contractile state of the myocardium, the preload of the ventricle (the end-diastolic volume and the resultant fiber length of the ventricles prior to onset of the contraction), the afterload applied to the ventricles (the impedance to LV ejection), and the heart rate.
Cardiac function may be inadequate as a result of alterations in any of these determinants. In most instances, the primary derangement is depression of myocardial contractility caused either by loss of functional muscle (due to myocardial infarction, etc) or by processes diffusely affecting the myocardium. However, the heart may fail as a pump because preload is excessively elevated, such as in valvular regurgitation, or when afterload is excessive, such as in aortic stenosis or in severe hypertension. Pump function may also be inadequate when the heart rate is too slow or too rapid. Whereas the normal heart can tolerate wide variations in preload, afterload, and heart rate, the diseased heart often has limited reserve for such alterations. Finally, cardiac pump function may be supranormal but nonetheless inadequate when metabolic demands or requirements for blood flow are excessive. This situation is termed high-output heart failure and, though uncommon, tends to be specifically treatable. Causes of high output include thyrotoxicosis, severe anemia, arteriovenous shunting (including dialysis fistulas), Paget's disease of bone, and thiamine deficiency (beriberi).
Manifestations of cardiac failure can also occur as a result of isolated or predominant diastolic dysfunction of the heart. In these cases, filling of the LV or RV is abnormal, either because myocardial relaxation is impaired or because the chamber is noncompliant ( stiff ) due to excessive hypertrophy or changes in composition of the myocardium. Even though contractility may be preserved, diastolic pressures are elevated and cardiac output may be reduced, potentially causing fluid retention, dyspnea, and exercise intolerance.
When the heart fails, a number of adaptations occur both in the heart and systemically. If the stroke volume of either ventricle is reduced by depressed contractility or excessive afterload, end-diastolic volume and pressure in that chamber will rise. This increases end-diastolic myocardial fiber length, resulting in a greater systolic shortening (Starling's law of the heart). If the condition is chronic, ventricular dilation will occur. Although this may restore resting cardiac output, the resulting chronic elevation of diastolic pressures will be transmitted to the atria and to the pulmonary and systemic venous circulation. Ultimately, increased capillary pressure may lead to transudation of fluid with resulting pulmonary or systemic edema. Reduced cardiac output, particularly if associated with reduced arterial pressure or perfusion of the kidneys, will also activate several neural and humoral systems. Increased activity of the sympathetic nervous system will stimulate myocardial contractility, heart rate, and venous tone; the latter change results in a rise in the effective central blood volume, which serves to further elevate preload. Though these adaptations are designed to increase cardiac output, they may themselves be deleterious. Thus, tachycardia and increased contractility may precipitate ischemia in patients with underlying CAD, and the rise in preload may worsen pulmonary congestion. Sympathetic nervous system activation also increases peripheral vascular resistance; this adaptation is designed to maintain perfusion to vital organs, but when it is excessive it may itself reduce renal and other tissue blood flow. Peripheral vascular resistance is also a major determinant of LV afterload, so that excessive sympathetic activity may further depress cardiac function.
One of the more important effects of lower cardiac output is reduction of renal blood flow and glomerular filtration rate, which leads to sodium and fluid retention. The renin-angiotensin-aldosterone system is also activated, leading to further increases in peripheral vascular resistance and LV afterload as well as sodium and fluid retention. Heart failure is associated with increased circulating levels of arginine vasopressin, which also serves as a vasoconstrictor and inhibitor of water excretion. Whereas release of atrial natriuretic peptide is increased in heart failure owing to the elevated atrial pressures, there is evidence of resistance to its natriuretic and vasodilating effects.
Myocardial failure is characterized by two hemodynamic derangements, and the clinical presentation is determined by their severity. The first is reduction in cardiac reserve, ie, the ability to increase cardiac output in response to increased demands imposed by exercise or even ordinary activity. The second abnormality, elevation of ventricular diastolic pressures, is primarily a result of the compensatory processes in systolic heart failure but is the primary derangement in diastolic heart failure.
P.396
Heart failure may be right sided or left sided. Patients with left heart failure have symptoms of low cardiac output and elevated pulmonary venous pressure; dyspnea is the predominant feature. Signs of fluid retention predominate in right heart failure, with the patient exhibiting edema, hepatic congestion, and, on occasion, ascites. Most patients exhibit symptoms or signs of both right- and left-sided failure, and LV dysfunction is the primary cause of RV failure. Surprisingly, some individuals with severe LV dysfunction will display few signs of left heart failure and appear to have isolated right heart failure. Indeed, they may be clinically indistinguishable from patients with cor pulmonale, who have right heart failure secondary to pulmonary disease.
Although this section primarily concerns cardiac failure due to systolic LV dysfunction, patients with diastolic heart failure experience many of the same symptoms and may be difficult to distinguish clinically. Diastolic pressures are elevated even though diastolic volumes are normal or small. These pressures are transmitted to the pulmonary and systemic venous systems, resulting in dyspnea and edema. The most frequent cause of diastolic cardiac dysfunction is LVH, commonly resulting from hypertension, but conditions such as hypertrophic or restrictive cardiomyopathy, diabetes, and pericardial disease can produce the same clinical picture. Although diuretics are often useful in these patients, the other therapies discussed in this section (digitalis, vasodilators, inotropic agents) may be inappropriate.
Causes & Prevention of Cardiac Failure
The syndrome of cardiac failure can be produced by many diseases. In developed countries, CAD with resulting myocardial infarction and loss of functioning myocardium (ischemic cardiomyopathy) is the most common cause. Systemic hypertension remains an important cause of CHF and, even more commonly in the United States, an exacerbating factor in patients with cardiac dysfunction due to other causes such as CAD. A number of other processes may present with dilated or congestive cardiomyopathy, which is characterized by LV or biventricular dilation and generalized systolic dysfunction. These are discussed elsewhere in this chapter, but the most common are alcoholic cardiomyopathy, viral myocarditis (including infections by HIV), and dilated cardiomyopathies with no obvious underlying cause (idiopathic cardiomyopathy). Rare causes of dilated cardiomyopathy include infiltrative diseases (hemochromatosis, sarcoidosis, amyloidosis, etc), other infectious agents, metabolic disorders, cardiotoxins, and drug toxicity. Valvular heart diseases particularly degenerative aortic stenosis and chronic aortic or mitral regurgitation are not infrequent causes of heart failure.
Because many of the processes leading to heart failure are of long standing and progress gradually, heart failure is often preventable by early detection of patients at risk and early intervention. The importance of these approaches is emphasized by guidelines that have incorporated a classification of heart failure that includes four stages (Table 10-10). Stage A includes patients at risk for developing heart failure (such as patients with hypertension or CAD without current or previous symptoms or identifiable structural abnormalities of the myocardium). In the majority of these patients, development of heart failure can be prevented with interventions such as the aggressive treatment of hypertension, modification of coronary risk factors, and reduction of excessive alcohol intake (Figure 10-1). Stage B includes patients who have structural heart disease but no current or previously recognized symptoms of heart failure. Examples include patients with previous myocardial infarction, other causes of reduced systolic function, LVH, or asymptomatic valvular disease. Both ACE inhibitors and -blockers prevent heart failure in the first two of these conditions, and more aggressive treatment of hypertension and early surgical intervention are effective in the latter two. Stages C and D include patients with clinical heart failure and the relatively small group of patients that has become refractory to the usual therapies, respectively. These are discussed below.
Table 10-10. Stages of heart failure. | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Prognosis
Once manifest, heart failure carries a poor prognosis. The 5-year survival rate is less than 50% overall. Mortality rates vary from < 5% per year in those with no or few symptoms to > 30% per year in those with severe and refractory symptoms. In general, men have a poorer prognosis than women because they are more likely to have CAD, which is associated with a higher mortality rate, and are less likely to have diastolic heart failure, which has a lower mortality rate. These figures emphasize the critical importance of early detection and intervention. The prognosis of heart failure has improved in the past two decades, probably at least in part because of the more widespread use of ACE inhibitors and -blockers, which markedly improve survival.
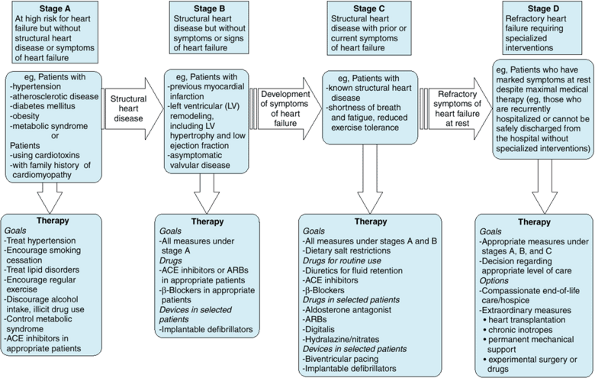 |
Figure 10-1. Stages in the evolution of heart failure and recommended therapy by stage. ACE = angiotensin-converting enzyme. (Reproduced, with permission, from Hunt SA et al: ACC/AHA 2005 Guidelines Update for the evaluation and management of chronic heart failure in the adult: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines [Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure]. ACC/AHA 2005 Guidelines Update for the diagnosis and management of chronic heart failure in the adult. 2005, American Heart, Inc. ) |
Brozena SC et al: The new staging system for heart failure. What every primary care physician should know. Geriatrics 2003; 58:31.
Gottdiener JS et al: Outcome of congestive heart failure in elderly patients: influence of left ventricular systolic function. Ann Intern Med 2002;137:631.
Hunt SA et al: ACC/AHA Guidelines for the evaluation and management of chronic heart failure in the adult: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to revise the 1995 Guidelines for the Evaluation and Management of Heart Failure). J Am Coll Cardiol 2001;38:2101.
Massie BM: Pathophysiology of heart failure. In: Cecil Textbook of Medicine, 21st ed. Saunders, 2001.
McMurray JJ et al: Epidemiology, aetiology, and prognosis of heart failure. Heart 2000;83:596.
Zile MR et al: New concepts in diastolic dysfunction and diastolic heart failure. Circulation 2002;105:1387, 1503.,
P.397
P.398
Clinical Findings
A. Symptoms
The symptoms of CHF have been discussed in part in earlier sections. The most common complaint is shortness of breath, chiefly exertional dyspnea at first and then progressing to orthopnea, paroxysmal nocturnal dyspnea, and rest dyspnea. A more subtle and often overlooked symptom of heart failure is a chronic nonproductive cough, which is often worse in the recumbent position. Nocturia due to excretion of fluid retained during the day and increased renal perfusion in the recumbent position is a common nonspecific symptom of heart failure. Patients with heart failure also complain of fatigue and exercise intolerance. These symptoms correlate poorly with the degree of cardiac dysfunction and result in part from changes in peripheral blood flow and blood flow to skeletal muscle, which are part of the syndrome of heart failure. Patients with right heart failure may experience right upper quadrant pain due to passive congestion of the liver, loss of appetite and nausea due to edema of the gut or impaired gastrointestinal perfusion, and peripheral edema.
Cardiac failure may present acutely in a previously asymptomatic patient. Causes include myocardial infarction, myocarditis, and acute valvular regurgitation due to endocarditis or other conditions. These patients usually present with pulmonary edema. The management of acute heart failure has been discussed under myocardial infarction and centers around initial stabilization with diuretics and parenteral vasodilators or inotropic agents.
Patients with episodic symptoms may be having LV dysfunction due to intermittent ischemia. This potentially reversible form of heart failure should be considered, especially in patients with angina pectoris and those with diabetes mellitus. Patients may also present with acute exacerbations of chronic, stable heart failure. Exacerbations are usually caused by alterations in therapy (or patient noncompliance), excessive salt and fluid intake, arrhythmias, excessive activity, pulmonary emboli, intercurrent infection, or progression of the underlying disease.
Patients with heart failure are often categorized by the New York Heart Association classification as class I (asymptomatic), class II (symptomatic with mild activity), class III (symptomatic with moderate activity), or class IV (symptomatic at rest). However, this classification has major limitations in that patient reports are highly subjective and in that symptoms vary from day to day. In any case, the classification is insufficiently sensitive to be useful in predicting outcomes or assessing the results of treatment.
B. Signs
Many patients with heart failure, including some with severe symptoms, appear comfortable at rest. Others will be dyspneic during conversation or minor activity, and those with long-standing severe heart failure may appear cachectic or cyanotic. The vital signs may be normal, but tachycardia, hypotension, and reduced pulse pressure may be present. Patients often show signs of increased sympathetic nervous system activity, including cold extremities and diaphoresis. Important peripheral signs of heart failure can be detected by examination of the neck, the lungs, the abdomen, and the extremities. RA pressure may be estimated through the height of the pulsations in the jugular venous system. In addition to the height of the venous pressure, abnormal pulsations such as regurgitant v waves should be sought. Examination of the carotid pulse may allow estimation of pulse pressure as well as detection of aortic stenosis. The thyroid examination is important, since occult hyperthyroidism and hypothyroidism are readily treatable causes of heart failure. In the lungs, crackles at the lung bases reflect transudation of fluid into the alveoli. Pleural effusions may cause bibasilar dullness to percussion. Expiratory wheezing and rhonchi may be signs of heart failure. Patients with severe right heart failure may have hepatic enlargement tender or nontender due to passive congestion. Systolic pulsations may be felt in tricuspid regurgitation. Sustained moderate pressure on the liver may increase jugular venous pressure (a positive hepatojugular reflux is an increase of > 1 cm). Ascites may also be present. Peripheral pitting edema is a common sign in patients with right heart failure and may extend into the thighs and abdominal wall.
The cardiac examination has been discussed. Cardinal signs in heart failure are a parasternal lift, indicating pulmonary hypertension; an enlarged and sustained LV impulse, indicating LV dilation and hypertrophy; a diminished first heart sound, suggesting impaired contractility; and S3 gallops originating in the LV and sometimes the RV. An S4 is usually present in diastolic heart failure. Murmurs should be sought to exclude primary valvular disease; secondary mitral regurgitation and tricuspid regurgitation murmurs are common in patients with dilated ventricles. In chronic heart failure, many of the expected signs of heart failure may be absent despite markedly abnormal cardiac function and hemodynamic measurements.
C. Laboratory Findings
A blood count may reveal anemia, a cause of high-output failure and an exacerbating factor in other forms of cardiac dysfunction. Biochemical studies may show renal insufficiency as a possible compounding factor. Renal function tests also determine whether cardiac failure is associated with prerenal azotemia. Serum electrolytes may disclose hypokalemia, which increases the risk of arrhythmias; hyperkalemia, which may limit the use of inhibitors of the renin-angiotensin system; or hyponatremia, an indicator of marked activation of the renin-angiotensin system and a poor prognostic sign. Thyroid function should be assessed in older patients to detect occult thyrotoxicosis or
P.399
myxedema. In unexplained cases, appropriate biopsies may lead to a diagnosis of amyloidosis, and additional assessment should include iron studies to exclude hemochromatosis. Myocardial biopsy may exclude specific causes of dilated cardiomyopathy but rarely reveals specific reversible diagnoses.
Assays of serum BNP or amino terminal pro-BNP can be a useful adjunct to the clinical history and physical examination in the diagnosis of heart failure. Measurement of serum BNP has been shown to add to clinical assessment in differentiating dyspnea due to heart failure from noncardiac causes. BNP is expressed primarily in the ventricles and is elevated when ventricular filling pressures are high. It is quite sensitive in patients with symptomatic heart failure whether due to systolic or to diastolic dysfunction but less specific in older patients, women, and patients with COPD. The roles of these assays in screening asymptomatic individuals or as a guide to management have not been established.
D. ECG and Chest Radiography
ECG may indicate an underlying or secondary arrhythmia, myocardial infarction, or nonspecific changes that often include low voltage, intraventricular conduction defects, LVH, and nonspecific repolarization changes. Chest radiographs provide information about the size and shape of the cardiac silhouette. Cardiomegaly is an important finding. Evidence of pulmonary venous hypertension includes relative dilation of the upper lobe veins, perivascular edema (haziness of vessel outlines), interstitial edema, and alveolar fluid. In acute heart failure, these findings correlate moderately well with pulmonary venous pressure. However, patients with chronic heart failure may show relatively normal pulmonary vasculature despite markedly elevated pressures. Pleural effusions are common and tend to be bilateral or right sided.
E. Additional Studies
Many studies have indicated that the clinical diagnosis of systolic myocardial dysfunction is often inaccurate. The primary confounding conditions are diastolic dysfunction of the heart with decreased relaxation and filling of the LV (particularly in hypertension and in hypertrophic states) and pulmonary disease. Because patients with heart failure usually have significant resting ECG abnormalities, stress imaging procedures such as perfusion scintigraphy or dobutamine echocardiography are often indicated.
The most useful test is the echocardiogram. This will reveal the size and function of both ventricles and of the atria. It will also allow detection of pericardial effusion, valvular abnormalities, intracardiac shunts, and segmental wall motion abnormalities suggestive of old myocardial infarction as opposed to more generalized forms of dilated cardiomyopathy.
Radionuclide angiography measures LV EF and permits analysis of regional wall motion. This test is especially useful when echocardiography is technically suboptimal, such as in patients with severe pulmonary disease. When myocardial ischemia is suspected as a cause of LV dysfunction, stress testing should be performed.
F. Cardiac Catheterization
In most patients with heart failure, clinical examination and noninvasive tests can determine LV size and function well enough to confirm the diagnosis. Left heart catheterization is necessary when significant valvular disease must be excluded and when the presence and extent of CAD must be determined. The latter is particularly important when LV dysfunction may be partially reversible by revascularization. The combination of angina or noninvasive evidence of significant myocardial ischemia with symptomatic heart failure is often an indication for coronary angiography if the patient is a potential candidate for revascularization. Right heart catheterization may be useful to select and monitor therapy in patients refractory to standard therapy.
Angeja BG et al: Evaluation and management of diastolic heart failure. Circulation 2003;107:659.
Cowie MR et al: BNP and congestive heart failure. Prog Cardiovasc Dis 2002;44:293.
Davies MK et al: ABC of heart failure. BMJ 2000;320:297.
Drazner MH et al: Prognostic importance of elevated jugular venous pressure and a third heart sound in patients with heart failure. N Engl J Med 2001;345:574.
Maisel AS et al: Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002;347:161.
Pharmacologic Treatment
The treatment of chronic heart failure is discussed here. Acute heart failure and pulmonary edema are discussed in the next section.
A. Correction of Reversible Causes
The major reversible causes of chronic heart failure include valvular lesions, myocardial ischemia, uncontrolled hypertension, arrhythmias (especially persistent tachycardias), alcohol- or drug-induced myocardial depression, intracardiac shunts, and high-output states. Calcium channel blockers, antiarrhythmic drugs, and nonsteroidal anti-inflammatory agents are important causes of worsening heart failure. Some metabolic and infiltrative cardiomyopathies may be partially reversible, or their progression may be slowed; these include hemochromatosis, sarcoidosis, and amyloidosis. Reversible causes of diastolic dysfunction include pericardial disease and LVH due to hypertension. Once it is established that there is no reversible component, the measures outlined below are appropriate.
B. Diuretic Therapy
Diuretics are the most effective means of providing symptomatic relief to patients with moderate to severe CHF.
P.400
Few patients with symptoms or signs of fluid retention can be optimally managed without a diuretic. However, excessive diuresis can lead to electrolyte imbalance and neurohormonal activation. A combination of a diuretic and an ACE inhibitor should be the initial treatment in most symptomatic patients. When fluid retention is mild, thiazide diuretics or a similar type of agent (hydrochlorothiazide, 25 100 mg; metolazone, 2.5 5 mg; chlorthalidone, 25 50 mg; etc) may be sufficient. These agents block sodium reabsorption in the cortical diluting segment at the terminal portion of the loop of Henle and in the proximal portion of the distal convoluted tubule. The result is natriuresis and kaliuresis. These agents also have weak carbonic anhydrase inhibitor activity, which results in proximal tubule inhibition of sodium reabsorption. Thiazide or related diuretics often provide better control of hypertension than short-acting loop agents.
The thiazides are generally ineffective when the glomerular filtration rate falls below 30 40 mL/min, a not infrequent occurrence in patients with severe heart failure. Metolazone maintains its efficacy down to a glomerular filtration rate of approximately 20 30 mL/min. Adverse reactions include hypokalemia and intravascular volume depletion with resulting prerenal azotemia, skin rashes, neutropenia and thrombocytopenia, hyperglycemia, hyperuricemia, and hepatic dysfunction.
Patients with more severe heart failure should be treated with one of the loop diuretics. These include furosemide (20 320 mg daily), bumetanide (1 8 mg daily), and torsemide (20 200 mg daily). These agents have a rapid onset and a relatively short duration of action. In patients with preserved renal function, two or more doses are preferable to a single larger dose. In acute situations or when gastrointestinal absorption is in doubt, they should be given intravenously. The loop diuretics inhibit chloride reabsorption in the ascending limb of the loop of Henle, which results in natriuresis, kaliuresis, and metabolic alkalosis. They are active even in severe renal insufficiency, but larger doses (up to 500 mg of furosemide or equivalent) may be required. The major adverse reactions include intravascular volume depletion, prerenal azotemia, and hypotension. Hypokalemia, particularly with accompanying digitalis therapy, is a major problem. Less common side effects include skin rashes, gastrointestinal distress, and ototoxicity (the latter more common with ethacrynic acid and possibly less common with bumetanide).
The potassium-sparing agents spironolactone, triamterene, and amiloride are often useful in combination with the loop diuretics and thiazides. Triamterene and amiloride act on the distal tubule to reduce potassium secretion. Their diuretic potency is only mild and not adequate for most patients with heart failure, but they may minimize the hypokalemia induced by more potent agents. Side effects include hyperkalemia, gastrointestinal symptoms, and renal dysfunction. Spironolactone is a specific inhibitor of aldosterone, which is often increased in CHF and has important effects beyond potassium retention (see below). Its onset of action is slower than the other potassium-sparing agents, and its side effects include gynecomastia. Combinations of potassium supplements or ACE inhibitors and potassium-sparing drugs can produce hyperkalemia but have been used with success in patients with persistent hypokalemia.
Patients with refractory edema may respond to combinations of a loop diuretic and thiazide-like agents. Metolazone, because of its maintained activity with renal insufficiency, is the most useful agent for such a combination. Extreme caution must be observed with this approach, since massive diuresis and electrolyte imbalances often occur; 2.5 mg of metolazone should be added to the previous dosage of loop diuretic. In many cases this is necessary only once or twice a week, but dosages up to 10 mg daily have been used in some patients.
C. Inhibitors of the Renin-Angiotensin-Aldosterone System
The renin-angiotensin-aldosterone system is activated early in the course of heart failure and plays an important role in the progression of this syndrome. Inhibition of this system with ACE inhibitors should be considered part of the initial therapy of this syndrome based on their favorable effects on prognosis.
1. ACE inhibitors
ACE inhibitors block the renin-angiotensin-aldosterone system by inhibiting the conversion of angiotensin I to angiotensin II, producing vasodilation by limiting angiotensin II-induced vasoconstriction, and decreasing sodium retention by reducing aldosterone secretion. Because ACE is also involved in the degradation of bradykinin, ACE inhibitors result in higher bradykinin levels, which in turn stimulate the synthesis of prostaglandins and nitric oxide. Experimental data and hemodynamic studies in patients indicate that these latter actions may be important. Although the other vasodilators tend to stimulate the renin-angiotensin system and often lose part of their effect due to the resulting fluid retention, tolerance to the ACE inhibitors is uncommon.
Many ACE inhibitors are available, and at least seven have been shown to be effective for the treatment of heart failure or the related indication of postinfarction LV dysfunction (see Table 11-8). ACE inhibitors reduce mortality by approximately 20% in patients with symptomatic heart failure and have been shown also to prevent hospitalizations, increase exercise tolerance, and reduce symptoms in these patients. As a result, ACE inhibitors should be part of first-line treatment of patients with symptomatic LV systolic dysfunction (EF < 40%), usually in combination with a diuretic. They are also indicated for the management of patients with reduced EFs without symptoms because they prevent the progression to clinical heart failure.
Because ACE inhibitors may induce significant hypotension, particularly following the initial doses, they must be started with caution. Hypotension is most
P.401
prominent in patients with already low BPs (systolic pressure < 100 mm Hg), hypovolemia, prerenal azotemia (especially if it is diuretic induced), and hyponatremia (an indicator of activation of the renin-angiotensin system). These patients should generally be started at low dosages (captopril 6.25 mg three times daily, enalapril 2.5 mg daily, or the equivalent), but other patients may be started at twice these dosages. Within several days (for those with the markers of higher risk) or at most 2 weeks, patients should be questioned about symptoms of hypotension, and both renal function and K+ levels should be monitored.
ACE inhibitors should be titrated to the dosages proved effective in clinical trials (captopril 50 mg three times daily, enalapril 10 mg twice daily, lisinopril 10 mg daily, or the equivalent) over a period of 1 3 months. Most patients will tolerate these doses. Asymptomatic hypotension is not a contraindication to up-titrating or continuing ACE inhibitors. Some patients exhibit increases in serum creatinine or K+, but they do not require discontinuation if the levels stabilize even at values as high as 3 mg/dL and 5.5 mEq/L, respectively. Renal dysfunction is more frequent in diabetics, older patients, and those with low systolic pressures, and these groups should be monitored more closely. The most common side effects of ACE inhibitors in heart failure patients are dizziness (often not related to the level of BP) and cough, though the latter is often due as much to heart failure or intercurrent pulmonary conditions as to the ACE inhibitor.
2. Angiotensin II receptor blockers
Another approach to inhibiting the renin-angiotensin-aldosterone system is the use of specific ARBs (see Table 11-8), which will block or decrease most of the effects of the system. In addition, because there are alternative pathways of angiotensin II production in many tissues, the receptor blockers may provide more complete system blockade.
However, these agents do not share the effects of ACE inhibitors on other potentially important pathways that produce increases in bradykinin, prostaglandins, and nitric oxide in the heart, blood vessels, and other tissues. The Valsartan in Heart Failure Trial (Val-HeFT) examined the efficacy of adding valsartan (titrated to a dose of 160 mg twice a day) to ACE inhibitor therapy. While valsartan did not reduce mortality, the composite of death or hospitalization for heart failure was significantly reduced. The CHARM trial randomized 7601 patients with chronic heart failure with or without LV systolic dysfunction and with or without background ACE inhibitor therapy to candesartan (titrated to 32 mg a day) or placebo. Among patients with an LV EF of < 40%, there was an 18% reduction in cardiovascular death or heart failure hospitalization and a statistically significant 12% reduction in all-cause mortality. The benefits were similar among patients on ACE inhibitors, including among patients on full-dose ACE inhibitors. Thus, ARBs, specifically candesartan or valsartan, provide important benefits as an alternative, and in addition, to ACE inhibitors in chronic heart failure.
3. Spironolactone
There is growing evidence that aldosterone may mediate some of the major effects of renin-angiotensin-aldosterone system activation, such as myocardial remodeling and fibrosis, as well as sodium retention and potassium loss at the distal tubules. Thus, spironolactone should be considered as a neurohormonal antagonist rather than narrowly as a potassium-sparing diuretic. The RALES trial compared spironolactone 25 mg daily with placebo in patients with advanced heart failure already receiving ACE inhibitors and diuretics and showed a 29% reduction in mortality as well as similar decreases in other clinical end points. Hyperkalemia was uncommon in this severe heart failure clinical trial population, which was maintained on high doses of diuretic, but hyperkalemia with spironolactone appears to be common in general practice. Potassium levels should be monitored closely during initiation of spironolcatone (after 1 and 4 weeks of therapy), particularly for patients with even mild degrees of renal insufficiency, and in patients receiving ACE inhibitors. Neither the efficacy nor the safety of spironolactone has been established in the large majority of patients with mild or moderate heart failure who are taking low doses of diuretics, though this agent may be considered in patients who require potassium supplementation. It is not known whether the more selective aldosterone inhibitor, eplerenone, is effective in improving outcome in chronic heart failure.
D. -Blockers
Although -blockers have traditionally been considered to be contraindicated in patients with heart failure because they may block the compensatory actions of the sympathetic nervous system, there is now strong evidence that these agents have important beneficial effects in this patient population. The mechanism of this benefit remains unclear, but it is likely that chronic elevations of catecholamines and sympathetic nervous system activity cause progressive myocardial damage, leading to worsening LV function and dilation. The primary evidence for this hypothesis is that over a period of 3 6 months, -blockers produce consistent substantial rises in EF (averaging 10% absolute increase) and reductions in LV size and mass.
Clinical trial results have been reported in nearly 14,000 patients (ranging from asymptomatic post-myocardial infarction LV dysfunction to severe heart failure with LV EFs < 35 40%) receiving ACE inhibitors and diuretics randomized to -blockers or placebo. Carvedilol, a nonselective 1- and 2-receptor blocker with additional weak -blocking activity, was the first -blocker approved for heart failure in the United States after showing a reduction in death and hospitalizations in four smaller studies with a total of nearly 1100 patients. Subsequently, trials with two 1-selective agents, bisoprolol (CIBIS II, with 2647 patients) and sustained-release metoprolol succinate (MERIT, with nearly 4000 patients), showed 35% reductions in mortality as well as fewer hospitalizations. Recently,
P.402
a trial using carvedilol in 2200 patients with severe (New York Heart Association [NYHA] class III/IV) heart failure was terminated ahead of schedule because of a 35% reduction in mortality. In these trials, there were reductions in sudden deaths and deaths from worsening heart failure, and benefits were seen in patients with underlying coronary disease and those with primary cardiomyopathies. In all these studies, the -blockers were generally well tolerated, with similar numbers of withdrawals in the active and placebo groups. This has led to a strong recommendation that stable patients (defined as having no recent deterioration or evidence of volume overload) with mild, moderate, and even severe heart failure should be treated with a -blocker unless there is a noncardiac contraindication. In the COPERNICUS trial, carvedilol was both well tolerated and highly effective in reducing both mortality and heart failure hospitalizations in a group of patients with severe (NYHA class III or IV) symptoms, but care was taken to ensure that they were free of fluid retention at the time of initiation. In this study, one death was prevented for every 13 patients treated for 1 year as dramatic an effect as has been seen with a pharmacologic therapy in the history of cardiovascular medicine. One trial comparing carvedilol and (short-acting) metoprolol tartrate (COMET) found significant reductions in all-cause mortality and cardiovascular mortality with carvedilol, and thus patients should be treated with extended-release metoprolol, bisoprolol, or carvedilol, but not short-acting metoprolol.
Because even apparently stable patients may deteriorate when -blockers are initiated, initiation must be done gradually and with great care. Carvedilol is initiated at a dosage of 3.125 mg twice daily and may be increased to 6.25, 12.5, and 25 mg twice daily at intervals of approximately 2 weeks. The protocols for sustained-release metoprolol use were started at 12.5 or 25 mg daily and doubled at intervals of 2 weeks to a target dose of 200 mg daily (using the Toprol XL sustained-release preparation). Bisoprolol was administered at a dosage of 1.25, 2.5, 3.75, 5, 7.5, and 10 mg daily, with increments at 1- to 4-week intervals. More gradual up-titration is often more convenient and may be better tolerated.
Patients should be instructed to monitor their weights at home as an indicator of fluid retention and to report any increase or change in symptoms immediately. Before each dose increase, the patient should be seen and examined to ensure that there has not been fluid retention or worsening of symptoms. If heart failure worsens, this can usually be managed by increasing diuretic doses and delaying further increases in -blocker doses, though downward adjustments or discontinuation is sometimes required. Carvedilol, because of its -blocking activity, may cause dizziness or hypotension. This can usually be managed by reducing the doses of other vasodilators and by slowing the pace of dose increases.
E. Digitalis Glycosides
The digitalis glycosides (primarily digoxin) are the only orally active positive inotropic agents currently available. They bind to the sodium-potassium ATPase on the sarcolemmal membrane, inhibiting the sodium pump and thereby increasing intracellular sodium. This facilitates sodium-calcium exchange, with a resultant increase in cytosolic calcium, which enhances contractile protein cross-bridge formation and force generation. The digitalis glycosides also have electrophysiologic effects that may be beneficial or deleterious in individual patients. The primary therapeutic effect is an enhancement of cardiac parasympathetic tone, which delays AV conduction and reduces sinus node automaticity, thereby decreasing the ventricular response in patients with atrial fibrillation and slightly slowing the rate of patients in sinus rhythm. However, the increase in intracellular calcium and sodium may enhance automaticity of latent pacemakers, increasing the excitability of ventricular myocytes and inducing ventricular arrhythmias, especially when hypokalemia or myocardial ischemia is present.
Although the digitalis glycosides were once the mainstay of treatment of CHF, their use in patients who are in sinus rhythm has declined because they lack the benefits of the neurohormonal antagonists on prognosis and because safety concerns persist. However, their efficacy in reducing the symptoms of heart failure has been established in at least four multicenter trials that have demonstrated that digoxin withdrawal is associated with worsening symptoms and signs of heart failure, more frequent hospitalizations for decompensation, and reduced exercise tolerance. This was also seen in the 6800-patient Digitalis Investigators Group (DIG) trial, though that study found no benefit (or harm) with regard to survival. A reduction in deaths due to progressive heart failure was balanced by an increase in deaths due to ischemic and arrhythmic events. Based on these results, digoxin should be used for patients who remain symptomatic when taking diuretics and ACE inhibitors as well as for patients with heart failure who are in atrial fibrillation and require rate control.
Digoxin, the only widely used digitalis preparation, has a half-life of 24 36 hours and is eliminated almost entirely by the kidneys. The oral maintenance dose may range from 0.125 mg three times weekly to 0.5 mg daily. It is lower in patients with renal dysfunction, in older patients, and in those with smaller lean body mass. Although a loading dose of 0.75 1.25 mg (depending primarily on lean body size) over 24 48 hours may be given if an early effect is desired, in most patients with chronic heart failure it is sufficient to begin with the expected maintenance dose (usually 0.125 0.25 mg daily). Amiodarone, quinidine, propafenone, and verapamil are among the drugs that may increase digoxin levels up to 100%. It is prudent to measure a blood level after 7 14 days (and at least 6 hours after the last dose was administered). Most of the positive inotropic effect is apparent with serum digoxin levels between 0.7 ng/mL and 1.2 ng/mL, and levels above this range may be associated with a higher risk of arrhythmias and lower survival rates, though clinically evident
P.403
toxicity is rare with levels below 1.8 ng/mL. Once an appropriate maintenance dose is established, subsequent levels are usually not indicated unless there is a change in renal function or medications that affects digoxin levels or a significant deterioration in cardiac status that may be associated with reduced clearance.
Digoxin toxicity has become less frequent as there has been a better appreciation of its pharmacology, but the therapeutic-to-toxic ratio is quite narrow. Symptoms of digitalis toxicity include anorexia, nausea, headache, blurring or yellowing of vision, and disorientation. Cardiac toxicity may take the form of AV conduction or sinus node depression; junctional, atrial, or ventricular premature beats or tachycardias; or ventricular fibrillation. Potassium administration (following serum potassium measurement, since severe toxicity may be associated with hyperkalemia) is usually indicated for the tachyarrhythmias even when levels are in the normal range, but may worsen conduction disturbances. Lidocaine or phenytoin may be useful for ventricular arrhythmias, as is overdrive pacing, but quinidine, amiodarone, and propafenone should be avoided because they will increase digoxin levels. Electrical cardioversion should be avoided if possible, as it may cause intractable ventricular fibrillation or cardiac standstill. Pacing is indicated for third-degree AV block (complete heart block) and symptomatic or severe block (heart rate < 40 beats/min) if they persist after treatment with atropine. Digoxin immune fab (ovine) is available for life-threatening toxicity or large overdoses, but it should be remembered that its half-life is shorter than that of digoxin and so repeat administration may be required.
F. Vasodilators
Agents that dilate arteriolar smooth muscle and lower peripheral vascular resistance reduce LV afterload. Medications that diminish venous tone and increase venous capacitance reduce the preload of both ventricles as their principal effect. Because most patients with moderate to severe heart failure have both elevated preload and reduced cardiac output, the maximum benefit of vasodilator therapy can be achieved by an agent or combination of agents with both actions. Many patients with heart failure have mitral or tricuspid regurgitation; agents that reduce resistance to ventricular outflow tend to redirect regurgitant flow in a forward direction.
Although vasodilators that are also neurohumoral antagonists specifically, the ACE inhibitors improve prognosis, such a benefit is less clear with the direct-acting vasodilators. The combination of hydralazine and isosorbide dinitrate has also improved survival, but to a lesser extent than ACE inhibitors. The A-HeFT trial studied hydralazine (75 mg) and isosorbide dinitrate (40 mg) three times a day in 1050 African Americans with NYHA class III or IV chronic heart failure, most of whom were treated with ACE inhibitors and -blockers. The primary endpoint was a clinical composite. The trial was stopped early because of a significant 43% reduction in all-cause mortality with hydralazine and nitrates. Whether the benefits of this approach are limited to African Americans, who may have a less active renin-angiotensin system and less available nitric oxide, is not known, but it is now prudent to use this combination in addition to other effective therapies in African Americans with severe heart failure.
The intravenous vasodilating drugs and their dosages have been discussed elsewhere in this chapter (in the section on complications in acute myocardial infarction).
1. Nitrates
Intravenous vasodilators (sodium nitroprusside or nitroglycerin) are used primarily for acute or severely decompensated chronic heart failure, especially when accompanied by hypertension or myocardial ischemia. If neither of the latter is present, therapy is best initiated and adjusted based on hemodynamic measurements. The starting dosage for nitroglycerin is generally about 10 mcg/min, which is titrated upward by 10 20 mcg/min (to a maximum of 200 mcg/min) until mean arterial pressure drops by 10%. Hypotension (BP < 100 mm Hg systolic) should be avoided. For sodium nitroprusside, the starting dosage is 0.3 0.5 mcg/kg/min with upward titration to a maximum dose of 10 mcg/kg/min.
Isosorbide dinitrate, 20 80 mg orally three times daily, has proved effective in several small studies. Nitroglycerin ointment, 12.5 50 mg (1 4 inches) every 6 8 hours, appears to be equally effective although somewhat inconvenient for long-term therapy. The nitrates are moderately effective in relieving shortness of breath, especially in patients with mild to moderate symptoms, but less successful probably because they have little effect on cardiac output in advanced heart failure. Nitrate therapy is generally well tolerated, but headaches and hypotension may limit the dose of all agents. The development of tolerance to long-term nitrate therapy is now generally acknowledged. This is minimized by intermittent therapy, especially if a daily 8- to 12-hour nitrate-free interval is used, but probably develops to some extent in most patients receiving these agents. Transdermal nitroglycerin patches have no sustained effect in patients with heart failure and should not be used for this indication.
2. Nesiritide
This agent, a recombinant form of human brain natriuretic peptide, is a potent vasodilator that reduces ventricular filling pressures and improves cardiac output. Its hemodynamic effects resemble those of intravenous nitroglycerin with a more predictable dose-response curve and a longer duration of action. In clinical studies, nesiritide (administered as 2 mcg/kg by intravenous bolus injection followed by an infusion of 0.01 mcg/kg/min, which may be up-titrated if needed) produced a rapid improvement in both dyspnea and hemodynamics. The primary adverse effect is hypotension, which may be symptomatic and sustained. Because most patients with acute heart failure respond well to conventional therapy, the role of nesiritide may be primarily in patients who
P.404
continue to be symptomatic after initial treatment with diuretics and nonparenteral nitrates.
3. Hydralazine
Oral hydralazine is a potent arteriolar dilator and markedly increases cardiac output in patients with CHF. However, as a single agent, it has not been shown to improve symptoms or exercise tolerance during long-term treatment. The combination of nitrates and oral hydralazine produces greater hemodynamic effects.
Hydralazine therapy is frequently limited by side effects. Approximately 30% of patients are unable to tolerate the relatively high doses required to produce hemodynamic improvement in heart failure (200 400 mg daily in divided doses). The major side effect is gastrointestinal distress, but headaches, tachycardia, and hypotension are relatively common. ARBs have largely supplanted the use of the hydralazine-isosorbide dinitrate combination in ACE-intolerant patients.
G. Positive Inotropic Agents
The digitalis derivatives are the only available oral inotropic agents in the United States. A number of other oral positive inotropic agents have been investigated for the long-term treatment of heart failure, but all have increased mortality without convincing evidence of improvement in symptoms. Intravenous agents, such as the 1-agonist dobutamine and the phosphodiesterase inhibitor milrinone, are sometimes used on a long-term or intermittent basis. The limited available data suggest that continuous therapy is also likely to increase mortality; intermittent inotropic therapy has never been evaluated in controlled trials, and its use is largely based on anecdotal experience. A recent randomized placebo-controlled trial of 950 patients evaluating intravenous milrinone in patients admitted for decompensated heart failure who had no definite indications for inotropic therapy showed no benefit in terms of survival, decreasing length of admission, or preventing readmission and significantly increased rates of sustained hypotension and atrial fibrillation. Thus, the role of positive inotropic agents appears to be limited to patients with symptoms and signs of low cardiac output (primarily hypoperfusion and deteriorating renal function) and those who do not respond to intravenous diuretics. In some cases, dobutamine or milrinone may help maintain patients who are awaiting cardiac transplantation.
H. Calcium Channel Blockers
First-generation calcium channel blockers may accelerate the progression of CHF. However, two trials with amlodipine in patients with severe heart failure showed that this agent was safe, though not superior to placebo. These agents should be avoided unless they are being utilized to treat associated angina or hypertension, and for these indications amlodipine is the drug of choice.
I. Anticoagulation
Patients with LV failure and reduced EFs are at somewhat increased risk for developing intracardiac thrombi and systemic arterial emboli. However, this risk appears to be primarily in patients who are in atrial fibrillation or who have large recent (within 3 6 months) myocardial infarctions. These groups should be anticoagulated. Other patients with heart failure have embolic rates of approximately two per 100 patient-years of follow-up, which approximates the rate of major bleeding, and routine anticoagulation does not appear warranted except in patients with prior embolic events or mobile LV thrombi.
J. Antiarrhythmic Therapy
Patients with moderate to severe heart failure have a high incidence of both symptomatic and asymptomatic arrhythmias. Although fewer than 10% of patients have syncope or presyncope resulting from ventricular tachycardia, ambulatory monitoring reveals that up to 70% of patients have asymptomatic episodes of nonsustained ventricular tachycardia. These arrhythmias indicate a poor prognosis independent of the severity of LV dysfunction, but many of the deaths are probably not arrhythmia related. -Blockers, because of their marked favorable effect on prognosis in general and on the incidence of sudden death specifically, should be initiated in these as well as all other patients with heart failure. Empiric antiarrhythmic therapy with amiodarone did not improve outcome in the SCD-HeFT trial, and most other agents are contraindicated because of their proarrhythmic effects in this population and their adverse effect on cardiac function.
K. Implantable Cardioverter Defibrillators
Randomized clinical trials have extended the indications for ICDs beyond patients with symptomatic or asymptomatic arrhythmias to the broad population of patients with chronic heart failure and LV systolic dysfunction. In the second Multicenter Automatic Defibrillator Implantation Trial (MADIT II), 1232 patients with prior myocardial infarction and an EF < 30% were randomized to an ICD or a control group. Mortality was 31% lower in the ICD group, which translated into nine lives saved for each 100 patients who received a device and were monitored for 3 years. The Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) reinforced and extended these results, showing a 23% relative (7.2% absolute) reduction in mortality over 5 years with a simple single-lead ICD in a population of patients with symptomatic chronic heart failure and an EF of 35%. These patients were well-managed with contemporary heart failure treatments, including -blockers. Based on these results, the United States Centers for Medicare and Medicaid Services have expanded reimbursement coverage to include patients with chronic heart failure and ischemic or nonischemic cardiomyopathy with an EF 35%.
Nonpharmacologic Treatment
A. Case Management, Diet, and Exercise Training
Thirty to 50 percent of CHF patients who are hospitalized will be readmitted within 3 6 months. Strategies
P.405
to prevent clinical deterioration, such as case management, home monitoring of weight and clinical status, and patient adjustment of diuretics, can prevent rehospitalizations and should be part of the treatment regimen of advanced heart failure.
Patients should routinely practice moderate salt restriction (2 2.5 g sodium or 5 6 g salt per day). More severe sodium restriction is usually difficult to achieve and unnecessary because of the availability of potent diuretic agents.
Exercise training improves activity tolerance in significant part by reversing the peripheral abnormalities associated with heart failure and deconditioning. In severe heart failure, restriction of activity may facilitate temporary recompensation. However, in stable patients, a prudent increase in activity or a regular exercise regimen can be encouraged. Indeed, a gradual exercise program is associated with diminished symptoms and substantial increases in exercise capacity.
B. Coronary Revascularization
Since underlying CAD is the cause of heart failure in the majority of patients, coronary revascularization may both improve symptoms and prevent progression. However, trials have not been performed in patients with symptomatic heart failure. Nonetheless, patients with angina who are candidates for surgery should be evaluated for revascularization, usually by coronary angiography. Noninvasive testing for ischemic but viable myocardium may be a more appropriate first step in patients with known coronary disease but no current clinical evidence of ischemia. The benefit of evaluating patients with heart failure of new onset without angina or prior myocardial infarction is limited. In general, bypass surgery is preferable to PTCA in the setting of heart failure because it provides more complete revascularization.
C. Biventricular Pacing (Resynchronization)
Many patients with heart failure due to systolic dysfunction have abnormal intraventricular conduction that results in dyssynchronous and hence inefficient contractions. Several studies have evaluated the efficacy of multisite pacing, using leads that stimulate the RV from the apex and the LV from the lateral wall via the coronary sinus. Patients with wide QRS complexes (generally 120 milliseconds), reduced EFs, and moderate to severe symptoms have been evaluated. Results from trials with up to 2 years of follow-up have shown an increase in EF, improvement in symptoms and exercise tolerance, and reduction in death and hospitalization. The COMPANION trial included 1520 patients with NYHA class III or IV heart failure, EF of 35%, and QRS duration 120 milliseconds. In addition to optimal medical therapy, resynchronization therapy with biventricular pacing with or without implantable defibrillator capability reduced death and hospitalization from any cause by about 20%. The CARE-HF trial randomized 813 similar patients, who also required mechanical evidence of dyssynchrony if QRS duration was 120 149 milliseconds, to resynchronization therapy. Over a mean follow-up of 29 months, death or hospitalization for cardiac cause was reduced by 37% and mortality was reduced by 36%. Thus, resynchronization therapy is indicated for patients with moderate to severe heart failure and LV dyssynchrony.
D. Cardiac Transplantation
Because of the poor prognosis of patients with advanced heart failure, cardiac transplantation has become widely used. Since the advent of cyclosporine immunosuppressive therapy and more careful screening of donor hearts, the survival of patients after cardiac transplantation has increased considerably. Many centers now have 1-year survival rates exceeding 80 90%, and 5-year survival rates above 70%. Infections, hypertension and renal dysfunction caused by cyclosporine, rapidly progressive coronary atherosclerosis, and immunosuppressant-related cancers have been the major complications. The high cost and limited number of donor organs require careful patient selection early in the course.
E. Other Surgical Treatment Options
Several surgical procedures for severe heart failure have received considerable publicity. Cardiomyoplasty is a procedure in which the latissimus dorsi muscle is wrapped around the heart and stimulated to contract synchronously with it. In ventricular reduction surgery, a large part of the anterolateral wall is resected to make the heart function more efficiently. Both approaches are too risky in end-stage patients and have not been shown to improve prognosis or symptoms in controlled studies, and for these reasons they have largely been dropped. Externally powered and implantable ventricular assist devices can be used in patients who require ventricular support either to allow the heart to recover or as a bridge to transplantation. The latest generation devices are small enough to allow patients unrestricted mobility and even discharge from the hospital. However, complications are frequent, including bleeding, thromboembolism, and infection, and the cost is very high, exceeding $200,000 in the initial 1 3 months.
Although 1-year survival was improved in a recent randomized trial, all patients died by 26 months.
F. Palliative Care
Despite the technologic advances of recent years, including cardiac resynchronization, implantable defibrillators, LV assist devices, and totally implantable artificial hearts, it should be remembered that many patients with chronic heart failure are elderly and have multiple comorbidities. Many of them will not experience meaningful improvements in survival with aggressive therapy, and the goal of management should be symptomatic improvement and palliation (see Chapter 5).
Adams KF Jr et al: B-type natriuretic peptide: from bench to bedside. Am Heart J 2003;145(2 Suppl):S34.
P.406
Albert NM et al: Improving the care of patients dying of heart failure. Cleve Clin J Med 2002;69:321.
Bardy GH; Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) Investigators: Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med 2005;352:225.
Bettencourt P: Brain natriuretic peptide (nesiritide) in the treatment of heart failure. Cardiovasc Drug Rev 2002;20:27.
Bradley DJ et al: Cardiac resynchronization and death from progressive heart failure: a meta-analysis of randomized controlled trials. JAMA 2003;289:730.
Bristow MR et al: Comparison of Medical Therapy, Pacing, and Defibrillation in Heart Failure (COMPANION) Investigators: Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med 2004;350:2140.
Cleland JG et al: The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med 2005; 352:1539.
Cuffe MS et al: Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA 2002;287:1541.
Eichhorn EJ et al: Digoxin. Prog Cardiovasc Dis 2002;44:251.
Fonarow GC: Pharmacologic therapies for acutely decompensated heart failure. Rev Cardiovasc Med 2002;3(Suppl 4):S18.
Hunt SA et al: ACC/AHA Guidelines for the evaluation and management of chronic heart failure in the adult: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1995 Guidelines for the Evaluation and Management of Heart Failure). J Am Coll Cardiol 2001;38:2101.
Kukin ML: Beta-blockers in chronic heart failure: considerations for selecting an agent. Mayo Clin Proc 2002;77:1199.
Moller JE et al: Effects of losartan and captopril on left ventricular systolic and diastolic function after acute myocardial infarction: results of the Optimal Trial in Myocardial Infarction with Angiotensin II Antagonist Losartan (OPTIMAAL) echocardiographic substudy. Am Heart J 2004;147:494.
Moss AJ et al: Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med 2002;346:877.
Mueller C et al: Use of B-type natriuretic peptide in the evaluation and management of acute dyspnea. N Engl J Med 2004;350:647.
Nohria A et al: Medical management of advanced heart failure. JAMA 2002;287:628.
Pepine CJ et al: A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003;290:2805.
Pfeffer MA et al: Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003;349:1893.
Taylor AL et al; African-American Heart Failure Trial Investigators: Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 2004;351:2049.
Williams M et al: Cardiac assist devices for end-stage heart failure. Heart Dis 2001;3:109.
Young JB et al; Candesartan in Heart failure Assessment of Reduction in Mortality and morbidity (CHARM) Investigators and Committees: Mortality and morbidity reduction with candesartan in patients with chronic heart failure and left ventricular systolic dysfunction: results of the CHARM low-left ventricular ejection fraction trials. Circulation 2004;110:2618.
Acute Heart Failure & Pulmonary Edema
![]() Essentials of Diagnosis
Essentials of Diagnosis
Acute onset or worsening of dyspnea at rest.
Tachycardia, diaphoresis, cyanosis.
Pulmonary rales, rhonchi; expiratory wheezing.
Radiograph shows interstitial and alveolar edema with or without cardiomegaly.
Arterial hypoxemia.
General Considerations
Typical causes of acute cardiogenic pulmonary edema include acute myocardial infarction or severe ischemia, exacerbation of chronic heart failure, acute volume overload of the LV (valvular regurgitation), and mitral stenosis. By far the most common presentation in developed countries is one of acute or subacute deterioration of chronic heart failure, precipitated by discontinuation of medications, excessive salt intake, myocardial ischemia, tachyarrhythmias (especially rapid atrial fibrillation), or intercurrent infection. Often in the latter group, there is preceding volume overload with worsening edema and progressive shortness of breath for which earlier intervention can usually avoid the need for hospital admission.
Clinical Findings
Acute pulmonary edema presents with a characteristic clinical picture of severe dyspnea, the production of pink, frothy sputum, and diaphoresis and cyanosis. Rales are present in all lung fields, as are generalized wheezing and rhonchi. Pulmonary edema may appear acutely or subacutely in the setting of chronic heart failure or may be the first manifestation of cardiac disease, usually acute myocardial infarction, which may be painful or silent. Less severe decompensations usually present with dyspnea at rest and rales and other evidence of fluid retention but without severe hypoxia.
A number of noncardiac conditions can also produce pulmonary edema. This occurs either because of imbalance in the Starling forces (either a decrease in plasma proteins or an increase in pulmonary venous pressure) or a functional or anatomic abnormality of the alveolar-capillary membrane. Causes include intravenous opioids, increased intracerebral pressure,
P.407
high altitude, sepsis, several medications, inhaled toxins, transfusion reactions, shock, and disseminated intravascular coagulation. These are distinguished from cardiogenic pulmonary edema by the clinical setting, the history, and the physical examination. Conversely, in most patients with cardiogenic pulmonary edema, an underlying cardiac abnormality can usually be detected clinically or by the ECG, chest radiograph, or echocardiogram.
The chest radiograph reveals signs of pulmonary vascular redistribution, blurriness of vascular outlines, increased interstitial markings, and, characteristically, the butterfly pattern of distribution of alveolar edema. The heart may be enlarged or normal in size depending on whether heart failure was previously present. Assessment of cardiac function by echocardiography is important, since a substantial proportion of patients has normal EFs with elevated atrial pressures due to diastolic dysfunction. In cardiogenic pulmonary edema, the PCWP is invariably elevated, usually over 25 mm Hg. In noncardiogenic pulmonary edema, the wedge pressure may be normal or even low.
Treatment
In full-blown pulmonary edema, the patient should be placed in a sitting position with legs dangling over the side of the bed; this facilitates respiration and reduces venous return. Oxygen is delivered by mask to obtain an arterial PO2 greater than 60 mm Hg. Noninvasive pressure support ventilation may improve oxygenation and prevent severe CO2 retention while pharmacologic interventions take effect. However, if respiratory distress remains severe, endotracheal intubation and mechanical ventilation may be necessary.
Morphine is highly effective in pulmonary edema and may be helpful in less severe decompensations when the patient is uncomfortable. The initial dosage is 2 8 mg intravenously (subcutaneous administration is effective in milder cases) and may be repeated after 2 4 hours. Morphine increases venous capacitance, lowering LA pressure, and relieves anxiety, which can reduce the efficiency of ventilation. However, morphine may lead to CO2 retention by reducing the ventilatory drive. It should be avoided in patients with opioid-induced pulmonary edema, who may improve with opioid-antagonists, and in those with neurogenic pulmonary edema.
Intravenous diuretic therapy (furosemide, 40 mg, or bumetanide, 1 mg or higher doses if the patient has been receiving long-term diuretic therapy) is usually indicated even if the patient has not exhibited prior fluid retention. These agents produce venodilation prior to the onset of diuresis.
Nitrate therapy accelerates clinical improvement by reducing both BP and LV filling pressures. Sublingual nitroglycerin or isosorbide dinitrate, topical nitroglycerin, or intravenous nitrates will ameliorate dyspnea rapidly prior to the onset of diuresis, and these agents are particularly valuable in patients with accompanying hypertension. Intravenous nesiritide (recombinant BNP), when given as a bolus followed by an infusion, improves dyspnea more rapidly than intravenous nitroglycerin, though this may reflect the cautious way in which nitroglycerin is up-titrated by many practitioners. This agent, as well as nitrates, may precipitate hypotension, especially since these agents are used in combination with multiple drugs that lower BP. In patients with low-output states particularly when hypotension is present positive inotropic agents are indicated. These approaches to treatment have been discussed previously.
Bronchospasm may occur in response to pulmonary edema and may itself exacerbate hypoxemia and dyspnea. Treatment with inhaled -adrenergic agonists or intravenous aminophylline may be helpful, but both may also provoke tachycardia and supraventricular arrhythmias.
In most cases, pulmonary edema responds rapidly to therapy. When the patient has improved, the cause or precipitating factor should be ascertained. In patients without prior heart failure, evaluation should include echocardiography and in many cases cardiac catheterization and coronary angiography. Patients with acute decompensation of chronic heart failure should be treated to achieve a euvolemic state and have their medical regimen optimized. Generally, an oral diuretic and an ACE inhibitor should be initiated, with efficacy and tolerability confirmed prior to discharge. In selected patients, early but careful initiation of -blockers in low doses should be considered.
Cotter G et al: Pulmonary edema: new insight on pathogenesis and treatment. Curr Opin Cardiol 2001;16:159.
Fonarow GC: Pharmacologic therapies for acutely decompensated heart failure. Rev Cardiovasc Med 2002;3(Suppl 4):S18.
Gandhi SK et al: The pathogenesis of acute pulmonary edema associated with hypertension. N Engl J Med 2001;344:17.
Jain P et al: Current medical treatment for the exacerbation of chronic heart failure resulting in hospitalization. Am Heart J 2003;145(2 Suppl):S3.
Young JB et al: Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA 2002;287:1531.
Myocarditis & the Cardiomyopathies
Acute Myocarditis
Acute myocarditis causes focal or diffuse inflammation of the myocardium. Most cases are infectious, caused by viral, bacterial, rickettsial, spirochetal, fungal, or parasitic agents; but toxins, drugs, and immunologic disorders can also cause myocarditis.
P.408
1. Infectious Myocarditis
![]() Essentials of Diagnosis
Essentials of Diagnosis
Often follows an upper respiratory infection.
May present with chest pain (pleuritic or nonspecific) or signs of heart failure.
ECG may show sinus tachycardia, other arrhythmias, nonspecific repolarization changes, and intraventricular conduction abnormalities.
Echocardiogram documents cardiomegaly and contractile dysfunction.
Myocardial biopsy, though not sensitive, may reveal a characteristic inflammatory pattern.
General Considerations
Cardiac dysfunction due to primary myocarditis is presumed to be caused by either an acute viral infection or a postviral immune response. Secondary myocarditis is the result of inflammation caused by nonviral pathogens, drugs, chemicals, physical agents, or inflammatory diseases such as systemic lupus erythematosus. The list of infectious causes of myocarditis is extensive and includes viruses with DNA and RNA cores. The coxsackie virus is the predominant agent, but many others have been implicated. Rickettsial myocarditis occurs with scrub typhus, Rocky Mountain spotted fever, and Q fever. Diphtheritic myocarditis is caused by the exotoxin and is often manifested by conduction abnormalities as well as heart failure.
Chagas' disease, caused by the insect-borne protozoan Trypanosoma cruzi, is a common form of myocarditis in Central and South America; the major clinical manifestations appear after a latent period of more than a decade. At this stage, patients present with cardiomyopathy, conduction disturbances, and sudden death. Associated gastrointestinal involvement (megaesophagus and megacolon) is the rule. Toxoplasmosis causes myocarditis that is usually asymptomatic but can lead to heart failure. Among parasitic infections, trichinosis is the most common cause of cardiac involvement. The potential for the HIV virus to cause myocarditis is now well recognized, though the prevalence of this complication is not known and it appears related to the level of viral load and CD4 count. In addition, other infectious causes of myocarditis are more common in patients with AIDS. A complete list of infectious causes of myocarditis is shown in Table 10-11.
Giant cell myocarditis is a rare idiopathic disorder characterized by giant cell and lymphocyte infiltration of the heart muscle. Patients usually die of ventricular arrhythmias or heart failure but occasionally respond to immunosuppressive therapy or early transplantation.
Table 10-11. Major causes of infectious myocarditis. | ||
|---|---|---|
|
Clinical Findings
A. Symptoms and Signs
Patients may present several days to a few weeks after the onset of an acute febrile illness or a respiratory infection or with heart failure without antecedent symptoms. The onset of heart failure may be gradual or may be abrupt and fulminant. Emboli may occur due to the procoagulant effect of cytokines combined with decreased myocardial contractility and blood pooling. Pleural-pericardial chest pain is common. Examination reveals tachycardia, gallop rhythm, and other evidence of heart failure or conduction defect. Many acute infections are subclinical, though they may present later as idiopathic cardiomyopathy or with ventricular arrhythmias. At times, the presentation may mimic an acute myocardial infarction with ST changes, positive cardiac markers, and regional wall motion abnormalities despite normal coronaries. Microaneurysms may also occur and may be associated with serious ventricular arrhythmias. Patients may present in a variety of ways with fulminant, subacute, or chronic myocarditis.
P.409
B. ECG and Chest Radiography
Nonspecific ST-T changes and conduction disturbances are common. Ventricular ectopy may be the initial and only clinical finding. Chest radiograph is nonspecific, but cardiomegaly is frequent, though not universal. Evidence for pulmonary venous hypertension is common and frank pulmonary edema may be present.
C. Diagnostic Studies
There is no specific laboratory study that is consistently present, though the white blood cell count is usually elevated and the sedimentation rate may increase. Troponin I levels are elevated in about one-third of patients, but CK-MB is elevated in only 10%. Echocardiography provides the most convenient way of evaluating cardiac function and can exclude many other processes. Gallium-67 scintigraphy may reveal increased cardiac uptake in acute or subacute myocarditis, but it is not very sensitive. MRI with gadolinium enhancement reveals spotty areas of injury throughout the myocardium. Paired serum viral titers and serologic tests for other agents may indicate the cause.
D. Endomyocardial Biopsy
Pathologic examinations may reveal a lymphocytic inflammatory response with necrosis, but the patchy distribution of abnormalities makes this relatively insensitive. By biopsy, the diagnosis of myocarditis has been established by the 1986 Dallas criteria. The diagnosis is dependent on describing the severity of an inflammatory infiltrate with necrosis and degeneration of adjacent myocytes. The type of infiltrate is dependent on the causal agent; usually this is lymphocytic in viral disease, but it may be neutrophilic, eosinophilic, giant cell, granulomatous, or mixed.
Treatment & Prognosis
Patients with fulminant myocarditis may present with acute cardiogenic shock. Their ventricles are usually not dilated, but thickened (possibly due to myoedema). There is a high death rate, but if the patients recover, they are usually left with no residual cardiomyopathy. Patients who present with subacute disease have a dilated cardiomyopathy and generally make an incomplete recovery. Those who present with chronic disease tend to have only mild dilation of the LV and eventually present with a more restrictive cardiomyopathy.
Specific antimicrobial therapy is indicated when an infecting agent is identified. All patients should receive standard heart failure therapy and have arrhythmias suppressed. Exercise should be limited during the recovery phase. Some believe digoxin should be avoided. Immunosuppressive therapy with corticosteroids and intravenous immunoglobulins may improve the outcome when the process is acute (< 6 months) and if the biopsy suggests ongoing inflammation. However, controlled trials have not been positive, so the value of routine myocardial biopsies in patients presenting with an acute myocarditic picture is uncertain; immunosuppressive therapy without histologic confirmation is clearly unwise, and there are few data to support its use. Patients with fulminant myocarditis require aggressive short-term support including an intra-aortic balloon pump or an LV assist device. Ongoing studies are addressing whether patients with giant cell myocarditis may be responsive to immunosuppressive agents, as a special case. Overall, if improvement does not occur, many patients may be eventual candidates for cardiac transplantation.
Barbaro G: Cardiovascular manifestations of HIV infection. Circulation 2002;106:1420.
Brady WJ et al: Myocarditis: emergency department recognition and management. Emerg Med Clin North Am 2004;22: 865.
Feldman AM et al: Myocarditis. N Engl J Med 2000;343: 1388.
Liu PP et al: Advances in the understanding of myocarditis. Circulation 2001;104:1076.
Magnani JW et al: Myocarditis: current trends in diagnosis and treatment. Circulation 2006;113:876.
Veinot JP: Diagnostic endomyocardial biopsy pathology general biopsy considerations, and its use for myocarditis and cardiomyopathy: a review. Can J Cardiol 2002;18:55.
Wijetunga M et al: Myocarditis in systemic lupus erythematosus. Am J Med 2002;113:419.
2. Drug-Induced & Toxic Myocarditis
A variety of medications, illicit drugs, and toxic substances can produce acute or chronic myocardial injury; the clinical presentation varies widely. Doxorubicin and other cytotoxic agents, emetine, and catecholamines (especially with pheochromocytoma) can produce a pathologic picture of inflammation and necrosis together with clinical heart failure and arrhythmias; toxicity of the first two is dose related. The phenothiazines, lithium, chloroquine, disopyramide, antimony-containing compounds, and arsenicals can also cause ECG changes, arrhythmias, or heart failure. Hypersensitivity reactions to sulfonamides, penicillins, and aminosalicylic acid as well as other drugs can result in cardiac dysfunction. Radiation can cause an acute inflammatory reaction as well as a chronic fibrosis of heart muscle, usually in conjunction with pericarditis.
The incidence of cocaine cardiotoxicity has increased markedly. Cocaine can cause coronary artery spasm, myocardial infarction, arrhythmias, and myocarditis. Because many of these processes are believed to be mediated by cocaine's inhibitory effect on norepinephrine reuptake by sympathetic nerves, -blockers have been used therapeutically. In documented coronary spasm, calcium channel blockers and nitrates may be effective.
Floyd JD et al: Cardiotoxicity of cancer therapy. J Clin Oncol 2005;23:7685.
P.410
Gharib MI et al: Chemotherapy-induced cardiotoxicity: current practice and prospects of prophylaxis. Eur J Heart Fail 2002;4:235.
Yeh ET et al: Cardiovascular complications of cancer therapy: diagnosis, pathogenesis, and management. Circulation 2004; 109:3122.
The Cardiomyopathies
The cardiomyopathies are a heterogeneous group of entities affecting the myocardium primarily and not associated with the major causes of cardiac disease, ie, ischemic heart disease, hypertension, pericardial disease, valvular disease, or congenital defects. Although some have specific causes, many cases are idiopathic. There is now general agreement on a classification based upon features of presentation and pathophysiology (Table 10-12).
Ardehali H et al: Endomyocardial biopsy plays a role in diagnosing patients with unexplained cardiomyopathy. Am Heart J 2004;147:919.
Bowles KR et al: Genetics of inherited cardiomyopathies. Expert Rev Cardiovasc Ther 2004;2:683.
Davies MJ: The cardiomyopathies: an overview. Heart 2000; 83:469.
Franz WM et al: Cardiomyopathies: from genetics to the prospect of treatment. Lancet 2001;358:1627.
Torpy JM et al: JAMA patient page. Cardiomyopathy JAMA 2004;292:2936.
1. Primary Dilated Cardiomyopathy
![]() Essentials of Diagnosis
Essentials of Diagnosis
Symptoms and signs of heart failure.
Examination often reveals cardiomegaly, elevated JVP, S3, S4, mitral and tricuspid regurgitation, low systemic BP and pulse width, occasionally pulsus alternans.
ECG may show low QRS voltage, nonspecific repolarization abnormalities, intraventricular conduction abnormalities.
Radiograph shows cardiomegaly.
Echocardiogram confirms LV dilation, thinning, and global dysfunction.
General Considerations
Dilated cardiomyopathies cause about 25% of all cases of CHF. It usually presents with symptoms and signs of CHF (most commonly dyspnea). Occasionally, symptomatic ventricular arrhythmias are the presenting event. LV dilation and systolic dysfunction (EF < 50%) are essential for diagnosis. Dilated cardiomyopathy occurs more often in blacks than whites and in
P.411
men more than women. A growing number of cardiomyopathies due to genetic abnormalities are being recognized, and these may represent up to 25 30% of cases. Often no cause can be identified, but chronic alcohol abuse and unrecognized myocarditis are probably frequent causes. Chronic tachycardia may also precipitate a dilated cardiomyopathy. Amyloidosis, sarcoidosis, hemochromatosis, and diabetes may rarely present as dilated cardiomyopathies, as well as the more classic restrictive picture. The RV may be primarily involved in arrhythmogenic RV dysplasia, an unusual cardiomyopathy with displacement of myocardial cells by adipose tissue, or in Uhl's disease, in which there is extreme thinning of the RV walls. Intraventricular thrombus is not uncommon. Histologically, the picture is one of extensive fibrosis unless a specific diagnosis is established. Myocardial biopsy is rarely useful in establishing the diagnosis, though occasionally the underlying cause (eg, sarcoidosis, hemochromatosis) can be discerned.
Table 10-12. Classification of the cardiomyopathies. | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Clinical Findings
A. Symptoms and Signs
In most patients, symptoms of heart failure develop gradually. Cardiomyopathy may be recognized because of asymptomatic cardiomegaly or ECG abnormalities, including arrhythmias. The initial presentation may be severe biventricular failure. The physical examination reveals rales, an elevated JVP, cardiomegaly, S3 gallop rhythm, often the murmurs of functional mitral or tricuspid regurgitation, peripheral edema, or ascites. In severe CHF, Cheyne-Stokes breathing, pulsus alternans, pallor, and cyanosis may be present.
B. ECG and Chest Radiography
The major findings are listed in Table 10-12. Sinus tachycardia is common. Other common abnormalities include left bundle branch block and ventricular or atrial arrhythmias. The chest radiograph reveals cardiomegaly, evidence for left and/or right heart failure, and pleural effusions (right > left).
C. Diagnostic Studies
An echocardiogram is indicated to exclude unsuspected valvular or other lesions and confirm the presence of dilated cardiomyopathy and reduced systolic function. Mitral Doppler inflow patterns also help in the diagnosis of diastolic dysfunction. Colorflow Doppler can reveal tricuspid or mitral regurgitation, and continuous Doppler can help define PA pressures. Exercise or pharmacologic stress myocardial perfusion imaging may suggest the possibility of underlying coronary disease. Radionuclide ventriculography provides a noninvasive measure of the EF and both RV and LV wall motion. Cardiac MRI is particularly helpful in infiltrative processes, such as sarcoidosis or hemochromatosis, and is probably the diagnostic study of choice for RV dysplasia. MRI can also help exclude an ischemic etiology by noting gadolinium enhancement consistent with myocardial scar. Cardiac catheterization is seldom of specific value unless myocardial ischemia or LV aneurysm is suspected. The serum ferritin is an adequate screening study for hemochromatosis. The erythrocyte sedimentation rate may be low due to liver congestion. The serum level of BNP or pro-BNP can be used to help quantitate the severity of CHF.
Treatment
Standard therapy for heart failure should include ACE inhibitor, -blockers, diuretics, and an aldosterone antagonist. Digoxin is a second-line drug but remains favored as an adjunct by some clinicians. Calcium channel blockers should generally be avoided. Sodium restriction is helpful, especially in acute cardiomyopathy. When atrial fibrillation is present, heart rate control is important if sinus rhythm cannot be established or maintained. Many patients may now be candidates for cardiac synchronization therapy with biventricular pacing and an implantable defibrillator. Few cases of cardiomyopathy are amenable to specific therapy for the underlying cause. Alcohol use should be discontinued. There is often marked recovery of cardiac function following a period of abstinence in alcoholic cardiomyopathy. Endocrine causes (thyroid dysfunction, acromegaly, and pheochromocytoma) should be treated. Immunosuppressive therapy is not indicated in chronic dilated cardiomyopathy. The management of CHF is outlined in the section on heart failure.
Prognosis
The prognosis of dilated cardiomyopathy without clinical heart failure is variable, with some patients remaining stable, some deteriorating gradually, and others declining rapidly. Once heart failure is manifest, the natural history is similar to that of other causes of heart failure, with an annual mortality around 11 13%. Arterial and pulmonary emboli are more common in dilated cardiomyopathy than in ischemic cardiomyopathy. Suitable candidates may benefit from long-term anticoagulation, and all patients with atrial fibrillation should be so treated. Some patients may be candidates for cardiac transplantation.
Burkett EL et al: Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 2005;45:969.
Corrado D et al: Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart 2000;83:588.
Felker GM et al: Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med 2000;342:1077.
Maisch B et al: Dilated cardiomyopathies as a cause of congestive heart failure. Herz 2002;27:113.
Mohan SB et al: Idiopathic dilated cardiomyopathy: a common but mystifying cause of heart failure. Cleve Clin J Med 2002;69:481.
P.412
Piano MR: Alcoholic cardiomyopathy: incidence, clinical characteristics, and pathophysiology. Chest 2002;121:1638.
Sen-Chowdhry S et al: Arrhythmogenic right ventricular cardiomyopathy: clinical presentation, diagnosis, and management. Am J Med 2004;117:685.
Wu LA et al: Current role of endomyocardial biopsy in the management of dilated cardiomyopathy and myocarditis. Mayo Clin Proc 2001;76:1030.
2. Hypertrophic Cardiomyopathy
![]() Essentials of Diagnosis
Essentials of Diagnosis
May present with dyspnea, chest pain, syncope.
Though outflow gradient is classic, symptoms are primarily related to diastolic dysfunction.
Examination shows sustained apical impulse, S4, systolic ejection murmur that increases with Valsalva.
ECG shows LVH, occasionally septal Q waves in the absence of infarction.
Echocardiogram shows hypertrophy, which is usually asymmetric, and enhanced contractility. Systolic anterior motion of the anterior mitral valve is present if there is outflow tract obstruction.
Echocardiography/Doppler confirms outflow tract gradient.
General Considerations
Myocardial hypertrophy unrelated to any pressure or volume overload reduces LV systolic stress, increases the EF, and can result in an empty ventricle at end-systole. The interventricular septum may be disproportionately involved (asymmetric septal hypertrophy), but in some cases the hypertrophy is localized to the apex (the apical form of hypertrophic cardiomyopathy). The LV outflow tract is often narrowed during systole between the bulging septum and an anteriorly displaced anterior mitral valve leaflet, causing a dynamic obstruction (hence the name idiopathic hypertrophic subaortic stenosis; IHSS). The obstruction is worsened by factors that increase myocardial contractility (sympathetic stimulation, digoxin, postextrasystolic beat) or that decrease LV filling (Valsalva's maneuver, peripheral vasodilators). The amount of obstruction is variable even from day to day. The consequence of the hypertrophy is elevated diastolic pressures rather than systolic dysfunction. The LV is usually more involved than the RV and the atria are frequently significantly enlarged. When the septum is primarily involved, the term asymmetric septal hypertrophy has been applied. Hypertrophic cardiomyopathy in some cases is inherited as an autosomal dominant trait with variable penetrance; the inherited form is caused by mutations of a number of genes, most of which code for myosin heavy chains or proteins regulating calcium handling. The prognosis is related to the specific gene mutation. These patients usually present in early adulthood. Elite athletes may demonstrate considerable hypertrophy that can be confused with hypertrophic cardiomyopathy, but generally diastolic dysfunction is not present. The apical variety is particularly common in those of Asian descent. A hypertrophic cardiomyopathy in the elderly (usually in association with hypertension) has also been defined as a distinct entity. Mitral annular calcification is often present.
Clinical Findings
A. Symptoms and Signs
The most frequent symptoms are dyspnea and chest pain. Syncope is also common and is typically postexertional, when diastolic filling diminishes and outflow obstruction increases. Arrhythmias are an important problem. Atrial fibrillation is a long-term consequence of chronically elevated LA pressures and is a poor prognostic sign. Ventricular arrhythmias are also common, and sudden death may occur, often in athletes after extraordinary exertion.
Features on physical examination are a bisferiens carotid pulse, triple apical impulse (due to the prominent atrial filling wave and early and late systolic impulses), and a loud S4. The JVP may reveal a prominent a wave due to reduced RV compliance. In cases with outflow obstruction, a loud systolic murmur is present along the left sternal border that increases with upright posture or Valsalva's maneuver and decreases with squatting. These maneuvers help differentiate the murmur of hypertrophic cardiomyopathy from that of aortic stenosis. Mitral regurgitation is frequently present as well.
B. ECG and Chest Radiography
LVH is nearly universal in symptomatic patients, though entirely normal ECGs are present in up to 25%, usually in those with localized hypertrophy. Exaggerated septal Q waves inferolaterally may suggest myocardial infarction. The chest radiograph is often unimpressive. Unlike aortic stenosis, the ascending aorta is not dilated.
C. Diagnostic Studies
The echocardiogram is diagnostic, revealing asymmetric LVH, systolic anterior motion of the mitral valve, early closing followed by reopening of the aortic valve, a small and hypercontractile LV, and delayed relaxation and filling of the LV during diastole. The septum is usually 1.3 1.5 times the thickness of the posterior wall. Septal motion tends to be reduced. Doppler ultrasound reveals turbulent flow and a dynamic gradient in the LV outflow tract and, commonly, mitral regurgitation. Abnormalities in the diastolic
P.413
filling pattern are present in 80% of patients. Myocardial perfusion imaging may suggest septal ischemia in the presence of normal coronary arteries. Cardiac MRI confirms the hypertrophy and contrast enhancement frequently reveals evidence for scar at the junction of the RV attachment to the septum. Cardiac catheterization confirms the diagnosis and assesses the presence of CAD. Frequently, coronary arterial bridging (squeezing in systole) occurs, especially of the septal arteries. Alcohol in small doses can be injected into these septal perforators to create a septal infarction that reduces the outflow tract gradient.
Treatment
-Blockers should be the initial drug in symptomatic individuals, especially when dynamic outflow obstruction is noted on the echocardiogram. The resulting slower heart rates assist with diastolic filling of the stiff LV. Dyspnea, angina, and arrhythmias respond in about 50% of patients. Calcium channel blockers, especially verapamil, have also been effective in symptomatic patients. Their effect is due primarily to improved diastolic function, but their vasodilating actions can also increase outflow obstruction. Disopyramide (Norpace) is also used because of its negative inotropic effects; it is usually used in addition to the other therapies. Diuretics are frequently necessary due to the high diastolic pressure and PCWP. Patients do best in sinus rhythm, and atrial fibrillation should be aggressively treated with antiarrhythmics. Dual-chamber pacing may prevent the progression of hypertrophy and obstruction. Nonsurgical septal ablation has been performed by injection of alcohol into septal branches of the left coronary artery with good results in small series of patients. Patients with malignant ventricular arrhythmias and unexplained syncope in the presence of a positive family history for sudden death are probably best managed with an implantable defibrillator. Excision of part of the outflow myocardial septum (myotomy-myomectomy) by surgeons experienced with the procedure has been successful in patients with severe symptoms. Some experts advocate mitral valve replacement, as this results in resolution of the gradient as well, and prevents associated mitral regurgitation.
Prognosis
The natural history of hypertrophic cardiomyopathy is highly variable. Several specific mutations are associated with a higher incidence of early malignant arrhythmias and sudden death, and definition of the genetic abnormality provides the best estimate of prognosis. Some patients remain asymptomatic for many years or for life. Sudden death, especially during exercise, may be the initial event. Indeed, hypertrophic cardiomyopathy is the pathologic feature most frequently associated with sudden death in athletes. Those athletes with marked hypertrophy are at greatest risk. Other patients have a history of gradually progressive symptoms. Pregnancy is generally well tolerated. Endocarditis prophylaxis is indicated. A final stage may be a transition into dilated cardiomyopathy in 5 10% of patients.
Abbas AE et al: Alcohol septal ablation for hypertrophic obstructive cardiomyopathy. J Interv Cardiol 2005;18:155.
Elliott P et al: Hypertrophic cardiomyopathy. Lancet 2004;363: 1881.
Ly HQ et al: Sudden death and hypertrophic cardiomyopathy: a review. Can J Cardiol 2005;21:441.
Maron BJ: Hypertrophic cardiomyopathy: a systematic review. JAMA 2002;287:1308.
Maron BJ: Risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Cardiol Rev 2002;10:173.
Maron MS et al: Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med 2003;348:295.
Qin JX et al: Outcome of patients with hypertrophic obstructive cardiomyopathy after percutaneous transluminal septal myocardial ablation and septal myectomy surgery. J Am Coll Cardiol 2001;38:1994.
3. Restrictive Cardiomyopathy
![]() Essentials of Diagnosis
Essentials of Diagnosis
Right heart failure tends to dominate over left heart failure.
Pulmonary hypertension present.
Amyloidosis is the most common cause.
Echocardiography is key to diagnosis. Rapid early filling is present with diastolic dysfunction. Normal or near normal EF.
MRI and cardiac catheterization are helpful. Myocardial biopsy can confirm.
Restrictive cardiomyopathy is characterized by impaired diastolic filling with preserved contractile function. The diastolic filling pattern in restrictive cardiomyopathy differs from other diseases with diastolic dysfunction in that the early diastolic filling is accentuated rather than inhibited. Once the rapid early filling occurs, then filling is restricted and the pressure waveform reflects this by rising rapidly, demonstrating a square root sign. The LV systolic function may be mildly depressed and the atria are enlarged; if present, hypertrophy of the interatrial septum is a helpful additional finding diagnostically. The condition is relatively uncommon, with the most frequent causes being amyloidosis. In Africa, endomyocardial fibrosis, a specific entity in which there is severe fibrosis of the endocardium, often with eosinophilia (L ffler's syndrome), is common. Other causes of restrictive cardiomyopathy are infiltrative cardiomyopathies (eg, hemochromatosis, carcinoid syndrome) and connective tissue diseases (eg, scleroderma).
P.414
Amyloidosis results from deposition of various proteins within the myocardium. Primary amyloidosis is caused by deposition of immunoglobulin light chains (AL) by monoclonal plasma cells, often as a consequence of multiple myeloma. The heart may be the only organ involved at times. Systemic amyloidosis is due to production of AA light chains. Familial amyloidosis results from production of a carrier protein called transthyretin. Systemic amyloidosis may affect any organ but is particularly associated with significant pulmonary and renal involvement and with a peripheral neuropathy (that often results in orthostatic hypotension).
Conduction disturbances are frequently present. Low voltages on the ECG combined with ventricular hypertrophy revealed by echocardiography are suggestive. Cardiac MRI presents a distinctive pattern in amyloidosis and is a useful screening test. The echocardiogram reveals a small thickened LV with bright myocardium, rapid early diastolic filling revealed by Doppler, and biatrial enlargement. Atrial septal thickening may also be evident. Rectal, abdominal fat, or gingival biopsies can confirm systemic involvement, but myocardial involvement may still be present if these are negative. Myocardial amyloidosis can be confirmed by myocardial biopsy.
Restrictive cardiomyopathy must be distinguished from constrictive pericarditis. The clinical differentiation is described below, but the key feature is that ventricular interaction is accentuated with respiration in constrictive pericarditis, and that interaction is absent in restrictive cardiomyopathy. Pulmonary pressure is invariably elevated in restrictive cardiomyopathy and is normal in uncomplicated constrictive pericarditis.
Unfortunately, little useful therapy is available for either the causative conditions or the restrictive cardiomyopathy itself. Diuretics can help, but excessive diuresis can produce worsening symptoms. As with most patients with severe right heart failure, loop diuretics, thiazides, and aldosterone antagonists are all useful. Digoxin may precipitate arrhythmias and generally should not be used. -Blockers help slow heart rates and improve filling. Corticosteroids may be helpful in sarcoidosis but relieve conduction abnormalities more often than heart failure. Chemotherapy for amyloidosis with alkylating agents or with interferon is sometimes used, but unless the underlying diagnosis is multiple myeloma, there is little cardiac improvement. Bone marrow stem cell replacement for amyloidosis is under investigation and some of the best results to date have been attributed to this process. Cardiac transplantation is an option in patients with primary cardiac amyloidosis. Liver transplantation has been used as well in the familial form in an attempt to prevent the production of transthyretin.
Ammash NM et al: Clinical profile and outcome of idiopathic restrictive cardiomyopathy. Circulation 2000;101:2490.
Asher CR et al: Diastolic heart failure: restrictive cardiomyopathy, constrictive pericarditis, and cardiac tamponade: clinical and echocardiographic evaluation. Cardiol Rev 2002;10:218.
Gertz MA et al: Primary systemic amyloidosis. Curr Treat Options Oncol 2002;3:261.
Goldstein JA: Cardiac tamponade, constrictive pericarditis, and restrictive cardiomyopathy. Curr Probl Cardiol 2004;29:503.
Hancock EW: Differential diagnosis of restrictive cardiomyopathy and constrictive pericarditis. Heart 2001;86:343.
Yazdani K et al: Differentiating constrictive pericarditis from restrictive cardiomyopathy. Rev Cardiovasc Med 2005;6:61.
Acute Rheumatic Fever & Rheumatic Heart Disease
![]() Essentials of Diagnosis
Essentials of Diagnosis
Uncommon in the United States (approximately 2 cases/100,000 population); more common (100 cases/100,000 population) in developing countries.
Peak incidence ages 5 15 years.
Diagnosis based on Jones criteria and confirmation of streptococcal infection.
May involve mitral and other valves acutely, rarely leading to heart failure.
General Considerations
Rheumatic fever is a systemic immune process that is a sequela to -hemolytic streptococcal infection of the pharynx. Pyodermic infections are not associated with rheumatic fever. Signs of rheumatic fever usually commence 2 3 weeks after infection but may appear as early as 1 week or as late as 5 weeks. In recent years, the disease has become quite uncommon in the United States, except in immigrants. However, there have been reports of new outbreaks in several regions of the United States. The peak incidence is between ages 5 and 15 years; rheumatic fever is rare before age 4 years or after age 40 years. Rheumatic carditis and valvulitis may be self-limited or may lead to slowly progressive valvular deformity. The characteristic lesion is a perivascular granulomatous reaction with vasculitis. The mitral valve is attacked in 75 80% of cases, the aortic valve in 30% (but rarely as the sole valve), and the tricuspid and pulmonary valves in under 5% of cases.
Clinical Findings
The diagnostic criteria first described by Jones were updated in 1992. The presence of two major criteria or
P.415
one major and two minor criteria establishes the diagnosis.
A. Major Criteria
1. Carditis
Carditis is most likely to be evident in children and adolescents. Any of the following suggests the presence of carditis: (1) pericarditis; (2) cardiomegaly, detected by physical signs, radiography, or echocardiography; (3) CHF, right- or left-sided the former perhaps more prominent in children, with painful liver engorgement due to tricuspid regurgitation; and (4) mitral or aortic regurgitation murmurs, indicative of dilation of a valve ring with or without associated valvulitis. The Carey-Coombs short mid-diastolic mitral murmur may be present.
In the absence of any of the above definitive signs, the diagnosis of carditis depends on the following less specific abnormalities: (1) ECG changes, including changing contour of P waves or inversion of T waves; (2) changing quality of heart sounds; and (3) sinus tachycardia, arrhythmia, or ectopic beats.
2. Erythema marginatum and subcutaneous nodules
Erythema marginatum begins as rapidly enlarging macules that assume the shape of rings or crescents with clear centers. They may be raised, confluent, and either transient or persistent.
Subcutaneous nodules are uncommon except in children. They are small ( 2 cm in diameter), firm, and nontender and are attached to fascia or tendon sheaths over bony prominences. They persist for days or weeks, are recurrent, and are indistinguishable from rheumatoid nodules.
3. Sydenham's chorea
Sydenham's chorea involuntary choreoathetoid movements primarily of the face, tongue, and upper extremities may be the sole manifestation; only 50% of cases have other overt signs of rheumatic fever. Girls are more frequently affected, and occurrence in adults is rare. This is the least common (3% of cases) but most diagnostic of the manifestations of rheumatic fever.
4. Polyarthritis
This is a migratory polyarthritis that involves the large joints sequentially. In adults, only a single joint may be affected. The arthritis lasts 1 5 weeks and subsides without residual deformity. Prompt response of arthritis to therapeutic doses of salicylates or nonsteroidal agents is characteristic.
B. Minor Criteria
These include fever, polyarthralgias, reversible prolongation of the PR interval, and an elevated erythrocyte sedimentation rate or CRP. Supporting evidence includes positive throat culture or rapid streptococcal antigen test and elevated or rising streptococcal antibody titer.
C. Laboratory Findings
There is nonspecific evidence of inflammatory disease, as shown by a rapid sedimentation rate. High or increasing titers of antistreptococcal antibodies (antistreptolysin O and anti-DNase B) are used to confirm recent infection; 10% of cases lack this serologic evidence.
Differential Diagnosis
Rheumatic fever may be confused with the following: rheumatoid arthritis, osteomyelitis, endocarditis, chronic meningococcemia, systemic lupus erythematosus, Lyme disease, sickle cell anemia, surgical abdomen, and many other diseases.
Complications
CHF occurs in severe cases. In the longer term, the development of rheumatic heart disease is the major problem. Other complications include arrhythmias, pericarditis with effusion, and rheumatic pneumonitis.
Treatment
A. General Measures
The patient should be kept at strict bed rest until the temperature returns to normal (without the use of antipyretic medications) and the sedimentation rate, plus the resting pulse rate, and the ECG have all returned to baseline.
B. Medical Measures
1. Salicylates
The salicylates markedly reduce fever and relieve joint pain and swelling. They have no effect on the natural course of the disease. Adults may require large doses of aspirin, 0.6 0.9 g every 4 hours; children are treated with lower doses. Toxicity includes tinnitus, vomiting, and gastrointestinal bleeding.
2. Penicillin
Penicillin (benzathine penicillin, 1.2 million units intramuscularly once, or procaine penicillin, 600,000 units intramuscularly daily for 10 days) is used to eradicate streptococcal infection if present. Erythromycin may be substituted (40 mg/kg/d).
3. Corticosteroids
There is no proof that cardiac damage is prevented or minimized by corticosteroids. A short course of corticosteroids (prednisone, 40 60 mg orally daily, with tapering over 2 weeks) usually causes rapid improvement of the joint symptoms and is indicated when response to salicylates has been inadequate.
Prevention of Recurrent Rheumatic Fever
The initial episode of rheumatic fever can usually be prevented by early treatment of streptococcal pharyngitis. (See Chapter 33.) Prevention of recurrent episodes is critical. Recurrences of rheumatic fever are most common in patients who have had carditis during their initial episode and in children, 20% of whom will have a second episode within 5 years. Recurrences are uncommon after 5 years following the first episode, and in patients over 25 years of age. Prophylaxis is usually discontinued after these times except in
P.416
groups with a high risk of streptococcal infection parents or teachers of young children, nurses, military recruits, etc. Secondary prevention of rheumatic fever depends on whether carditis has occurred. If there is no evidence for carditis, preventive therapy can be stopped at age 21 years. If carditis has occurred but there is no residual valvular disease, it can be stopped at 10 years after the episode. If carditis has occurred with residual valvular involvement, it should be continued for 10 years after the last episode or until age 40 years if the patient is in a situation in which reexposure would be expected.
A. Penicillin
The preferred method of prophylaxis is with benzathine penicillin G, 1.2 million units intramuscularly every 4 weeks. Oral penicillin (200,000 250,000 units twice daily) is less reliable.
B. Alternatives for Penicillin-Allergic Patients
If the patient is allergic to penicillin, sulfadiazine (or sulfisoxazole), 1 g daily, or erythromycin, 250 mg orally twice daily, may be substituted. The macrolide azithromycin is similarly effective against group A streptococcal infection. If the patient has not had an immediate hypersensitivity (anaphylactic-type) reaction to penicillin, then cephalosporin may also be used.
Prognosis
Initial episodes of rheumatic fever may last months in children and weeks in adults. The immediate mortality rate is 1 2%. Persistent rheumatic carditis with cardiomegaly, heart failure, and pericarditis implies a poor prognosis; 30% of children thus affected die within 10 years after the initial attack. After 10 years, two-thirds of patients will have detectable valvular abnormalities (usually thickened valves with limited mobility), but significant symptomatic valvular heart disease or persistent cardiomyopathy occurs in less than 10% of patients with a single episode. In developing countries, acute rheumatic fever occurs earlier in life, recurs more frequently, and the evolution to chronic valvular disease is both accelerated and more severe.
Rheumatic Heart Disease
Chronic rheumatic heart disease results from single or repeated attacks of rheumatic fever that produce rigidity and deformity of valve cusps, fusion of the commissures, or shortening and fusion of the chordae tendineae. Stenosis or insufficiency results, and the two often coexist. The mitral valve alone is affected in 50 60% of cases; combined lesions of the aortic and mitral valves occur in 20%; pure aortic lesions are less common. Tricuspid involvement occurs in about 10% of cases but only in association with mitral or aortic disease and is thought to be more common when recurrent infections have occurred. The pulmonary valve is rarely affected. A history of rheumatic fever is obtainable in only 60% of patients with rheumatic heart disease.
Recurrences of acute rheumatic fever can be prevented (see above). The patient should also receive prophylactic antibiotics preceding dental extraction, urologic and surgical procedures, etc, to prevent endocarditis (Table 33-4). With mitral valve disease, it is important to identify the onset of atrial fibrillation to institute anticoagulation. The important findings in each of the major valve lesions are summarized in Table 10-1.
Carapetis JR et al: Acute rheumatic fever. Lancet 2005;366:155.
Cilliers AM et al: Anti-inflammatory treatment for carditis in acute rheumatic fever. Cochrane Database Syst Rev 2003; (2):CD003176.
Guilherme L et al: Rheumatic fever: from sore throat to autoimmune heart lesions. Int Arch Allergy Immunol 2004;134: 56.
McDonald M et al: Acute rheumatic fever: a chink in the chain that links the heart to the throat? Lancet Infect Dis 2004;4:240.
Narula J et al: Diagnosis of active rheumatic carditis. Circulation 1999;100:1576.
Rullan E et al: Rheumatic fever. Curr Rheumatol Rep 2001; 3:445.
Stollerman GH: Rheumatic fever in the 21st century. Clin Infect Dis 2001;33:806.
Diseases of the Pericardium
Acute Pericarditis
Anatomic & Physiologic Considerations
The pericardium consists of two layers: the inner visceral layer, which is attached to the epicardium, and an outer parietal layer. About 50 mL of serous fluid is normally present and provides lubrication between the two layers. The pericardial reflection encompasses the heart and great vessels. The pericardium stabilizes the heart in anatomic position and reduces contact between the heart and the surrounding structures. It is composed of fibrous tissue, and although it will permit moderate changes in cardiac size, it cannot stretch rapidly enough to accommodate rapid dilation of the heart or accumulation of fluid without increasing intrapericardial (and, therefore, intracardiac) pressure.
The pericardium is often involved by processes that affect the heart, but it may also be affected by diseases of adjacent tissues and may itself be a primary site of disease.
Inflammatory Pericarditis
Acute (< 2 weeks) inflammation of the pericardium may be infectious in origin or may be due to systemic diseases
P.417
(autoimmune syndromes, uremia), neoplasm, radiation, drug toxicity, hemopericardium, postcardiac surgery, or contiguous inflammatory processes in the myocardium or lung. In many of these conditions, the pathologic process involves both the pericardium and the myocardium.
The presentation and course of inflammatory pericarditis depend on its cause, but all syndromes are often (not always) associated with chest pain, which is usually pleuritic and postural (relieved by sitting). The pain is substernal but may radiate to the neck, shoulders, back, or epigastrium. Dyspnea may also be present. A pericardial friction rub is characteristic, with or without evidence of fluid accumulation or constriction (see below). Fever and leukocytosis are often present. The ECG usually shows generalized ST and T wave changes and may manifest a characteristic progression beginning with diffuse ST elevation, followed by a return to baseline and then to T wave inversion. Atrial injury is often present and manifested by PR depression especially in the limb leads. The chest radiograph is frequently normal, but may show cardiac enlargement if fluid has collected, as well as signs of related pulmonary disease. Mass lesions and enlarged lymph nodes may suggest a neoplastic process. The echocardiogram is often normal or reveals only a trivial amount of fluid during the acute inflammatory process.
Viral Pericarditis
Viral infections (especially infections with coxsackieviruses and echoviruses but also influenza, Epstein-Barr, varicella, hepatitis, mumps, and HIV viruses) are the most common cause of acute pericarditis and probably are responsible for many cases classified as idiopathic. Males usually under age 50 years are most commonly affected. Pericardial involvement often follows upper respiratory infection. The diagnosis is usually clinical, but rising viral titers in paired sera may be obtained for confirmation. Cardiac enzymes may be slightly elevated, reflecting a myocarditic component. The differential diagnosis is primarily with myocardial infarction.
Treatment is generally symptomatic. Aspirin (650 mg every 3 4 hours) or other nonsteroidal agents (eg, indomethacin, 100 150 mg daily in divided doses) are usually effective. Corticosteroids may be beneficial in unresponsive cases. In general, symptoms subside in several days to weeks. The major early complication is tamponade, which occurs in less than 5% of patients. There may be recurrences in the first few weeks or months. Rare patients will continue to experience recurrences chronically. These patients may require long-term anti-inflammatory medications, either corticosteroids or colchicine. Rarely, acute pericarditis may lead to constrictive pericarditis, when pericardial resection may be necessary (see below).
Tuberculous Pericarditis
Tuberculous pericarditis has become rare in developed countries but remains common in other areas. It results from direct lymphatic or hematogenous spread; clinical pulmonary involvement may be absent or minor, although associated pleural effusions are common. The presentation tends to be subacute, but nonspecific symptoms (fever, night sweats, fatigue) may be present for days to months. One to 8 percent of patients with pulmonary tuberculosis develop pericardial involvement. Pericardial effusions are usually small or moderate but may be large. The diagnosis can be inferred if acid-fast bacilli are found elsewhere. The yield of organisms by pericardiocentesis is low; pericardial biopsy has a higher yield but may also be negative, and pericardiectomy may be required. Standard antituberculous drug therapy is usually successful (see Chapter 9), but constrictive pericarditis can occur.
Other Infectious Pericarditides
Bacterial pericarditis has become rare and usually results from direct extension from pulmonary infections. Pneumococci can cause a primary pericardial infection. Symptoms and signs of bacterial pericarditis are similar to those of other types of inflammatory pericarditides, but patients appear toxic and are often critically ill. Borrelia burgdorferi, the organism responsible for Lyme disease, can also cause myopericarditis. If bacterial pericarditis is suspected on clinical grounds, diagnostic pericardiocentesis may be of value.
Uremic Pericarditis
Uremic pericarditis is a common complication of renal failure. The pathogenesis is uncertain; it occurs both with untreated uremia and in otherwise stable dialysis patients. In uremic patients not on dialysis, the incidence correlates roughly with the level of blood urea nitrogen (BUN) and creatinine. The pericardium is characteristically shaggy, and the effusion is hemorrhagic and exudative. Uremic pericarditis can present with or without symptoms; fever is absent. The pericarditis usually resolves with the institution of or with more aggressive dialysis. Tamponade is fairly common, and partial pericardiectomy (pericardial window) may be necessary. Whereas anti-inflammatory agents may relieve the pain and fever associated with uremic pericarditis, indomethacin and systemic corticosteroids do not affect its natural history. Most uremic patients with pericarditis respond to intensive dialysis, although patients who have progressed to tamponade do so less well and usually require drainage.
Neoplastic Pericarditis
Spread of adjacent lung cancer as well as invasion by breast cancer, renal cell carcinoma, Hodgkin's disease, and lymphomas are the most common neoplastic processes involving the pericardium and have become the most frequent causes of pericardial tamponade in many countries. Often the process is painless, and the presenting symptoms relate to hemodynamic compromise
P.418
or the primary disease. Pericardial effusions develop over a long period of time and may become quite huge (> 2 L). The diagnosis can occasionally be made by cytologic examination of the effusion or by pericardial biopsy, but it may be difficult to establish clinically if the patient has received mediastinal radiation within the previous year. MRI and CT scan can visualize neighboring tumor when present. The prognosis with neoplastic effusion is dismal, with only a small minority surviving 1 year. If it is compromising the clinical comfort of the patient, the effusion is initially drained percutaneously. Early attempts at ballooning the pericardium from a subxiphoid approach have been mostly abandoned in favor of surgical approaches. A pericardial window, either by a subxiphoid approach or via video-assisted thoracic surgery, allows for partial pericardiectomy. Instillation of chemotherapeutic agents or tetracycline may occasionally be used to reduce the recurrence rate.
Postmyocardial Infarction or Postcardiotomy Pericarditis (Dressler's Syndrome)
Pericarditis may occur 2 5 days after infarction due to an inflammatory reaction to transmural myocardial necrosis. It usually presents as a recurrence of pain with pleural-pericardial features. A rub is often audible, and repolarization changes may be confused with ischemia. Large effusions are uncommon, and spontaneous resolution usually occurs in a few days. Aspirin or nonsteroidal agents in the dosages given in the section on viral pericarditis provide symptomatic relief.
Dressler's syndrome occurs weeks to several months after myocardial infarction or open heart surgery, may be recurrent, and probably represents an autoimmune syndrome. Patients present with typical pain, fever, malaise, and leukocytosis. Occasionally, the syndrome will occur within days of surgery. The sedimentation rate is high. Large pericardial effusions and accompanying pleural effusions are frequent. Rarely, other symptoms of an autoimmune disorder, such as joint pain and fever, may occur. Tamponade is rare with Dressler's syndrome after myocardial infarction but not when it occurs postoperatively. Nonsteroidal anti-inflammatory agents given for 2 4 weeks are usually effective. In more severe cases, corticosteroids should be given in rapidly tapering doses. Relapses do occur and may require slow withdrawal of anti-inflammatory therapy over several months. Colchicine may be required for months to help prevent recurrences.
Radiation Pericarditis
Radiation can initiate a fibrinous and fibrotic process in the pericardium, presenting as subacute pericarditis or constriction. The clinical onset is usually within the first year but may be delayed for many years. Radiation pericarditis usually follows treatments of more than 4000 cGy delivered to ports including more than 30% of the heart. Symptomatic therapy is the initial approach, but recurrent effusions and constriction often require surgery.
Other Causes of Pericarditis
These include connective tissue diseases, such as lupus erythematosus and rheumatoid arthritis, drug-induced pericarditis (minoxidil, penicillins), and myxedema. Myxedema pericardial effusions usually are characterized by the presence of cholesterol crystals.
Imazio M et al: Management, risk factors, and outcomes in recurrent pericarditis. Am J Cardiol 2005;96:736.
Lee PJ et al: Cardiovascular effects of radiation therapy: practical approach to radiation therapy-induced heart disease. Cardiol Rev 2005;13:80.
Ross AM et al: Acute pericarditis. Evaluation and treatment of infectious and other causes. Postgrad Med 2004;115: 67.
Talreja DR et al: Constrictive pericarditis in 26 patients with histologically normal pericardial thickness. Circulation 2003; 108:1852.
Troughton RW et al: Pericarditis. Lancet 2004;363:717.
Pericardial Effusion
Pericardial effusion can develop during any of the processes previously discussed. The speed of accumulation determines the physiologic importance of the effusion. Because the pericardium stretches, large effusions (> 1000 mL) that develop slowly may produce no hemodynamic effects. Smaller effusions that appear rapidly can cause tamponade. Tamponade is characterized by elevated intrapericardial pressure (> 15 mm Hg), which restricts venous return and ventricular filling. As a result, the stroke volume and pulse pressure fall, and the heart rate and venous pressure rise. Shock and death may result.
Clinical Findings
A. Symptoms and Signs
Pericardial effusions may be associated with pain if they occur as part of an acute inflammatory process or may be painless, as is often the case with neoplastic or uremic effusion. Dyspnea and cough are common, especially with tamponade. Other symptoms may result from the primary disease.
A pericardial friction rub may be present even with large effusions. In cardiac tamponade, tachycardia, tachypnea, a narrow pulse pressure, and a relatively preserved systolic pressure are characteristic. Pulsus paradoxus a greater than 10 mm Hg decline in systolic pressure during inspiration due to further impairment of LV filling is the classic finding, but it may also occur with obstructive lung disease. Central venous pressure is elevated and there is no evident y descent in the RA, RV, or LV hemodynamic tracings. Edema or ascites are rarely present; these signs favor a more chronic process.
P.419
B. Laboratory Findings
Laboratory tests tend to reflect the underlying processes (see causes of pericarditis above).
C. Diagnostic Studies
Chest radiograph can suggest effusion by an enlarged cardiac silhouette with a globular configuration but may appear normal. The ECG often reveals nonspecific T wave changes and low QRS voltage. Electrical alternans is present uncommonly but is pathognomonic. It is due to the heart swinging within the large effusion. Echocardiography is the primary method for demonstrating pericardial effusion and is quite sensitive. If tamponade is present, the high intrapericardial pressure may collapse lower pressure cardiac structures, such as the RA and RV. In tamponade, the normal inspiratory reduction in LV filling is accentuated due to RV/LV interaction and there is a > 25% reduction in mitral inflow velocities. Cardiac CT and MRI also demonstrate pericardial fluid and lesions. Diagnostic pericardiocentesis or biopsy is often indicated for microbiologic and cytologic studies; a pericardial biopsy may be performed relatively simply through a small subxiphoid incision.
Treatment
Small effusions can be followed clinically by careful observations of the JVP and by testing for a paradoxical pulse. Serial echocardiograms are indicated if no intervention is immediately contemplated. When tamponade is present, urgent pericardiocentesis is required. Because the pressure-volume relationship in the pericardial fluid is curvilinear and upsloping, removal of a small amount of fluid often produces a dramatic fall in the intrapericardial pressure and immediate hemodynamic benefit; but complete drainage with a catheter is preferable. Continued or repeat drainage may be indicated, especially in malignant effusions. Pericardial windows via video-assisted thorascopy have been particularly effective in preventing recurrences.
Additional therapy is determined by the nature of the primary process. Recurrent effusion in neoplastic disease and uremia, in particular, may require partial pericardiectomy as noted earlier.
Burgess LJ et al: Role of biochemical tests in the diagnosis of large pericardial effusions. Chest 2002;121:495.
Rienmuller R et al: CT and MR imaging of pericardial disease. Radiol Clin North Am 2004;42:587.
Shabetai R: Pericardial effusion: haemodynamic spectrum. Heart 2004;90:255.
Soler-Soler J et al: Management of pericardial effusion. Heart 2001;86:235.
Tsang TS: Outcomes of primary and secondary treatment of pericardial effusion in patients with malignancy. Mayo Clin Proc 2000;75:248.
Wang ZJ et al: CT and MR imaging of pericardial disease. Radiographics 2003;23 Spec No:S167.
Constrictive Pericarditis
Inflammation can lead to a thickened, fibrotic, adherent pericardium that restricts diastolic filling and produces chronically elevated venous pressures. In the past, tuberculosis was the most common cause of constrictive pericarditis, but the process now more often occurs after radiation therapy, cardiac surgery, or viral pericarditis; histoplasmosis is another uncommon cause.
The principal symptoms are slowly progressive dyspnea, fatigue, and weakness. Chronic edema, hepatic congestion, and ascites are usually present. Ascites often seems out of proportion to the degree of peripheral edema. The examination reveals these signs and a characteristically elevated jugular venous pressure with a rapid y descent. This can be detected at bedside by careful observation of the jugular pulse and noting an apparent increased pulse wave at the end of systole (due to accentuation of the v wave by the rapid y descent). Kussmaul's sign a failure of the JVP to fall with inspiration is also a frequent finding. The apex may actually retract with systole and a pericardial knock may be heard in early diastole. Pulsus paradoxus is unusual. Atrial fibrillation is common.
The chest radiograph may show normal heart size or cardiomegaly. Pericardial calcification is best seen on the lateral view and is uncommon. It rarely involves the LV apex, and finding of calcification at the LV apex is more consistent with myocardial aneurysm. Echocardiography rarely demonstrates a thickened pericardium. A septal bounce reflecting the rapid early filling is common, though. RV/LV interaction may be demonstrated by a reduction in the mitral inflow pattern of > 25%, much as in tamponade. Cardiac CT and MRI are only occasionally helpful. Pericardial thickening of > 4 mm must be present to establish the diagnosis, yet no pericardial thickening is demonstrated in 20 25% of patients with constrictive pericarditis. At times constrictive pericarditis is extremely difficult to differentiate from restrictive cardiomyopathy. When unclear, the use of noninvasive testing and cardiac catheterization is required to sort out the difference. As a generality, the pulmonary pressure is lower in constriction and there must be signs of RV/LV interaction (reduced LV filling pattern with inspiration) on echocardiography/Doppler. In constrictive pericarditis, because of the need to demonstrate RV/LV interaction, cardiac catheterization should include simultaneous measurement of both the LV and RV. Hemodynamically, patients with constriction have equalization of end-diastolic pressures throughout their cardiac chambers, there is rapid early filling then an abrupt increase in diastolic pressure ( square-root sign), the RV end-diastolic pressure is more than one-third the systolic pressure, simultaneous measurements of RV and LV systolic pressure reveal a discordance with inspiration (the RV rises as the LV falls), and there is usually a Kussmaul's sign (failure of the RA pressure to fall with inspiration). The width of the RV pressure tracing may also be narrow in expiration
P.420
and greater during inspiration, reflecting the wide variation in filling of the RV with respiration. In restrictive cardiomyopathy, the LV diastolic pressure is usually greater than the RV diastolic pressure by 5 mm Hg, there is pulmonary hypertension, and simultaneous measurements of the RV and LV systolic pressure reveal a concordant drop in both with inspiration. Initial treatment consists of diuresis. As in other disorders of right heart failure, the diuresis should be aggressive, using loop diuretics (torsemide if bowel edema is suspected), thiazides, and aldosterone antagonists (especially if ascites is present). Surgical pericardiectomy should be done when diuretics are unable to control symptoms. Pericardiectomy removes the pericardium between the phrenic nerves only, however, and most patients still require diuretics after the procedure, though symptoms are usually dramatically improved. Morbidity and mortality after pericardiectomy are high (up to 15%) and are greatest in those with the most disability prior to the procedure. For that reason, most experts recommend earlier rather than later pericardiectomy if symptoms are present.
Asher CR et al: Diastolic heart failure: restrictive cardiomyopathy, constrictive pericarditis, and cardiac tamponade: clinical and echocardiographic evaluation. Cardiol Rev 2002;10: 218.
Bertog SC et al: Constrictive pericarditis: etiology and cause-specific survival after pericardiectomy. J Am Coll Cardiol 2004; 43:1445.
Hoit BD: Management of effusive and constrictive pericardial heart disease. Circulation 2002;105:2939.
Nishimura RA: Constrictive pericarditis in the modern era: a diagnostic dilemma. Heart 2001;86:619.
Sagrista-Sauleda J et al: Effusive-constrictive pericarditis. N Engl J Med 2004;350:469.
Wang A et al: Clinical problem-solving. Undercover and overlooked. N Engl J Med 2004;351:1014.
Pulmonary Hypertension & Pulmonary Heart Disease
Primary Pulmonary Hypertension
The normal pulmonary bed offers about one-tenth as much resistance to blood flow as the systemic arterial system. Pulmonary hypertension is defined as mild if the mean PA pressure is > 20 mm Hg, moderate if > 30 mm Hg, and severe if > 45 mm Hg. As opposed to the systemic circulation, the pulmonary resistance is influenced by local vascular mediators much more than the adrenergic nervous system, even though both - and -receptors are present on pulmonary vascular smooth muscle. The normal pulmonary endothelial cell maintains the vascular smooth muscle in a state of relaxation. Pulmonary vasoconstriction is mediated by hypoxia and by endothelin and angiotensin II. Vasodilation can be affected by certain prostaglandins, endothelin receptor blockers, smooth muscle relaxants and by nitric oxide.
Primary pulmonary hypertension is defined as pulmonary hypertension and elevated PVR in the absence of other disease of the lungs or heart. Its cause is unknown, though there are clear genetic patterns that have been identified, and it likely represents a derangement in one or more of the biologic pathways described above. Pathologically, it is characterized by diffuse narrowing of the pulmonary arterioles. Circumstantial evidence suggests that unrecognized recurrent pulmonary emboli or in situ thrombosis may play a role in some cases. However, the latter may well be an exacerbating factor (precipitated by local endothelial injury) rather than a cause of the syndrome. Primary pulmonary hypertension must be distinguished from other causes of severe secondary pulmonary hypertension, such as systemic sclerosis, HIV-related pulmonary hypertension, cirrhosis, and congenital heart defects (primarily those with a shunt distal to the level of the tricuspid valve). Rarely, pulmonary venous disease (pulmonary veno-occlusive disease) or peripheral PA stenosis may be present. Left heart disease, in particular mitral stenosis, or any reason for an elevated LA pressure must be excluded as well. Table 10-13 includes clinical disorders causing pulmonary hypertension.
The laboratory evaluation of primary pulmonary hypertension must exclude a secondary cause. A hypercoagulable state should be sought and chronic pulmonary emboli excluded (usually by lung scan or contrast CT). The chest radiograph helps exclude a primary pulmonary etiology evidence for patchy edema may raise the suspicion of pulmonary veno-occlusive disease. The ECG is generally consistent with RVH and RA enlargement. Echocardiography/Doppler demonstrates an enlarged RV and RA at times they may be huge and hypocontractile. Severe pulmonic or tricuspid regurgitation may be present. Septal flattening is consistent with pulmonary hypertension. Doppler interrogation of the tricuspid regurgitation jet helps provide an estimate of RV systolic pressure. Pulmonary function tests help exclude other disorders, though primary pulmonary hypertension may present with a reduced carbon monoxide diffusing capacity of the lung (DLCO) and severe desaturation (particularly if a PFO has been stretched open and a right-to-left shunt is present). Chest CT demonstrates enlarged pulmonary arteries and excludes other causes (such as emphysema or interstitial lung disease). Pulmonary angiography (or MR angiography or CT angiography) reveals loss of the smaller acinar pulmonary vessels and tapering of the larger ones. Catheterization allows measurement of pulmonary pressures and testing for vasoreactivity using a variety of agents, including 100% oxygen, adenosine, epoprostenol, and nitric oxide. A positive response is one that decreases the pulmonary mean pressure by > 25% (ideally below 30 mm Hg).
The clinical picture is similar to that of pulmonary hypertension from other causes. Patients characteristically young women present with evidence of right heart failure that is usually progressive, leading to
P.421
death in 2 8 years. This is a decidedly different prognosis than patients with Eisenmenger physiology due to a left-to-right shunt; 40% of Eisenmenger patients are alive 25 years after the diagnosis has been made. Patients have manifestations of low cardiac output, with weakness and fatigue, as well as edema and ascites as right heart failure advances. Peripheral cyanosis is present, and syncope on effort may occur.
Table 10-13. Causes of pulmonary hypertension. | ||
|---|---|---|
|
Until recently, there was no therapy for primary pulmonary hypertension, except lung transplantation. Currently, a variety of therapeutic options are being investigated. Some authorities advocate long-term oral anticoagulation and this is given in most patients with primary pulmonary hypertension. Supplemental oxygen, particularly at night, appears to improve symptoms and helps reduce pulmonary pressures. Diuretics help with right heart edema. Calcium channel blockers, at times in large doses, have been used with mixed results. Some patients respond well, especially if they are proven responders to vasodilators. However, calcium channel blockers can worsen RV function by their negative inotropic effects.
Other agents are now available for direct treatment of pulmonary hypertension. Epoprostenol (Flolan) is administered by continuous infusion and has been shown to improve functional capacity and survival. Analogs of epoprostenol are also available, including trepostinol (Remodulin), beraprost, and iloprost. Treprostinil is given by subcutaneous injection, beraprost by mouth, and iloprost by inhalation. Endothelin antagonists are also available orally, such as bosentan (Tracleer) and sitaxentan (Thelin). Finally, phosphodiesterase inhibitors, such as sildenafil, are also being carefully investigated. The cost of all of these agents, except for sildenafil and calcium channel blockers, is quite high.
Pulmonary transplantation is a viable option in selected centers, though the operative mortality is high (around 20 25%) and 2-year survival only about 55%.
Chatterjee K et al: Pulmonary hypertension: hemodynamic diagnosis and management. Arch Intern Med 2002;162:1925.
McLauglin VV: Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002;15:1477.
Olschewski H et al: Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002;347:322.
Rubin LJ et al: Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:806.
Rubin LJ et al: Evaluation and management of the patient with pulmonary arterial hypertension. Ann Intern Med 2005;143:282.
Sitbon O et al: Primary pulmonary hypertension: current therapy. Prog Cardiovasc Dis 2002;45:115.
Pulmonary Heart Disease (Cor Pulmonale)
![]() Essentials of Diagnosis
Essentials of Diagnosis
Symptoms and signs of chronic bronchitis and pulmonary emphysema.
Elevated jugular venous pressure, parasternal lift, edema, hepatomegaly, ascites.
ECG shows tall, peaked P waves (P pulmonale), right axis deviation, and RVH.
Chest radiograph: Enlarged RV and PA.
Echocardiogram or radionuclide angiography excludes primary LV dysfunction.
General Considerations
The term cor pulmonale denotes RV hypertrophy and eventual failure resulting from pulmonary disease and attendant hypoxia or from pulmonary vascular disease. Its clinical features depend upon both the primary underlying disease and its effects on the heart.
P.422
Cor pulmonale is most commonly caused by COPD. Less frequent causes include pneumoconiosis, pulmonary fibrosis, kyphoscoliosis, primary pulmonary hypertension, repeated episodes of subclinical or clinical pulmonary embolization, Pickwickian syndrome, schistosomiasis, and obliterative pulmonary capillary or lymphangitic infiltration from metastatic carcinoma.
Clinical Findings
A. Symptoms and Signs
The predominant symptoms of compensated cor pulmonale are related to the pulmonary disorder and include chronic productive cough, exertional dyspnea, wheezing respirations, easy fatigability, and weakness. When the pulmonary disease causes RV failure, these symptoms may be intensified. Dependent edema and right upper quadrant pain may also appear. The signs of cor pulmonale include cyanosis, clubbing, distended neck veins, RV heave or gallop (or both), prominent lower sternal or epigastric pulsations, an enlarged and tender liver, and dependent edema.
B. Laboratory Findings
Polycythemia is often present in cor pulmonale secondary to COPD. The arterial oxygen saturation is often below 85%; PCO2 may or may not be elevated.
C. ECG and Chest Radiography
The ECG may show right axis deviation and peaked P waves. Deep S waves are present in lead V6. Right axis deviation and low voltage may be noted in patients with pulmonary emphysema. Frank RVH is uncommon except in primary pulmonary hypertension. The ECG often mimics myocardial infarction; Q waves may be present in leads II, III, and aVF because of the vertically placed heart, but they are rarely deep or wide, as in inferior myocardial infarction. Supraventricular arrhythmias are frequent and nonspecific.
The chest radiograph discloses the presence or absence of parenchymal disease and a prominent or enlarged RV and PA.
D. Diagnostic Studies
Pulmonary function tests usually confirm the underlying lung disease. The echocardiogram should show normal LV size and function but RV and RA dilation. Perfusion lung scans are rarely of value, but, if negative, they help to exclude chronic pulmonary emboli, an occasional cause of cor pulmonale. Multislice CT has replaced pulmonary angiography as the most specific method of diagnosis for the pulmonary emboli.
Differential Diagnosis
In its early stages, cor pulmonale can be diagnosed on the basis of radiologic, echocardiographic, or ECG evidence. Catheterization of the right heart will establish a definitive diagnosis but is more often performed to exclude left-sided heart failure, which may in some patients be an inapparent cause of right-sided failure. Differential diagnostic considerations relate primarily to the specific pulmonary disease that has produced RV failure (see above).
Treatment
The details of the treatment of chronic pulmonary disease (chronic respiratory failure) are discussed in Chapter 9. Otherwise, therapy is directed at the pulmonary process responsible for right heart failure. Oxygen, salt and fluid restriction, and diuretics are mainstays, with combination diuretic therapy (loop diuretics, thiazides and spironolactone) often useful, as described above for other causes of right heart failure.
Prognosis
Compensated cor pulmonale has the same prognosis as the underlying pulmonary disease. Once congestive signs appear, the average life expectancy is 2 5 years, but survival is significantly longer when uncomplicated emphysema is the cause.
Lehrman S et al: Primary pulmonary hypertension and cor pulmonale. Cardiol Rev 2002;10:265.
Morrison LK et al: Utility of a rapid B-natriuretic peptide assay in differentiating congestive heart failure from lung disease in patients presenting with dyspnea. J Am Coll Cardiol 2002; 39:202.
Vizza CD et al: Right and left ventricular dysfunction in patients with severe pulmonary disease. Chest 1998;113:576.
Neoplastic Diseases of the Heart
Primary cardiac tumors are rare and constitute only a small fraction of all tumors that involve the heart or pericardium. The most common primary tumor is atrial myxoma; it comprises about 50% of all tumors in adult case series. It is generally attached to the atrial septum and is more likely to affect the LA than the RA. Familial myxomas occur as part of the Carney complex that consists of myxomas, pigmented skin lesions, and endocrine neoplasia. Patients with myxoma can present with the characteristics of a systemic illness, with obstruction of blood flow through the heart, or with signs of peripheral embolization. The characteristics include fever, malaise, weight loss, leukocytosis, elevated sedimentation rate, and emboli (peripheral or pulmonary, depending on the location of the tumor). This is often confused with infective endocarditis, lymphoma, other cancers, or autoimmune diseases. In other cases, the tumor may grow to considerable size and produce symptoms by obstructing mitral flow. Episodic pulmonary edema (classically occurring when an upright posture is assumed) and signs of low output may result.
P.423
Physical examination may reveal a diastolic sound related to motion of the tumor ( tumor plop ) or a diastolic murmur similar to that of mitral stenosis. Right-sided myxomas may cause symptoms of right-sided failure. The diagnosis is established by echocardiography or by pathologic study of embolic material. Cardiac MRI is useful as an adjunct only. Contrast angiography is frequently not necessary. Surgical excision is usually curative, though recurrences do occur and serial echocardiographic follow-up, on at least a yearly basis, is recommended.
The second most common cardiac tumors are valvular papillary fibroelastomas and atrial septal lipomas. These tend to be benign and usually require no therapy. Other primary cardiac tumors include rhabdomyomas (that often appear multiple in both the RV and LV), fibrous histiocytomas, hemangiomas, and a variety of unusual sarcomas. The diagnosis may be supported by an abnormal cardiac contour on radiograph. Echocardiography is usually helpful but may miss tumors infiltrating the ventricular wall. It is likely that MRI will prove useful as well.
Metastases from malignant tumors can also affect the heart. Most often this occurs in malignant melanoma, but other tumors involving the heart include bronchogenic carcinoma, carcinoma of the breast, the lymphomas, renal cell carcinoma, and, in patients with AIDS, Kaposi's sarcoma. These are often clinically silent but may lead to pericardial tamponade, arrhythmias and conduction disturbances, heart failure, and peripheral emboli. The diagnosis is often made by echocardiography, but cardiac MRI and CT scanning are also helpful. ECG may reveal regional Q waves. The prognosis is dismal; effective treatment is not available. On rare occasions, surgical resection or chemotherapy is warranted.
Bennett KR et al: The Carney complex: unusual skin findings and recurrent cardiac myxoma. Arch Dermatol 2005;141:916.
Butany J et al: Cardiac tumours: diagnosis and management. Lancet Oncol 2005;6:219.
Restrepo CS et al: CT and MR imaging findings of benign cardiac tumors. Curr Probl Diagn Radiol 2005;34:12.
Reynen K et al: Metastases to the heart. Ann Oncol 2004;15:375.
Roberts WC: Primary and secondary neoplasms of the heart. Am J Cardiol 1997;80:671.
Sun JP et al: Clinical and echocardiographic characteristics of papillary fibroelastomas: a retrospective and prospective study in 162 patients. Circulation 2001;103:2687.
Cardiac Involvement in Miscellaneous Systemic Diseases
The heart may be involved in a number of systemic syndromes. Many of these have been mentioned briefly in this chapter. The pericardium, myocardium, heart valves, and coronary arteries may be involved either singly or in various combinations. In most cases the cardiac manifestations are not the dominant feature, but in some it is the primary cause of symptoms and may be fatal.
The most common type of myocardial involvement is an infiltrative cardiomyopathy, such as systemic amyloidosis, sarcoidosis, hemochromatosis, Fabry's or glycogen storage disease. These result in a restrictive cardiomyopathy (see above). Cardiac calcinosis can occur in hyperparathyroidism (usually the secondary form) and in primary oxalosis. A number of muscular dystrophies can cause a cardiomyopathic picture (particularly Duchenne's, less frequently myotonic dystrophy, and several rarer forms). Involvement of the heart in Duchenne's dystrophy results in a focal cardiomyopathy of the posterior wall; the classic ECG has prominent anterior precordial forces. In addition to LV dysfunction and heart failure, all of these conditions frequently cause conduction abnormalities, which may be the presenting or only feature. The myocardium may also be involved in inflammatory and autoimmune diseases. It is commonly affected in polymyositis and dermatomyositis, but usually this is subclinical. Systemic lupus erythematosus, scleroderma, and mixed connective tissue disease may cause myocarditis, but these more commonly involve the pericardium, coronary arteries, or valves. Several endocrinopathies, including acromegaly, thyrotoxicosis, myxedema, and pheochromocytoma, can produce dilated cardiomyopathies that resolve when the underlying disease is appropriately treated.
Pericardial involvement is quite common in many of the connective tissue diseases. Systemic lupus erythematosus may present with pericarditis, and pericardial involvement is not uncommon (but is less frequently symptomatic) in active rheumatoid arthritis, systemic sclerosis, and mixed connective tissue disease. Endocardial involvement takes the form of patchy fibrous predominantly on the right side or inflammatory or sclerotic changes of the heart valves. Carcinoid heart disease results from the layering of plaque-like material over the tricuspid valve, RV endocardium, and pulmonic valve and presents with right heart failure due to tricuspid and pulmonic regurgitation. The hypereosinophilic syndromes involve the endocardium, leading to restrictive cardiomyopathy. A variety of arthritic syndromes are associated with aortic valvulitis or aortitis with resulting aortic regurgitation. These include ankylosing spondylitis, rheumatoid arthritis, and Reiter's syndrome. Disorders of collagen (Marfan syndrome is the most frequent, followed by Ehlers-Danlos syndrome) often affect the ascending aorta, with resulting aneurysmal dilation and aortic regurgitation. Mitral valve prolapse is also a common finding in these disorders.
Almost any vasculitic syndrome can involve the coronary arteries, leading to myocardial infarction. This is most common with polyarteritis nodosa and systemic lupus erythematosus. Two vasculitic syndromes have a particular predilection for the coronary arteries Kawasaki's disease and Takayasu's disease. Kawasaki's disease can result in coronary aneurysms,
P.424
occasionally of huge size. Takayasu's disease affects the great vessels more than the coronaries and smooth, tapering lesions are usually seen, particularly at the vessel ostia. In these, myocardial infarction may be the presenting symptom.
Biondi M: Effects of subclinical thyroid dysfunction on the heart. Ann Intern Med 2002;137:904.
Kulke MH et al: Carcinoid tumors. N Engl J Med 1999;340:858.
Magnani JW et al: Myocarditis: current trends in diagnosis and treatment. Circulation 2006;113:876.
Moder KG et al: Cardiac involvement in systemic lupus erythematosus. Mayo Clin Proc 1999;74:275.
Riboldi P et al: Cardiac involvement in systemic autoimmune diseases. Clin Rev Allergy Immunol 2002;23:247.
Steen V: The heart in systemic sclerosis. Curr Rheumatol Rep 2004;6:137.
Wijetunga M et al: Myocarditis in systemic lupus erythematosus. Am J Med 2002;113:419.
Traumatic Heart Disease
Penetrating wounds to the heart are, of course, usually lethal unless surgically repaired. Stab wounds to the RV occasionally lead to hemopericardium without progressing to tamponade.
Blunt trauma is a more frequent cause of cardiac injuries, particularly outside of the emergency department setting. This type of injury is quite frequent in motor vehicle accidents and may occur with any form of chest trauma, including CPR efforts. The most common injuries are myocardial contusions or hematomas. Other forms of nonischemic cardiac injury include metabolic injury due to burns, electrical current, or sepsis. These may be asymptomatic (particularly in the setting of more severe injuries) or may present with chest pain of a nonspecific nature or, not uncommonly, with a pericardial component. Elevations of cardiac enzymes are frequent but the levels do not correlate with prognosis. Echocardiography may reveal an akinetic segment or pericardial effusion. Pericardiocentesis is warranted if tamponade is evident. Heart failure is uncommon if there are no associated cardiac or pericardial injuries, and conservative management is usually sufficient.
Severe trauma may also cause cardiac or valvular rupture. Cardiac rupture may involve any chamber, but survival is most likely if injury is to one of the atria or the RV. Hemopericardium or pericardial tamponade is the usual clinical presentation, and surgery is almost always necessary. Mitral and aortic valve rupture may occur during severe blunt trauma the former presumably if the impact occurs during systole and the latter if during diastole. Patients reach the hospital in shock or severe heart failure. Immediate surgical repair is essential. The same types of injuries may result in transection of the aorta, either at the level of the arch or distal to the takeoff of the left subclavian artery. Transthoracic echocardiography and TEE are the most helpful and immediately available diagnostic techniques.
Blunt trauma may also result in damage to the coronary arteries. Acute or subacute coronary thrombosis is the most common presentation. The clinical syndrome is one of acute myocardial infarction with attendant ECG, enzymatic, and contractile abnormalities. Emergent revascularization is sometimes feasible, either by the percutaneous route or by coronary artery bypass surgery. LV aneurysms are common outcomes of traumatic coronary occlusions. Coronary artery dissection or rupture may also occur in the setting of blunt cardiac trauma.
Bansal MK et al: Myocardial contusion injury: redefining the diagnostic algorithm. Emerg Med J 2005;22:465.
Geddes LA et al: Evolution of our knowledge of sudden death due to commotio cordis. Am J Emerg Med 2005;23:67.
Harada H et al: Traumatic coronary artery dissection. Ann Thorac Surg 2002;74:236.
Lindstaedt M et al: Acute and long-term clinical significance of myocardial contusion following blunt thoracic trauma: results of a prospective study. J Trauma 2002;52:479.
Schultz JM et al: Blunt cardiac injury. Crit Care Clin 2004;20:57.
Wall MJ Jr et al: Trauma to cardiac valves. Curr Opin Cardiol 2002;17:188.
The Cardiac Patient & Surgery
Patients with known or suspected cardiac disease undergoing general surgery present a common management problem. Anesthesia and surgery are often associated with marked fluctuations of heart rate and BP, changes in intravascular volume, myocardial ischemia or depression, arrhythmias, decreased oxygenation, increased sympathetic nervous system activity, and alterations in medical regimens and pharmacokinetics. Even with careful monitoring and management, the perioperative period can be very stressful to cardiac patients.
The risk of surgery in patients with heart disease depends primarily on three factors: the type of operation, the nature of the heart disease, and the degree of preoperative stability. The type of anesthesia is less important, though halothane, enflurane, and barbiturates are more severe myocardial depressants, whereas opioids have little depressive effect. Spinal and epidural anesthesia were previously thought to be preferable in patients with heart disease, but this has not proved to be the case.
The highest-risk procedures are surgery of the aorta and vascular procedures, in part because these patients often have associated severe coronary disease but also
P.425
because marked BP and volume changes are common. Major abdominal and thoracic surgery are also associated with substantial cardiovascular risk, particularly in older patients with associated cardiovascular disease.
Numerous studies have evaluated the excess risk of surgery in patients with various cardiac diseases. Recent (within 3 months) myocardial infarction, unstable angina, CHF, and significant aortic stenosis are associated with substantial increases in operative morbidity and mortality rates. Any degree of instability in these conditions magnifies the potential risk. Stable angina, particularly in an inactive individual, is also associated with a higher operative risk. Although less common, cyanotic congenital heart disease and severe primary or secondary pulmonary hypertension pose great risks during major surgery. In patients with any of these problems, the risk-to-benefit ratio of the planned surgery should be carefully examined. If the procedure is necessary but elective, consideration should be given to delaying it until full recovery postinfarction and correction or optimal stabilization of the other conditions are achieved. Hypertension should be at least moderately controlled. Patients with severe angina should have increased medical therapy initiated or be considered for revascularization before noncardiac surgery. Symptomatic arrhythmias, nonsustained ventricular tachycardia, or high-grade AV block and cardiac failure should be treated optimally.
Clinical assessment provides the most useful guidance in determining the risk of noncardiac surgery. Important indicators of high risk have been discussed above. A multifactorial cardiac risk index is included in Chapter 3 (Table 3-4). Patients with known but clinically stable heart disease, such as angina pectoris or prior myocardial infarction, are at intermediate risk, particularly for major operations such as vascular surgery. If a history or symptoms of heart failure are present, assessment of LV function can be very helpful in perioperative management. Although frequently advocated, further noninvasive testing for myocardial ischemia for the purpose of risk stratification is probably overutilized. Tests such as stress myocardial perfusion scintigraphy or dobutamine echocardiography should be reserved for situations in which the results may alter patient management. There is no evidence that prophylactic revascularization by either PCI or coronary artery bypass surgery alters long-term outcome in patients undergoing noncardiac surgical procedures without the usual indications for PCI or CABG. Only in the case of major vascular operations is perioperative mortality and morbidity high enough that prophylactic PCI or CABG should be considered.
However, it should be noted that many patients undergoing surgery have not had recent medical follow-up, and this may be an appropriate opportunity to perform a more complete evaluation. Thus, stress testing may be indicated for selected patients with symptomatic angina or prior myocardial infarction with a view to instituting more comprehensive medical management or performing coronary revascularization to reduce long-term (rather than perioperative) mortality and morbidity. At the least, such patients should not be discharged without a plan for an appropriate follow-up and institution of antihyperlipidemic, aspirin, and -blocker therapy as indicated.
Once the decision to operate is made, careful management is essential. Most cardiac medications should be continued preoperatively and postoperatively. In patients judged to be at high risk or medium risk, -blockers should be initiated preoperatively unless contraindicated. If practical, oral therapy with atenolol or metoprolol should be started several days prior to surgery with the dosage gradually increased to 100 mg in single or divided doses. Otherwise, 15 mg of metoprolol or 10 mg of atenolol may be administered intravenously in 5 mg increments separated by 5 10 minutes prior to induction. These doses should be repeated every 12 hours or more frequently if excessive tachycardia occurs until oral therapy can be commenced. Monitoring is an important prophylactic measure in high-risk individuals; hemodynamic monitoring can facilitate early intervention in patients with heart failure, severe valve disease, or easily induced myocardial ischemia. Excessive hypertension, hypotension, and myocardial ischemia should be identified and appropriately treated using rapidly acting agents. TEE can also be used for intraoperative monitoring of ischemia, but its value has never been established in well-designed studies. Ischemic events, whether symptomatic or silent, should be vigorously treated.
Auerbach AD et al: Beta-blockers and reduction of cardiac events in noncardiac surgery: scientific review. JAMA 2002;287:1435.
Boersma E et al: Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001;285:1865.
Devereaux PJ et al: How strong is the evidence for the use of perioperative beta blockers in non-cardiac surgery? Systematic review and meta-analysis of randomised controlled trials. BMJ 2005;331:313.
Eagle KA et al: ACC/AHA guideline update for perioperative cardiovascular evaluation for noncardiac surgery executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1996 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). J Am Coll Cardiol 2002;39:542.
Fleisher LA et al: Clinical practice. Lowering cardiac risk in non-cardiac surgery. N Engl J Med 2001;345:1677.
Hernandez AF et al: Preoperative evaluation for major noncardiac surgery: focusing on heart failure. Arch Intern Med 2004; 164:1729.
The Cardiac Patient & Pregnancy
The management of cardiac disease in pregnancy is discussed in detail in the references listed below. Only a few major points can be covered in this brief section.
P.426
Cardiovascular Changes During Pregnancy
Normal physiologic changes during pregnancy can exacerbate symptoms of underlying cardiac disease even in previously asymptomatic individuals. Maternal blood volume rises progressively until the end of the sixth or seventh month. Stroke volume increases over the same time course as a result of the volume change and an increase in EF. The latter reflects predominantly a decline in peripheral resistance due to vasodilation and the low-resistance shunting through the placenta. The heart rate tends to rise in the third trimester to further increase the cardiac output as the stroke volume levels out. Overall, cardiac output increases by 30 50%; systolic BP tends to rise slightly or remain unchanged, but diastolic pressure falls significantly. Venocaval compression of the IVC from the gravid uterus can lead to reduced venous return and a lower cardiac output in the supine position.
High cardiac output causes alterations in the cardiac examination. A third heart sound is prominent and normal, and a pulmonary flow murmur is common. ECG changes include rate-related decreases in PR and QT intervals, a leftward axis shift, inferior Q waves due to the more horizontal position of the heart, and nonspecific ST-T wave changes. Normal echocardiographic findings include slightly increased chamber sizes, functional valvular regurgitation, and occasionally small pericardial effusions.
Management of Preexisting Conditions
The physiologic changes imposed by pregnancy can cause cardiac decompensation in patients with any significant cardiac abnormality, but the most severe problems are encountered in patients with valvular stenosis (especially mitral and aortic stenosis), congenital or acquired abnormalities associated with pulmonary hypertension or right-to-left shunting, CHF due to any cause, coronary heart disease, and hypertension. Valvular insufficiency or left-to-right shunting often diminishes because of the fall in peripheral resistance and is better tolerated than other lesions.
Mitral stenosis becomes more hemodynamically severe because of the increase in diastolic flow and the rate-related shortening of diastole. LA pressures rise, and dyspnea or pulmonary edema can occur in previously asymptomatic individuals. The onset of atrial fibrillation often leads to acute decompensation. Patients with moderate to severe mitral or aortic stenosis should have the condition corrected prior to becoming pregnant if possible. Patients who become symptomatic can undergo successful surgery, preferably in the third trimester, although balloon valvuloplasty is the treatment of choice if the mitral valve is amenable. Coarctation of the aorta is usually well tolerated unless severe, when perfusion of the gravid uterus and upper extremity hypertension become an issue. Patients with symptoms or significant upper extremity hypertension before pregnancy should have corrective surgery before embarking on a pregnancy. Patients with severe pulmonary hypertension and cyanotic congenital heart disease and those with severe aortic stenosis are at extremely high risk and should avoid pregnancy. Balloon valvuloplasty of severe aortic stenosis is feasible and preferable to surgical intervention during pregnancy if possible. Patients with most right-sided lesions do well with pregnancy, including those with ASD, pulmonic stenosis, pulmonic regurgitation, operated tetralogy of Fallot, and Ebstein's anomaly. Those with an RV that supports the systemic circulation can usually tolerate pregnancy unless symptomatic heart failure has already occurred. Most adult patients with a systemic RV have transposition of the great vessels that has been surgically corrected or is congenitally corrected (L-transposition of the great vessels). Patients with hypertrophic cardiomyopathy generally do well, though the risk of pregnancy is higher than normal.
In Marfan syndrome, there are two issues, the risk of transmission of the disease to the child and the risk of cardiovascular complications with pregnancy. Marfan patients tolerate pregnancy well if the aorta is 4.0 cm or less. If the aorta is significantly dilated and/or there has been prior dissection, the pregnancy may not be tolerated. If the Marfan patient becomes pregnant, then physical activity should be limited and -blockers used throughout the pregnancy. Cesarean section is favored over vaginal delivery.
Asymptomatic arrhythmias should be closely observed unless underlying heart disease is present, in which case they should be treated with drugs. Paroxysmal supraventricular arrhythmias are quite common. Patients with Wolff-Parkinson-White syndrome may have more problems during pregnancy. Therapy is similar to that required for nonpregnant women. Issues related to pregnancy and the use of anticoagulation are discussed above under Prosthetic Valve Management.
Preexisting systemic hypertension is usually well tolerated and controllable, though the fetal morbidity rate is slightly increased. The incidence of preeclampsia and eclampsia (see Chapter 18) is increased.
Hydralazine and methyldopa are the antihypertensive agents for which there has been the greatest experience during pregnancy. Diuretics have also been used frequently, but concern has been raised that intravascular hypovolemia might impair uterine blood flow. Nonetheless, these agents are relatively safe. More recently, there has been considerable use of the combined - -blocker labetalol and of calcium channel blockers, which have proved to be effective and safe to both the mother and fetus. On the other hand, atenolol has been associated with lower fetal weights. ACE inhibitors and angiotensin II blockers are contraindicated in pregnancy because of the risk of fetal injury. -Blockers may retard fetal growth, but experience with them has been generally favorable. Little is known about the safety of most other antihypertensive agents.
P.427
A comprehensive review of the safety of drugs in pregnancy and during breast-feeding can be found at http://www.perinatology.com/exposures/druglist.htm.
Cardiovascular Complications of Pregnancy
Pregnancy-related hypertension (eclampsia and preeclampsia) is discussed in Chapter 18.
Cardiomyopathy of Pregnancy (Peripartum Cardiomyopathy)
In approximately one of 4000 15,000 patients, dilated cardiomyopathy develops in the final month of pregnancy or within 6 months after delivery. The cause is unclear, but immune and viral causes have been postulated. The disease occurs more frequently in women over age 30 years, is generally related to the first or second pregnancy, and is associated with gestational hypertension and drugs used to stop uterine contractions. The course of the disease is variable; many cases improve or resolve completely over several months, but others progress to refractory heart failure. About 60% of patients make a complete recovery. Immunosuppressive therapy has been advocated, but few supportive data are available. Recently, -blockers have been administered judiciously to these patients, with at least anecdotal success. Some advocate anticoagulation because of an increased risk for thrombotic events. Recurrence in subsequent pregnancies is common, particularly if cardiac function has not recovered.
Coronary Artery & other Vascular Abnormalities
There have been a number of reports of myocardial infarction during pregnancy. It is known that pregnancy predisposes to dissection of the aorta and other arteries, perhaps because of the accompanying connective tissue changes. The risk may be particularly high in patients with Marfan or Ehlers-Danlos syndromes. However, coronary artery dissection is responsible for only a minority of the infarctions; most are caused by atherosclerotic CAD or coronary emboli. Most of the events occur near term or shortly following delivery, and paradoxical emboli through a PFO has been implicated in some cases. Clinical management is essentially similar to that of other patients with acute infarction, unless there is a connective tissue disorder. If nonatherosclerotic dissection is present, coronary intervention is risky as further dissection can be aggravated. In most instances, conservative management is warranted.
Special Problems
Prophylaxis for Infective Endocarditis
Although there is no universal agreement, many authorities recommend antibiotic prophylaxis during labor for patients at risk for endocarditis, especially if forceps delivery is anticipated or episiotomy is performed. Ampicillin (2 g intravenously or intramuscularly) plus gentamicin (1.5 mg/kg intravenously or intramuscularly [up to 80 mg]) followed by amoxicillin, 1.5 g orally every 6 hours, is the recommended regimen.
Management of Labor
Although vaginal delivery is usually well tolerated, unstable patients (including patients with severe hypertension and worsening heart failure) should have planned cesarean section. An increased risk of aortic rupture has been noted during delivery in patients with coarctation of the aorta and severe aortic root dilation with Marfan syndrome, and vaginal delivery should be avoided in these conditions. For most patients, even those with congenital heart disease, vaginal delivery is preferred.
Elkayam U et al: Maternal and fetal outcomes of subsequent pregnancies in women with peripartum cardiomyopathy. N Engl J Med 2001;344:1567.
Elkayam U et al: Valvular heart disease and pregnancy part I: native valves. J Am Coll Cardiol 2005;46:223.
Elkayam U et al: Valvular heart disease and pregnancy: part II: prosthetic valves. J Am Coll Cardiol 2005;46:403.
Ginsberg JS et al: Use of antithrombotic agents during pregnancy. Chest 2001;119:122S.
Head CE et al: Congenital heart disease in pregnancy. Postgrad Med J 2005;81:292.
Lupton M et al: Cardiac disease in pregnancy. Curr Opin Obstet Gynecol 2002;14:137.
Pearson GD et al: Peripartum cardiomyopathy: NHLBI workshop recommendations and review. JAMA 2000;283:1183.
Report of the National High Blood Pressure Education Program Working Group on High Blood Pressure in Pregnancy. Am J Obstet Gynecol 2000;183:S1.
Sliwa K et al: Outcome of subsequent pregnancy in patients with documented peripartum cardiomyopathy. Am J Cardiol 2004;93:1441.
Tidswell M: Peripartum cardiomyopathy. Crit Care Clin 2004; 20:777.
Cardiovascular Screening of Athletes
The sudden death of a competitive athlete inevitably becomes an occasion for local if not national publicity. On each occasion, the public and the medical community ask whether such events could be prevented by more careful or complete screening. Although each event is tragic, it must be appreciated that there are approximately 5 million competitive athletes at the high school level or above in any given year. The number of cardiac deaths occurring during athletic participation is unknown, but estimates at the high school level range from one in 300,000 to one in 100,000 participants. Death rates among more mature athletes increase as the prevalence of CAD rises. These numbers
P.428
highlight the problem of how to screen individual participants. Even an inexpensive test such as an ECG would generate an enormous cost if required of all athletes, and it is likely that few at-risk individuals would be detected. Echocardiography, either as a routine test or as a follow-up examination for abnormal ECGs, would be prohibitively expensive except for the elite professional athlete. Thus, the most feasible approach is that of a careful medical history and cardiac examination performed by personnel aware of the conditions responsible for most sudden deaths in competitive athletes. In a series of 158 athletic deaths in the United States between 1985 and 1995, hypertrophic cardiomyopathy (36%) and coronary anomalies (19%) were by far the most frequent underlying conditions. LV hypertrophy was present in another 10%, ruptured aorta (presumably due to Marfan syndrome or cystic medial necrosis) in 6%, myocarditis or dilated cardiomyopathy in 6%, aortic stenosis in 4%, and arrhythmogenic RV dysplasia in 3%. In addition, commotio cordis, or sudden death due to direct myocardial injury, may occur. More common in children, it may occur even after a minor direct blow to the heart; it is thought to be due to the precipitation of a premature ventricular contraction just prior to the peak of the T wave on ECG.
It is likely that a careful family and medical history and cardiovascular examination will identify some individuals at risk. A family history of premature sudden death or cardiovascular disease or of any of these predisposing conditions should mandate further workup, including an ECG and echocardiogram. Symptoms of chest pain, syncope, or near-syncope also warrant further evaluation. A Marfan-like appearance, significant elevation of BP or abnormalities of heart rate or rhythm, and pathologic heart murmurs or heart sounds should also be investigated before clearance for athletic participation is given. Such an evaluation is recommended before participation at the high school and college levels and every 2 years during athletic competition.
Stress-induced syncope or chest pressure may be the first clue to an anomalous origin of a coronary artery. Anatomically, this lesion occurs most often when the left anterior descending artery arises from the right coronary cusp and traverses between the aorta and pulmonary trunks. The slit-like orifice that results from the angulation at the vessel origin is thought to cause ischemia when the aorta and pulmonary arteries enlarge during rigorous exercise.
The toughest distinction may be in sorting out the healthy athlete with LVH from the athlete with hypertrophic cardiomyopathy. In general, the healthy athlete's heart is less likely to have an unusual pattern of LVH, LA enlargement, an abnormal ECG, an LV cavity < 45 mm in diameter at end-diastole, an abnormal diastolic filling pattern, a family history of hypertrophic cardiomyopathy, and the athlete is more likely to be male than the individual with hypertrophic cardiomyopathy.
Selective use of routine ECG and stress testing is recommended in men above age 40 years and women above age 50 years who continue to participate in vigorous exercise and at earlier ages when there is a positive family history for premature CAD, hypertrophic cardiomyopathy, or multiple risk factors.
Corrado D et al: Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998;339:364.
Hosey RG: Sudden cardiac death. Clin Sports Med 2003;22:51.
Maron BJ et al: Working Groups of the American Heart Association Committee on Exercise, Cardiac Rehabilitation, and Prevention; Councils on Clinical Cardiology and Cardiovascular Disease in the Young: Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004;109:2807.
Pelliccia A et al: Clinical significance of abnormal electrocardiographic patterns in trained athletes. Circulation 2000;102: 278.
Pfister GC et al: Preparticipation cardiovascular screening for US collegiate student-athletes. JAMA 2000;283:1597.
Pluim BM et al: The athlete's heart. A meta-analysis of cardiac structure and function. Circulation 2000;101:336.
Seto CK: Preparticipation cardiovascular screening. Clin Sports Med 2003;22:23.
EAN: 2147483647
Pages: 49
- ERP Systems Impact on Organizations
- Challenging the Unpredictable: Changeable Order Management Systems
- ERP System Acquisition: A Process Model and Results From an Austrian Survey
- Distributed Data Warehouse for Geo-spatial Services
- Development of Interactive Web Sites to Enhance Police/Community Relations