5 - Serum Calcium and Phosphate
Editors: Schrier, Robert W.
Title: Manual of Nephrology, 6th Edition
Copyright 2005 Lippincott Williams & Wilkins
> Table of Contents > 5 - The Patient with Disorders of Serum Calcium and Phosphate
5
The Patient with Disorders of Serum Calcium and Phosphate
Robert F. Reilly
Disorders of Serum Calcium
The majority of calcium in the body is in the form of hydroxyapatite in bone (99%). Although a small fraction of total body calcium is contained in the extracellular fluid (ECF), only the concentration of ionized calcium in the ECF is physiologically active and regulated. Approximately 60% of calcium in the ECF is ultrafiltrable and exists either free in solution as ionized calcium (50%) or complexed to anions such as citrate, phosphate, sulfate, and bicarbonate (10%). The remaining 40% is bound to proteins (primarily albumin).
Figure 5-1 illustrates calcium fluxes between the ECF, intestine, kidney, and bone. Net intestinal absorption of calcium amounts to about 200 mg of the normal dietary intake of 800 to 1,000 mg. In the steady state, this net intestinal absorption is matched by urinary excretion. As a result, 10,600 mg of the approximately 10,800 mg (98%) of calcium that is filtered daily is reabsorbed.
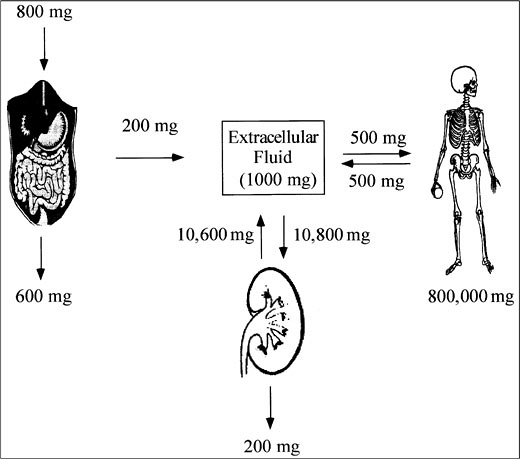 |
Figure 5-1. Calcium Homeostasis. |
Calcium regulation. Plasma-ionized calcium is regulated through a complex and coordinated interplay of parathyroid hormone (PTH) and 1,25(OH)2 vitamin D3 (calcitriol) in intestine, bone, and kidney. The parathyroid gland senses the ECF ionized calcium concentration via a calcium-sensing receptor. High concentrations of ECF calcium stimulate the receptor and activate second messenger pathways that, in turn, inhibit the release of PTH. Low concentrations of ECF calcium stimulate PTH secretion and production and increase parathyroid gland mass. The parathyroid gland responds quickly (within minutes) to alterations in ionized calcium concentration. An inverse sigmoid relationship exists between ECF calcium concentration and PTH secretion, with a nonsuppressible component present even at high plasma calcium concentration. The amount of hormone stored is enough to support basal secretion for 6 hours and stimulated secretion for 2 hours.
In bone, PTH in the presence of permissive amounts of calcitriol stimulates reabsorption by increasing osteoclast number and activity. In intestine, PTH enhances calcium and phosphate absorption indirectly by promoting the formation of calcitriol. In kidney, PTH augments distal tubular reabsorption of calcium, stimulates the formation of calcitriol in the proximal tubule, and decreases proximal tubular reabsorption of phosphate and bicarbonate.
Calcitriol is produced in proximal tubule via 1- hydroxylation of 25(OH)vitamin D3 (calcidiol). The calcitriol biosynthetic pathway is illustrated in Figure 5-2. Principal stimulators of 1- hydroxylase are PTH and hypophosphatemia. The major function of calcitriol is to enhance the availability of calcium and phosphate for new bone formation and the prevention of symptomatic hypocalcemia and hypophosphatemia. In intestine and kidney, calcitriol increases the production of calcium-binding proteins (calbindins) that aid in the transcellular movement of calcium; this may be the rate-limiting step in transepithelial calcium flux. In bone, calcitriol potentiates the actions of PTH, stimulates osteoclastic reabsorption, and induces the differentiation of monocytes into osteoclasts.
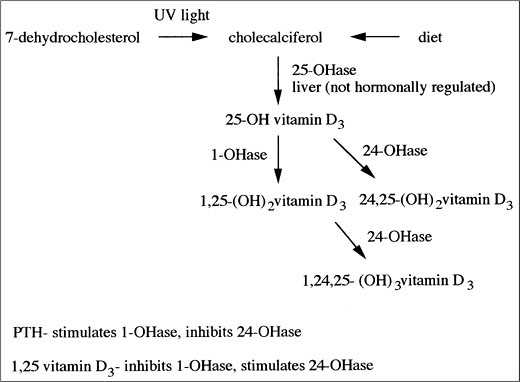
Figure 5-2. Vitamin D Metabolism. (PTH, Parathyroid Hormone; UV, Ultraviolet.)
In the parathyroid gland, calcitriol binds to its receptor, leading to a decrease in PTH production. The PTH gene promoter contains regions that bind the calcitriol receptor. Binding results in a dramatic decrease in PTH expression. Calcitriol is the most potent suppressor of PTH gene transcription.
Hypercalcemia
Etiology. Three basic pathophysiologic mechanisms contribute to hypercalcemia: increased calcium absorption from the gastrointestinal tract, decreased renal calcium excretion, and increased bone resorption of calcium. The most common etiologies of hypercalcemia are listed in Table 5-1.
Table 5-1. Etiologies of Hypercalcemia
Hyperparathyroidism Malignancy Vitamin A and D intoxication Milk-alkali syndrome Thyrotoxicosis Granulomatous disease Immobilization Paget's disease Thiazide diuretic intake Addison's disease Pheochromocytoma Lithium intake Renal failure Post renal transplant Familial hypocalciuric hypercalcemia Increased calcium absorption from the gastrointestinal tract plays a primary role in the hypercalcemia of the milk-alkali syndrome, vitamin D intoxication, and granulomatous disorders.
Milk-alkali syndrome is the result of the ingestion of excess amounts of calcium and alkali. In the past, the most common sources of calcium and alkali were milk and sodium bicarbonate, respectively. Today this syndrome most often occurs in elderly women consuming excess amounts of calcium carbonate or citrate for the treatment of osteoporosis. Patients present with the classic triad of hypercalcemia, metabolic alkalosis, and renal insufficiency. Treatment of hypercalcemia in these patients is often complicated by hypocalcemia resulting from sustained PTH suppression.
Hypercalcemia in renal failure is uncommon, except in patients treated with calcium and vitamin D supplements. This disorder and the milk-alkali syndrome illustrate the important concept that hypercalcemia from ingestion of excessive amounts of dietary calcium alone does not occur in the absence of renal impairment.
Vitamin D intoxication also results in hypercalcemia. Calcium is
P.63
absorbed primarily in the small intestine, and this process is stimulated by calcitriol.Hypercalcemia may also be secondary to granulomatous disorders, such as sarcoidosis. Activated macrophages produce calcitriol, which leads to increased intestinal absorption of dietary calcium. Hypercalciuria is seen more commonly than hypercalcemia. Lymphomas occasionally cause hypercalcemia via the same mechanism.
Increased calcium resorption from bone plays a primary role in hypercalcemia resulting from primary and secondary hyperparathyroidism, malignancy, hyperthyroidism, immobilization, Paget's disease, and vitamin A toxicity.
Primary hyperparathyroidism is the most frequent cause of hypercalcemia, with an estimated prevalence of 1 per 10,000. The underlying pathology is most often a solitary adenoma (80%). Among the remainder, 15% to 20% have diffuse hyperplasia, and approximately one-half of these have a familial syndrome [multiple endocrine neoplasia (MEN) type I, associated with pituitary adenomas and islet cell tumors, or MEN type II, associated with medullary carcinoma of the thyroid and pheochromocytoma]. Multiple adenomas are uncommon, and parathyroid carcinoma is rare (occurring in less than 1%). Hypercalcemia results from increased calcium reabsorption from bone, increased calcium absorption from intestine mediated by calcitriol, and increased distal tubular renal calcium reabsorption. In primary hyperparathyroidism, the hypercalcemia is often mild, asymptomatic, and identified on routine blood chemistries in the outpatient setting. Patients most commonly present in the fifth and sixth decades. Women are affected two to three times more frequently than men, and two-thirds of all cases occur in postmenopausal women.
Secondary hyperparathyroidism leads to hypercalcemia after renal transplantation when plasma phosphate, vitamin D metabolism, and renal function improve, but PTH secretion remains high secondary to increased parathyroid mass. In most patients, the hypercalcemia resolves during the first year following transplantation.
Malignancy is the second most common cause of hypercalcemia. Hypercalcemia of malignancy results from several pathophysiologic mechanisms: the overproduction of PTH-related peptide (PTHrP), local reabsorption of bone around sites of tumor infiltration (mediated via a variety of cytokines and osteolytic prostaglandins), and the production of calcitriol (e.g., with lymphomas). Patients with squamous cell cancer of the lung, breast cancer, multiple myeloma, and renal cell carcinoma are at highest risk. Hypercalcemia due to tumoral production of PTHrP is often referred to as humoral hypercalcemia of malignancy (HHM). PTHrP has 70% amino acid identity to the first 13 amino acids of PTH, binds to the PTH receptor, and has biological activity similar, but not identical to PTH. HHM often presents with severe hypercalcemia (calcium concentration greater than 14 mg per dL) in a patient with either a known history of malignancy or evidence of malignancy at initial presentation. PTHrP is immunologically distinct from PTH and is not detected by standard PTH assays, but specific assays for PTHrP are commercially available. Assays that measure the C-terminal fragment of PTHrP may show an increase when used in pregnant patients or patients with chronic kidney disease. Median survival from the onset of hypercalcemia with HHM is only 3 months. Squamous cell tumors, renal cell carcinomas, and most breast neoplasms produce PTHrP. The diagnoses of primary hyperparathyroidism and malignancy are not mutually exclusive. An increased incidence of primary hyperparathyroidism has been reported in patients with malignancy.
Multiple myeloma is associated with hypercalcemia and localized osteolytic lesions in the skeleton. Approximately 30% of myeloma patients experience hypercalcemia at some time during the course of their disease. Bone destruction occurs as a consequence of the release of interleukin-6, interleukin-1, and tumor necrosis factor by malignant plasma cells. Bony lesions demonstrate a marked increase in osteoclastic resorption without manifestations of increased bone formation, in contrast to metastatic lesions of breast and prostate
P.65
cancer, which generally show some increase in bone formation and radionuclide uptake at sites of increased osteoblastic activity.Hyperthyroidism results in mild hypercalcemia in 10% to 20% of patients as a result of increased bone turnover.
Immobilization and Paget's disease also cause hypercalcemia, although this is more likely in children. In adults, hypercalciuria is more common than hypercalcemia.
Rare causes of hypercalcemia include lithium use (mild; interferes with the calcium sensing receptor); thiazide diuretic use (occult primary hyperparathyroidism should be suspected); pheochromocytoma; primary adrenal insufficiency; and a rare genetic disorder, familial hypocalciuric hypercalcemia (FHH).
FHH is an autosomal dominant disorder caused by a mutation in the calcium-sensing receptor. It presents with mild hypercalcemia early in life, hypocalciuria, and a normal or slightly increased PTH concentration in the absence of signs or symptoms of hypercalcemia. As a result of the mutation, the calcium-sensing receptor is less sensitive to plasma calcium concentration, and a higher than normal calcium concentration is required to suppress PTH. One should be aware of FHH, because this condition is often misdiagnosed as primary hyperparathyroidism, and patients may be inappropriately subjected to neck exploration. FHH may account for a small percentage of patients who undergo surgery for primary hyperparathyroidism in whom no adenoma is found.
P.64
Signs and symptoms of hypercalcemia are related to the severity and rate of rise in plasma-ionized calcium concentration. Mild hypercalcemia is generally asymptomatic and often incidentally discovered on routine blood chemistries, as is the case in many patients with primary hyperparathyroidism. In contrast, severe hypercalcemia is often associated with neurologic and gastrointestinal symptoms. The patient may present with a wide range of central nervous system symptoms, from mild mental status changes to stupor and coma. Gastrointestinal symptoms include constipation, anorexia, nausea, and vomiting. Abdominal pain may result from hypercalcemia-induced peptic ulcer disease or pancreatitis. As discussed in Chapter 2, Hypernatremia (section I.C.2.a), hypercalcemia results in polyuria and secondary polydipsia that leads to ECF volume contraction, a reduction in the glomerular filtration rate (GFR), and an elevation in blood urea nitrogen (BUN) and creatinine concentrations. Hypercalcemia also potentiates the cardiac effects of digitalis toxicity.
Diagnosis. The most common causes of hypercalcemia are primary hyperparathyroidism and malignancy. These two disorders make up more than 90% of all cases. Initial evaluation includes a history and physical examination. Use of calcium supplements, antacids, vitamin preparations, and over-the-counter medications should be determined. A chest x-ray should be obtained to rule out pulmonary malignancies and granulomatous disorders.
Initial laboratory examination includes measurement of electrolytes, BUN, creatinine, and phosphorus; serum protein electrophoresis; and a 24-hour urine test for calcium and creatinine. The presence of a high serum chloride concentration and a low serum phosphorus concentration in a ratio greater than 33 to 1 is suggestive of primary hyperparathyroidism resulting from PTH's effect of decreasing proximal tubular phosphate reabsorption. A low serum chloride concentration, a high serum bicarbonate concentration, and elevated BUN and creatinine concentrations are characteristic of milk-alkali syndrome. A monoclonal spike on either serum protein electrophoresis or urine protein electrophoresis is suggestive of multiple myeloma or light chain disease. A low serum phosphorus concentration is found in primary hyperparathyroidism and HHM. The 24-hour urinary calcium excretion is low in hypercalcemia caused by the milk-alkali syndrome, use of thiazide diuretics, or FHH.
P.66
As a general rule, primary hyperparathyroidism is the etiology in asymptomatic outpatients with a serum calcium concentration of less than or equal to 11 mg per dL, whereas malignancy is often the cause in symptomatic patients with an abrupt onset of disease and serum calcium concentration of greater than or equal to 14 mg per dL.
Intact PTH concentration is obtained after the initial evaluation is completed. The most common cause of an elevated PTH concentration is primary hyperparathyroidism, although an elevated PTH concentration may also be seen with lithium use and FHH. Occasionally, in primary hyperparathyroidism, the PTH concentration will be inappropriately within the normal range compared to the increase in serum calcium. In all other conditions, PTH will be suppressed by hypercalcemia.
If no obvious malignancy is present and the PTH concentration is not increased, then the possibility of vitamin D intoxication or granulomatous disease should be evaluated further with an analysis of calcidiol and calcitriol concentration. An increased calcidiol concentration is seen with the ingestion of either vitamin D or calcidiol. An elevated calcitriol concentration is observed with calcitriol ingestion, granulomatous disease, lymphoma, and primary hyperparathyroidism.
As a final step, if the calcitriol concentration is increased without apparent cause, occult granulomatous disease can be evaluated with a hydrocortisone suppression test. After the administration of 40 mg of hydrocortisone every 8 hours for 10 days, the hypercalcemia will resolve if it is the result of granulomatous disease.
Treatment of hypercalcemia varies depending on the severity of the serum calcium elevation. It is directed at increasing urinary calcium excretion, inhibiting bone resorption, and decreasing intestinal calcium absorption.
Urinary calcium excretion is increased by expanding the ECF volume and, subsequently, administering loop diuretics. Calcium reabsorption in the proximal tubule is passive and parallels sodium reabsorption. ECF volume contraction, therefore, increases proximal sodium reabsorption and helps maintain hypercalcemia. Patients with hypercalcemia are often volume contracted. Hypercalcemia decreases sodium reabsorption in the thick ascending limb of the loop of Henle via the activation of the calcium sensing receptor, and it also antagonizes the effects of antidiuretic hormone. In the setting of a reduced GFR, higher doses of loop diuretics may be required. In the presence of little or no renal function and severe hypercalcemia, hemodialysis is indicated. If hypercalcemia is moderate, volume expansion and loop diuretics may be the only therapy required.
An agent that inhibits bone resorption is often required when hypercalcemia is moderate or severe. In the acute setting, calcitonin is often helpful because of its rapid onset of action (2 to 4 hours). Calcitonin inhibits osteoclastic bone reabsorption and increases renal calcium excretion. It reduces serum calcium concentration, however, by only 1 to 2 mg per dL, and tachyphylaxis often develops with repeated use. For these reasons, calcitonin should not be used as the sole agent to inhibit bone resorption.
Bisphosphonates are the agents of choice for the management of hypercalcemia due to bone resorption. These analogues of inorganic pyrophosphate are selectively concentrated in bone, where they interfere with osteoclast attachment and function. Bisphosphonates have a slow onset (2 to 3 days) and a long duration of action (several weeks). Etidronate was the first bisphosphonate approved for the treatment of hypercalcemia. The serum calcium concentration begins to fall on day 2 with etidronate and reaches a nadir on day 7. The hypocalcemic effect may be prolonged for several weeks. If serum calcium falls rapidly within the first 48 hours, the drug should be discontinued to avoid hypocalcemia. Etidronate can be given intravenously at a dosage of
P.67
7.5 mg per kg over 4 hours for 3 consecutive days. A single intravenous dose of 30 mg per kg as an infusion over 24 hours in 1 L of normal saline, however, may be more effective.Pamidronate is more potent than etidronate and is the most widely used bisphosphonate in the treatment of hypercalcemia. It is given at a dose of either 60 or 90 mg intravenously over 4 hours. If serum calcium concentration is less than or equal to 13.5 mg per dL, 60 mg is given. If serum calcium concentration is greater than 13.5 mg per dL, 90 mg is administered. Serum calcium concentration gradually falls over the ensuing 2 to 4 days. A single dose is usually effective for 1 to 2 weeks. In most patients, serum calcium concentration normalizes after 7 days. The dose of both etidronate and pamidronate must be adjusted for renal function.
Plicamycin (mithramycin) can be used except in patients with severe hepatic, renal, and marrow disorders. Its effect begins in 12 hours and peaks at 48 hours. The dose can be repeated at 3- to 7-day intervals. Its side-effect profile (nausea, hepatotoxicity, proteinuria, and thrombocytopenia) has led to decreased enthusiasm for its use.
Gallium nitrate inhibits bone resorption by unknown mechanisms and is an additional agent that can be employed to treat hypercalcemia of malignancy. It is administered as a continuous infusion at a dose of 100 to 200 mg per m2 for 5 consecutive days. Gallium nitrate should not be administered to patients wih serum creatinine concentrations above 2.5 mg per dL, and it is probably best reserved for patients who have not responded to more conventional agents. A summary of treatment options is shown in Table 5-2.
Table 5-2. Treatment of Hypercalcemia
Drug Dosage Normal saline 2 4 L/d initially Furosemide 20 160 mg i.v. q8h after volume expansion Salmon calcitonin 4 IU/kg s.c. q12h Etidronate disodium 7.5 mg/kg i.v. over 4 hr q.d. for 3 7 d, 30 mg/kg i.v.
over 24 hrs single dosePamidronate disodium 60 90 mg i.v. over 4 hr Plicamycin 25 g/kg i.v. over 4 hr q.d. for 3 4 d Corticosteroids 200 300 mg hydrocortisone i.v. q.d. for 3 5 d Gallium nitrate 100 200 mg/m2 for 5 d
Measures to decrease intestinal absorption of calcium are often employed in outpatients with mild disease. Corticosteroids may be helpful in vitamin D intoxication, granulomatous disease, and certain neoplasms (lymphoma, myeloma). Alternatives to corticosteroids include ketoconazole and hydroxychloroquine. Oral phosphate can be administered, providing the patient does not have an elevated serum phosphorus concentration or renal failure. Oral phosphate, however, often causes diarrhea and only lowers the serum calcium concentration by about 1 mg per dL.
Whether to surgically remove a solitary parathyroid adenoma remains a difficult management issue. The following criteria for surgical intervention were suggested at a 1991 National Institutes of Health consensus conference: a total serum calcium concentration that is greater than 1 mg per dL above the upper limit of the normal range; evidence of overt bone disease; cortical bone mineral density that is more than 2 standard deviations (SD) below the adjusted mean for age, sex, and race; reduction in renal function by more than 30%; a history of nephrolithiasis or nephrocalcinosis; urinary calcium excretion greater than 400 mg per day; and
P.68
an episode of acute symptomatic hypercalcemia. It is estimated that approximately 50% of patients meet these criteria.With the advent of minimally invasive parathyroid surgery, these criteria are likely to be liberalized. The adenoma is initially localized with sestaMIBI scanning. If a solitary hot spot is identified, the adenoma is resected under local anesthesia. Parathyroid hormone concentration is measured intraoperatively. Its concentration should fall within minutes if the adenoma is successfully resected, given the short half-life of PTH (4 minutes). If the PTH concentration remains elevated, then the patient is placed under general anesthesia and the opposite side of the neck explored. The combination of sestaMIBI scanning and intraoperative PTH concentration measurement results in the successful removal of solitary adenomas in the vast majority of cases.
Hypocalcemia
Etiology. True hypocalcemia is the result of decreased calcium absorption from the gastrointestinal tract or decreased calcium resorption from bone. Given that 98% of total body calcium is contained within the skeleton, sustained hypocalcemia cannot occur without an abnormality of either PTH or calcitriol action in bone.
Total plasma calcium is composed of three components: ionized calcium (50%), complexed calcium (10%), and protein-bound calcium (40%). True hypocalcemia is present only when the ionized calcium concentration is reduced. The reference range for ionized calcium concentration is 4.2 to 5.0 mg per dL (1.05 to 1.25 mmol per L). Therefore, whenever a low total serum calcium concentration is observed, this value must be compared with the serum albumin concentration. For every decrease of 1 g per dL in serum albumin concentration from its normal concentration of 4 g per dL, a decrease of 0.8 mg per dL in total serum calcium concentration can be expected. Thus, for each fall of 1 g per dL in serum albumin concentration, 0.8 mg per dL must be added to the total serum calcium concentration. The binding of calcium to albumin is affected by ECF pH. Acidemia increases and alkalemia decreases the ionized calcium concentration. Ionized calcium concentration increases approximately 0.2 mg per dL for each 0.1 decrease in pH. These correction factors are only general guidelines and should not be used as a substitute for the direct measurement of serum-ionized calcium concentration if clinical suspicion warrants.
True hypocalcemia is caused by decreased PTH secretion, end-organ resistance to PTH, or disorders of vitamin D metabolism. Occasionally, hypocalcemia occurs acutely as a result of either the extravascular deposition of calcium or the intravascular binding of calcium. The most common etiologies of true hypocalcemia are illustrated in Table 5-3.
Table 5-3. Etiologies of Hypocalcemia
Hypoparathyroidism Idiopathic HAM syndrome Familial Post surgery hungry bone syndrome Infiltrative disorders Pseudohypoparathyroidism 1A, 1B, 2 Hypomagnesemia Defects in vitamin D metabolism Nutritional Malabsorption Drugs Liver disease Renal disease Vitamin D-dependent rickets Miscellaneous Tumor lysis syndrome Osteoblastic metastases Acute pancreatitis Toxic shock syndrome Sepsis HAM, hypoparathyroidism, adrenal insufficiency, and mucocutaneous candidiasis. Hypoparathyroidism is caused by a wide variety of acquired and inherited diseases that result from the impaired synthesis and release of PTH or from peripheral tissue resistance to PTH.
The most common cause of idiopathic hypoparathyroidism is polyglandular autoimmune syndrome type I, characterized by chronic mucocutaneous candidiasis and primary adrenal insufficiency. Occasionally, pernicious anemia, diabetes mellitus, vitiligo, and autoimmune thyroid disease also are associated. Mucocutaneous candidiasis often presents first in early childhood and is followed several years later by hypoparathyroidism. Adrenal insufficiency appears in adolescence. The combination of hypoparathyroidism, adrenal insufficiency, and mucocutaneous candidiasis is referred to as the HAM syndrome.
Familial hypocalcemia results from activating mutations in the calcium-sensing receptor that increase its sensitivity to calcium.
Parathyroid and radical neck surgery can result in a loss of glandular tissue. Surgical removal of parathyroid tissue in secondary or tertiary hyperparathyroidism is often complicated by severe hypocalcemia
P.69
due to the remineralization of bone, the so-called hungry bone syndrome.Hypocalcemia also occurs after thyroid surgery (5% of cases); in approximately 0.5% of these patients, the hypocalcemia is permanent. Risk factors for the development of permanent hypocalcemia include removal of three or more parathyroid glands; postoperative PTH concentration of less than or equal to 12 pg per mL; total serum calcium concentration of less than or equal to 8 mg per dL after 1 week of oral calcium supplementation; and a serum phosphorus concentration of less than or equal to 4 mg per dL after 1 week of calcium supplementation.
Transient hypoparathyroidism may occur after the removal of a parathyroid adenoma.
Infiltrative disorders such as hemochromatosis, Wilson's disease, and infection with human immunodeficiency virus (HIV) can also decrease PTH secretion.
Severe hypomagnesemia is the most common cause of hypoparathyroidism. Magnesium deficiency results in end-organ resistance to PTH and a decrease in PTH secretion. Patients with hypocalcemia as a result of hypomagnesemia do not respond to calcium or vitamin D replacement until the magnesium deficit is replaced.
A variety of rare genetic disorders cause end-organ resistance to PTH, including pseudohypoparathyroidism types I and II. Patients with pseudohypoparathyroidism are classified on the basis of the response of nephrogenous cyclic adenosine monophosphate to PTH administration. A decreased response is indicative of type I, and a normal response is indicative of type II.
Defects in vitamin D metabolism also cause hypocalcemia. Etiologies include decreased intake of vitamin D, malabsorption, drugs, liver disease, kidney disease, and vitamin D dependent rickets. Nutritional vitamin D deficiency is uncommon in the United States as a result of the supplementation of milk and other food products. It can occur, however, in poorly nourished patients with little sun exposure. Because vitamin D is fat-soluble, vitamin D deficiency can be seen in gastrointestinal
P.70
malabsorption from any cause. Anticonvulsants induce vitamin D deficiency through mechanisms that are not clear; this generally occurs in patients with additional predisposing factors, such as poor nutrition and decreased sun exposure. Phenobarbital enhances the hepatic metabolism of vitamin D and calcidiol. Vitamin D deficiency can result from hepatocellular disease if the disease is severe enough to impair the 25-hydroxylation of vitamin D to calcidiol. Chronic kidney disease impairs the 1- hydroxylation of calcidiol to calcitriol. Vitamin D-dependent rickets is a result of either the impaired hydroxylation of calcidiol to calcitriol (type I) or end-organ resistance to calcitriol (type II). Type I patients respond to physiologic doses of calcitriol. Patients with type II disease have dramatically increased concentrations of calcitriol, respond poorly to calcitriol therapy, and have mutations in the vitamin D receptor.Less common causes of hypocalcemia include the tumor lysis syndrome, osteoblastic metastases, acute pancreatitis, toxic shock syndrome, and sepsis. The acute addition or release of phosphate into the extracellular space may cause hypocalcemia via a variety of mechanisms. Calcium and phosphate may precipitate in tissues, although the exact tissue in which the deposition occurs has never been identified. In addition, phosphate infusion increases the rate of bone formation and inhibits PTH-induced bone resorption, which then decreases calcium concentration.
As is the case for hypercalcemia, the signs and symptoms of hypocalcemia depend not only on the degree of hypocalcemia, but also on the rate of decline of the serum calcium concentration. The threshold at which symptoms develop also depends on the serum pH and whether concomitant hypomagnesemia, hypokalemia, or hyponatremia is present. Symptoms of neuromuscular excitability predominate. The patient may complain of circumoral and distal extremity paresthesias or of carpopedal spasm. Central nervous system manifestations include mental status changes, irritability, and seizures. On physical examination, hypotension, bradycardia, laryngeal spasm, and bronchospasm may be present. Chvostek's and Trousseau's signs should be checked. Chvostek's sign is a facial twitch elicited by tapping on the facial nerve just below the zygomatic arch with the mouth slightly open. A positive sign is occasionally observed in normal patients. Trousseau's sign is the development of wrist flexion, metacarpophalangeal joint flexion, hyperextended fingers, and thumb flexion after a sphygmomanometer cuff is inflated around the arm to 20 mmHg above systolic pressure for a duration of 3 minutes.
Diagnosis. The differential diagnosis of true hypocalcemia is often straightforward, and a diagnostic algorithm is shown in Figure 5-3. The most common causes are magnesium deficiency, kidney failure, and complications of parathyroid surgery.
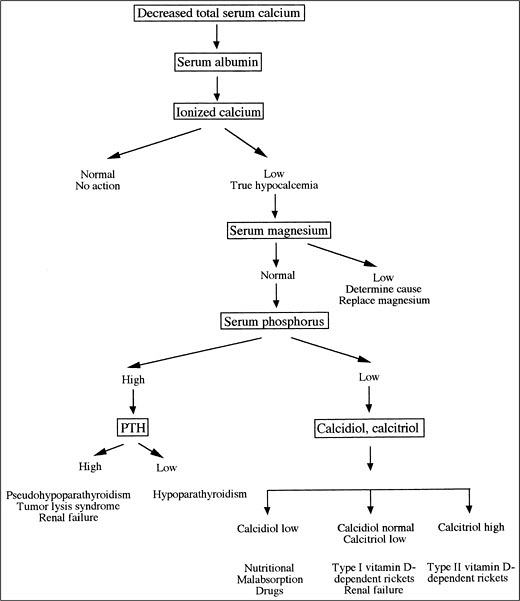
Figure 5-3. Evaluation of Hypocalcemia. (PTH, Parathyroid Hormone.)
The first step in the evaluation of the patient with a decreased total serum calcium concentration is to examine the serum albumin and, if necessary, measure the ionized serum calcium concentration. If true hypocalcemia is documented, then blood analysis should be obtained for BUN, creatinine, magnesium, and phosphorus concentration, and a 24-hour urine sample should be collected to test phosphorus and creatinine excretion.
The second step is to evaluate the serum magnesium concentration. Hypomagnesemia is the most common cause of hypocalcemia in hospitalized patients. A high index of suspicion should be present in patients with a history of steatorrhea, diarrhea, or chronic alcoholism. These patients generally have severe hypomagnesemia, and the hypocalcemia does not correct until magnesium losses are replaced. It frequently requires several days for the serum calcium concentration to correct after magnesium deficiency is reversed.
Serum and urinary phosphorus concentrations are evaluated next. Hyperphosphatemia in the absence of kidney failure suggests a diagnosis of either hypoparathyroidism or pseudohypoparathyroidism. Measuring the PTH concentration can
P.71
differentiate these disorders. In primary hypoparathyroidism, PTH is low; in pseudohypoparathyroidism, PTH is increased. A decrease in serum phosphorus concentration indicates a defect in vitamin D metabolism. Hypocalcemia results in secondary hyperparathyroidism that, in turn, reduces proximal tubular reabsorption of phosphate and thus results in phosphate wasting. Therefore, the fractional excretion (FE) of phosphate is expected to be high (more than 5%). In hypophosphatemia, the kidney has an extraordinary ability to conserve phosphate, and, in extrarenal disorders, the FE of phosphate is below 1%. If phosphaturia is noted, then calcidiol and calcitriol concentration should be measured. Calcidiol concentration is reduced with malabsorption, liver disease, and phenobarbital. Calcitriol concentration is reduced in kidney failure and increased in type II vitamin D-dependent rickets.
The management of hypocalcemia is dependent on both its severity and cause.
In an emergency situation in which hypocalcemia is suspected and seizures, tetany, hypotension, or cardiac arrhythmias are present, intravenous calcium should be administered (100 to 300 mg over 10 to 15 minutes) before the results of the serum calcium concentration return from the clinical laboratory. Patients with symptomatic hypocalcemia or a total serum calcium concentration corrected for albumin of less than or equal to 7.5 mg per dL should be initially managed with parenteral calcium. Chronic, mild hypocalcemia, as seen in the outpatient setting, can be treated with oral calcium supplements, to which a vitamin D preparation may be added if necessary.
Acute symptomatic hypocalcemia is treated with intravenous calcium. In the absence of seizures, tetany, or cardiac arrhythmias, an infusion of 15 mg per kg of elemental calcium given over 4 to 6 hours raises the total serum calcium by 2 to 3 mg per dL. Calcium gluconate (10%) is supplied in 10-mL ampules and contains 94 mg of elemental calcium. The first ampule can be administered over several minutes, followed by a constant infusion begun at a rate of 0.5 to 1.0 mg per kg per hour, with adjustment in the rate based on serial determinations of serum calcium concentration. Calcium gluceptate (10%) provides 90 mg of elemental calcium in a 5-mL ampule. Calcium chloride has higher bioavailability, and 272 mg of elemental calcium is contained in each 10-mL ampule. Treatment of hypocalcemia is ineffective in the presence of hypomagnesemia. In the setting of metabolic acidosis, hypocalcemia should be corrected before acidosis is reversed.
Patients with hypoparathyroidism are managed with calcium and vitamin D supplements. The serum calcium concentration should be maintained at the lower limit of normal. Oral elemental calcium, 1 to 3 g per day, is usually sufficient. A variety of oral calcium preparations are available, some of which are shown in Table 5-4. Calcium is best absorbed when taken between meals. Calcium citrate is more soluble than calcium carbonate, especially in patients who require H2 blockers or proton pump inhibitors. In the presence of severe hyperphosphatemia, calcium supplementation should be delayed, if possible, until the serum phosphorus concentration is reduced below 6 mg per dL using phosphate binders. Severe hypocalcemia, however, may need to be treated despite hyperphosphatemia.
Table 5-4. Oral Calcium Preparations
Preparation Formulation Elemental calcium per tablet Calcium carbonate Tums 500 mg 200 mg Rolaids 550 mg 220 mg Os-cal 1,250 mg 500 mg Calcium citrate Citracal 950 mg 200 mg Calcium lactate 650 mg 85 mg Calcium gluconate 1,000 mg 90 mg Calcitriol is the most potent of the vitamin D preparations and has the fastest onset and shortest duration of action, but is also the most expensive. A dose of 0.5 to 1.0 g per day is usually required. As one moves back up the metabolic pathway to calcidiol, cholecalciferol, and ergocalciferol, cost decreases and duration of action increases. These agents, however, may be less efficacious in the presence of kidney or liver disease.
Patients with hypoparathyroidism have decreased distal tubular calcium reabsorption as a result of a lack of PTH. Therefore, the increase
P.73
in filtered calcium load that results from calcium and vitamin D replacement can lead to hypercalciuria, nephrolithiasis, and nephrocalcinosis. If urinary calcium excretion exceeds 350 mg per day despite a serum calcium concentration in the low normal range, sodium intake should be restricted; if this is not effective, a thiazide diuretic should be added.
P.72
P.62
Disorders of Serum Phosphorus
Overview. Approximately two-thirds of total plasma phosphorus is organic phosphorus (phospholipids), and one-third is inorganic. Clinical chemistry laboratories assay only the inorganic fraction. The reference range is 2.8 to 4.5 mg per dL (0.89 to 1.44 mmol per L). Approximately 85% of inorganic phosphorus is free and circulates as either HPO4(-2) or H2PO4(-1). The ratio of these two ions depends upon ECF pH. Of the remainder, 10% is protein-bound, and 15% is complexed with either calcium or magnesium.
As is the case for calcium, the majority of total body phosphate is contained within the skeleton (80%). Of the remainder, about 10% is within skeletal muscle and viscera. Only a small fraction of the phosphate pool is inorganic and available for the synthesis of adenosine triphosphate (ATP). The average Western diet contains 800 to 1,400 mg of phosphate per day, of which approximately 80% is absorbed in small intestine. The majority is absorbed passively, but an active calcitriol-regulated component exists. PTH and calcitriol, through their effects in bone, intestine, and kidney, regulate phosphorus concentration. The major regulator of serum phosphorus concentration is renal phosphate excretion. In kidney, phosphate is reabsorbed primarily in the proximal tubule (80%), where it is cotransported with sodium across the luminal membrane. The sodium-phosphate cotransporter is upregulated in response to phosphate depletion, and, under these circumstances, the kidney is capable of reducing the FE of phosphate to very low levels.
Phosphate regulation. PTH acts directly in bone to increase phosphate entry into the ECF and indirectly in intestine by stimulating the production of calcitriol. The majority of dietary phosphate is reabsorbed in the small intestine, but a component of unregulated secretion is present in the colon (100 to 200 mg per day). PTH reduces the proximal tubular reabsorption of phosphate in the kidney. The net effect is to increase plasma calcium concentration while keeping serum phosphorus concentration constant. Calcitriol's major roles are to enhance the availability of calcium and phosphate for new bone formation and to defend the ECF from hypocalcemia and hypophosphatemia. PTH and hypophosphatemia stimulate calcitriol production in proximal tubule; although, the kidney is the primary regulator of serum phosphorus concentration. Hypophosphatemia causes the insertion of sodium-phosphate cotransporters into the luminal membrane of the proximal tubule, whereas PTH results in their removal. The ability of PTH to remove sodium-phosphate cotransporters from apical membrane is blunted in chronic phosphate depletion.
Hyperphosphatemia
Etiology. Hyperphosphatemia can result from renal failure, an acute phosphate load from either exogenous or endogenous sources, or increased proximal tubular reabsorption of phosphate. Etiologies are shown in Table 5-5.
Table 5-5. Etiologies of Hyperphosphatemia
Decreased renal excretion Acute renal failure Chronic renal failure Acute phosphate load Tumor lysis syndrome Rhabdomyolysis Bowel infarction Severe hemolysis Vitamin D intoxication Increased renal phosphate reabsorption Hypoparathyroidism Acromegaly Thyrotoxicosis Drugs bisphosphonates Tumoral calcinosis Pseudohyperphosphatemia Renal failure is the underlying cause in 90% or more of cases. As GFR begins to decline, the FE of phosphate increases. Once GFR falls below 30 mL per minute, however, phosphorus reabsorption is maximally suppressed, and the FE cannot increase further. As a result, renal excretion can no longer keep pace with dietary intake, and the serum phosphorus concentration rises. A new steady state is eventually established, albeit at a higher serum phosphorus concentration.
A sudden, massive phosphate load may result in an increase in serum phosphorus concentration. Phosphate may be released from either the
P.74
intracellular space, as is the case in tumor lysis syndrome or rhabdomyolysis, or it can be ingested and absorbed, as in vitamin D intoxication. Tumor lysis syndrome most commonly is seen with the treatment of rapidly growing malignancies such as leukemias and lymphomas. It can occur after the treatment of solid tumors such as small cell carcinoma, breast cancer, and neuroblastoma. Risk factors for tumor lysis syndrome in patients with solid tumors include pretreatment renal impairment, an increased lactate dehydrogenase level, and hyperuricemia. Increased lactate dehydrogenase levels and hyperuricemia are indicators of a large tumor burden.Primary increases in tubular phosphate reabsorption are less common. They can occur in hypoparathyroidism; in acromegaly, as a result of direct stimulation of insulin-like growth factor (IGF) on phosphate transport; with bisphosphonates, via a direct effect on renal phosphate reabsorption; and in tumoral calcinosis. Tumoral calcinosis is caused by an abnormality in the proximal tubule that leads to an increase in phosphate reabsorption.
Many of the signs and symptoms of an acute rise in serum phosphorus are secondary to concomitant hypocalcemia due to the deposition of calcium in soft tissues and a resultant fall in ECF ionized calcium concentration. Hyperphosphatemia can also cause hypocalcemia by decreasing the activity of 1- hydroxylase and calcitriol formation.
Diagnosis. Clinically unexplained, persistent hyperphosphatemia should raise the suspicion of pseudohyperphosphatemia, the most common cause of which is paraproteinemia. No consistent relationship of immunoglobulin type or subclass has been identified. This is a method-dependent artifact, and paraprotein interference may be a general problem in spectrophotometric assays. If paraproteinemia is absent, the cause is generally acute or chronic renal failure.
Treatment of hyperphosphatemia is aimed at reducing the intestinal absorption of phosphate. This is accomplished through the use of oral phosphate-binding drugs such as calcium carbonate, calcium acetate, sevelamer hydrochloride, and aluminum hydroxide. These agents should be administered with meals. Aluminum hydroxide may be used in the short term, but chronic use in patients with kidney disease should be avoided because of the potential for aluminum toxicity. In patients with coexistent hypocalcemia, it is preferable to lower the serum phosphorus below 6 mg per dL, if
P.75
possible, before treating the hypocalcemia to avoid the potential complication of metastatic calcification from calcium phosphate coprecipitation.
Hypophosphatemia
Etiology. Hypophosphatemia may result from the redistribution of phosphate from the extracellular to the intracellular space, a decrease in intestinal absorption of phosphate, or a decrease in renal reabsorption of phosphate. The differential diagnosis is presented in Table 5-6.
Table 5-6. Etiologies of Hypophosphatemia
Decreased dietary intake Alcoholism Phosphate-binding agents Shift of phosphate into the intracellular fluid Respiratory alkalosis Refeeding Diabetic ketoacidosis Hungry bone syndrome Increased renal excretion Hyperparathyroidism Vitamin D deficiency X-linked hypophosphatemic rickets Autosomal dominant hypophosphatemic rickets Fanconi's syndrome Drugs acetazolamide Osmotic diuresis Oncogenic osteomalacia Respiratory alkalosis and the refeeding syndrome are the most common causes of a shift of phosphate from the ECF to the ICF in hospitalized patients. Respiratory alkalosis causes a rise in intracellular pH that stimulates phosphofructokinase, the rate-limiting step in glycolysis. This results in severe hypophosphatemia with serum phosphorus concentrations of less than 0.5 to 1.0 mg per dL. Intracellular shifts are also seen with the treatment of diabetic ketoacidosis and in hungry bone syndrome, which occurs after parathyroidectomy for secondary hyperparathyroidism. In hungry bone syndrome, serum calcium and phosphorus concentration fall dramatically in the postoperative period, although clinically, hypocalcemia is more of a management issue than hypophosphatemia.
Decreased dietary intake is an unusual cause of hypophosphatemia because oral intake almost always exceeds gastrointestinal losses, and the kidney is capable of reclaiming nearly all the filtered load of phosphate. In general, decreased intake must be combined with increased gastrointestinal losses (e.g., diarrhea) or the use of phosphate binders for hypophosphatemia to result.
Increased urinary phosphate excretion occurs in primary hyperparathyroidism, secondary hyperparathyroidism due to defects in vitamin D metabolism, Fanconi's syndrome, osmotic diuresis, acetazolamide use, and oncogenic osteomalacia. Oncogenic osteomalacia is a rare disorder associated with mesenchymal tumors; it is characterized by hypophosphatemia, phosphaturia, decreased calcitriol concentration, normal calcidiol concentration, and clinical and histologic evidence of osteomalacia. A considerable delay may occur between the presentation of the syndrome and the discovery of the tumor. The tumor produces a humoral factor that interferes with the proximal tubular reabsorption of phosphate and the production of calcitriol; this interference resolves on removal of the tumor. The humoral factor was identified as fibroblast growth factor 23 (FGF23).
P.76
FGF23 is present in the circulation of normal individuals, consistent with a physiologic role in regulating serum phosphorus. Animal studies have shown that FGF23 is phosphaturic. When administered in vivo, it induces hypophosphatemia, suppresses 1,25(OH)2 vitamin D3 concentration and leads to osteomalacia. Two inherited renal phosphate wasting disorders, autosomal dominant hypophosphatemic rickets (ADHR) and X-linked hypophosphatemia (XLH), are the result of defects in FGF23 metabolism. Missense mutations in FGF23 cause ADHR.
ADHR is characterized by hypophosphatemia, renal phosphate wasting, short stature, and bony deformities. In ADHR, mutations at a proteolytic cleavage site prevent its cleavage and inactivation. In vivo studies showed that biologic activity is limited to full-length FGF23 (251 amino acids). The enzyme responsible for the cleavage of FGF23 has not been identified. One report suggested that Phex, a cell surface metalloprotease, may cleave FGF23, but this has not been confirmed.
XLH is characterized by renal phosphate wasting, hypophosphatemia, growth retardation, defective calcification of cartilage and bone, and resistance to phosphate and vitamin D repletion. Inactivating mutations of Phex cause XLH. Phex is a member of a family of zinc-dependent cell surface proteases that cleave small peptides such as endothelin. It is expressed predominantly in cartilage, bone, and teeth. Its physiologically relevant substrate has yet to be identified. Although it has been postulated that Phex cleaves and inactivates FGF23, the large size of FGF23 251 amino acids makes this less likely. Other intermediate small-molecular-weight substrates likely link Phex function to FGF23.
Signs and symptoms. Hypophosphatemia results in a variety of clinical sequelae. The correction of moderate hypophosphatemia improves diaphragmatic function in patients with acute respiratory failure. In patients with severe hypophosphatemia, failure to wean from mechanical ventilation until repletion of phosphate was demonstrated. In vitro hypophosphatemia causes a leftward shift in the oxygen dissociation curve. Neuromuscular symptoms include paresthesias, tremor, muscle weakness, and altered mental status; severe hypophosphatemia increases red cell fragility, which can lead to hemolysis and decreases chemotaxis, phagocytosis, and bacterial killing by white cells, with an increased susceptibility to infection as the possible result.
Diagnosis. FE of phosphate or 24-hour urinary phosphate excretion can be used to distinguish among the pathophysiologic mechanisms of hypophosphatemia. If the kidney is responding appropriately to decreased intestinal absorption or redistribution of phosphate into cells, FE of phosphate is below 5%, and the 24-hour urine phosphate excretion is less than 100 mg per day. When the kidney is the cause of hypophosphatemia, the FE of phosphate is greater than 5% and the 24-hour urine excretion contains more than 100 mg of phosphate per day. In this case, a urinalysis for glycosuria, PTH concentration to rule out hyperparathyroidism, and the measurement of calcidiol and calcitriol concentrations are indicated.
Treatment is indicated for severe hypophosphatemia (less than or equal to 1 mg per dL) or symptoms. It is complicated by the fact that phosphate is largely an intracellular ion, and that serum phosphorus concentration is not a reliable indicator of total body phosphate stores. Hypophosphatemia is often associated with potassium and magnesium depletion. Phosphate repletion should be undertaken with extreme caution in the rare patient with renal insufficiency; the safest mode of therapy is oral, and hypophosphatemia usually can be corrected with 1,000 mg of phosphate per day. Alternative forms of oral phosphate replacement are listed in Table 5-7. Diarrhea is the most common complication.
Table 5-7. Oral Phosphate Preparations
Preparation Dosage Contents K-phos-neutral 2 tablets, b.i.d. or t.i.d. 250 mg phosphate, 12 mEq
sodium, 2 mEq potassium
per tabletFleets Phospho-Soda 5 mL b.i.d. 149 mg phosphate, 6 mEq
sodium per mLNeutra-Phos-K 1 2 capsules, b.i.d. or t.i.d. 250 mg phosphate, 14 mEq
potassium per capsuleK-phos 2 tablets, t.i.d. or q.i.d. 114 mg phosphate, 3.68
mEq potassium per tabletIntravenous replacement carries the risk of hypocalcemia and hyperphosphatemia and is only warranted in patients with severe symptomatic hypophosphatemia. Sodium phosphate should be used unless the serum
P.77
potassium is less than 4 mEq per L. Serum concentrations of phosphorus, calcium, magnesium, potassium, and urine output should be carefully monitored during intravenous replacement. Once serum phosphorus concentration has increased to more than 1 mg per dL, the patient should be switched to an oral preparation. The administration of doses larger than 0.32 mmol per kg over a 12-hour period is rarely warranted.
Suggested Readings
Brooks MJ, Melnik G. The refeeding syndrome: an approach to understanding its complications and preventing its occurrence. Pharmacotherapy 1995;15:713 726.
Bugg NC, Jones JA. Hypophosphatemia. Anaesthesia 1998;53:895 902.
Bushinsky DA. Calcium. Lancet 1998;352:306 311.
Crook M. Phosphate: an abnormal anion? Br J Hosp Med 1994;52:200 203.
Econs MJ, Francis F. Positional cloning of the PEX gene: new insights into the pathophysiology of X-linked hypophosphatemic rickets. Am J Physiol 1997;273:F489 F498.
Fiaschi-Taesch NM, Stewart AF. Minireview: parathyroid hormone-related protein as an intracrine factor trafficking mechanisms and functional consequences. Endocrinology 2003;144:407 411.
Fiorino AS. Hypercalcemia and alkalosis due to the milk-alkali syndrome: a case report and review. Yale J Biol Med 1996;69:517 523.
Guise TA, Mundy GR. Evaluation of hypocalcemia in children and adults. J Clin Endocrinol Metab 1995;80:1473 1478.
Lourwood DL. The pharmacology and therapeutic utility of bisphosphonates. Pharmacotherapy 1998;18:779 789.
Mundy GR, Guise TA. Hypercalcemia of malignancy. Am J Med 1997;103:134 145.
Potts JT Jr., Fradkin JE, Aurbach JD, Bilezikian JP, Raisz LG. Proceedings of the NIH consensus development conference on diagnosis and management of asymptomatic primary hyperparathyroidism. J Bone Miner Res 1991;6:S1 S165.
Quarles, LD. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab 2003;285:E1 E9.
Tenenhouse HS, Murer H. Disorders of renal tubular phosphate transport. J Am Soc Nephrol 2003;14:240 248.
Reber PM, Heath HH III. Hypocalcemic emergencies. Med Clin North Am 1995;79:93 106.
Weisinger JR. Magnesium and phosphorus. Lancet 1998;352:391 396.
Zahrani AA, Levine MA. Primary hyperparathyroidism. Lancet 1997;349:1233 1236.
- Chapter III Two Models of Online Patronage: Why Do Consumers Shop on the Internet?
- Chapter XI User Satisfaction with Web Portals: An Empirical Study
- Chapter XIII Shopping Agent Web Sites: A Comparative Shopping Environment
- Chapter XVII Internet Markets and E-Loyalty
- Chapter XVIII Web Systems Design, Litigation, and Online Consumer Behavior