59 - Postsurgical Empyema
Editors: Shields, Thomas W.; LoCicero, Joseph; Ponn, Ronald B.; Rusch, Valerie W.
Title: General Thoracic Surgery, 6th Edition
Copyright 2005 Lippincott Williams & Wilkins
> Table of Contents > Volume I - The Lung, Pleura, Diaphragm, and Chest Wall > Section XII - Thoracic Trauma > Chapter 71 - Barotrauma and Inhalation Injuries
Chapter 71
Barotrauma and Inhalation Injuries
Kenneth J. Woodside
Lucinda Miller
Akhil Bidani
Joseph B. Zwischenberger
The parenchyma of the lung can be damaged by a number of processes. Ventilator-induced lung injury is the cascade of pathophysiology induced by positive-pressure ventilation that can induce or aggravate lung inflammation. The term barotrauma is used to signify damage induced by pressure applied to the airways and alveoli. Similarly, volutrauma is damage induced by excessive air volume in the airways and alveoli that causes alveolar overdistention and stretching. Inhalation injury usually occurs during a fire, as the culmination of upper airway thermal damage, inhaled toxins and gases, and systemic manifestations of injury. Each of these parenchymal injuries causes decreased gas exchange efficacy and pulmonary compliance.
BAROTRAUMA
Barotrauma may indicate damage at the cellular level, such as the injury to the parenchyma seen in adult respiratory distress syndrome (ARDS), or may indicate extraalveolar air caused by airway disruption, such as that seen with pneumothorax or subcutaneous emphysema. Although the etiology of the airway damage is similar, the pathophysiology manifests as distinct clinical presentations.
Extraalveolar Air Associated Barotrauma Etiology and Management
Common manifestations of extraalveolar air include pneumothoraces and subcutaneous emphysema. Subcutaneous emphysema usually is not clinically significant itself, but is an indicator of airway injury that may have other manifestations and requires prompt evaluation. Pneumothoraces can cause significant impairment of ventilation, gas exchange, and cardiac function, requiring prompt diagnosis and treatment. Parker and co-workers (1993) reported that pressure- and volume-related alveolar rupture is the most common cause of pneumothorax in ventilated patients. Macklin (1939) and Macklin and Macklin (1944) reported that any sudden increase in alveolar pressure may result in a gradient sufficient to disrupt the alveolar wall. Once ventilator-applied pressure ruptures the alveoli, air dissects along the loose connective tissues of the bronchovascular bundle or the interlobular septa medially toward the hilum and peripherally toward the visceral pleura. Because the mediastinal pleura is relatively weak and thin compared with the visceral pleura, air often dissects toward the hilum, where it can enter the mediastinum and form a pneumomediastinum. A continuum of fascial planes connects the soft tissue compartments of the neck, mediastinum, and retroperitoneum, allowing decompression of the pneumothorax into these compartments with subsequent formation of a new air pocket. With subsequent rupture of the mediastinal pleura, air enters the pleural space and forms a pneumothorax.
Several factors associated with barotrauma and volutrauma increase the risk for pneumothorax development. Gammon and colleagues (1992) found that high positive end-expiratory pressure (PEEP) values and high minute ventilations increase the pneumothorax risk. Also, Albelda and co-workers (1983) and Pierson (1988) showed that peak inspiratory pressures [(PIPs) >40 to 50 cm H2O] and high tidal volumes, respectively, increase gross barotrauma risk. Furthermore, Peterson and Baier (1983) reported a 43% incidence of extrapulmonary air type barotrauma when PIP exceeded 70 cm H2O in a retrospective study of 171 patients. In patients with PIPs between 50 and 70 cm H2O, extrapulmonary air type barotrauma rates were only 8%. In patients with PIPs of less than 50 cm H2O, there were no cases of extrapulmonary air.
Unfortunately, human studies have not distinguished the damaging effects of excessive airway pressure versus inspired volume as the cause of alveolar hyperinflation and injury, because direct measurement of lung volume is difficult in ventilated patients. However, Colebatch and Ng (1991) retrospectively examined 14 men who developed pneumothoraces during shallow water diving and compared their pulmonary mechanics with 34 healthy nonsmokers
P.973
and 10 age-matched healthy male divers. Those who had developed pneumothoraces had stiffer airways and smaller air spaces than the other group, suggesting that such mechanics would increase the likelihood of interstitial gas dissection and resulting barotrauma. These mechanics may be analogous to patients with ARDS, who have markedly decreased compliance and smaller thickened alveoli, with an increased risk for pneumothoraces.
Placement of a tube thoracostomy is often required for drainage of a pneumothorax, due to present or potential respiratory or cardiac compromise. Mechanically ventilated patients and those with persistent air leak require tube thoracostomy, because there is a high risk for tension pneumothorax development. Pneumothoraces with a tension component impair cardiac venous return and ventricular filling, and may first require urgent needle decompression. Drainage can be accomplished with traditional chest tubes or image-guided catheter techniques, as recently reviewed by Woodside and colleagues (2003). Placement and management of these tube thoracostomies are described in detail in Chapter 56. For barotrauma-related pneumothorax, tube thoracostomy management is similar to that of pneumothoraces from other etiologies. Although high airway pressures theoretically increase the magnitude of the air leak, evidence from trials is sparse. Cerfolio and co-workers (2001) found that air leak magnitude is associated with persistent air leaks in patients undergoing elective pulmonary resections. Although minimizing airway pressures may shorten the duration of the air leak for mechanically ventilated patients with barotrauma-related pneumothoraces, rigorous data are lacking in this subset of patients, as recently reviewed by Loran and associates (2002). However, Rice and Kirby (1992) found similar air leak durations for pulmonary resection patients extubated on the first postoperative day versus those extubated on subsequent days.
Parenchymal Barotrauma and Volutrauma
Ventilator-induced lung injury was first recognized as a distinct type of barotrauma by Webb and Tierney (1974). Rats ventilated with high airway pressure (45 cm H2O) developed alveolar edema, hypoxemia, and decreased lung compliance, and usually died within 1 hour. They suggested that the interstitial edema was caused by pulmonary interdependence and that the alveolar edema was caused from depletion or inactivation of surfactant. Later, Peevy and co-workers (1990) suggested that peak airway pressure was the most important mechanism of injury. In an isolated perfused rabbit lung model, they examined various methods of ventilation. Specifically, they ventilated four sets of lungs with either low or high gas flow rates while holding the PIP either low (27 cm H2O) or high (50 cm H2O). They found that the high PIP group had significant microvascular injuries. Using geometric and mathematical modeling to evaluate mean airway pressure, Marini and Ravenscraft (1992) found that, under conditions of passive inflation, mean arterial pressure correlated with arterial oxygenation, hemodynamic performance, and barotrauma.
Elevated PIP, high mandatory respiratory rates, oxygen toxicity, and alveolar overdistention probably account for ventilator-associated barotrauma. Dreyfuss and colleagues (1985, 1988) found that intermittent positive pressure hyperventilation with high inflation pressures produced pulmonary microvascular injury and capillary permeability edema that probably could be partially reversed with PEEP in a rodent model. Large-animal models by Tsuno and co-workers (1990, 1991) demonstrated that regional pressure related overdistention results in a pattern of diffuse alveolar damage that is histologically similar to clinical ARDS, with similar pulmonary mechanics. In a sleep model of ARDS induced by moderately high airway pressures, Tsuno and colleagues (1990) described progressive deterioration in total static lung compliance, functional residual capacity, and arterial blood gases, with radiologic abnormalities emerging over time. At necropsy, there was severe pulmonary atelectasis, increased wet lung weight, and increased minimum surface tension of saline lung lavage fluid. In a later study (1991), Tsuno and co-workers analyzed the histopathologic changes that occurred in the lungs of baby pigs after mechanical ventilation at a PIP of 40 cm H2O for approximately 1 day. They described a number of changes consistent with early ARDS, including alveolar hemorrhage, alveolar neutrophil and lymphocyte infiltration, alveolar macrophage and type II pneumocyte proliferation, interstitial congestion and thickening, emphysematous changes, and hyaline membrane formation. Further mechanical ventilation led to the additional finding of exudative organization, similar to that seen in late ARDS. Control animals ventilated at 18 cm H2O did not have any significant histologic findings.
Ultimately, volutrauma, rather than barotrauma, probably accounts for the majority of ARDS-like changes. Using a rabbit model, Hernandez and associates (1989) found that chest wall restriction limited high airway pressure induced lung injury in immature rabbits. Specifically, they found that inspiratory volume limitation with a full-body plaster cast abrogated capillary permeability increases by PIP, suggesting that the volume distention, rather than the high PIP, produces the capillary damage. Dreyfuss and co-workers (1988) also showed an effect of overdistention (volutrauma). They demonstrated that mechanically ventilated rats at high pressure and low volume did not demonstrate capillary permeability changes, whereas those at low pressure and high volume had significant pulmonary edema and permeability, suggesting the predominant role of volume in injury etiology. The Acute Respiratory Distress Syndrome Network Trial (2000) confirmed this finding for human patients. Furthermore, because ARDS is a nonuniform disease, as noted by Gattinoni and associates (1987), there may be furtherance of injury because preferential ventilation of normal alveoli leads to overdistention as they try to
P.974
accommodate the increased tidal volume. This overdistention, in turn, may lead to further stretch injury.
Interestingly, the lung injury described by Dreyfuss and co-workers (1988) was markedly reduced by the application of 10 cm H2O of PEEP, suggesting that the unequal forces generated by repeated recruitment and derecruitment of lung alveolar units may be partially responsible for parenchymal damage (Fig. 71-1). This subset of volutrauma is sometimes termed atelectotrauma (Fig. 71-2). PEEP acts as a pneumatic stent, preventing alveolar collapse at end expiration and minimizing shear forces associated with repeated opening and closing of collapsed alveolar units, as reported by Hudson (1998). PEEP is not, however, always appropriate. Ranieri and associates (1991) examined the effects of PEEP on ARDS patients. Observing the pressure-volume loops on zero end-expiratory pressure (ZEEP) and PEEP, they found two distinct populations of patients. The first group demonstrated evidence of alveolar recruitment, with upward concavity of the pressure-volume curve during ventilation with ZEEP, which increased with PEEP. The second group demonstrated an upward convexity that was further aggravated with PEEP, suggesting volume displacement without alveolar recruitment and increased susceptibility to barotrauma.
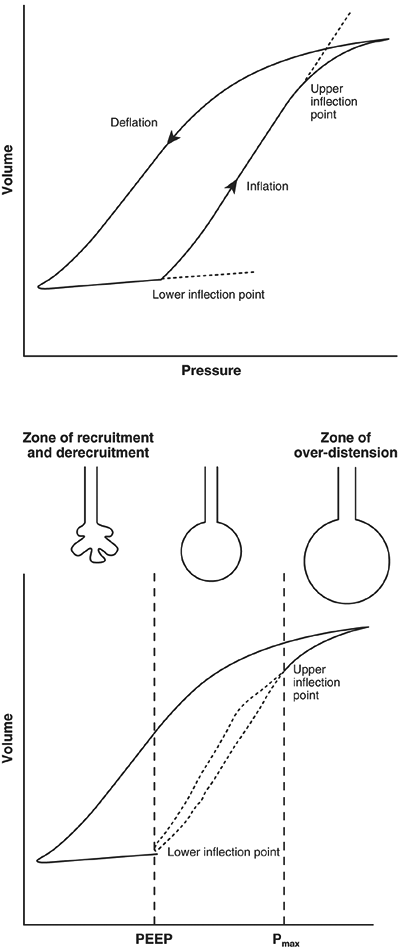 |
Fig. 71-1. Idealized pulmonary pressure-volume plot for a patient with acute lung injury, which demonstrates the relationship between PEEP, alveolar collapse, and alveolar overdistention. From Pinhu L, et al: Ventilator-associated lung injuries. Lancet 361:332, 2003. With permission. |
The mechanism of barotrauma and volutrauma is a subject of intense investigation. Kawano and colleagues (1987) demonstrated immune involvement with a neutropenic rabbit model. They applied a saline lung lavage, followed by controlled mechanical ventilation with tidal volumes of 12 mL/kg, to three groups of rabbits. The normopenic controls developed high permeability pulmonary edema with hyaline membrane formation. The second group of rabbits was rendered neutropenic with nitrogen mustard pretreatment, and lacked these pulmonary changes. The third group was also rendered neutropenic, but was retransfused with granulocytes before pulmonary lavage, and had similar findings to the normopenic group. In humans, Ranieri and co-workers (1999, 2000) demonstrated that concentrations of plasma and bronchoalveolar cytokines were significantly lower in patients receiving a protective lung strategy when compared with conventional mechanical ventilation. In addition, the Acute Respiratory Distress Syndrome Network trial (2000) demonstrated lower plasma levels of interleukin-6 in patients treated with protective ventilation. Current studies suggest that the physiologic changes seen with barotrauma and volutrauma involve both a mechanical and immune etiology for the development of lung injury. Recent recommendations from the National Heart, Lung, and Blood Institute Working Group on Acute Lung Injury and ARDS, which were summarized by Matthay and associates (2003), suggest that an improved understanding of the genetics of individual responses is required for a more comprehensive understanding of such a multifactorial disease process.
Prevention and Management
Prevention and management strategies for volutrauma are similar; the mechanical ventilation settings that contribute to the initiation of pneumothoraces and ARDS also worsen it. Ventilator strategies should target reducing airway pressures and volumes to avoid exacerbating volutrauma and thereby reducing endogenous inflammation. In a meta-analysis, Bidani and colleagues (1994) concluded that permissive hypercapnia allows avoidance
P.975
of overdistention through pressure or volume limitation and may improve outcome. Slutsky, writing for the American College of Chest Physicians Consensus Conference (1993), recommended that airway pressures should be limited to reduce tidal volumes under conditions of lung overdistention, while accepting the increase in arterial Pco2 levels. Hickling and associates (1990) suggested that lower tidal volumes of 5 to 8 mL/kg will prevent excessive alveolar distention. Low levels of PEEP and supplemental oxygen can be added to improve hypoxemia. Amato and co-workers (1998) directly compared mechanical ventilation with 6 mL/kg to 12 mL/kg of tidal volume. Seven percent of the patients in the former group had clinical evidence of barotrauma, compared with 42% in the latter group (with mortality rates of 38% and 71%, respectively). More recently, the Acute Respiratory Distress Syndrome Network (2000) released their multicenter, randomized, and controlled trial comparing 6 and 12 mL/kg of tidal volume, with plateau pressures below 30 and 50 cm H2O, respectively. The trial was terminated after 861 patients because the outcomes in the low tidal volume group were clearly superior. Specifically, mortality was 31% in the low tidal volume group and 40% in the higher tidal volume group (p = 0.007). Other techniques, such as inverse-ratio ventilation, high-frequency jet ventilation, and extracorporeal life support (ECLS), along with pharmacologic manipulation with agents such as activated protein C, show promise for treatment of severe established ARDS and systemic inflammatory response syndrome, rather than prevention and treatment of volutrauma, and are described in more detail in Chapter 72.
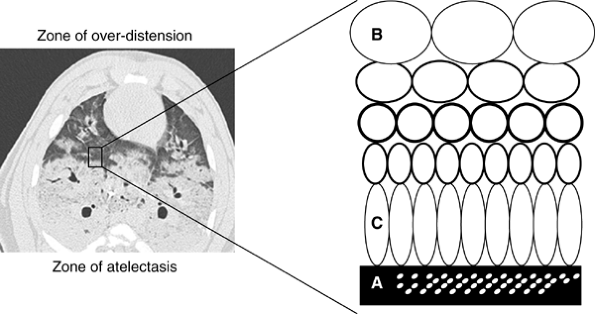 |
Fig. 71-2. Atelectotrauma. The interface between collapsed and consolidated lung (A) and overdistended lung units (B) is heterogeneous and unstable. Depending on ambient conditions, this region is prone to cyclic recruitment and derecruitment and localized asymmetric stretch of lung units (C) immediately apposed to regions of collapsed lung. From Pinhu L, et al: Ventilator-associated lung injuries. Lancet 361:332, 2003. With permission. |
INHALATION INJURIES
The most common pulmonary injury associated with burns is smoke inhalation. In fact, Ryan and co-workers (1998) described inhalation injury, along with advanced age (>60 years) and greater than 40% total body surface area (TBSA) burn, as one of the three most important risk factors for the determination of mortality following burn. Herndon and colleagues (1987) established a 33% incidence of inhalation injury with major burn. Furthermore, Demling (1989) stated that these injuries account for 50% of burn deaths at the burn scene. Therefore, an understanding of this malady is of particular importance for improved patient outcome.
Pathophysiology
Inhalation injury rarely implies direct pulmonary burn, and is also distinct from pulmonary edema resulting from overresuscitation. The upper airways absorb most of the heat, often resulting in swelling and possible obstruction of the upper airway. However, the lower airway usually receives
P.976
adequately cooled air or gas. The usual etiology of lower airway injury is inhalation of smoke or chemical irritants released during the fire, and may occur even in the absence of a cutaneous burn. Zikria (1975) and Dowell (1971) and their co-workers, as well as Traber and Herndon (1990), showed that the airway and lungs are initially damaged from incomplete products of combustion, most importantly aldehydes and oxides of sulfur and nitrogen. The injury is notable both for inhomogeneous distribution within a patient and variable severity between patients. The physicochemical properties of the causative agent, amount of smoke inhaled, and preexisting diseases that might decrease the resistance of the recipient determine the site and degree of injury. Because these patients often require intubation for airway protection, the synergy of inhalation injury with volutrauma can result in a severe ARDS-like state. Unlike traditional ARDS, this injury initially results from direct damage to the lungs, with additional damage occurring from the continued inflammation.
Fires often liberate numerous biologically harmful chemicals that can be inhaled. Common household items and home building materials produce a variety of toxic chemicals when burned (Table 71-1). Davies (1986) identified numerous chemicals in the blood of inhalation victims who died at the scene of fires. Of particular note, carbon monoxide is ubiquitous in burning buildings and binds avidly to hemoglobin with over 200 times the affinity of oxygen, causing severe tissue hypoxia. Hydrogen cyanide is also a product of incomplete combustion of plastics and blocks oxidative metabolism, with multiple delayed dose-dependent effects. Liberated ammonia combines with water on contact with mucosa to form ammonium hydroxide, a strong alkali that produces liquefaction necrosis of the tissue. Aldehydes, produced by combustion of wood, can initiate severe pulmonary edema, with exposure to greater than 30 parts per million producing death in 10 minutes. Hydrogen chloride released in the combustion of plastics produces glottic and pulmonary edema. Nitrogen dioxide produced from combustion of cellulose can cause severe respiratory epithelial damage and pulmonary edema, as well as the formation of methemoglobin, within a few hours of exposure. Carbonyl-chloride, produced in the combustion of polyvinyl chloride, causes necrosis of epithelium and pulmonary edema. However, with only a low concentration in most fires, it typically only produces only chest tightness and mucosal irritation. These compounds act together to increase mortality. In particular, Prien and Traber (1988) reported increased mortality with carbon monoxide and hydrogen cyanide exposure. Furthermore, Moore (1991) and Pitt (1979) and their colleagues found that this synergism exacerbates tissue hypoxia and acidosis and decreases cerebral oxygen consumption and metabolism.
In a dramatic experiment, Woolley (1984) reported on a reenactment of a fire that occurred in the Stardust Nightclub in Dublin in 1981. A portion of the nightclub was recreated and monitored during a controlled fire. Within minutes of starting the fire, smoke production exceeded 1,000 m3/min and visibility decreased to less than 1 m. Near the fire itself, the oxygen concentration declined to only 2%, with a 3% carbon monoxide level. Multiple toxic chemicals were present in lethal concentrations, including hydrogen cyanide and hydrogen chloride. Even near the exit, the oxygen concentration averaged only 13% and other toxic chemicals remained in lethal concentrations.
The extent of inhalation damage produced depends on ignition source, temperature, concentration, and solubility of the toxic gases generated. The more caustic materials, such as acrolein and other aldehydes, damage the airway and initiate inflammation of the bronchi and parenchyma. Although carbon monoxide and cyanide rarely damage the
P.977
airway directly, they affect gas exchange. Carbon monoxide toxicity remains one of the most frequent immediate causes of death after smoke-induced inhalation injury. While a low or normal carbon monoxide level does not rule out the presence of inhalation injury, inhalation of a 0.1% carbon monoxide mixture may result in generation of a carboxyhemoglobin level as high as 50%. Toxic symptoms manifest at carboxyhemoglobin levels greater than 20%, and death may occur at levels greater than 60%. In fact, carbon monoxide has an affinity for hemoglobin that is 200 to 250 times that of oxygen, so patients should receive 100% oxygen until their carbon monoxide level returns to 0%. The oxygen-hemoglobin dissociation curve loses its sigmoid shape and shifts to the left, thus further impairing tissue oxygen availability. Goldbaum and associates (1976) demonstrated that competitive inhibition of the cytochrome oxidase enzyme system by carbon monoxide probably results in decreased ability of oxidative efficacy in the mitochondria.
Table 71-1. Origin of Selected Toxic Compounds Other than CO or CO2 | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
Hinder (1997) and Abdi (1991) and their associates found a marked increase in tracheobronchial blood flow. Furthermore, Isago (1991) and Kramer (1989) and their co-workers noted increased capillary permeability and edema in the tracheobronchial tree after smoke inhalation. Ahn and colleagues (1990) described epithelial shedding, congestion, regional emphysema, and progressive separation of the epithelium with formation of pseudomembranous casts with resultant partial or complete airway obstruction. Furthermore, Abdi and associates (1995) found that the cyclooxygenase inhibitor ibuprofen attenuates changes in vascular resistance, and Kimura and colleagues (1988) noted a parallel decrease in the lung lymph flow seen after smoke inhalation injury, suggesting a significant immunologic involvement in this process.
Once initiated, immunologic aggravation of inhalation injury is difficult to control. Traber (1985) and Linares (1989) and their co-workers demonstrated that leukocytes and the production of oxygen radicals and proteolytic enzymes induced lung injury after smoke inhalation. Basadre and associates (1988) noted that pulmonary damage and changes in pulmonary arterial resistance and lymph flow were significantly attenuated in sheep depleted of leukocytes by nitrogen mustard. Furthermore, the production of oxygen radicals, as indicated by plasma-conjugated dienes, and the consumption of antiprotease, as measured by 2-macroglobulin levels in lung lymph, were not changed in these animals, whereas both of these variables were elevated in the smoke inhalation group. Ahn and associates (1990) postulated that pretreatment with allopurinol, a xanthine oxidase inhibitor, also attenuated many of these changes, indicating an ischemia-reperfusion phenomenon.
Several forms of pulmonary dysfunction may result from the complex pathophysiologic reactions to inhalation injury. Hypoxia, ventilation-perfusion mismatching, increased airway resistance, decreased pulmonary compliance, and increased pulmonary vascular resistance may result from the initial release of vasoactive substances from the damaged epithelium, paralleling traditional ARDS. Similarly, sloughed necrotic epithelium, serous exudate, blood cells, and mucus form casts that may cause atelectasis and exacerbate ventilation-perfusion mismatching and hypoxia. In studying percussive ventilation, Cioffi and colleagues (1989) described air trapping that occurs distal to obstruction caused by casts, resulting in volutrauma. Alternatively, total occlusion of the airways by casts can produce atelectasis and increase the risk for pneumonia. Decreased compliance and increased airway resistance may lead to elevated airway pressures and volutrauma. Furthermore, mean airway pressures exceeding the mucosal capillary perfusion pressure may result in ischemia of the already damaged tracheobronchial mucosa, compounding the initial epithelial insult. Increasing amounts of mucosal slough, interstitial edema, and decreasing compliance lead to further elevation in ventilatory pressures and worsening volutrauma, as well as further increases in the likelihood of the development of pneumonia.
Diagnosis
Patients often present with carbonaceous material around the nose and mouth or inside the oropharynx, making the diagnosis of smoke inhalation simple. Other times manifestations may be subtle, and only a high index of suspicion and invasive testing lead to the diagnosis. Because hyperemia, edema, superficial mucosal sloughing, and ulceration of the tracheal mucosa are often present before arterial blood gas abnormalities, deteriorating pulmonary function test results or respiratory failure signal the presence of inhalation injury. This pathophysiology is dynamic and results in variable presentations, as described by Clark and Nieman (1988). Patients with severe cases of inhalation injury often die at the scene. Some patients may respond initially to resuscitation, only to die later of severe hypoxia. Patients with less severe cases may enter the hospital in respiratory distress that is caused by airway edema and bronchorrhea. These patients may have stridor, wheezing, and decreased breath sounds. The mildest group may appear relatively comfortable, only to deteriorate within the first 24 hours as they develop severe respiratory compromise. These differences probably reflect the differential progression of disease.
A high index of suspicion for inhalation injury is essential to avoid missed injuries. Clark and colleagues (1989) noted a majority (56%) of patients with inhalation injury present with some combination (usually three or more) of history and physical findings indicating inhalation injury (Table 71-2). Fiberoptic bronchoscopy and intravenous xenon 133 (133Xe) ventilation scanning, which identify regions of complete or incomplete small airway obstruction secondary to inhalation injury, can be used to establish the diagnosis. To confirm injury, particularly in mild cases, one must rely on laboratory evaluation and bronchoscopy. Arterial blood gases are usually diagnostic. Although the Pco2 may be normal,
P.978
carboxyhemoglobin and cyanmethemoglobin levels may be elevated. Mildly elevated carboxyhemoglobin may not be diagnostic, because it is often elevated in smokers. Elevated cyanmethemoglobin levels, however, are diagnostic. Fitzpatrick and Cioffi (1995) found that the combination of bronchoscopy and 133Xe scanning is 93% accurate in the diagnosis of smoke inhalation injury. Fiberoptic bronchoscopy also allows immediate institution of therapy when severe inhalation injury or airway obstruction is diagnosed, and is rapidly becoming the standard of care.
Table 71-2. Signs and Symptoms Suggesting Possible Inhalation Injury | |
|---|---|
|
Management
In the management of inhalation injury, one must constantly reassess the patency of the airway and the adequacy of ventilation. According to Clark (1992), patients surviving the acute accident scene with isolated smoke inhalation injury usually experience illness for only several days and then begin to improve, allowing cautious withdrawal of support. Haponik and Summer (1987) noted that as many as 50% of surviving inhalation victims require intubation. Bronchoscopy is rarely required to determine the need for intubation, as it is often required for airway protection. Because of the mucosal edema that is present, high inspired oxygen concentrations may help to improve arterial oxygenation. Because carbon monoxide has such a great affinity for hemoglobin, a high Po2 is required to displace it.
Medical management of inhalation injury stresses the use of measures to avoid prolonged intubation and positive-pressure ventilation, and if ventilatory support is necessary, permissive hypercapnia and borderline adequacy of oxygenation are preferable to attaining arbitrarily normal blood gases at the cost of inflicting volutrauma. The end points of fluid resuscitation are not changed by the presence of an inhalation injury; a urine output of 30 to 50 mL/h in adults or 0.5 to 1.0 mL/kg per hour in children is the best indication that appropriate fluid resuscitation is being administered to the patient. Several retrospective clinical reviews by Scheulen and Munster (1982), as well as Navar (1985), Hughes (1989) and Herndon (1988) and their associates, demonstrated that coexisting inhalation injury increases the fluid resuscitation requirements of patients with cutaneous thermal injury, particularly in the first 24 hours, by up to twofold in comparison to patients without inhalation injury. A prospective animal study by Herndon and colleagues (1986) demonstrated that inadequate fluid resuscitation after smoke inhalation injury was detrimental. Limiting fluid resuscitation for pulmonary protection may actually lead to the opposite, with increased extravascular lung fluid, hypoxia, and decrements in lung function, along with the risks for inadequate tissue perfusion, renal failure, and possible death from hypovolemia.
Therapeutic coughing, chest physiotherapy, early ambulation, airway suctioning, and pharmacologic agents, all of which are effective in mobilizing and removing retained secretions, are essential to respiratory management of patients with smoke inhalation injury. Reactive airway disease must be treated, usually with 2-agonists. Tracheobronchial suctioning is required for the removal of debris that cannot be cleared by patients with an incapacitated mucociliary apparatus and ineffective cough. Adequate humidification of inspired gas is essential to help limit inspissation of mucus in the injured airways. Despite adequate humidification, inspissation of secretions still occurs in many patients with severe inhalation injury. In experimental ovine models of inhalation injury, heparin administered systemically, as demonstrated by Cox and colleagues (1993), or by nebulizer, as demonstrated by Brown (1988) and Murakami (2002) and their co-workers, prevented or decreased the formation of tracheobronchial casts, along with decreased lung edema and cellular infiltrates. In human pediatric burn victims with inhalation injury, Desai and colleagues (1998) reported a reduction in mortality with aerosolized heparin and N-acetylcystine therapy. Recently, Murakami and associates (2003) demonstrated that recombinant antithrombin III attenuates pulmonary inflammation in an ovine model of inhalation injury with pneumonia. They suggested that the decrease in antithrombin activity that occurs during sepsis induces severe airway obstruction that is abrogated by exogenous antithrombin.
In two separate prospective clinical trials conducted by Levine and co-workers (1978) and Moylan and Alexander (1978), corticosteroids were not beneficial in altering morbidity or mortality after inhalation injury and may be associated with a higher rate of infection-related complications; therefore, they should be avoided unless required for other medical conditions. Fiberoptic bronchoscopy, although labor intensive and expensive, is effective when all other therapies fail to remove secretions. Common treatment algorithms for pathophysiologic events resulting from mild and moderate inhalation injury are listed in Table 71-3.
Unfortunately, florid respiratory failure is present in some victims. Patients with marginal oxygenation who fail to improve
P.979
rapidly are treated vigorously, as alveolar volume loss and total atelectasis is such a prominent feature in this illness. Many techniques can be borrowed from ARDS treatment, although the pathophysiology can be quite different from traditional (indirect) ARDS. However, as in traditional ARDS, it is easier to prevent collapse than it is to restore alveolar volume. Cioffi and co-workers (1991) showed that prophylactic use of high-frequency percussive ventilation reduces the incidence of pneumonia and death in adults with inhalation injury. Cortiella and associates (1999) demonstrated similar findings in pediatric burn patients with inhalation injury. Patients treated with high-frequency percussive ventilation had lower pneumonia rates, better pressure flow (P/F) ratios, lower PIPs, and less work of breathing than those on conventional mechanical ventilation.
Table 71-3. Treatment Algorithms for Mild and Moderate Inhalation Injury | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
There are a number of therapies which may have some promise in animal models, although human trials are few. Many of these experimental trials occur in ovine models, because sheep have lung physiology that is similar to that of humans and have an easily accessed pulmonary vascular and thoracic duct. Alpard and colleagues (2000) developed a model of inhalation injury where a sheep receives concurrent 40% TBSA cutaneous full-thickness flame burns while inhaling 24 to 48 breaths of smoke through a tracheostomy, reliably resulting in a severe respiratory distress syndrome with P/F ratios of less than 200 within 48 hours. Alpard (1999) and Schmalstieg (2001) and their co-workers demonstrated improved P/F ratios, coupled with reductions in ventilator settings and near-total carbon dioxide removal in an ovine model of inhalation injury with the use of arteriovenous carbon dioxide removal (AVCO2R), which was associated with decreased interleukin-8 expression and decreased neutrophil infiltration in the lung parenchyma, supporting the benefit of reduced ventilatory requirements and alveolar stretching in inhalation injury. One of us (JBZ) and associates (2002) recently reported a reduction in inhalation injury mortality with the use of a paracorporeal artificial lung prototype in a lethal dose 90% to 100% smoke/burn ARDS ovine model. Clinical trials using AVCO2R and other ECLS techniques in burn patients with severe lung injury are ongoing.
REFERENCES
Abdi S, et al: Lung edema formation following inhalation injury: role of the bronchial blood flow. J Appl Physiol 71:727, 1991.
Abdi S, et al: Effects of ibuprofen on airway vascular response to cotton smoke injury. Eur J Pharmacol 293:475, 1995.
Acute Respiratory Distress Syndrome Network: Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 342: 1301, 2000.
Ahn SY, et al: Effects of allopurinol on smoke inhalation in the ovine model. J Appl Physiol 68:228, 1990.
Albelda SM, et al: Ventilator-induced subpleural air cysts: clinical, radiographic, and pathologic significance. Am Rev Respir Dis 127:360, 1983.
Alpard SK, et al: Reduced ventilator pressure and improved P/F ratio during percutaneous arteriovenous carbon dioxide removal for severe respiratory failure. Ann Surg 230:215, 1999.
Alpard SK, et al: New clinically relevant sheep model of severe respiratory failure secondary to combined smoke inhalation/cutaneous flame burn injury. Crit Care Med 28:1469, 2000.
Amato MB, et al: Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 338:347, 1998.
Basadre JO, et al: The effect of leukocyte depletion on smoke inhalation injury in sheep. Surgery 104:208, 1988.
Bidani A, et al: Permissive hypercapnia in acute respiratory failure. JAMA 272:957, 1994.
Brown M, et al: Dimethylsulfoxide with heparin in the treatment of smoke inhalation injury. J Burn Care Rehabil 9:22, 1988.
Cerfolio RJ, Bass C, Katholi CR: Prospective randomized trial compares suction versus water seal for air leaks. Ann Thorac Surg 71:1613, 2001.
Cioffi WG Jr, et al: High-frequency percussive ventilation in patients with inhalation injury. J Trauma 29:350, 1989.
Cioffi WG Jr, et al: Prophylactic use of high-frequency percussive ventilation in patients with inhalation injury. Ann Surg 213:575, 1991.
Clark WR, Bonaventura M, Myers W: Smoke inhalation and airway management at a regional burn unit: 1974 1983. Part I: Diagnosis and consequences of smoke inhalation. J Burn Care Rehabil 10:52, 1989.
Clark WR Jr: Smoke inhalation: diagnosis and treatment. World J Surg 16: 24, 1992.
Clark WR Jr, Nieman GF: Smoke inhalation. Burns Incl Therm Inj 14:473, 1988.
Colebatch HJ, Ng CK: Decreased pulmonary distensibility and pulmonary barotrauma in divers. Respir Physiol 96:293, 1991.
P.980
Cortiella J, Micak R, Herndon D: High frequency percussive ventilation in pediatric patients with inhalation injury. J Burn Care Rehabil 20:232, 1999.
Cox CS Jr, et al: Heparin improves oxygenation and minimizes barotrauma after severe smoke inhalation in an ovine model. Surg Gynecol Obstet 176:339, 1993.
Davies JWL: Toxic chemicals versus lung tissue an aspect of inhalation injury revisited. J Burn Care Rehabil 7:213, 1986.
Demling RH: Management of the burn patient. In Schumaker HB, et al (eds): Textbook of Critical Care. 2nd Ed. Philadelphia: WB Saunders, 1989.
Desai MH, et al: Reduction in mortality in pediatric patients with inhalation injury with aerosolized heparin/N-acetylcystine therapy. J Burn Care Rehabil 19:210, 1998.
Dowell AR, Kilburn KH, Pratt PC: Short-term exposure to nitrogen dioxide. Effects on pulmonary ultrastructure, compliance, and the surfactant system. Arch Intern Med 128:74, 1971.
Dreyfuss D, et al: Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am Rev Respir Dis 132:880, 1985.
Dreyfuss D, et al: High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis 137:1159, 1988.
Fitzpatrick JC, Cioffi WG Jr: Diagnosis and treatment of inhalation injury. In Herndon DN (ed): Total Burn Care. Philadelphia: WB Saunders, 1995, pp. 184 192.
Gammon RB, Shin MS, Buchalter SE: Pulmonary barotrauma in mechanical ventilation: patterns and risk factors. Chest 102:568, 1992.
Gattinoni L, et al: Pressure-volume curve of total respiratory system in acute respiratory failure. Computed tomographic scan study. Am Rev Respir Dis 136:730, 1987.
Goldbaum LR, Orellano T, Dergal E: Mechanism of the toxic action of carbon monoxide. Ann Clin Lab Sci 6:372, 1976.
Haponik ER, Summer WR: Respiratory complications in burn patients: diagnosis and management of inhalation injury. J Crit Care 2:121, 1987.
Hernandez LA, et al: Chest wall restriction limits high airway pressure-induced lung injury in young rabbits. J Appl Physiol 66:2364, 1989.
Herndon DN, Traber DL, Traber LD: The effect of resuscitation on inhalation injury. Surgery 100:248, 1986.
Herndon DN, et al: Pulmonary injury in burned patients. Surg Clin North Am 67:31, 1987.
Herndon DN, et al: Inhalation injury in burned patients: effects and treatment. Burns Incl Therm Inj 14:349, 1988.
Hickling KG, Henderson SJ, Jackson R: Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intens Care Med 16:372, 1990 Hinder F, et al: Inhalation injury increases the anastomotic bronchial blood flow in the pouch model of the left ovine lung. Shock 8:131, 1997.
Hudson LD: Protective ventilation for patients with acute respiratory distress syndrome. N Engl J Med 338:385, 1998.
Hughes KR, et al: Fluid requirements of patients with burns and inhalation injuries in an intensive care unit. Intensive Care Med 15:464, 1989.
Isago T, et al: Analysis of pulmonary microvascular permeability after smoke inhalation. J Appl Physiol 71:1403, 1991.
Kawano T, et al: Effect of granulocyte depletion in a ventilated surfactant-depleted lung. J Appl Physiol 62:27, 1987.
Kimura R, et al: Ibuprofen reduces the lung lymph flow changes associated with inhalation injury. Circ Shock 24:183, 1988.
Kramer GC, et al: Effects of inhalation injury on airway blood flow and edema formation. J Burn Care Rehabil 10:45, 1989.
Levine BA, et al: Prospective trials of dexamethasone and aerosolized gentamicin in the treatment of inhalation in the burned patient. J Trauma 18:188, 1978.
Linares HA, Herndon DN, Traber DL: Sequence of morphologic events in experimental smoke inhalation. J Burn Care Rehabil 10:27, 1989.
Loran DB, et al: Predictors of alveolar air leaks. Chest Surg Clin North Am 12:477, 2002.
Macklin CC: Transport of air along sheaths of pulmonic blood vessels from alveoli to mediastinum: clinical implications. Arch Intern Med 64: 913, 1939.
Macklin MT, Macklin CC: Malignant interstitial emphysema of the lungs and mediastinum as an important occult complication in many respiratory diseases and other conditions: an interpretation of the clinical literature in the light of laboratory experiment. Medicine 23:281, 1944.
Marini JJ, Ravenscraft SA: Mean airway pressure: physiologic determinants and clinical importance. Part II. Clinical implications. Crit Care Med 20:1604, 1992.
Matthay MA, et al: Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute Working Group. Am J Respir Crit Care Med 167:1027, 2003.
Moore SJ, Ho IK, Hume AS: Severe hypoxia produced by concomitant intoxication with sublethal doses of carbon monoxide and cyanide. Toxicol Appl Pharmacol 109:412, 1991.
Moylan JA, Alexander LG Jr: Diagnosis and treatment of inhalation injury. World J Surg 2:185, 1978.
Murakami K, et al: Heparin nebulization attenuates acute lung injury in sepsis following smoke inhalation in sheep. Shock 18:236, 2002.
Murakami K, et al: Recombinant antithrombin attenuates pulmonary inflammation following smoke inhalation and pneumonia in sheep. Crit Care Med 31:577, 2003.
Navar PD, Saffle JR, Warden GD: Effect of inhalation injury on fluid resuscitation requirements after thermal burn. Am J Surg 150:716, 1985.
Parker JC, Hernandez LA, Peevy KJ: Mechanisms of ventilator-induced lung injury. Crit Care Med 21:131, 1993.
Peevy KG, et al: Barotrauma and microvascular injury in lungs of nonadult rabbits: effect of ventilation pattern. Crit Care Med 18:634, 1990.
Peterson GW, Baier H: Incidence of pulmonary barotrauma in a medical ICU. Crit Care Med 11:67, 1983.
Pierson DJ: Alveolar rupture during mechanical ventilation: role of PEEP, peak airway pressure, and distending volume. Respir Care 33:472, 1988.
Pinhu L, et al: Ventilator-associated lung injury. Lancet 361:332, 2003.
Pitt BR, et al: Interaction of carbon monoxide and cyanide on cerebral circulation and metabolism. Arch Environ Health 34:345, 1979.
Prien T, Traber DL: Toxic smoke compounds and inhalation injury a review. Burns Incl Therm Inj 14:451, 1988.
Ranieri VM, et al: The effects of positive end expiratory pressure on alveolar recruitment and gas exchange in patients with the adult respiratory distress syndrome. Am Rev Respir Dis 144:544, 1991.
Ranieri VM, et al: Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 282:54, 1999.
Ranieri VM, et al: Mechanical ventilation as a mediator of multisystem organ failure in acute respiratory distress syndrome. JAMA 284:43, 2000.
Rice TW, Kirby TTJ: Prolonged air leak. Chest Surg Clin North Am 2:803, 1992.
Ryan CM, et al: Objective estimates of the probability of death from burn injuries. N Engl J Med 338:362, 1998.
Scheulen JJ, Munster AM: The Parkland formula in patients with burns and inhalation injury. J Trauma 22:869, 1982.
Schmalstieg FC, et al: Interleukin-8, aquaporin-1, and inducible nitric oxide synthase in smoke and burn injured sheep treated with percutaneous carbon dioxide removal. ASAIO J 47:365, 2001.
Slutsky AS: Mechanical ventilation. American College of Chest Physicians' Consensus Conference. Chest 104:1833, 1993.
Traber DL, Herndon DN: Pathophysiology of smoke inhalation. In Haponik EF, M nster AM (eds): Respiratory Sequelae of Burns. New York: McGraw-Hill, 1990, pp. 61 71.
Traber DL, et al: Pulmonary edema and compliance changes following smoke inhalation. J Burn Care Rehabil 6:490, 1985.
Tsuno K, Prato P, Kolobow T: Acute lung injury from mechanical ventilation at moderately high airway pressures. J Appl Physiol 69:956, 1990.
Tsuno K, et al: Histopathologic pulmonary changes from mechanical ventilation at high peak airway pressures. Am Rev Respir Dis 143:1115, 1991.
Webb HH, Tierney DF: Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis 110:556, 1974.
Woodside KJ, et al: Pneumothorax in patients with acute respiratory distress syndrome: pathophysiology, detection, and treatment. J Intens Care Med 18:9, 2003.
Woolley WB: The Stardust Disco fire: Dublin 1981: studies of combustion products during simulation experiments. Fire Safe J 7:267, 1984.
Zikria BA, et al: What is clinical smoke poisoning? Ann Surg 181:151, 1975.
Zwischenberger JB, et al: The paracorporeal artifical lung improves 5-day outcomes from lethal smoke/burn-induced acute respiratory distress syndrome in sheep. Ann Thorac Surg 74:1011, 2002.
EAN: 2147483647
Pages: 203
- Chapter V Consumer Complaint Behavior in the Online Environment
- Chapter VI Web Site Quality and Usability in E-Commerce
- Chapter IX Extrinsic Plus Intrinsic Human Factors Influencing the Web Usage
- Chapter XI User Satisfaction with Web Portals: An Empirical Study
- Chapter XIV Product Catalog and Shopping Cart Effective Design